Simultaneous Chronoamperometric Sensing of Ascorbic Acid and Acetaminophen at a Boron-Doped Diamond Electrode

Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
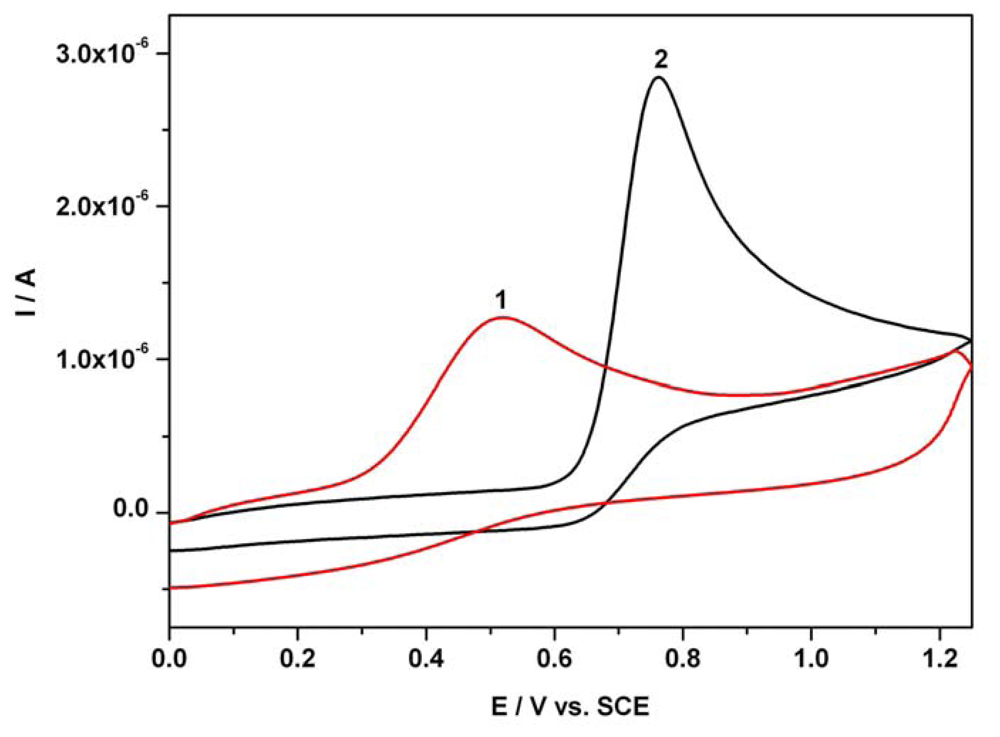
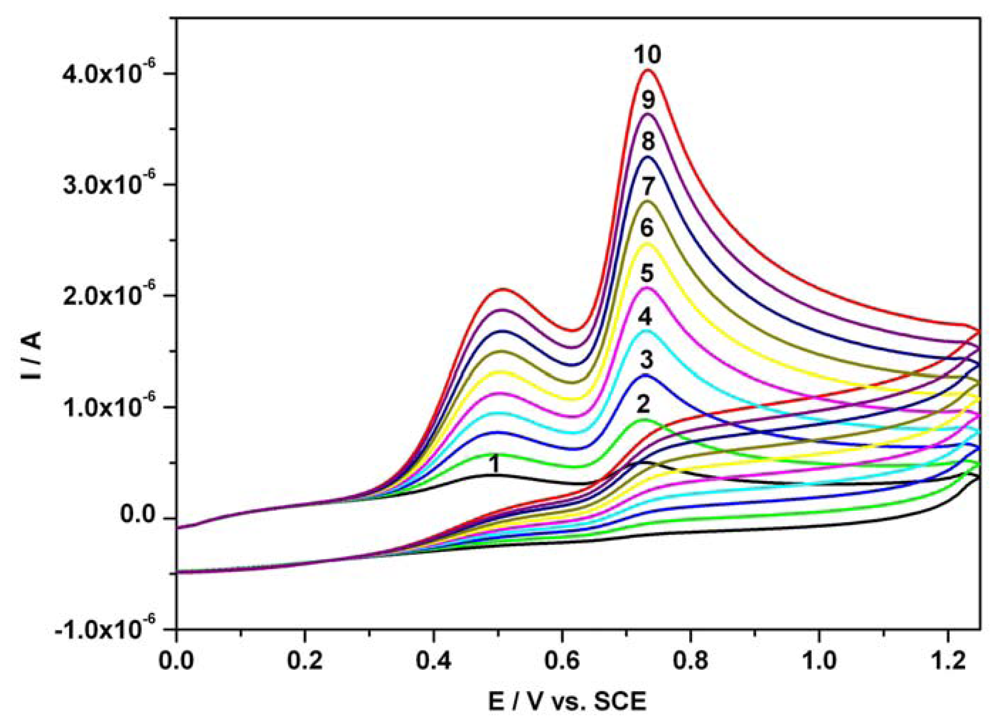
