
Theranostic Value of Multimers: Lessons Learned from Trimerization of Neurotensin Receptor Ligands and Other Targeting Vectors

Abstract
:1. Introduction
2. Results
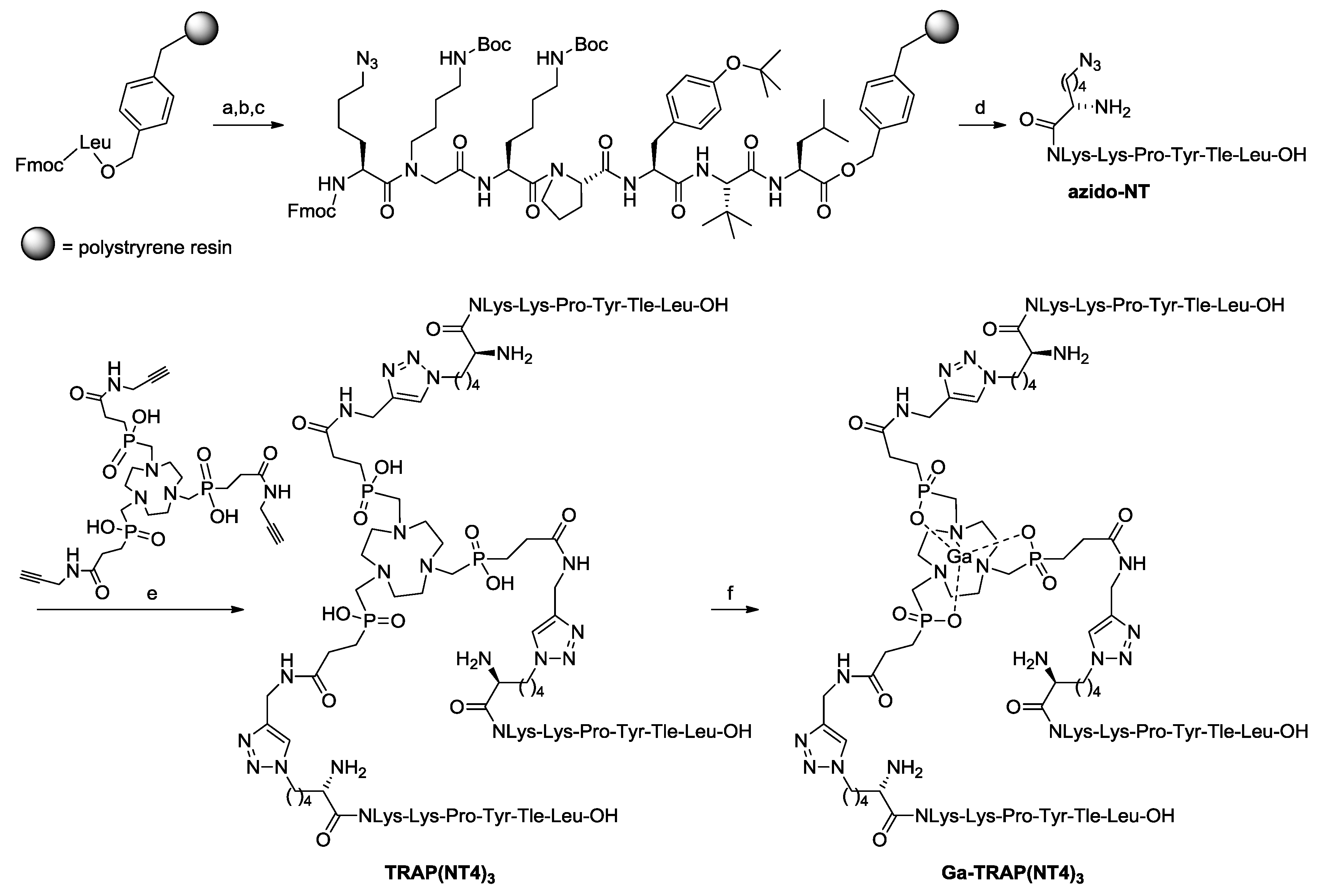
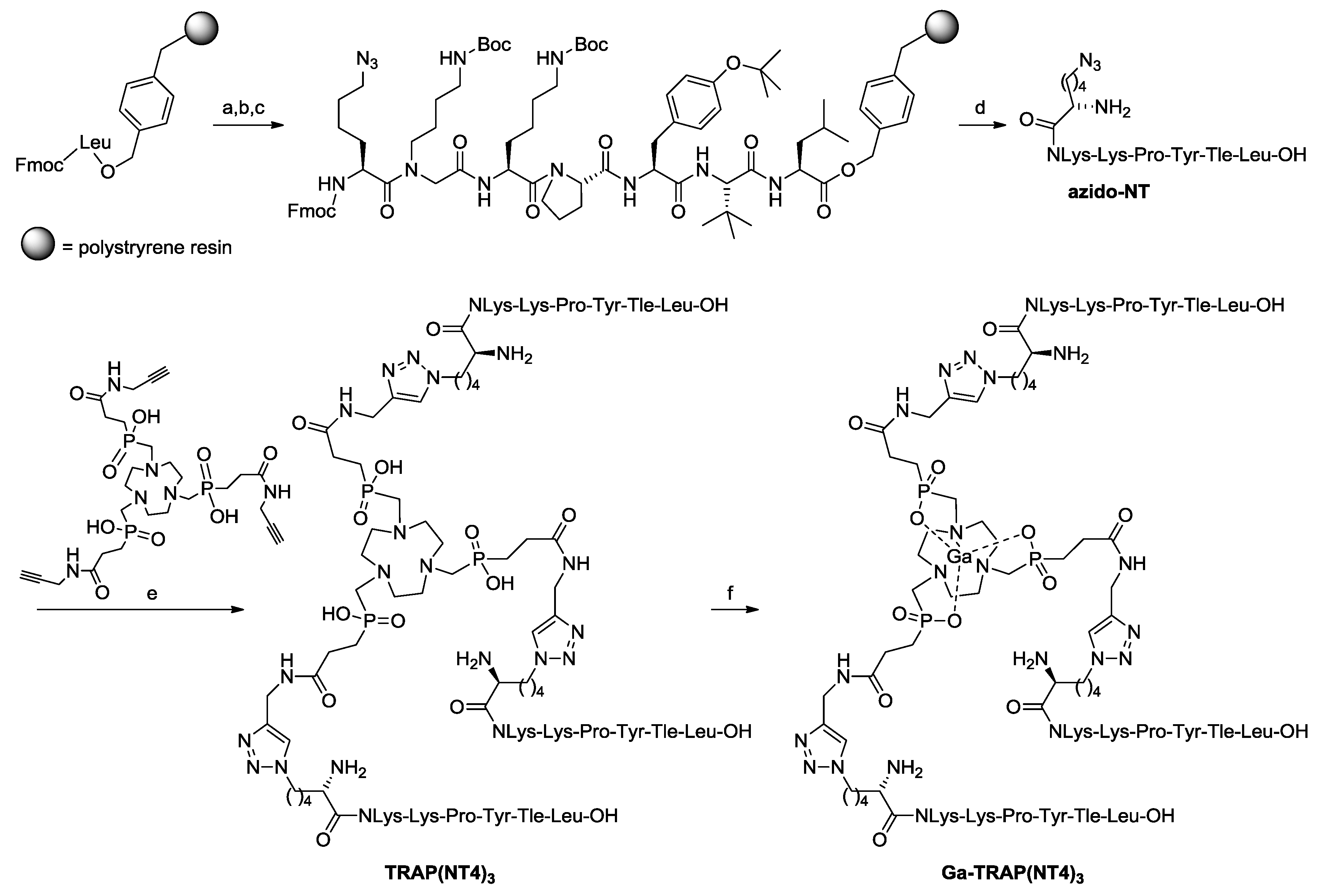
2.1. Syntheses and Radiosyntheses
2.2. In Vitro Evaluation
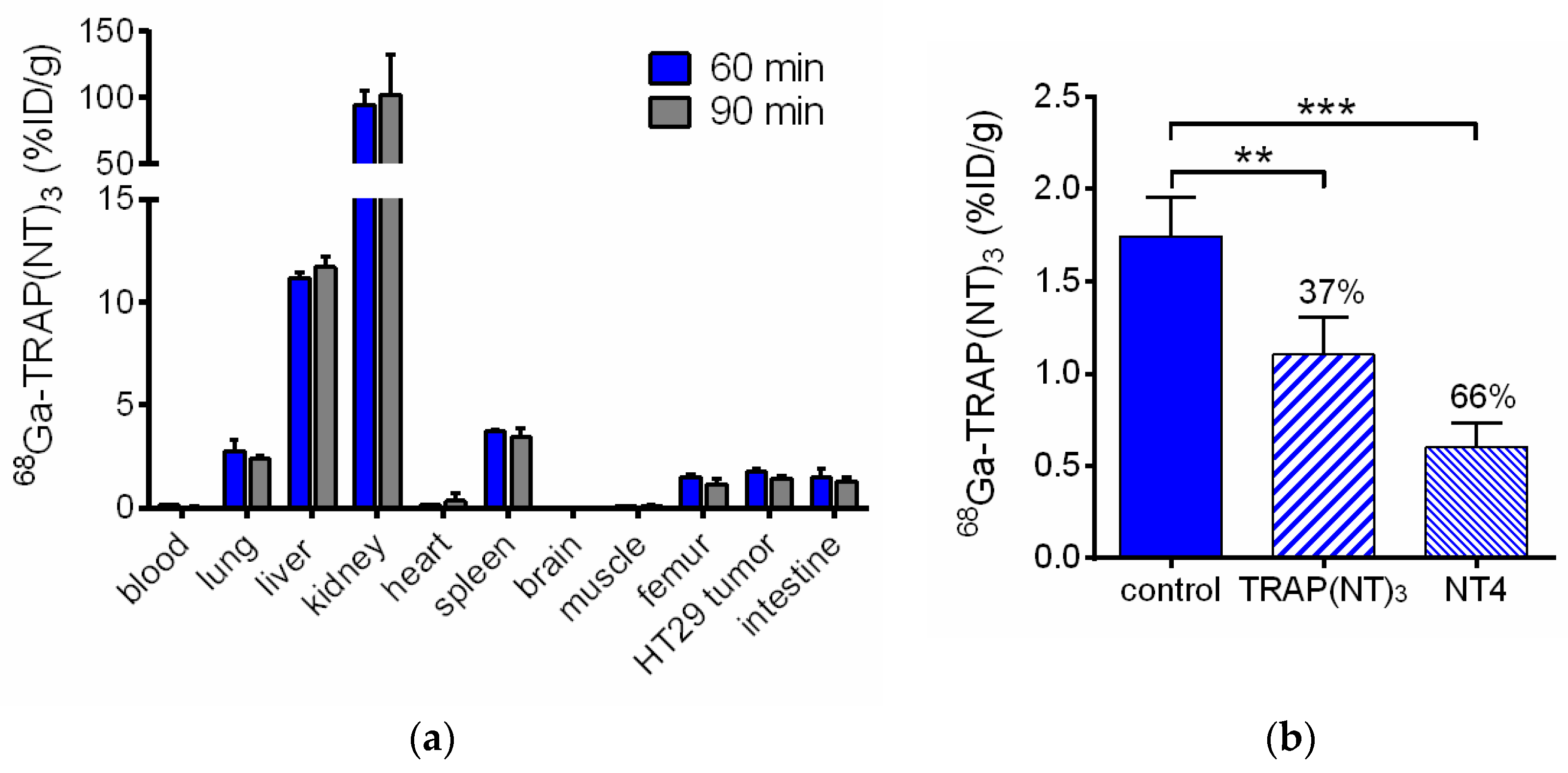
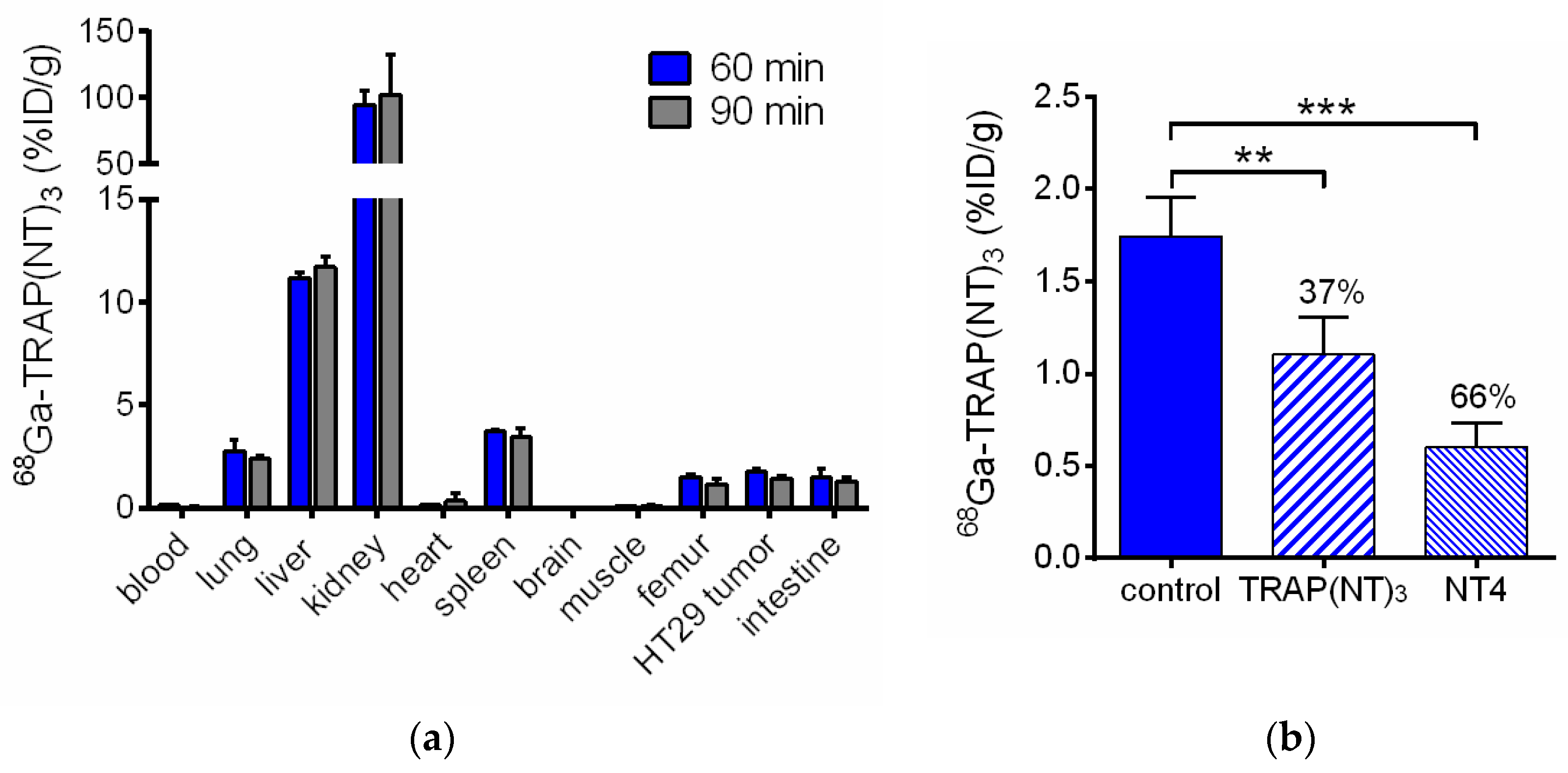
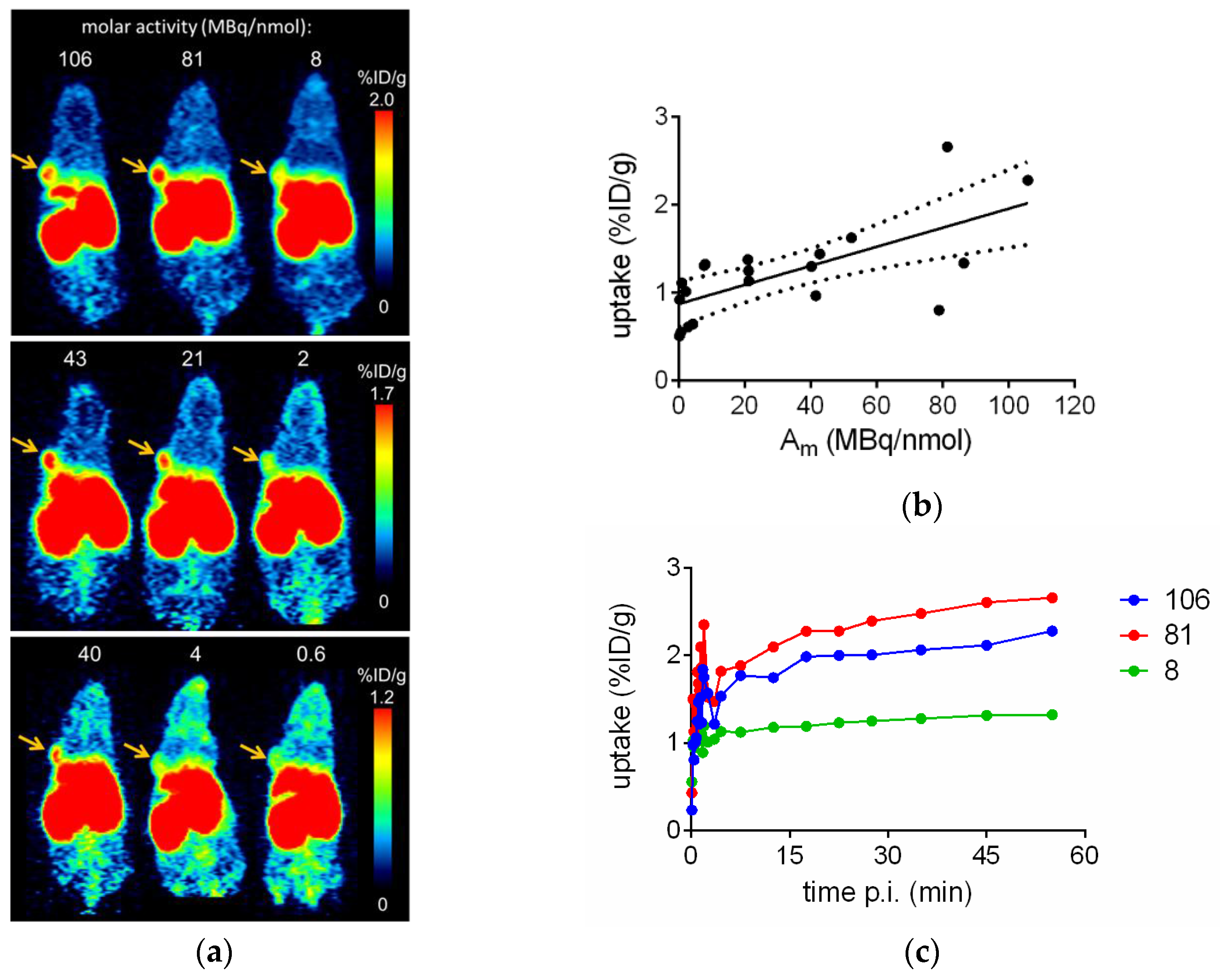
2.3. In Vivo Evaluation of [68Ga]Ga-TRAP(NT4)3
3. Discussion
3.1. Characteristics of the Neurotensin Ligand Trimer [68Ga]Ga-TRAP(NT4)3
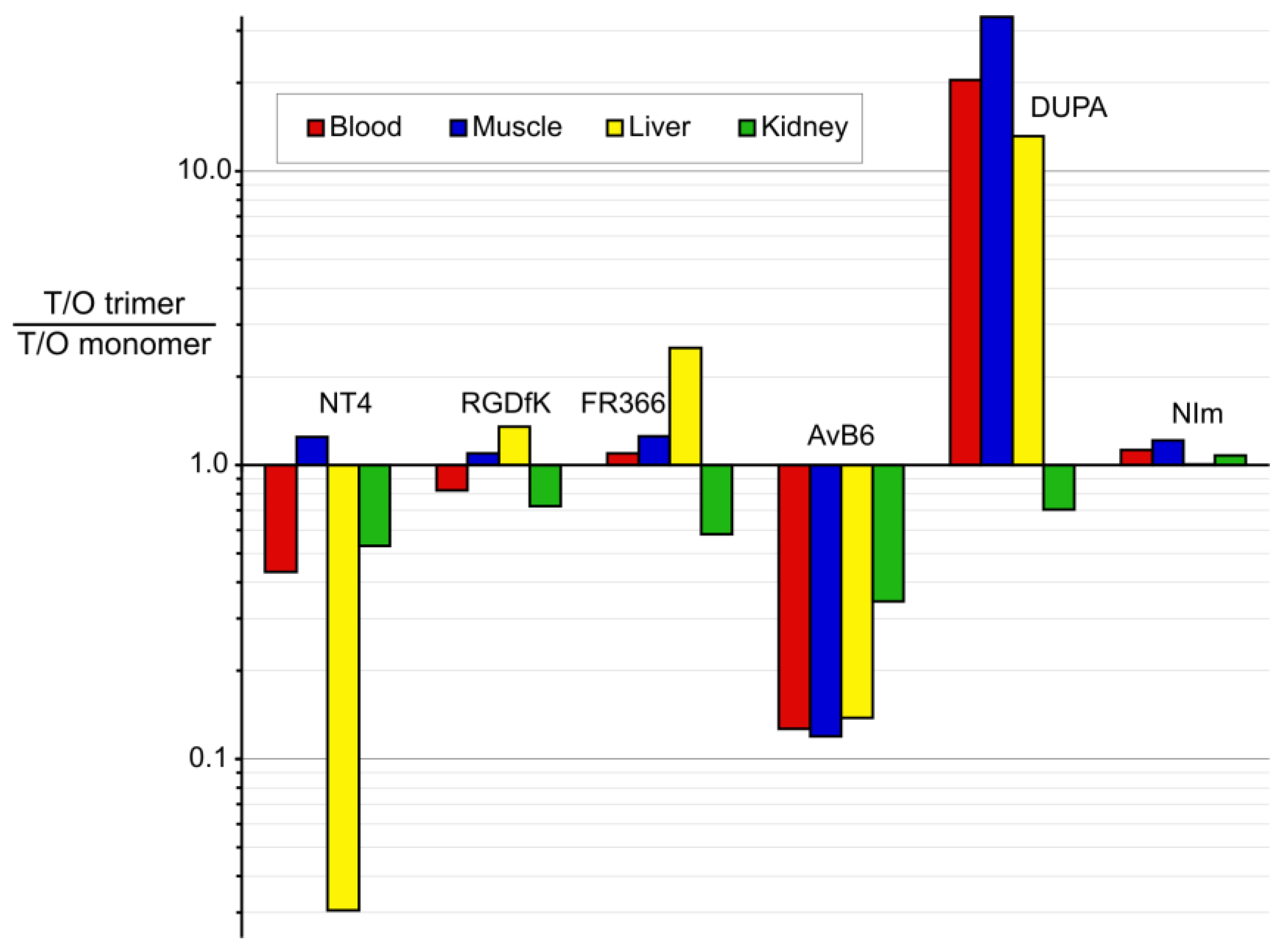
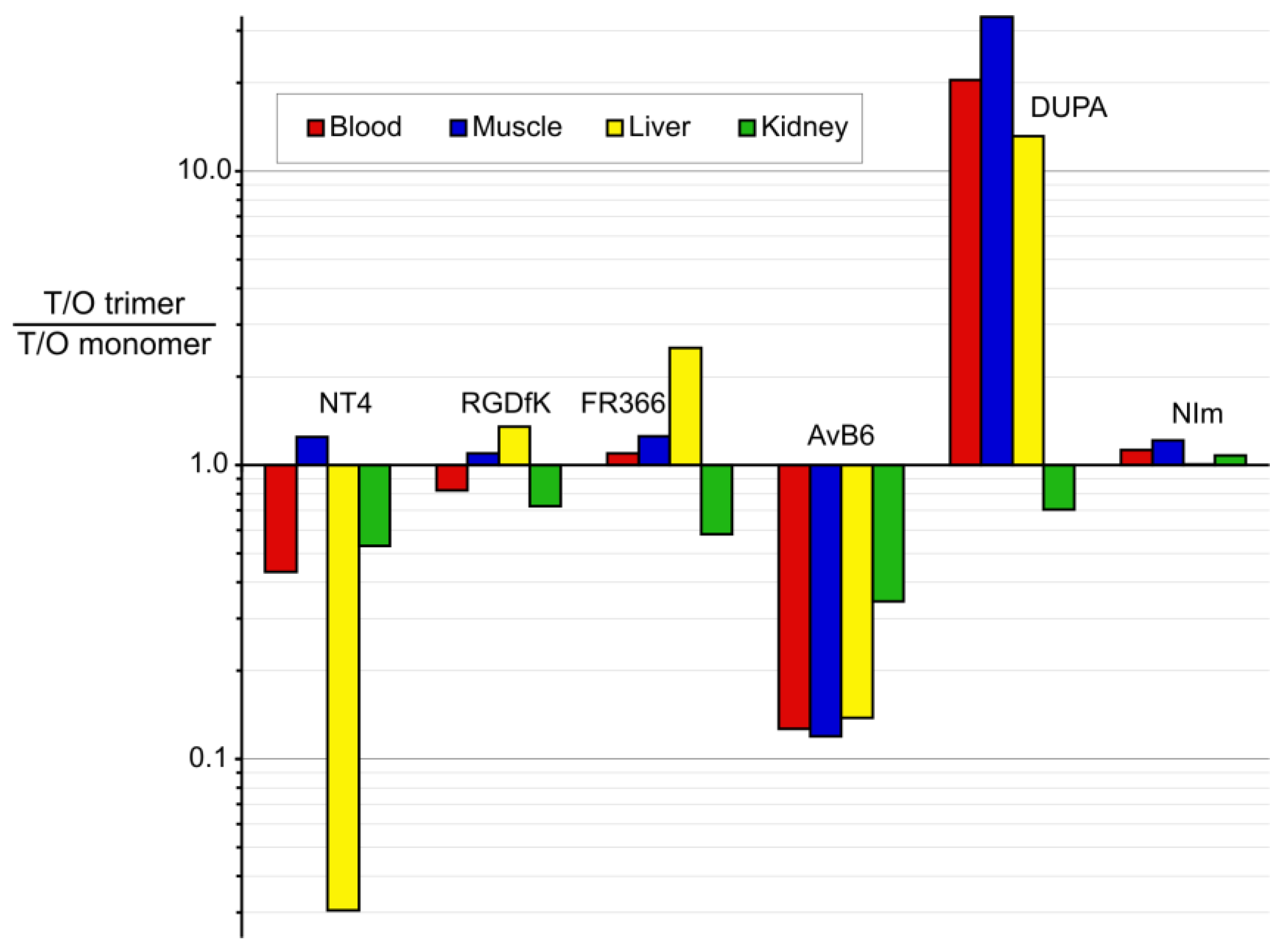
3.2. Putting Trimerization into Perspective: Comparison of Various Ligand Monomers and Their TRAP Trimers
4. Materials and Methods
4.1. General
4.2. Synthesis of H-Nle(6-N3)-NLys-Lys-Pro-Tyr-Tle-Leu-OH (azido-NT)
4.3. Synthesis of TRAP(NT4)3
4.4. Synthesis of Ga-TRAP(NT4)3
4.5. Radiosynthesis of [68Ga]Ga-TRAP(NT4)3
4.6. Receptor Binding Assays
4.7. Cell Culture
4.8. Internalization and Efflux
4.9. Animal Model
4.10. Biodistribution Studies
4.11. Small Animal PET Imaging
5. Conclusions
- trimerization invariably effected enhancement of target affinities and tumor uptakes;
- trimerization had a highly variable influence on tumor-to-organ ratios, ranging from substantial improvement to strong deterioration; and
- there is no correlation of in vitro data (affinity, log D) with in vivo performance.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dupouy, S.; Mourra, N.; Gompel, A.; Alifano, M.; Forgez, P. The potential use of the neurotensin high affinity receptor 1 as a biomarker for cancer progression and as a component of personalized medicine in selective cancers. Biochimie 2011, 93, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.; Waser, B.; Friess, H.; Büchler, M.; Laissue, J. Neurotensin receptors: A new marker for human ductal pancreatic adenocarcinoma. Gut 1998, 42, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Alifano, M.; Souazé, F.; Dupouy, S.; Camilleri-Broët, S.; Younes, M.; Ahmed-Zaïd, S.-M.; Takahashi, T.; Cancellieri, A.; Damiani, S.; Boaron, M. Neurotensin receptor 1 determines the outcome of non–small cell lung cancer. Clin. Cancer Res. 2010, 16, 4401–4410. [Google Scholar] [CrossRef] [PubMed]
