2-Deoxystreptamine Conjugates by Truncation–Derivatization of Neomycin

Abstract
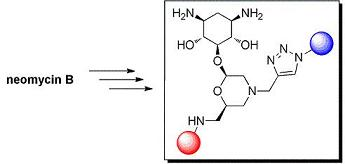
:
Introduction
Results and Discussion
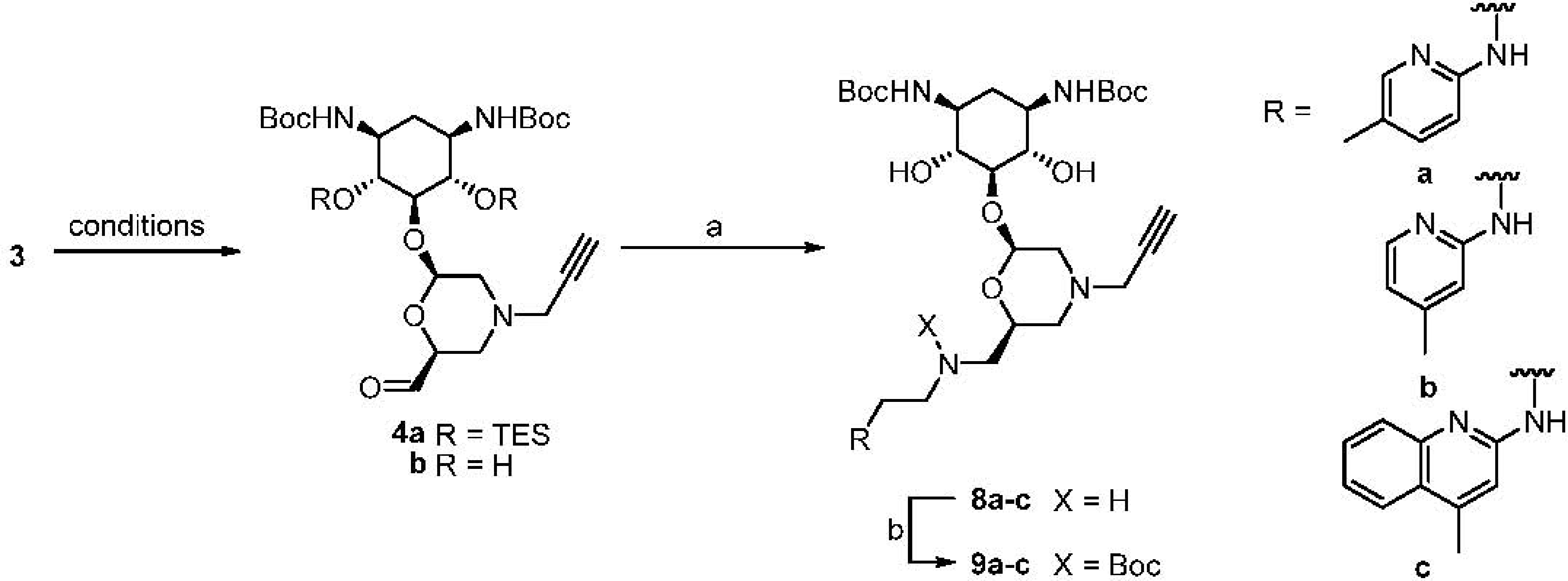
Synthesis of 5-O-Morpholino-2-Deoxystreptamine
Synthesis and Conjugation of Aminopyridines and Aminoquinolines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | Product | Yield |
|---|---|---|---|
| 1 | i) TESOTf, Et3N, CH2Cl2, r.t., 3 h ii) (COCl)2, DMSO, CH2Cl2, -78 °C→0°C | 4a | mixture |
| 2 | TCCA, TEMPO, CH3CN, r.t., 16 h | 4b | no reaction |
| 3 | IBX, DMF, r.t.→80 °C, 16 h | 4b | <10% conversion |
| 4 | DMP, DMF, r.t., 16 h | 4b | 60% conversion |
Dimerization and Deprotection of the Conjugates
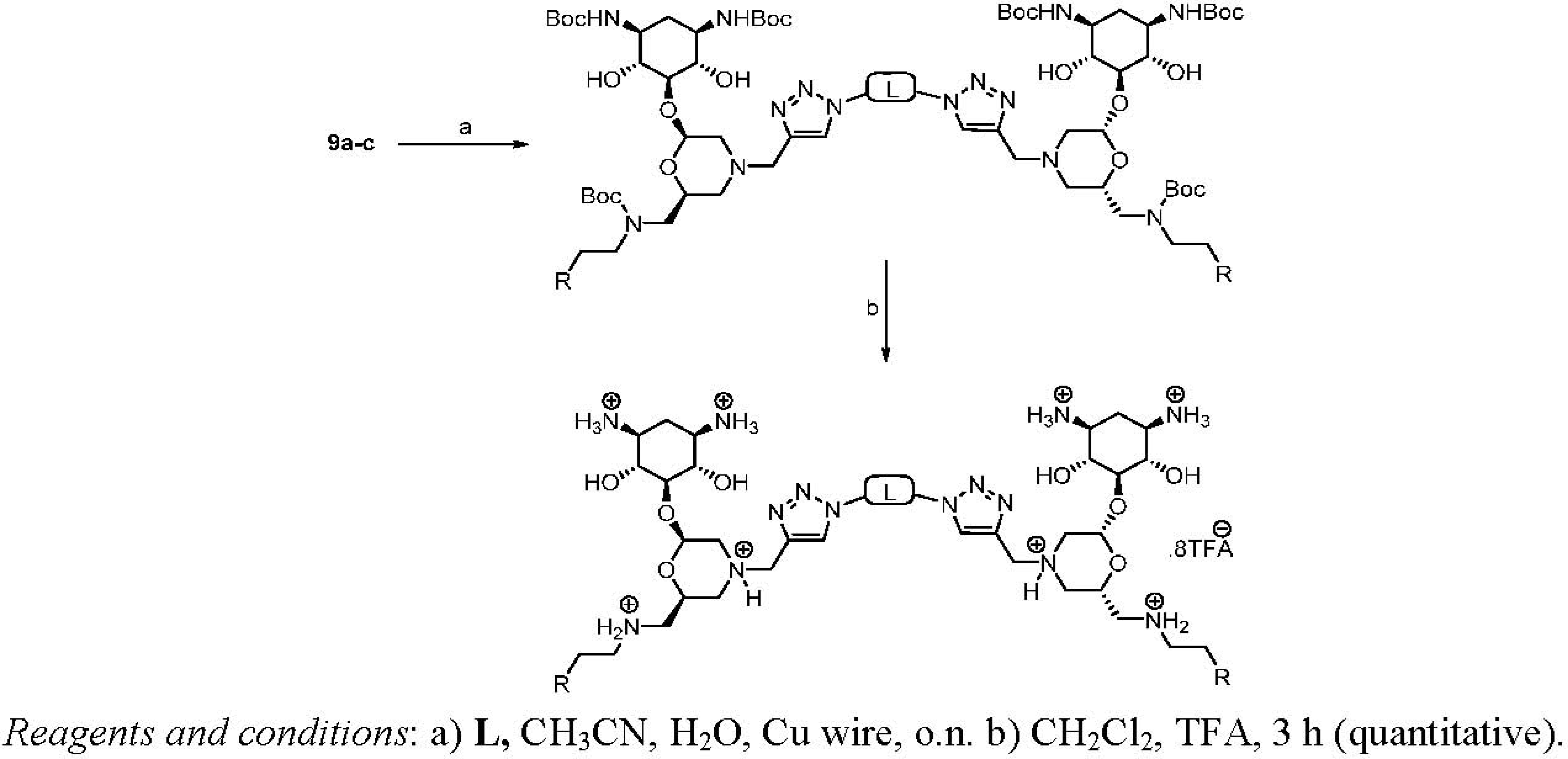
| Entry | Linker (L) | Starting | Product | Yield |
| material | ||||
| 1 |  | 9a | 9aA | 63% |
| 2 | 9b | 9bA | 73% | |
| 3 | 9c | 9cA | 60% | |
| 4 |  | 9a | 9aB | 57% |
| 5 | 9b | 9bB | 65% | |
| 6 | 9c | 9cB | 58% | |
| 7 |  | 9a | 9aC | 61% |
| 8 | 9b | 9bC | 66% | |
| 9 | 9c | 9cC | 69% | |
| 10 |  | 9a | 9aD | 60% |
| 11 | 9b | 9bD | 65% | |
| 12 | 9c | 9cD | 66% | |
| 13 |  | 9a | 9aE | 87% |
| 14 | 9b | 9bE | 92% | |
| 15 | 9c | 9cE | 90% |
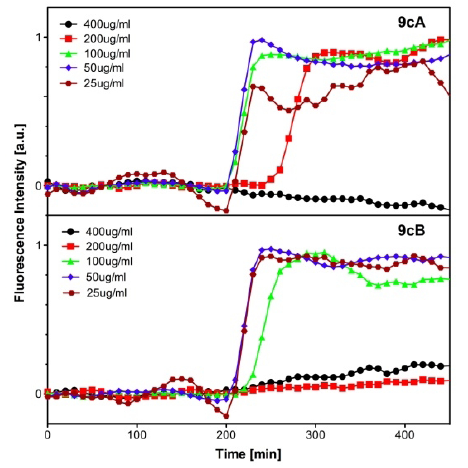
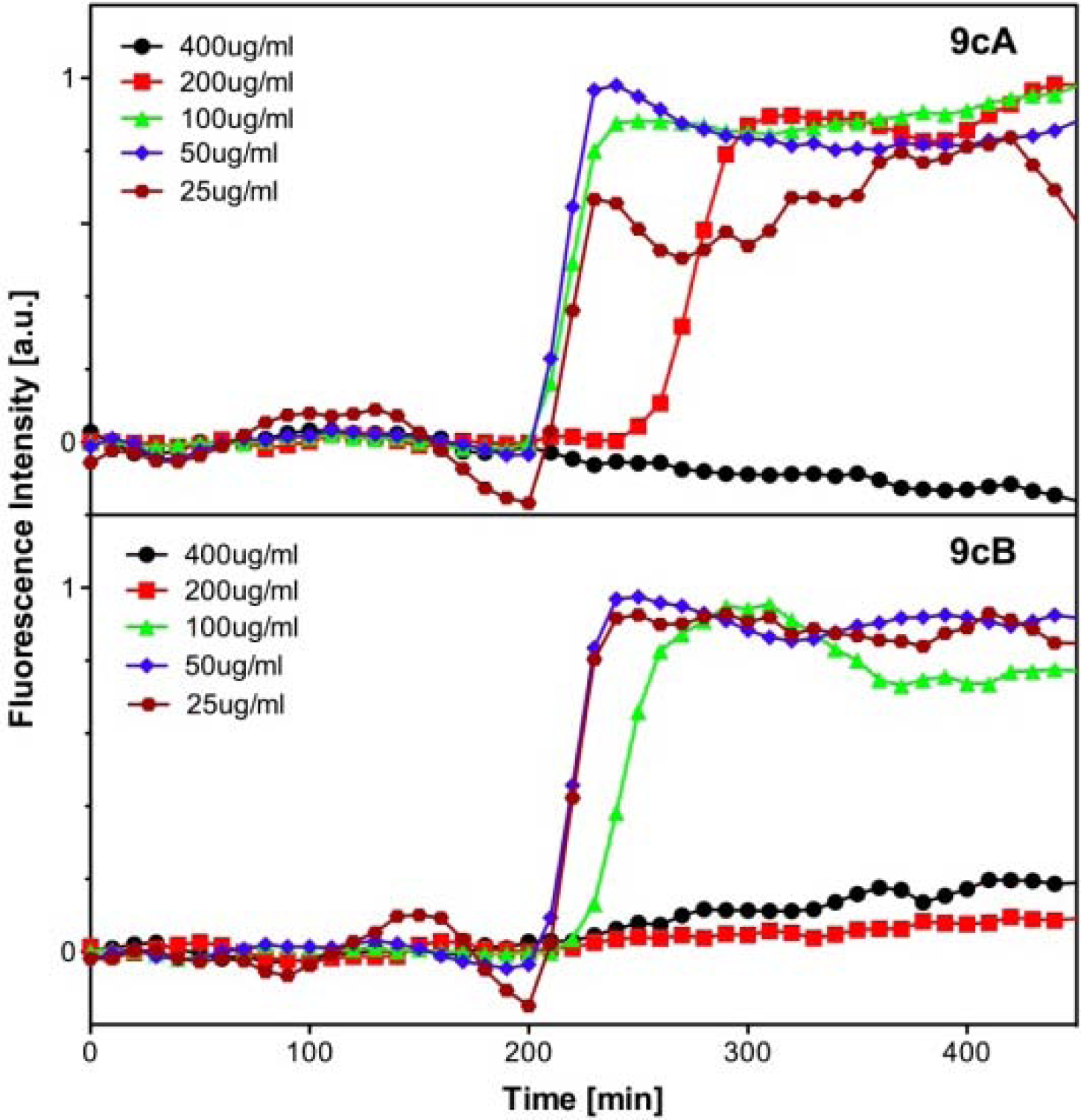
Evaluation of Antibacterial Activity

| Entry | bacterial strain | MIC (μg/ mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| kanamycin | s.m. | linker A | linker B | linker C | linker D | linker E | ||
| 1 | E. coli | 10 | 9a | 200–400 | 200–400 | 400–800 | 400–800 | 400–800 |
| 2 | -do- | 10 | 9b | 400–800 | 400–800 | 400–800 | 400–800 | 400–800 |
| 3 | -do- | 10 | 9c | 200–400 | 100–200 | 200–400 | 400–800 | 200–400 |
Second Generation Monomeric Ligands
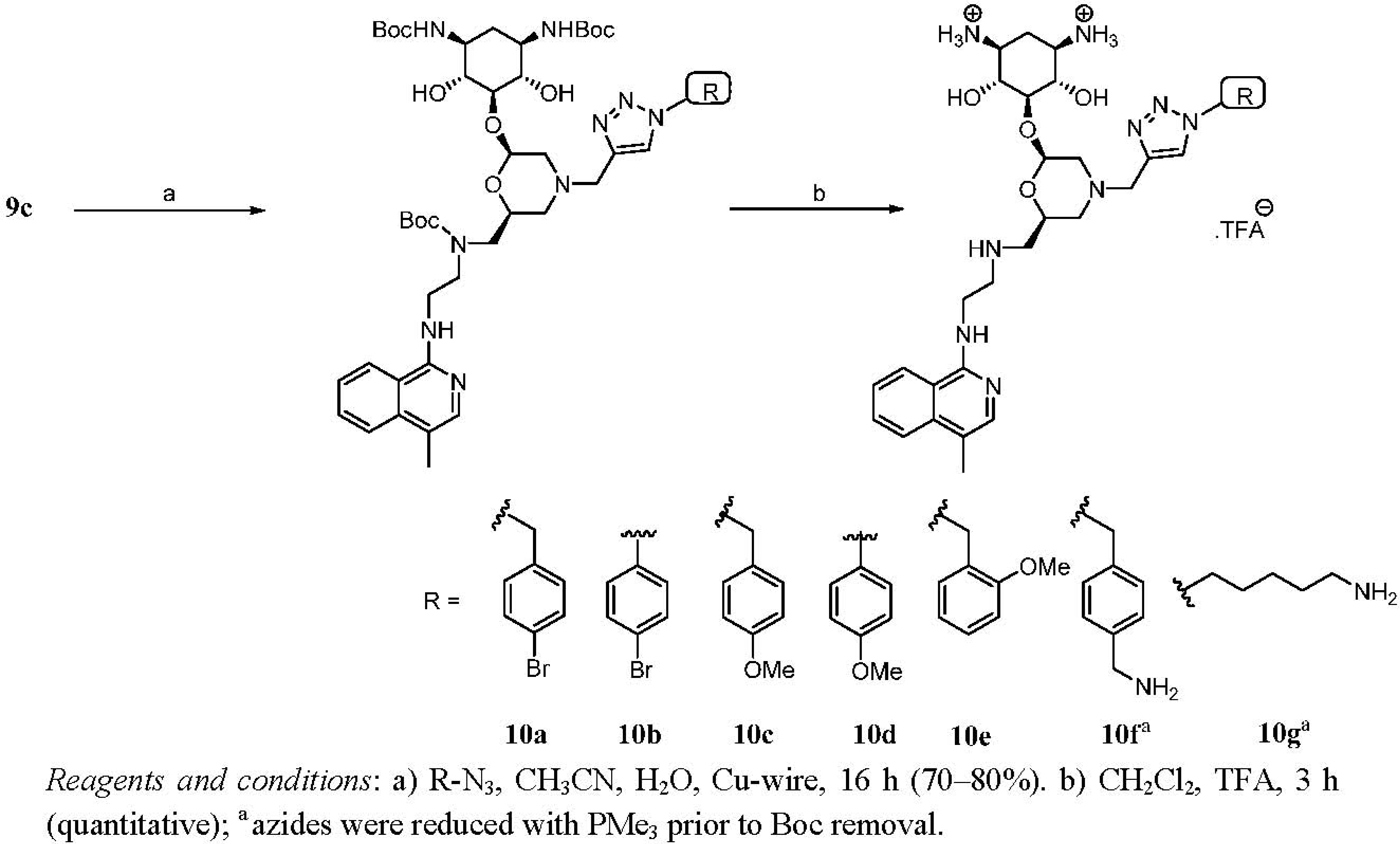
| Entry | bacterial strains | MIC (μg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| kanamycin | 10a | 10b | 10c | 10d | 10e | 10f | 10g | 9cB | 9cE | ||
| 1 | E. coli | 10 | 100-200 | 100-200 | 400-800 | 400-800 | 400-800 | 400-800 | 400-800 | - | - |
| 2 | E. coli kanr | 400–800 | 400-800 | 200-400 | >800 | - | - | - | - | >800 | >800 |
Experimental Section
General
Hexa-N-(tert-butoxycarbonyl) neomycin B (1) [49]
5-O-(β-d-ribofuranosyl)-2-deoxystreptamine (2)
5-O-(N-propyn-1-yl -morpholino)-2-deoxystreptamine (3)
2-(2-Aminoethylamino)-4-methylpyridine (5)
2-(2-Aminoethylamino)-5-methylpyridine (6)
2-(2-Aminoethylamino)-4-methylquinoline (7)
