Antitumor Activity of Some Prenylated Xanthones


Abstract
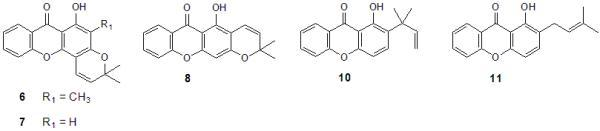
:
1. Introduction
2. Results and Discussion
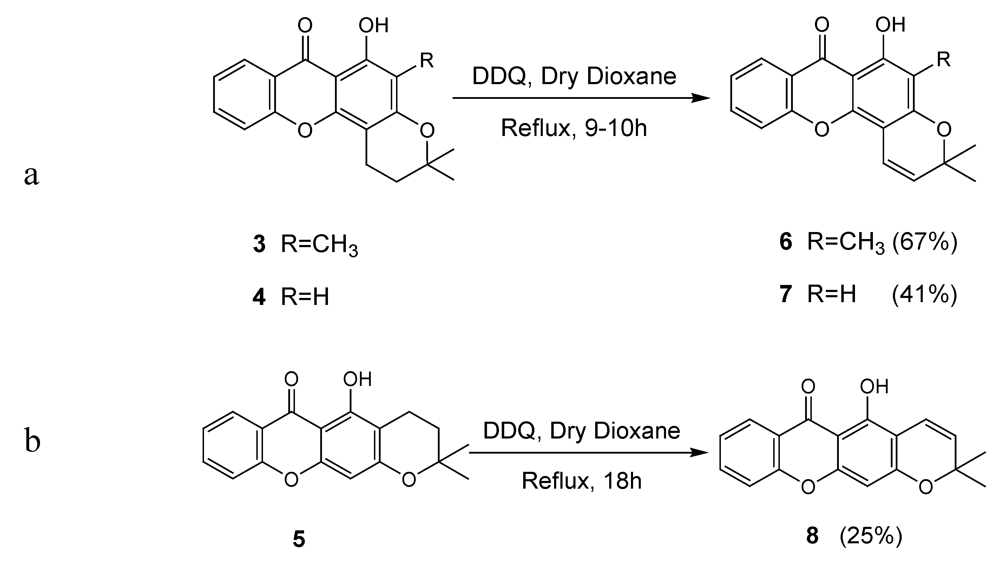
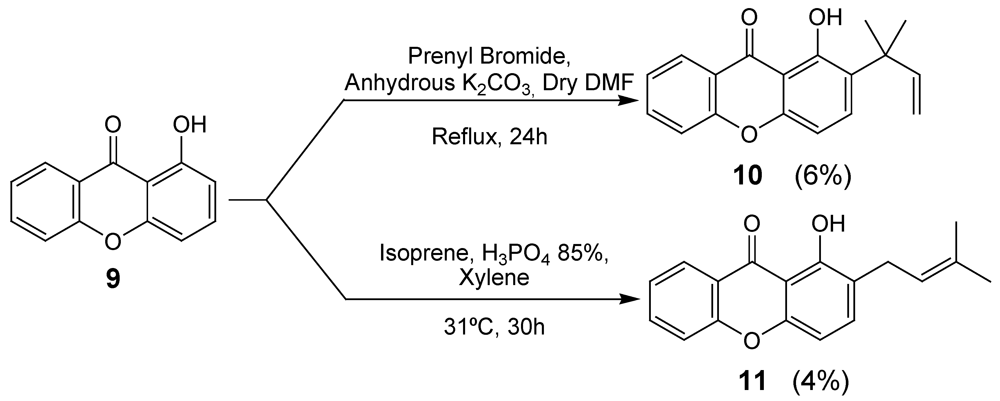
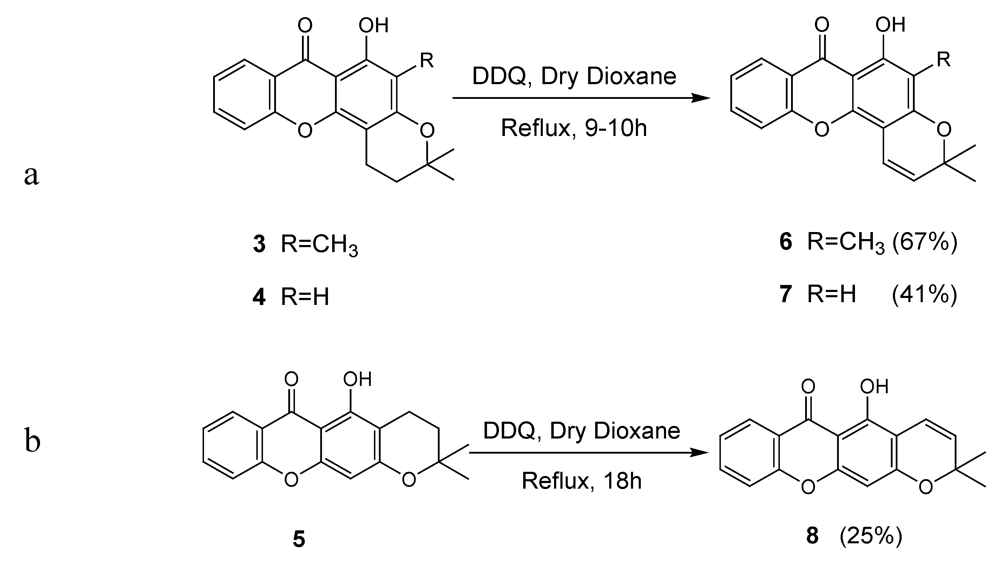
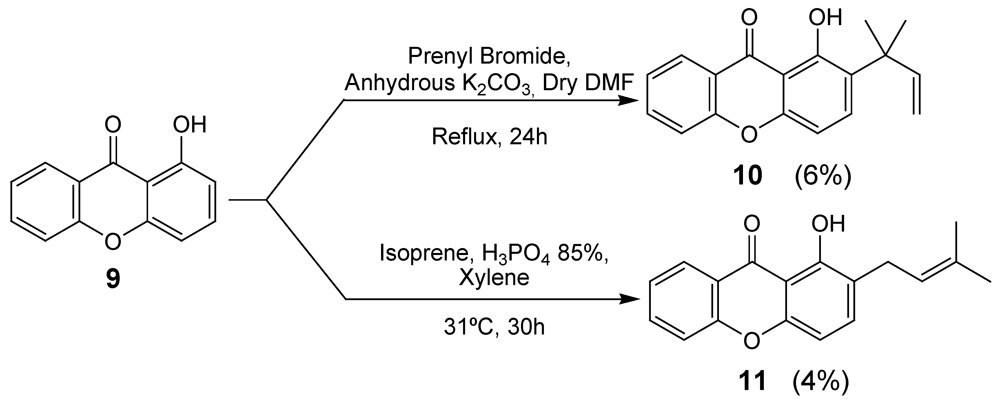
2.1. Synthesis of prenylated derivatives
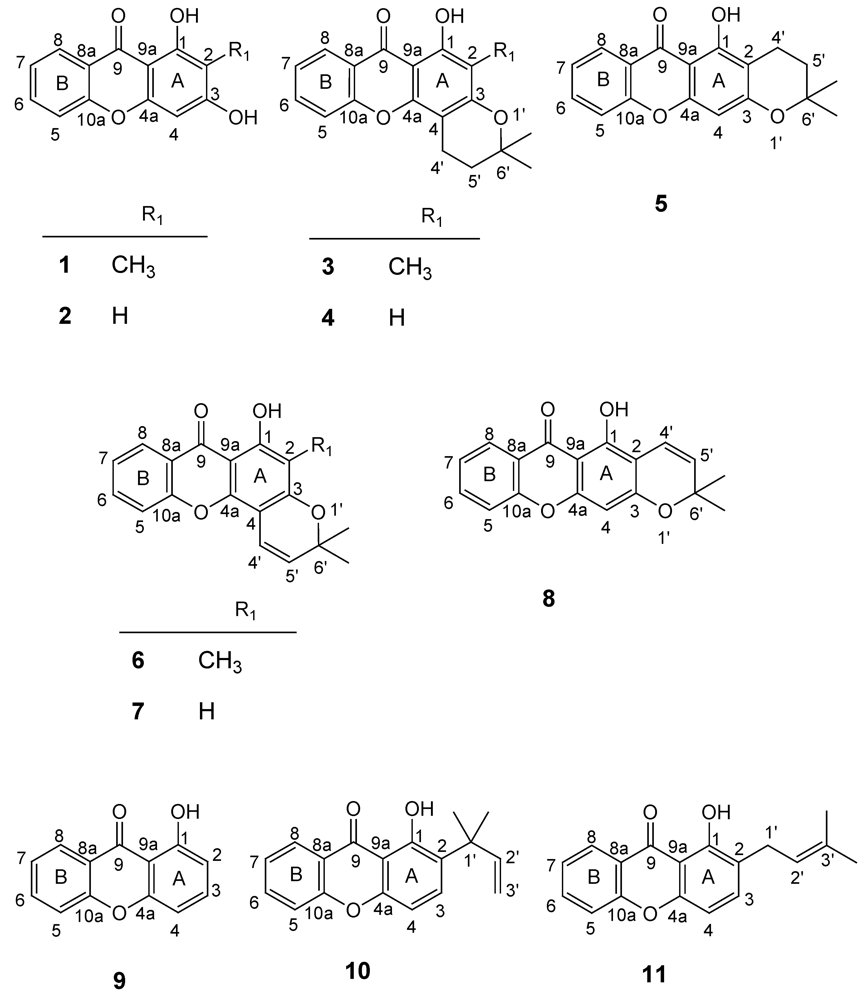
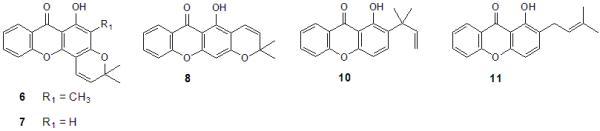
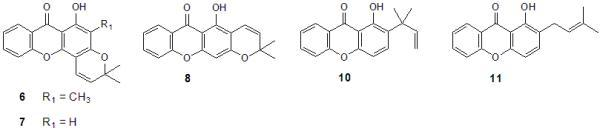
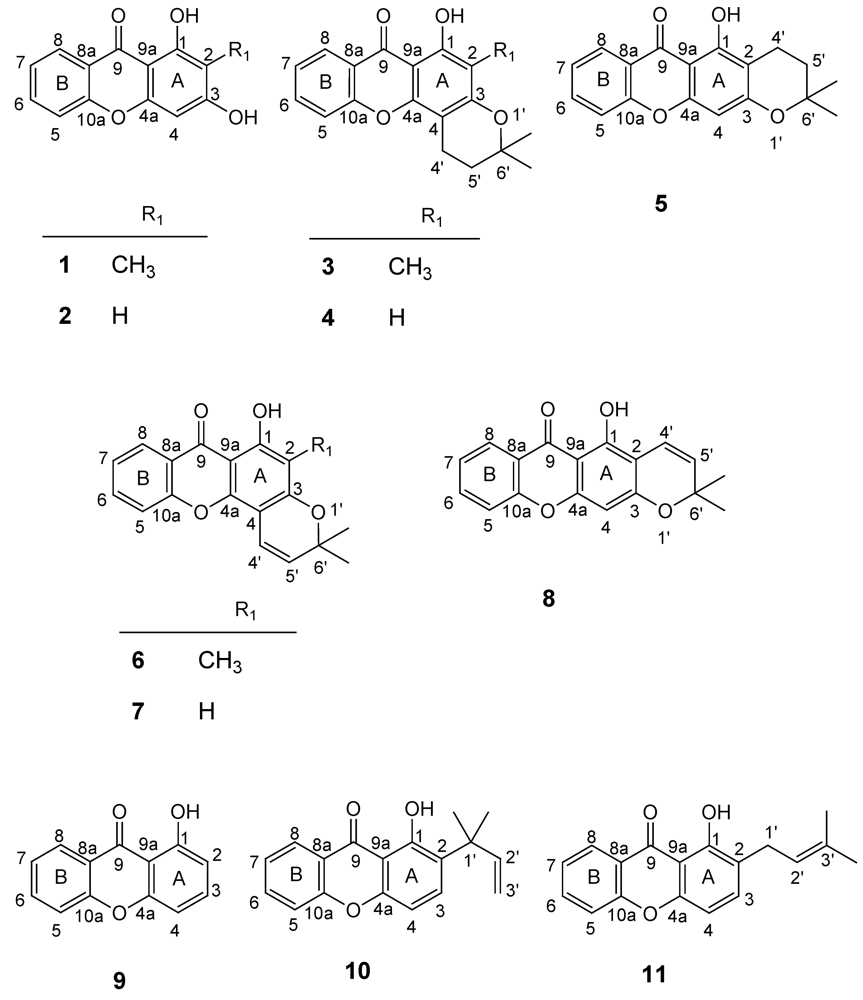
2.2. Structural elucidation of the prenylated xanthones
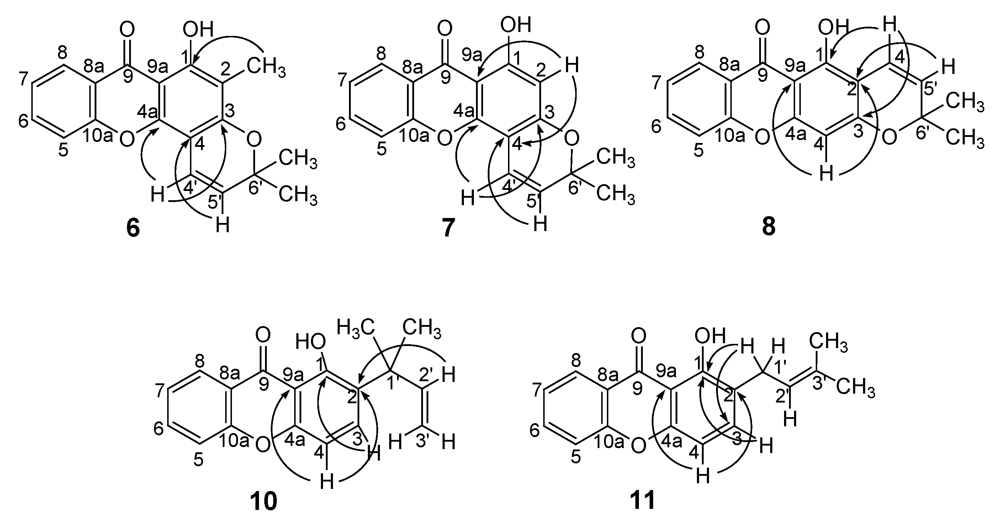
2.3. Biological Activity studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GI50 (µM) | ||
|---|---|---|---|
| MCF-7 | NCI-H460 | SF-268 | |
| Results are given in concentrations that were able to cause 50% of cell growth inhibition (GI50) after a continuous exposure of 48h and represent means of ±SEM of 3 independent experiments performed in duplicate and carry out independently. aResults published elsewhere [3,4]. bResults of one or two experiments performed in duplicate. Doxorubicin was used as positive control, GI50: MCF-7 = 42.8±8.2 nM; NCI-H460 = 94.0±8.7 nM; SF-268 = 93.0±7.0 nM. ND = not determined. | |||