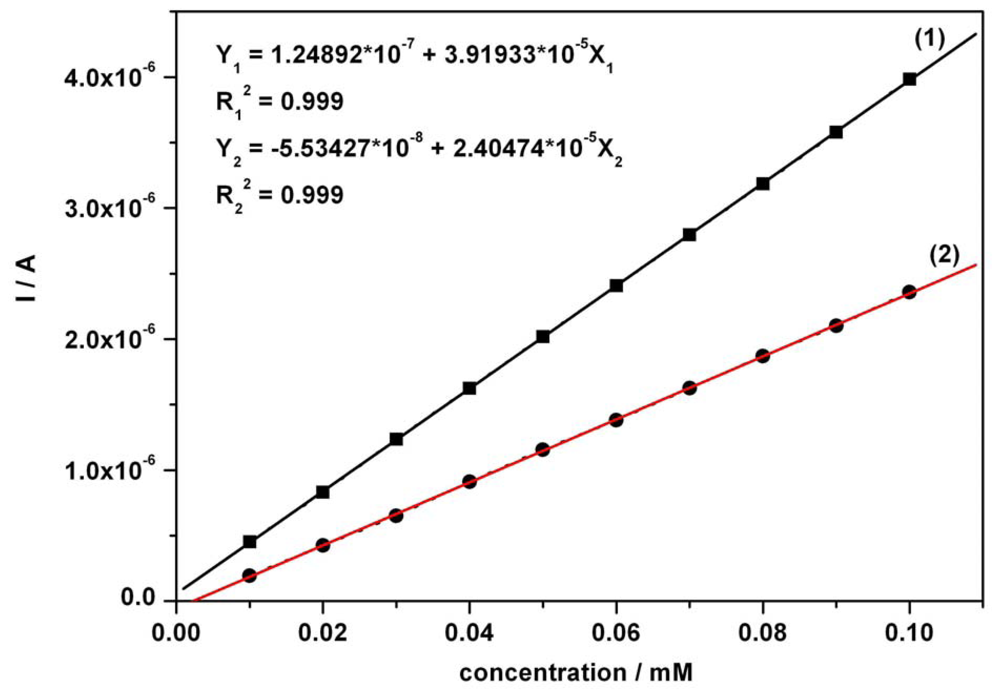
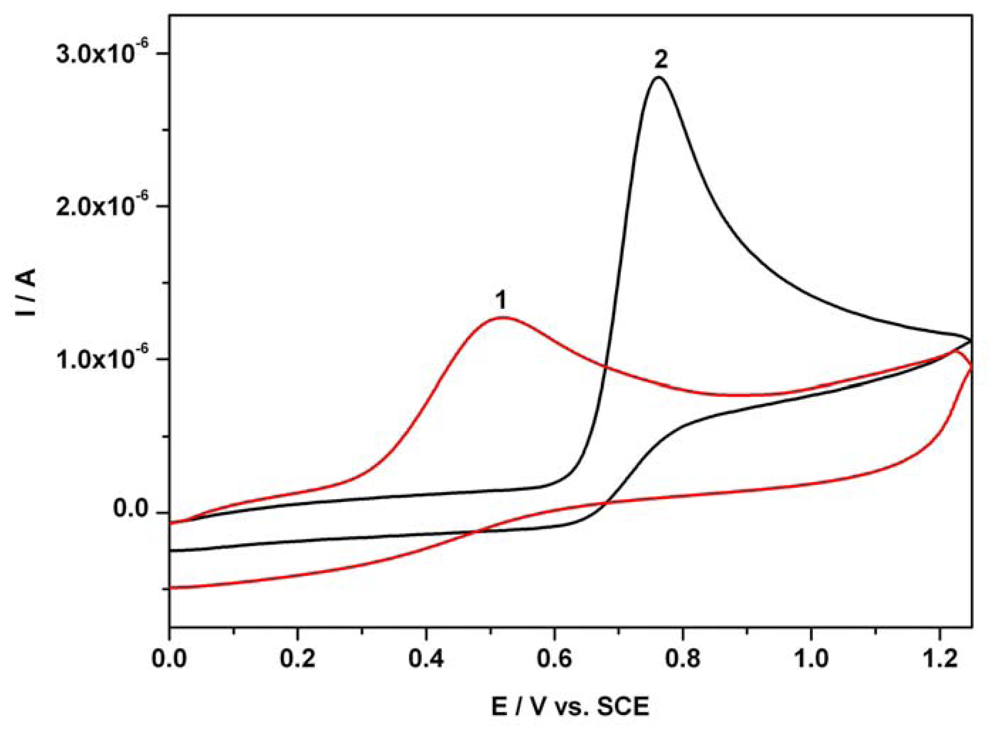
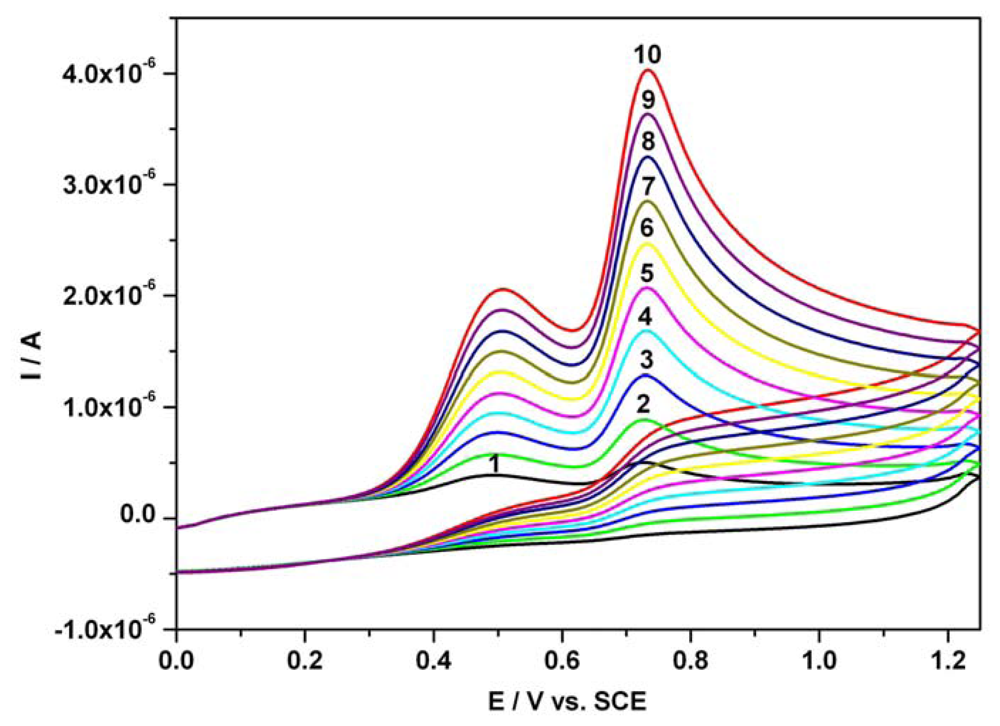