5-O-(N-propyn-1-yl-2-(methylamino-N-ethylamino-N -4-methylpyridin-2-yl)-morpholino)-2-deoxystreptamine 8a
5-O-(N-propyn-1-yl-2-(methylamino-N-ethylamino-N -5-methylpyridin-2-yl)-morpholino)-2-deoxystreptamine 8b
5-O-(N-propyn-1-yl-2-(methylamino-N-ethylamino-N-4-methylquinolin-2-yl)-morpholino)-2-deoxystreptamine 8c
General procedure for Boc-protection of 9a-c
3.3. General procedure for the synthesis of bis-azides A-D
1-Bromo-4-(azidomethyl)bromobenzene
1-Azido-4-bromobenzene
1-(Azidomethyl)-4-methoxybenzene
1-Azido-4-methoxybenzene
1-(Azidomethyl)-2-methoxybenzene
General Procedure for Copper (I)-Catalyzed Azido-Alkyne Cycloaddition (CuAAC) (Compounds 9aA-10g), exemplified for compound 9aA
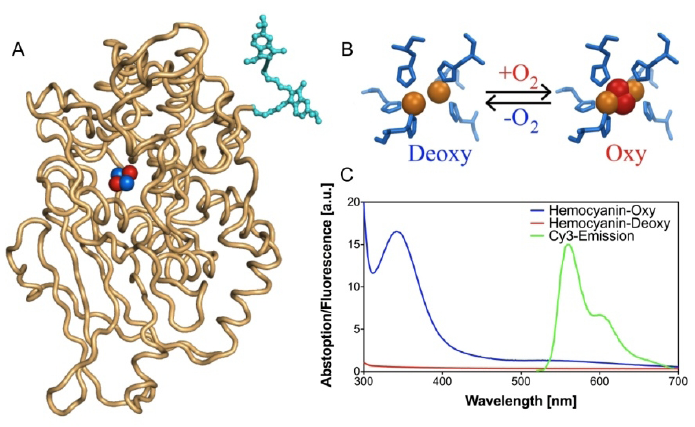
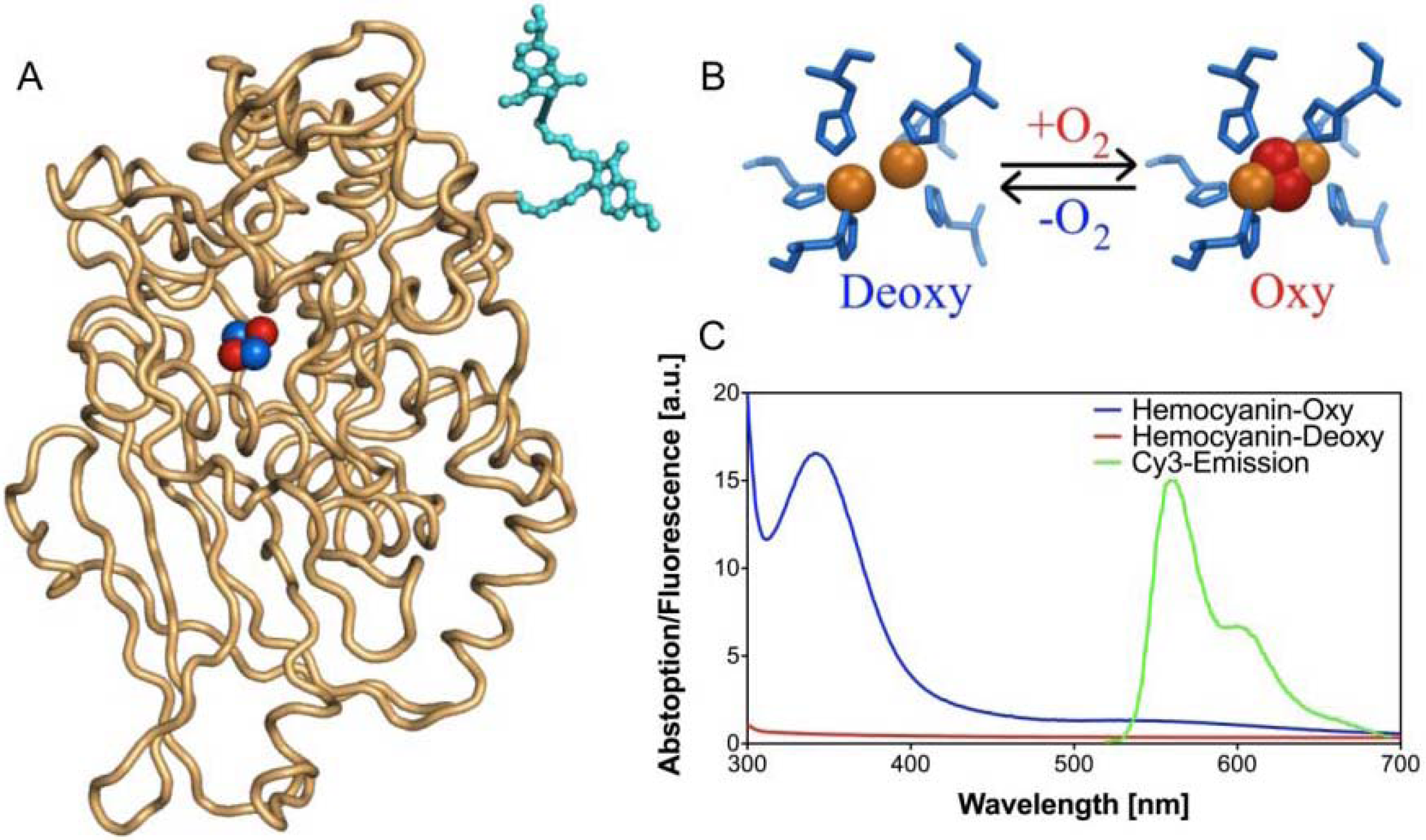
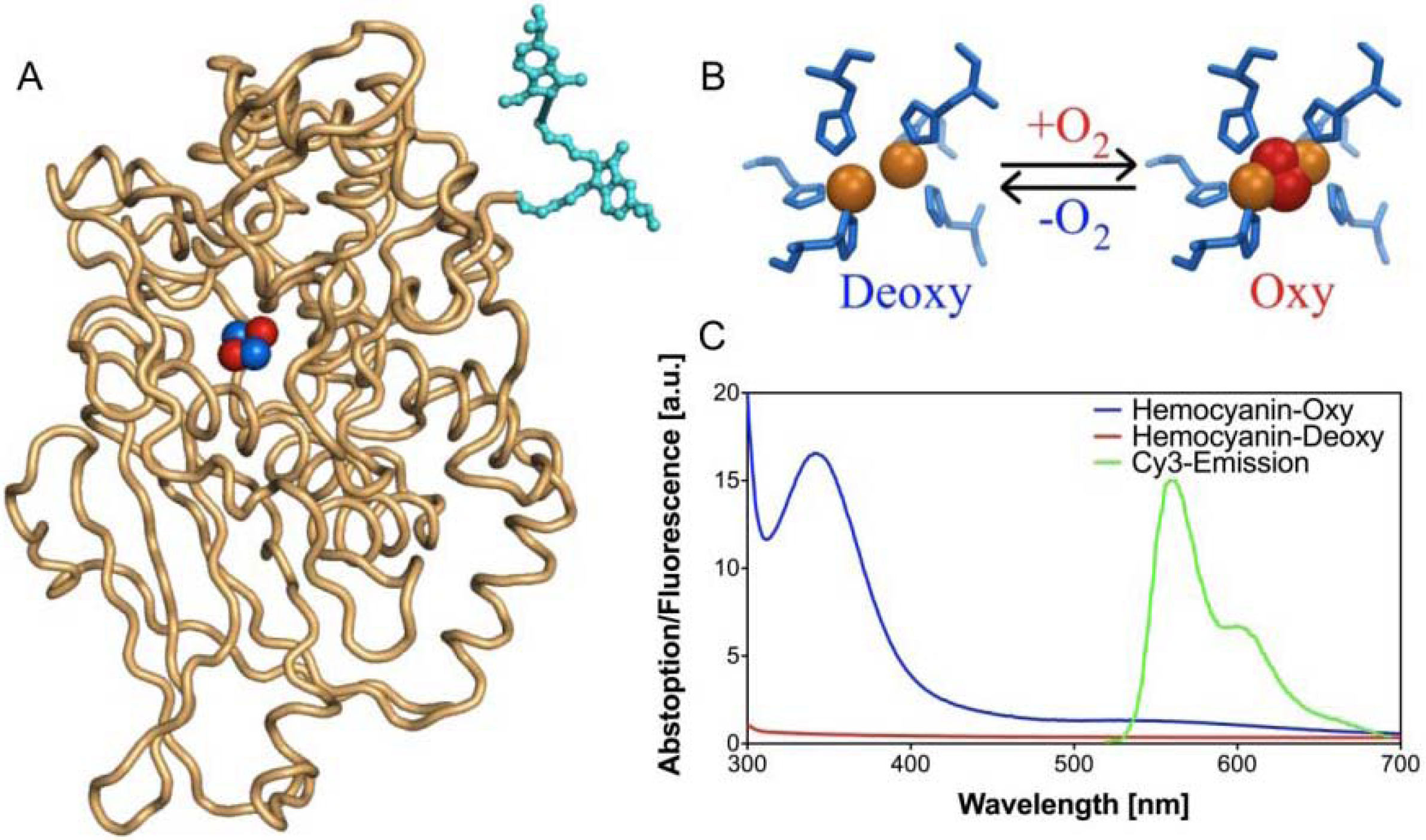
FRET-based oxygen-sensitive assay [71]
Conclusions
Acknowledgements
References and Notes
- Zembower, T.R.; Noskin, G.A.; Postelnick, M.J.; Nguyen, C.; Peterson, L.R. The Utility of Aminoglycosides in an Era of Emerging During of Resistance. Int. J. Antimicrob. Agents 1998, 10, 95–105. [Google Scholar]
- Recht, M.I.; Douthwaite, S.; Puglisi, J.D. Basis for Prokaryotic Specificity of Action of Aminoglycoside Antibiotics. EMBO J. 1999, 18, 3133–3138. [Google Scholar] [PubMed]
- Beaucaire, G. The role of aminoglycosides in Modern Therapy. J. Chemother. 1995, 7, 111–123. [Google Scholar] [PubMed]
- Zhao, F.; Zhao, Q.; Blount, K.F.; Han, Q.; Tor, Y.; Hermann, T. Molecular Recognition of RNA by Neomycin and a Restricted Neomycin Derivative. Angew. Chem. Int. Ed. 2005, 44, 5329–5334. [Google Scholar]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–497. [Google Scholar]
- Zembower, T.R.; Noskin, G.A.; Postelnick, M.J.; Nguyen, C.; Peterson, L.R. The Utility of Aminoglycosides in an Era of Emerging During of Resistance. Int. J. Antimicrob. Agents 1998, 10, 95–105. [Google Scholar] [PubMed]
- Nagai, J.; Takano, M. Molecular Aspects of Renal Handling of Aminoglycoside and Strategies for Preventing the Nephrotoxicity. Drug Metab. Pharmacokinet. 2004, 19, 159–170. [Google Scholar] [PubMed]
- Hayashi, S.F.; Norcia, L.J.; Seibel, S.B.; Silvia, A.M. Structure-activity Relationships of Hygromycin A and its Analogs: Protein Synthesis Inhibition Activity in a Cell-Free System. J. Antibiot. 1997, 50, 514–521. [Google Scholar] [PubMed]