- Souazé, F.; Dupouy, S.; Viardot-Foucault, V.; Bruyneel, E.; Attoub, S.; Gespach, C.; Gompel, A.; Forgez, P. Expression of neurotensin and NT1 receptor in human breast cancer: A potential role in tumor progression. Cancer Res. 2006, 66, 6243–6249. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.L.; Burns, J.E.; Maitland, N.J. Altered expression of neurotensin receptors is associated with the differentiation state of prostate cancer. Cancer Res. 2010, 70, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Morgat, C.; Mishra, A.K.; Varshney, R.; Allard, M.; Fernandez, P.; Hindié, E. Targeting neuropeptide receptors for cancer imaging and therapy: Perspectives with bombesin, neurotensin, and neuropeptide-Y receptors. J. Nucl. Med. 2014, 55, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Martinez-Fong, D.; Trédaniel, J.; Forgez, P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy. Front. Endocrinol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Aronin, N.; Carraway, R.E.; Ferris, C.F.; Hammer, R.A.; Leeman, S.E. The stability and metabolism of intravenously administered neurotensin in the rat. Peptides 1982, 3, 637–642. [Google Scholar] [CrossRef]
- Lee, Y.C.; Uttenthal, L.O.; Smith, H.A.; Bloom, S.R. In vitro degradation of neurotensin in human plasma. Peptides 1986, 7, 383–387. [Google Scholar] [CrossRef]
- Orwig, K.S.; Lassetter, M.R.; Hadden, M.K.; Dix, T.A. Comparison of N-Terminal Modifications on Neurotensin(8–13) Analogues Correlates Peptide Stability but Not Binding Affinity with in Vivo Efficacy. J. Med. Chem. 2009, 52, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Sparr, C.; Purkayastha, N.; Yoshinari, T.; Seebach, D.; Maschauer, S.; Prante, O.; Hübner, H.; Gmeiner, P.; Kolesinska, B.; Cescato, R.; et al. Syntheses, Receptor Bindings, in vitro and in vivo Stabilities and Biodistributions of DOTA-Neurotensin(8–13) Derivatives Containing β-Amino Acid Residues—A Lesson about the Importance of Animal Experiments. Chem. Biodivers. 