| 1a | 21.9 ± 0.4 | 20.6 ± 0.9 | 33.4 ± 0.2 |
| 2a | 50.8 ± 2.2 | 37.9 ± 2.9 | 61.4 ± 5.2 |
| 3a | 18.4 ± 1.9 | >160 | >160 |
| 4a | >160 | >160 | >160 |
| 5a | 88.6 ± 12.9 | >160 | >160 |
| 6 | >150b | >150b | ND |
| 7 | >150 | >150b | ND |
| 8 | >150b | >150b | ND |
| 9a | >200 | ND | ND |
| 10 | 55b | >150b | ND |
| 11 | 88b | ND | ND |
3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of Pyranoxanthones 6-8
3.3. Synthesis of Prenylated Xanthone 10
3.4. Synthesis of prenylated xanthone 11
3.5. Tumor cell growth assay
4. Conclusions
Acknowledgements
References and Notes
- Pinto, M.; Castanheiro, R. Natural Prenylated Xanthones: Chemistry and Biological Activities. In Natural Products: Chemistry, Biochemistry and Pharmacology, 1st; Brahmachari, G., Ed.; Narosa Publishing House PVT. LTD: New Dehli, India, 2009; pp. 520–676. [Google Scholar]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar]
- Castanheiro, R.A.P.; Pinto, M.M.M.; Silva, A.M.S.; Cravo, S.M.M.; Gales, L.; Damas, A.M.; Nazareth, N.; Nascimento, M.S.J.; Eaton, G. Dihydroxyxanthones Prenylated Derivatives: Synthesis, Structure Elucidation and Growth Inhibitory Activity on Human Tumor Cell Lines with Improvement of Selectivity for MCF-7. Bioorg. Med. Chem. 2007, 15, 6080–6088. [Google Scholar]