- Hotta, K.; Zhu, C.B.; Ogata, T.; Sunada, A.; Ishikawa, J.; Mizuno, S.; Kondo, S. Enzymatic 2'-N-Acetylation of Arbekacin and Antibiotic Activity of its Product. J. Antibiot. 1996, 49, 458–464. [Google Scholar] [PubMed]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular Genetics of Aminoglycoside Resistance Genes and familial Relationships of the Aminoglycoside-Modifying Enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [PubMed]
- Rai, R.; Chen, H.N.; Czyryca, P.G.; Li, J.; Chang, C.W.T. Design and Synthesis of Pyrankacin: A Pyranmycin Class of Broad-Spectrum Aminoglycoside Antibiotic. Org. Lett. 2006, 8, 887–889. [Google Scholar] [PubMed]
- Wang, J.; Li, J.; Chen, H.N.; Chang, H.; Tanifum, C.T.; Liu, H.H.; Czyryca, P.G.; Chang, C.W.T. Glycodiversification for the Optimization of the Kanamycin Class Aminoglycosides. J. Med. Chem. 2005, 48, 6271–6285. [Google Scholar] [PubMed]
- Ding, Y.; Hofstadler, S.A.; Swayze, E.E.; Risen, L.; Griffey, R.H. Design and Synthesis of Paromomycin-Related Heterocycle-Substituted Aminoglycoside Mimetics Based on a Mass Spectrometry RNA-Binding Assay. Angew. Chem. Int. Ed. 2003, 42, 3409–3412. [Google Scholar]
- Kudyba, I.; Fernandez, D.P.; Bottger, E.C.; Vasella, A. Synthesis of Paromomycin Derivatives Modified at C(5'') to Selectively Target Bacterial rRNA. Carbohydr. Res. 2007, 342, 499–519. [Google Scholar] [PubMed]
- Francois, B.; Szychowski, J.; Adhikari, S.S.; Pachamuthu, K.; Swayze, E.E.; Griffey, R.H.; Migawa, M.T.; Westhof, E.; Hanessian, S. Antibacterial Aminoglycosides with a Modified Mode of Binding to the Ribosomal-RNA Decoding Site. Angew. Chem. Int. Ed. 2004, 43, 6735–6738. [Google Scholar]
- Fridman, M.; Blakhov, V.; Yaron, S.; Baasov, T. A New Class of Branched Aminoglycosides: Pseudo-Pentasaccharide Derivatives of Neomycin B. Org. Lett. 2003, 5, 3575–3578. [Google Scholar] [PubMed]
- Roestamadji, J.; Grapsas, I.; Mobashery, S. Mechanism-based Inactivation of Bacterial Aminoglycoside 3' Phosphotransferases. J. Am. Chem. Soc. 1995, 117, 80–84. [Google Scholar]
- Hanessian, S.; Tremblay, M.; Swayze, E.E. Tobramycin Analouges with C-5 Aminoalkyl Ether Chains Intended to Mimic Rings III and IV of Paromomycin. Tetrahedron 2003, 59, 983–993. [Google Scholar]
- Fridman, M.; Blakhov, V.; Lee, L.V.; Liang, F.S.; Wong, C.H.; Baasov, T. Dual Effect of Synthetic Aminoglycosides: Antibacterial Activity Against Bacillus Anthracis and Inhibition of Anthrax Lethal Factor. Angew. Chem. Int. Ed. 2005, 44, 447–452. [Google Scholar]