3.1. Cyclic anodic voltammetry data
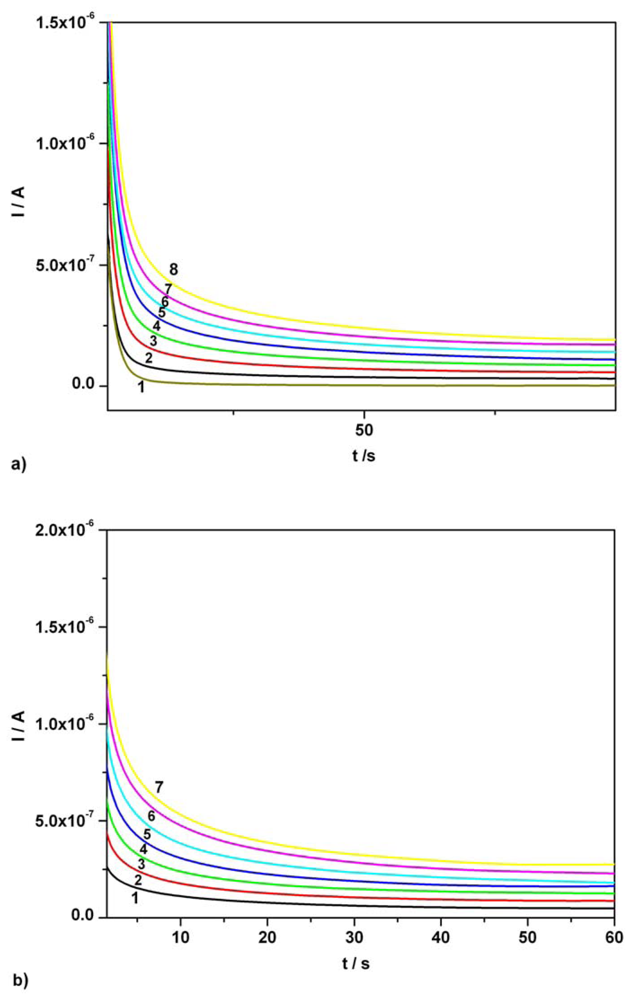
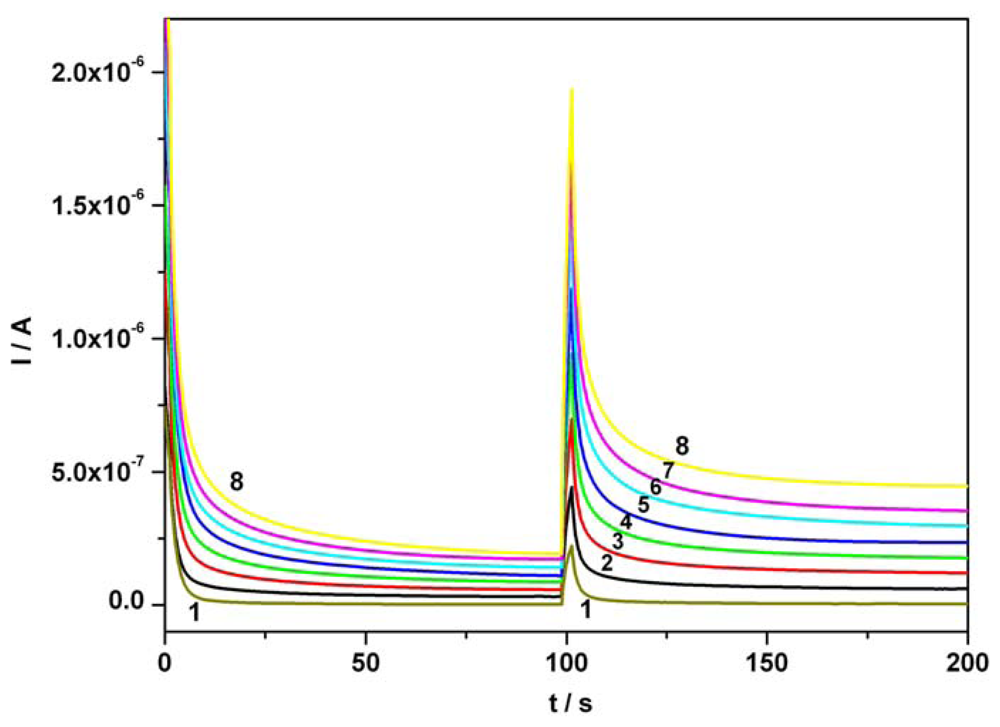