- Pedro, M.M.; Cerqueira, F.; Sousa, M.E.; Nascimento, M.S.J.; Pinto, M.M.M. Xanthones as Inhibitors of Growth of Human Cancer Cell Lines and Their Effects on the Proliferation of Human Lymphocytes In Vitro. Bioorg. Med. Chem. 2002, 10, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.-K.; Yu, H.-J.; Ho, C.-T.; Don, M.-J. Synthesis of Naturally Occurring Rubilactone, Mollugin, and Dihydromollugin of Rubia cordifolia. J. Chin. Chem. Soc. 2001, 48, 77–79. [Google Scholar]
- Pisco, L.; Kordian, M.; Peseke, K.; Feist, H.; Michalik, D.; Estrada, E.; Carvalho, J.; Hamilton, G.; Rando, D.; Quincoces, J. Synthesis of compounds with antiproliferative activity as analogues of prenylated natural products existing in Brazilian propolis. Eur. J. Med. Chem. 2006, 41, 401–407. [Google Scholar]
- Helboe, P.; Arends, P. Xanthone Studies VI. Synthesis of Jacareubin, Isojacareubin and some hydroxyxanthones with allylic substituents. Arch. Pharm. Chemi. Sci. Ed. 1973, 1, 69–75. [Google Scholar]
- Ahluwalia, V.K.; Arora, K.K.; Jolly, R.S. Acid-catalysed Condensation of Isoprene with Phenols. Formation of 2,2-Dimethylchromans. J. Chem. Soc. Perkin Trans. I 1982, 335–338. [Google Scholar]