- Ye, X.S.; Zhang, L.H. Aminoglycoside Mimetics as Small-molecule Drugs Targeting RNA. Curr. Med. Chem. 2002, 9, 929–939. [Google Scholar] [PubMed]
- Arya, D.P. Design, Chemical Synthesis and Antibacterial Activity of Kanamycin and Neomycin Class Aminoglycoside Antibiotics. In Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery; Wiley: New Jersey, USA, 2007. [Google Scholar]
- Kirk, S.R.; Luedtke, N.W.; Tor, Y. Neomycin-Acridine Conjugate: A Potent Inhibitor of Rev-RRE Binding. J. Am. Chem. Soc. 2000, 122, 980–981. [Google Scholar]
- Kaiser, M.; Sainlos, M.; Lehn, J.; Bombard, S.; Teulade-Fichou, M. Aminoglycoside-quinacridine Conjugates: Towards Recognition of the P6.1 Element of Telomerase RNA. Chem. Bio. Chem. 2006, 7, 321–329. [Google Scholar]
- Blount, K.F.; Tor, Y. A Tale of Two Targets: Differential RNA Selectivity of Nucleobase-Aminoglycoside Conjugates. Chem. Bio. Chem. 2006, 7, 1612–1621. [Google Scholar]
- Charles, I.; Xi, H.; Arya, D.P. Sequence-specific Targeting of RNA with an Oligonuleotide-Neomycin Conjugate. Bioconjug. Chem. 2007, 18, 160–169. [Google Scholar] [PubMed]
- Ahn, D.; Yu, J. Library Construction of Neomycin-dipeptide Heteroconjugates and Selection Against RRE RNA. Bioorg. Med. Chem. 2005, 13, 1177–1183. [Google Scholar] [PubMed]
- Hyun, S.; Lee, K.H.; Yu, J. A Strategy for the Design of Selective RNA Binding Agents, Preparations and RRE RNA Binding Affinities of a Neomycin-peptide Nucleic Acid Heteroconjugate Library. Bioorg. Med. Chem. Lett. 2006, 16, 4757–4759. [Google Scholar] [PubMed]
- Lee, J.; Kwon, M.; Lee, K.H.; Jeong, S.; Hyun, S.; Shin, K.J.; Yu, J. An Approach to Enhance Specificity Against RNA Targets Using Heteroconjugates of Aminoglycosides and Chloramphenicol (or Linezolid). J. Am. Chem. Soc. 2004, 126, 1956–1957. [Google Scholar] [PubMed]
- Park, W.K.C.; Auer, M.; Jaksche, H.; Wong, C.H. Rapid Combinatorial Synthesis of Aminoglycoside Antibiotics Mimetics: Use of a Polyethylene glycol-linked Amine and Neamine Derived Aldehyde in Multicomponent Condensation as a Strategy for the Discovery of New Inhibitors of the HIV RNA Rev Response Element. J. Am. Chem. Soc. 1996, 118, 10150–10155. [Google Scholar]
- Greenberg, W.A.; Priestley, E.S.; Sears, P.S.; Alper, P.B.; Rosenbohn, C.; Hendrix, M.; Hung, S.C.; Wong, C.H. Design and Synthesis of New Aminoglycoside Antibiotics Containing Neamine as an Optimal Core: Correlation of Antibiotic Activity with in Vitro Inhibition of Translation. J. Am. Chem. Soc. 1999, 121, 6527–6541. [Google Scholar]