2013, 10, 2101–2121. [Google Scholar] [CrossRef] [PubMed]
- Mascarin, A.; Valverde, I.E.; Mindt, T.L. Structure-Activity Relationship Studies of Amino Acid Substitutions in Radiolabeled Neurotensin Conjugates. ChemMedChem 2016, 11, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Bruehlmeier, M.; Garayoa, E.G.; Blanc, A.; Holzer, B.; Gergely, S.; Tourwe, D.; Schubiger, P.A.; Blauenstein, P. Stabilization of neurotensin analogues: Effect on peptide catabolism, biodistribution and tumor binding. Nucl. Med. Biol. 2002, 29, 321–327. [Google Scholar] [CrossRef]
- Charron, C.; Hickey, J.; Nsiama, T.; Cruickshank, D.; Turnbull, W.; Luyt, L. Molecular imaging probes derived from natural peptides. Nat. Prod. Rep. 2016, 33, 761–800. [Google Scholar] [CrossRef] [PubMed]
- Rahmim, A.; Zaidi, H. PET versus SPECT: Strengths, limitations and challenges. Nucl. Med. Commun. 2008, 29, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Einsiedel, J.; Hocke, C.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Synthesis of a 68Ga-labeled peptoid-Peptide hybrid for imaging of neurotensin receptor expression in vivo. ACS Med. Chem. Lett. 2010, 1, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and glycosylation of peptides using click chemistry: A general approach to 18F-glycopeptides as effective imaging probes for positron emission tomography. Angew. Chem. Int. Ed. Engl. 2010, 49, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Prante, O. 18F- and 68Ga-Labeled Neurotensin Peptides for PET Imaging of Neurotensin Receptor 1. J. Med. Chem. 2016, 59, 6480–6492. [Google Scholar] [CrossRef] [PubMed]