- Fernandes, E.G.R.; Silva, A.M.S.; Cavaleiro, J.A.S.; Silva, F.M.; Borges, M.F.M.; Pinto, M.M. 1H and 13C NMR Spectroscopy of Mono-, Di-, Tri-, and Tetrasubstituted Xanthones. Magn. Reson. Chem. 1998, 36, 305–309. [Google Scholar]
- Pinto, M.M.M.; Polónia, J. Synthesis of New Xanthones, I. Helv. Chim. Acta 1974, 57, 2613–2618. [Google Scholar]
- Grover, P.K.; Shah, G.D.; Shah, R.C. Xanthones. Part IV. A New Synthesis of Hydroxyxanthones and Hydroxybenzophenones. J. Chem. Soc. 1955, 3982–3985. [Google Scholar]
- Pankajamani, K.S.; Seshadri, T.R. Synthetic Experiments in the Benzopyrone Series: Part XLVI- Application on the Nencki Reaction in the Synthesis of Xanthones. J. Sci. Industr. Res. 1954, 13B, 396–400. [Google Scholar]
- Kolokythas, G.; Kostakis, I.K.; Pouli, N.; Marakos, P.; Kousidou, O.C.; Tzanakakis, G.N.; Karamanos, N.K. Design and synthesis of new pyranoxanthenones bearing a nitro group or an aminosubstituted side chain on the pyran ring. Evaluation of their growth inhibitory activity in breast cancer cells. Eur. J. Med. Chem. 2007, 42, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Subba Rao, G.S.R.; Raghavan, S. Synthetic studies on morellin. Part 4: Synthesis of 2,2-dimethyl- 12-[3-methylbut-2-enyl]-2H,6H-pyrano[3,2-b]xanthen-6-one. J. Indian Inst. Sci. 2001, 81, 393–401. [Google Scholar]