- Nunns, C.L.; Spence, L.A.; Slater, M.J.; Berrisford, D.J. Synthesis of Neamine Libraries for RNA Recognition Using Solution Phase Chemistry. Tetrahedron Lett. 1999, 40, 9341–9345. [Google Scholar]
- Ryu, D.H.; Tan, C.H.; Rando, R.R. Synthesis of (+), (-) Neamine and Their Positional Isomers as Potential Antibiotics. Bioorg. Med. Chem. Lett. 2003, 13, 901–903. [Google Scholar] [PubMed]
- Minowa, N.; Akiyama, Y.; Hiraiwa, Y.; Maebashi, K.; Usui, T.; Ikeda, D. Synthesis and Antibacterial Activity of Novel Neamine Derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 6351–6354. [Google Scholar] [PubMed]
- Michael, K.; Wang, H.; Tor, Y. Enhanced RNA Binding of Dimerized Aminoglycosides. Bioorg. Med. Chem. 1999, 7, 1361–1371. [Google Scholar] [PubMed]
- Wang, H.; Tor, Y. Dimeric Aminoglycosides: Design, Synthesis and RNA Binding. Bioorg. Med. Chem. Lett. 1997, 7, 1951–1956. [Google Scholar]
- Sucheck, S.J.; Wong, A.L.; Koeller, K.M.; Boehr, D.D.; Draker, K.A.; Sears, P.; Wright, G.D.; Wong, C.H. Design of Bifunctional Antibiotics that Target Bacterial rRNA and Inhibit Resistance-causing Enzymes. J. Am. Chem. Soc. 2000, 122, 5230–5231. [Google Scholar]
- Tok, J.B.H.; Huffman, G.R. Enhanced Binding of Aminoglycoside Dimer to a ”Dimerized” A-site rRNA Construct. Bioorg. Med. Chem. Lett. 2000, 10, 1593–1595. [Google Scholar] [PubMed]
- Tok, J.B.H.; Dunn, L.J.; Des Jean, R.C. Binding of Dimeric Aminoglycosides to the HIV-1 Rev Response Element (RRE) RNA Construct. Bioorg. Med. Chem. Lett. 2001, 11, 1127–1131. [Google Scholar] [PubMed]
- Tok, J.B.H.; Fenker, J. Novel Synthesis and RNA-binding Properties of Aminoglycoside Dimers Conjugated via a Naphthalene diimide-based Intercalator. Bioorg. Med. Chem. Lett. 2001, 11, 2987–2991. [Google Scholar] [CrossRef] [PubMed]
- Leudtke, N.W.; Liu, Q.; Tor, Y. RNA-ligand Interactions: Affinity and Specificity of Aminoglycoside Dimers and Acridine Conjugates to the HIV-1 Rev Response Element. Biochemistry 2003, 42, 11391–11403. [Google Scholar] [PubMed]
- Riguet, E.; Desire, J.; Boden, O.; Ludwig, V.; Gobel, M.; Bailly, C.; Decout, J.L. Neamine Dimers Targeting the HIV-1 TAR RNA. Bioorg. Med. Chem. Lett. 2005, 15, 4651–4655. [Google Scholar] [PubMed]