- Röhrich, A.; Bergmann, R.; Kretzschmann, A.; Noll, S.; Steinbach, J.; Pietzsch, J.; Stephan, H. A novel tetrabranched neurotensin(8–13) cyclam derivative: Synthesis, 64Cu-labeling and biological evaluation. J. Inorg. Biochem. 2011, 105, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hultsch, C.; Berndt, M.; Bergmann, R.; Wuest, F. Radiolabeling of multimeric neurotensin(8–13) analogs with the short-lived positron emitter fluorine-18. Appl. Radiat. Isot. 2007, 65, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Reich, D.; Vagner, A.; Weineisen, M.; Toth, I.; Wester, H.J.; Notni, J. A shortcut to high-affinity Ga-68 and Cu-64 radiopharmaceuticals: One-pot click chemistry trimerisation on the TRAP platform. Dalton Trans. 2015, 44, 11137–11146. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Pohle, K.; Wester, H.J. Be spoilt for choice with radiolabelled RGD peptides: Preclinical evaluation of 68Ga-TRAP(RGD)3. Nucl. Med. Biol. 2013, 40, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Pohle, K.; Wester, H.J. Comparative gallium-68 labeling of TRAP-, NOTA-, and DOTA-peptides: Practical consequences for the future of gallium-68-PET. EJNMMI Res. 2012, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Simecek, J.; Schulz, M.; Notni, J.; Plutnar, J.; Kubicek, V.; Havlickova, J.; Hermann, P. Complexation of metal ions with TRAP (1,4,7-triazacyclononane phosphinic acid) ligands and 1,4,7-triazacyclononane-1,4,7-triacetic acid: Phosphinate-containing ligands as unique chelators for trivalent gallium. Inorg. Chem. 2012, 51, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Simecek, J.; Hermann, P.; Wester, H.J. TRAP, a Powerful and Versatile Framework for Gallium-68 Radiopharmaceuticals. Chem. Eur. J. 2011, 17, 14718–14722. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, J.; Hübner, H.; Hervet, M.; Harterich, S.; Koschatzky, S.; Gmeiner, P. Peptide backbone modifications on the C-terminal hexapeptide of neurotensin. Bioorg. Med. Chem. Lett. 2008, 18, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Maschauer, S.; Ruckdeschel, T.; Tripal, P.; Haubner, R.; Einsiedel, J.; Hübner, H.; Gmeiner, P.; Kuwert, T.; Prante, O. In vivo monitoring of the antiangiogenic effect of neurotensin receptor-mediated radiotherapy by small-animal positron emission tomography: A pilot study. Pharmaceuticals 2014, 7, 464–481. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Hermann, P.; Havlickova, J.; Kotek, J.; Kubicek, V.; Plutnar, J.; Loktionova, N.; Riss, P.J.; Rösch, F.; Lukes, I. A triazacyclononane-based bifunctional phosphinate ligand for the preparation of multimeric 68Ga tracers for positron emission tomography. Chem. Eur. J. 2010, 16, 7174–7185. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Simecek, J.; Wester, H.J. Phosphinic acid functionalized polyazacycloalkane chelators for radiodiagnostics and radiotherapeutics: Unique characteristics and applications. ChemMedChem 2014, 9, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Wester, H.J. A Practical Guide on the Synthesis of Metal Chelates for Molecular Imaging and Therapy by Means of Click Chemistry. Chem. Eur. J. 2016, 22, 11500–11508. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Maschauer, S.; Hübner, H.; Gmeiner, P.; Prante, O. Synthesis and evaluation of a 18F-labeled diarylpyrazole glycoconjugate for the imaging of NTS1-positive tumors. J. Med. Chem. 2013, 56, 9361–9365. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, W.; Fan, W.; Brusnahan, S.; Garrison, J. Investigation of the Biological Impact of Charge Distribution on a NTR1-Targeted Peptide. Bioconj. Chem. 2016, 27, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Hübner, H.; Schellhorn, T.; Gienger, M.; Schaab, C.; Kaindl, J.; Leeb, L.; Clark, T.; Möller, D.; Gmeiner, P. Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun. 2016, 7, 12298. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Reich, D.; Maltsev, O.V.; Kapp, T.G.; Steiger, K.; Hoffmann, F.; Esposito, I.; Weichert, W.; Kessler, H.; Wester, H.-J. In Vivo PET imaging of the “cancer integrin” αvβ6 using gallium-68 labelled cyclic RGD nonapeptides. J. Nucl. Med. 2017, 57. [Google Scholar] [CrossRef]