- Mesquita, A.A.L.; Corrêa, D.B.; Gottlieb, O.R; Magalhães, M.T. Methods for the Structural Investigation of Xanthones. Part II. Location of the Hydroxyl Groups by Ultra-Violet and Visible Spectroscopy. Anal. Chim. Acta 1968, 42, 311–323. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigrowolff, A.; Graygoodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer I. 1991, 83, 757–766. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokessch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer I. 1990, 82, 1107–1112. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Castanheiro, R.A.P.; Silva, A.M.S.; Campos, N.A.N.; Nascimento, M.S.J.; Pinto, M.M.M. Antitumor Activity of Some Prenylated Xanthones. Pharmaceuticals 2009, 2, 33-43. https://doi.org/10.3390/ph2020033
Castanheiro RAP, Silva AMS, Campos NAN, Nascimento MSJ, Pinto MMM. Antitumor Activity of Some Prenylated Xanthones. Pharmaceuticals. 2009; 2(2):33-43. https://doi.org/10.3390/ph2020033
Chicago/Turabian StyleCastanheiro, Raquel A.P., Artur M.S. Silva, Naïr A.N. Campos, Maria S.J. Nascimento, and Madalena M.M. Pinto. 2009. "Antitumor Activity of Some Prenylated Xanthones" Pharmaceuticals 2, no. 2: 33-43. https://doi.org/10.3390/ph2020033