- Liang, C.H.; Romero, A.; Rabuka, D.; Sgarbi, P.W.M.; Marby, K.A.; Duffield, J.; Yao, S.; Cheng, M.L.; Ichikawa, Y.; Sears, P.; Hu, C.; Hwang, S.B.; Shue, Y.K.; Sucheck, S.J. Structure-activity Releationship of Bivalent Aminoglycosides and Evaluation of Their Microbiological Activities. Bioorg. Med. Chem. Lett. 2005, 15, 2123–2128. [Google Scholar] [PubMed]
- Chen, G.H.; Pan, P.; Chen, Y.; Meng, X.B.; Li, Z.J. Selective Deprotection of the Cbz Amine Protecting Group for the Facile Synthesis of Kanamycin A Dimers Linked at N-3'' Position. Tetrahedron 2009, 65, 5922–5927. [Google Scholar]
- Busscher, G.F.; Rutjes, F.P.J.T.; van Delft, F.L. 2-Deoxystreptamine: Central Scaffold of Aminoglycoside Antibiotics. Chem. Rev. 2005, 205, 775–792. [Google Scholar]
- Busscher, G.F.; Rutjes, F.P.J.T.; van Delft, F.L. Synthesis of a Protected Enantiomerically Pure 2-Deoxystreptamine Derivative from d-Allylglycine. Tetrahedron Lett. 2004, 69, 3629–3632. [Google Scholar]
- Busscher, G.F.; Groothuys, S.; de Gelder, R.; Rutjes, F.P.J.T.; van Delft, F.L. Efficient Preparation of a 1,3-Diazidocyclitol as a Versatile 2-Deoxystreptamine Precursor. J. Org. Chem. 2004, 69, 4477–4481. [Google Scholar] [PubMed]
- van den Broek, S.A.M.W.; Gruijters, B.W.T.; Rutjes, F.P.J.T.; van Delft, F.L.; Blaauw, R.H. A Short and Scalable Route to Orthogonally O-Protected 2-Deoxystreptamine. J. Org. Chem. 2007, 72, 3577–3580. [Google Scholar] [PubMed]
- Aslam, M.W.; Busscher, G.F.; Weiner, D.P.; de Gelder, R.; Rutjes, F.P.J.T.; van Delft, F.L. Fully Orthogonally Protected 2-Deoxystreptamine from Kanamycin. J. Org. Chem. 2008, 73, 5131–5134. [Google Scholar] [PubMed]
- Michael, K.; Wang, H.; Tor, Y. Enhanced RNA-Binding of Dimerized Aminoglycosides. Bioorg. Med. Chem. 1999, 7, 1361–1371. [Google Scholar] [PubMed]
- Yu, L.; Oost, T.K.; Schkeryantz, J.M.; Yang, J.; Janowick, D.; Fesik, S.W. Discovery of Aminoglycoside Mimetics by NMR-based Screening of Escherichia Coli A-site RNA. J. Am. Chem. Soc. 2003, 125, 4444–4450. [Google Scholar] [PubMed]
- Rodriguez, A.; Nomen, M.; Spur, B.W.; Godfroid, J.J. Selective Oxidation of Primary Silyl Ethers and its Application to the Synthesis of Natural Products. Tetrahedron Lett. 1999, 40, 5161–5164. [Google Scholar]
- Luca, L.D.; Giacomelli, G.; Masala, S.; Porcheddu, A. Trichloroisocynauric/TEMPO Oxidation of Alcohols under Mild Conditions: A Close Investigation. J. Org. Chem. 2003, 68, 4999–5001. [Google Scholar] [PubMed]