- Weineisen, M.; Simecek, J.; Schottelius, M.; Schwaiger, M.; Wester, H.J. Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. EJNMMI Res. 2014, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Bacher, L.; Fischer, G.; Litau, S.; Schirrmacher, R.; Wängler, B.; Baller, M.; Wängler, C. Improving the stability of peptidic radiotracers by the introduction of artificial scaffolds: Which structure element is most useful? J. Label. Compd Radiopharm. 2015, 58, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Simecek, J.; Notni, J.; Kapp, T.G.; Kessler, H.; Wester, H.J. Benefits of NOPO as chelator in gallium-68 peptides, exemplified by preclinical characterization of 68Ga-NOPO-c(RGDfK). Mol. Pharm. 2014, 11, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Steiger, K.; Hoffmann, F.; Reich, D.; Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Kessler, H.; Wester, H.J. Complementary, Selective PET Imaging of Integrin Subtypes α5β1 and αvβ3 Using 68Ga-Aquibeprin and 68Ga-Avebetrin. J. Nucl. Med. 2016, 57, 460–466. [Google Scholar] [CrossRef] [PubMed]
- D‘Alessandria, C.; Pohle, K.; Rechenmacher, F.; Neubauer, S.; Notni, J.; Wester, H.J.; Schwaiger, M.; Kessler, H.; Beer, A.J. In vivo biokinetic and metabolic characterization of the 68Ga-labelled α5β1-selective peptidomimetic FR366. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Steiger, K.; Hoffmann, F.; Reich, D.; Schwaiger, M.; Kessler, H.; Wester, H.J. Variation of Specific Activities of 68Ga-Aquibeprin and 68Ga-Avebetrin Enables Selective PET Imaging of Different Expression Levels of Integrins α5β1 and αvβ3. J. Nucl. Med. 2016, 57, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Seelam, S.R.; Lee, J.Y.; Lee, Y.S.; Hong, M.K.; Kim, Y.J.; Banka, V.K.; Lee, D.S.; Chung, J.K.; Jeong, J.M. Development of 68Ga-labeled multivalent nitroimidazole derivatives for hypoxia imaging. Bioorg. Med. Chem. 2015, 23, 7743–7750. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (NTS1, nM) | Ki (NTS2, nM) |
|---|---|---|