- More, J.D.; Finney, N.S. A Simple and Advantageous Protocol for the Oxidation of Alcohols with O-iodoxybenzoic Acid (IBX). Org. Lett. 2002, 4, 3001–3003. [Google Scholar] [PubMed]
- Dess, D.B.; Martin, J.C. Readially Accessible12-1-5 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-triazoles by Regiospecific Copper (I)-catalyzed 1,3-dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper (I)-catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar]
- Jawalekar, A.M.; Cremers, N.M.J.G.O.; van der Marel, H.S.O.G.A.; Rutjes, F.P.J.T.; van Delft, F.L. Conjugation of Nucleosides and Oligonucleotides by [3+2] Cycloaddition. J. Org. Chem. 2008, 73, 287–290. [Google Scholar] [PubMed]
- Wang, Y.; Killian, J.; Hamasaki, K.; Rando, R.R. RNA Molecules That Specifically and Stoichiometrically Bind Aminoglycoside Antibiotics with High Affinities. Biochemistry 1996, 35, 12338–12346. [Google Scholar] [PubMed]
- Wang, Y.; Hamasaki, K.; Rando, R.R. Specificity of Aminoglycoside Binding to RNA Construct Derived from the 16S rRNA Decoding Region and the HIV-RRE Activator Region. Biochemistry 1997, 36, 768–779. [Google Scholar] [PubMed]
- Wang, Y.; Hamasaki, K.; Rando, R.R. Specificity in the Binding of Aminoglycoside to HIV-RRE RNA. Biochemistry 1999, 38, 8548–8554. [Google Scholar] [PubMed]
- Ryu, D.H.; Rando, R.R. Aminoglycoside Binding to Human and Bacterial A-site rRNA Decoding Region Construct. Bioorg. Med. Chem. 2001, 9, 2601–2608. [Google Scholar] [PubMed]
- Ryu, D.H.; Litovchick, A.; Rando, R.R. Stereospecificity of Aminoglycoside-Ribosomal Interactions. Biochemistry 2002, 41, 10499–10509. [Google Scholar] [PubMed]
- Luedtke, N.W.; Liu, Q.; Tor, Y. RNA-ligand Interactions: Affinity and Specificity of Aminoglycoside Dimers and Acridine Conjugates to the HIV-1 Rev Response Element. Biochemistry 2003, 42, 11391–11403. [Google Scholar] [PubMed]
- Xiao, G.; Kumar, A.; Li, K.; Rigl, C.T.; Bajic, M.; Davis, T.M.; Boykin, D.W.; Wilson, W.D. Inhibition of the HIV-1 Rev-RRE Complex Formation by Unfused Aromatic Cations. Bioorg. Med. Chem. 2001, 9, 1097–1113. [Google Scholar] [PubMed]
- Arya, D.P.; Coffee, R.L.; Xue, L. From Triplex to B-form Duplex Stabalization: Reversal of Target Selectivity by Aminoglycoside Dimers. Bioorg. Med. Chem. Lett. 2004, 14, 4643–4646. [Google Scholar] [PubMed]
- Kaul, M.; Barbieri, C.M.; Pilch, D.S. Fluorescence-based Approach for Detecting and Characterizing Antibiotic-induced Conformational Changes in Ribosomal RNA: Comparing Aminoglycoside Binding to Prokaryotic and Eukaryotic Ribosomal RNA Sequences. J. Am. Chem. Soc. 2004, 126, 3447–3453. [Google Scholar] [PubMed]