| NT(8–13) | 0.29 ± 0.03 2 | 1.40 ± 0.11 2,3 |
| Pra-NLys-Lys-Pro-Tyr-Tle-Leu-OH (NT4) | 4.6 ± 0.64 | 51 ± 10 |
| TRAP(NT4)3 | 0.47 ± 0.05 | 0.26 ± 0.05 |
| Ga-TRAP(NT4)3 | 0.12 ± 0.03 | 0.21 ± 0.05 |
| Organ | 60 min | 90 min | 60 min Blocking 1 | 60 min Blocking 2 |
|---|---|---|---|---|
| blood | 0.13 ± 0.02 | 0.04 ± 0.02 | 0.07 ± 0.01 | 0.40 ± 0.01 |
| lung | 2.76 ± 0.53 | 2.37 ± 0.21 | 1.61 ± 0.42 | 6.06 ± 1.19 |
| liver | 11.18 ± 0.30 | 11.71 ± 0.48 | 8.30 ± 0.87 | 11.21 ± 2.27 |
| kidney | 94.55 ± 10.84 | 102.37 ± 29.88 | 96.26 ± 8.83 | 99.91 ± 15.52 |
| heart | 0.13 ± 0.02 | 0.35 ± 0.37 | 0.15 ± 0.11 | 0.45 ± 0.13 |
| spleen | 3.72 ± 0.09 | 3.45 ± 0.38 | 2.20 ± 0.39 | 3.75 ± 0.81 |
| brain | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.06 ± 0.07 | 0.07 ± 0.02 |
| muscle | 0.09 ± 0.03 | 0.11 ± 0.08 | 0.10 ± 0.05 | 0.16 ± 0.03 |
| femur | 1.51 ± 0.12 | 1.17 ± 0.21 | 0.74 ± 0.14 | 1.72 ± 0.12 |
| HT29 tumor | 1.74 ± 0.21 | 1.44 ± 0.13 | 1.10 ± 0.20 | 0.60 ± 0.13 |
| intestine | 1.50 ± 0.42 | 1.31 ± 0.20 | 0.49 ± 0.08 | 0.49 ± 0.07 |
| Ga-Monomer | NODAGA-PEG6-NT4 | NOPO-RGD | NODAGA-FR366 | TRAP(AvB6)1 (Avebehexin) | DOTAGA-DUPA | TRAP(NIm)1 | ||
|---|---|---|---|---|---|---|---|---|
| Ga-Trimer | TRAP(NT4)3 | TRAP(RGD)3 (Avebetrin) | TRAP(FR366)3 (Aquibeprin) | TRAP(AvB6)3 | TRAP(DUPA)3 | TRAP(NIm)3 | ||
| Targeting vector | NTS1-selective peptoid (Pra-NLys-Lys-Pro-Tyr-Tle-Leu-OH (NT4)) | c(RGDfK) | α5β1-integrin- selective peptoid FR366 | c(FRGDLAF-p[NMe]K) | DUPA-Pep (EuK-C8-Phe-Phe) | Nitro -imidazole | ||
| Target | NTS1 | NTS2 | αvβ3 integrin | α5β1 integrin | αvβ6 integrin | PSMA | hypoxia | |
| Affinity IC50 or #Ki (nM) | Monomer | 20 # | 87 # | 1.1 | 1.3 * | 0.26 | 36 | n/a |
| Trimer | 0.12 # | 0.20 # | 0.22 | 0.083 | 0.023 | 2 | n/a | |
| factor | 166 | 435 | 5 | 16 | 11 | 18 | n/a | |
| log D pH 7.4 | Monomer | −4.1 | −4.6 | −3.9 | −3.7 | −3.6 ± 0.2 | n/a | |
| Trimer | −3.7 ± 0.1 | −3.9 | −4.2 | −1.7 | −2.9 ± 0.1 | −3.3 | ||