- Verhelst, S.H.; Michiels, P.J.; van der Marel, G.A.; van Boeckel, C.A.; van Boom, J.H. Surface Plasmon Resonance Evaluation of Various Aminoglycoside-RNA Hairpin Interactions Reveal Low Degree of Selectivity. Chem. Bio. Chem. 2004, 5, 937–942. [Google Scholar]
- Agnelli, F.; Sucheck, S.J.; Marby, K.A.; Rabuka, D.; Yao, S.L.; Sears, P.S.; Liang, F.S.; Wong, C.H. Dimeric Aminoglycosides as Antibiotics. Angew. Chem. Int. Ed. 2004, 43, 1562–1566. [Google Scholar]
- Swayze, E.E.; Jefferson, E.A.; Sannes-Lowery, K.A.; Blyn, L.B.; Risen, L.M.; Arakawa, S.; Osgood, S.A.; Hofstadler, S.A.; Griffey, R.H. SAR by MS: A Ligand Based Technique for Drug Lead Discovery Against Structured RNA Targets. J. Med. Chem. 2002, 45, 3816–3819. [Google Scholar] [PubMed]
- Kaul, M.; Barbieri, C.M.; Pilch, D.S. Aminoglycoside-induced Reduction in Nucleotide Mobility at the Ribosomal RNA A-site as a Potentially Key Determinant of Antibacterial Activity. J. Am. Chem. Soc. 2006, 128, 1261–1271. [Google Scholar] [PubMed]
- Strianese, M.; Zauner, G.; Tepper, A.W.J.W.; Bubacco, L.; Breukink, E.; Aartsma, T.J.; Canters, G.W.; Tabares, L.C. A Protein-based Oxygen Biosensor for High-throughput Mointoring of Cell Growth and Cell Viability. Anal. Biochem. 2009, 385, 242–248. [Google Scholar] [PubMed]
- Zauner, G.; Strianes, M.; Bubacco, L.; Aartsma, T.J.; Tepper, A.W.J.W.; Canters, G.W. Tryptophan-to-dye Fluorescence Energy Transfer Applied to Oxygen Sensing by Using Type-3 Copper Proteins. Chem. Eur. J. 2007, 13, 7085–7090. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aslam, M.W.; Tabares, L.C.; Andreoni, A.; Canters, G.W.; Rutjes, F.P.J.T.; Van Delft, F.L. 2-Deoxystreptamine Conjugates by Truncation–Derivatization of Neomycin. Pharmaceuticals 2010, 3, 679-701. https://doi.org/10.3390/ph3030679
Aslam MW, Tabares LC, Andreoni A, Canters GW, Rutjes FPJT, Van Delft FL. 2-Deoxystreptamine Conjugates by Truncation–Derivatization of Neomycin. Pharmaceuticals. 2010; 3(3):679-701. https://doi.org/10.3390/ph3030679
Chicago/Turabian StyleAslam, M. Waqar, Leandro C. Tabares, Alessio Andreoni, Gerard W. Canters, Floris P.J.T. Rutjes, and Floris L. Van Delft. 2010. "2-Deoxystreptamine Conjugates by Truncation–Derivatization of Neomycin" Pharmaceuticals 3, no. 3: 679-701. https://doi.org/10.3390/ph3030679