| Xenograft/Time p.i. | HT29/60 min | M21/120 min | M21/90 min | H2009/90 min | LNCaP/60 min | CT26/60 min | ||
| Tumor %ID/g | Monomer | 1.55 | 1.4 | 0.64 | 0.65 | 2.0 ± 0.2 | 0.33 | |
| Trimer | 1.74 | 4.6 | 2.4 | 0.92 ± 0.08 | 6.7 ± 1.9 | 0.47 | ||
| Blood %ID/g | Monomer | 0.05 | 0.04 | 0.07 | 0.17 | 2.5 ± 0.14 | 0.41 | |
| Trimer | 0.13 | 0.16 | 0.24 | 1.9 ± 0.15 | 0.41 ± 0.18 | 0.52 | ||
| Muscle %ID/g | Monomer | 0.10 | 0.22 | 0.04 | 0.06 | 1.2 ± 0.13 | 0.17 | |
| Trimer | 0.09 | 0.66 | 0.12 | 0.71 ± 0.10 | 0.12 ± 0.03 | 0.20 | ||
| Liver %ID/g | Monomer | 0.3 | 1.6 | 0.32 | 0.36 | 2.0 ± 0.17 | 0.24 | |
| Trimer | 11 | 3.9 | 0.48 | 3.7 ± 0.14 | 0.51 ± 0.10 | 0.34 | ||
| Kidney %ID/g | Monomer | 45 | 1.9 | 1.2 | 4.3 | 29 ± 7 | 2.06 | |
| Trimer | 95 | 8.6 | 8.0 | 17.7 ± 6.5 | 138 ± 11 | 2.72 | ||
| Remarks | All data for high molar activities (1–2 GBq/µmol) | Trimer data estimated from PET ROI analysis | Monomer data estimated from PET ROI analysis | |||||
| References | [18], this work | [22,37] | [38,39,40] | [34] | [21] | [41] | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maschauer, S.; Einsiedel, J.; Reich, D.; Hübner, H.; Gmeiner, P.; Wester, H.-J.; Prante, O.; Notni, J. Theranostic Value of Multimers: Lessons Learned from Trimerization of Neurotensin Receptor Ligands and Other Targeting Vectors. Pharmaceuticals 2017, 10, 29. https://doi.org/10.3390/ph10010029
Maschauer S, Einsiedel J, Reich D, Hübner H, Gmeiner P, Wester H-J, Prante O, Notni J. Theranostic Value of Multimers: Lessons Learned from Trimerization of Neurotensin Receptor Ligands and Other Targeting Vectors. Pharmaceuticals. 2017; 10(1):29. https://doi.org/10.3390/ph10010029
Chicago/Turabian StyleMaschauer, Simone, Jürgen Einsiedel, Dominik Reich, Harald Hübner, Peter Gmeiner, Hans-Jürgen Wester, Olaf Prante, and Johannes Notni. 2017. "Theranostic Value of Multimers: Lessons Learned from Trimerization of Neurotensin Receptor Ligands and Other Targeting Vectors" Pharmaceuticals 10, no. 1: 29. https://doi.org/10.3390/ph10010029