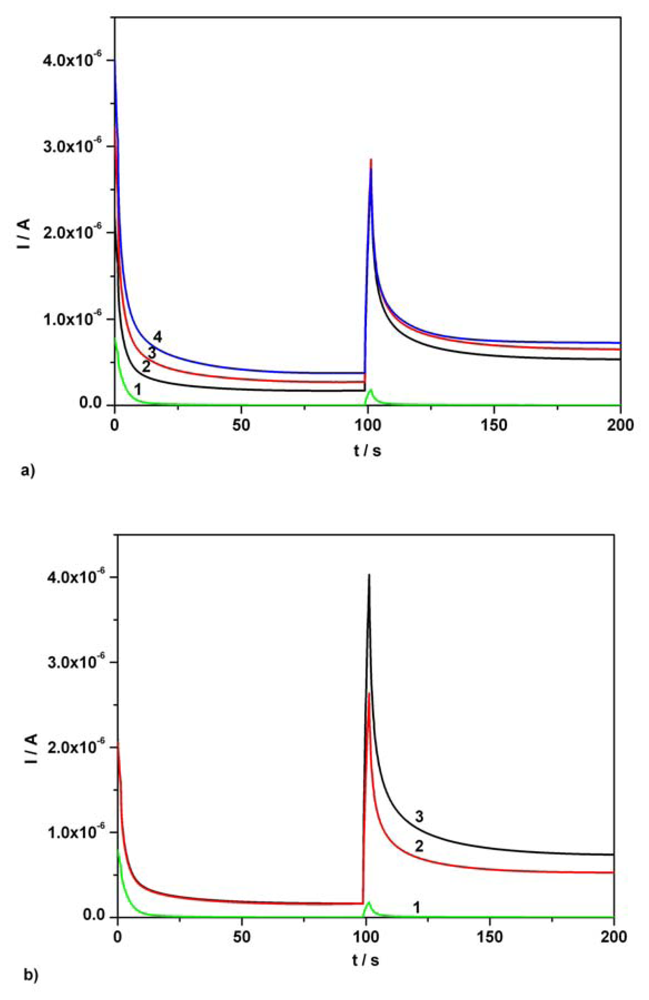
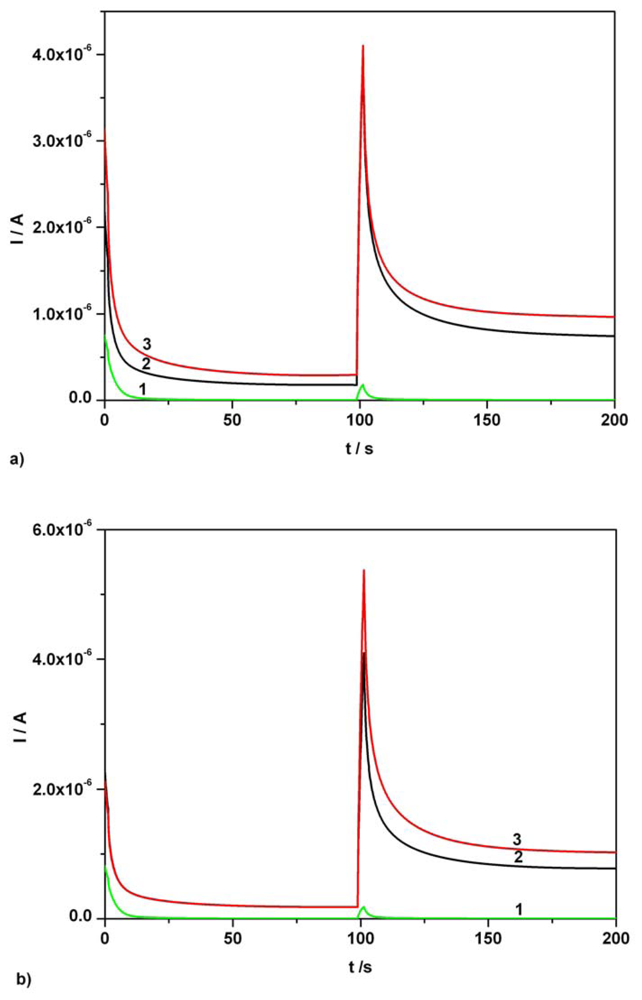
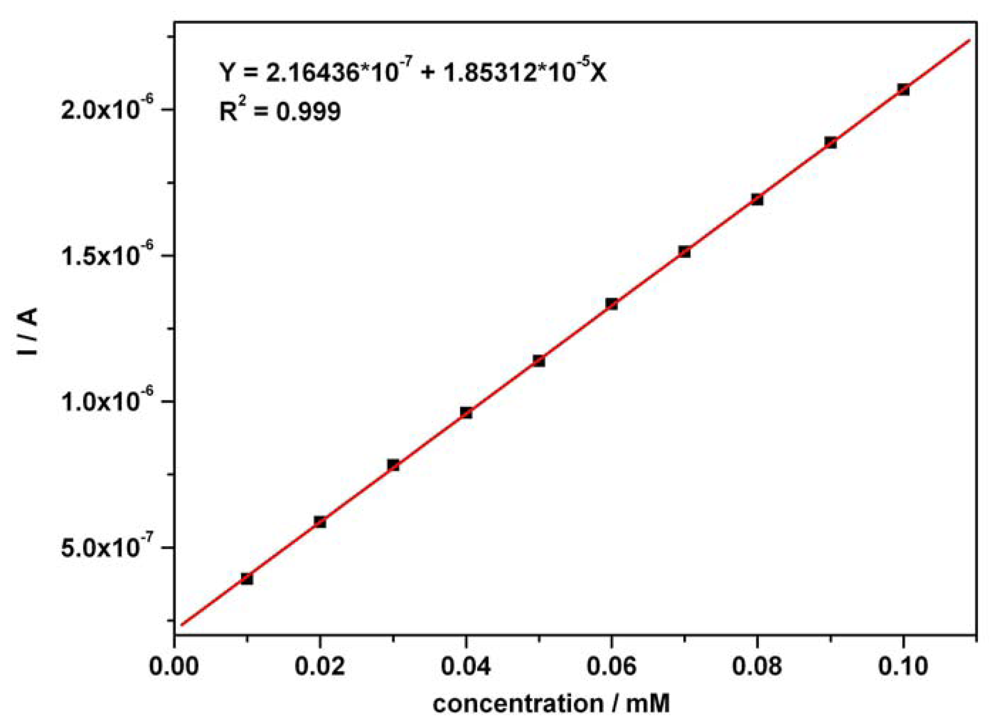
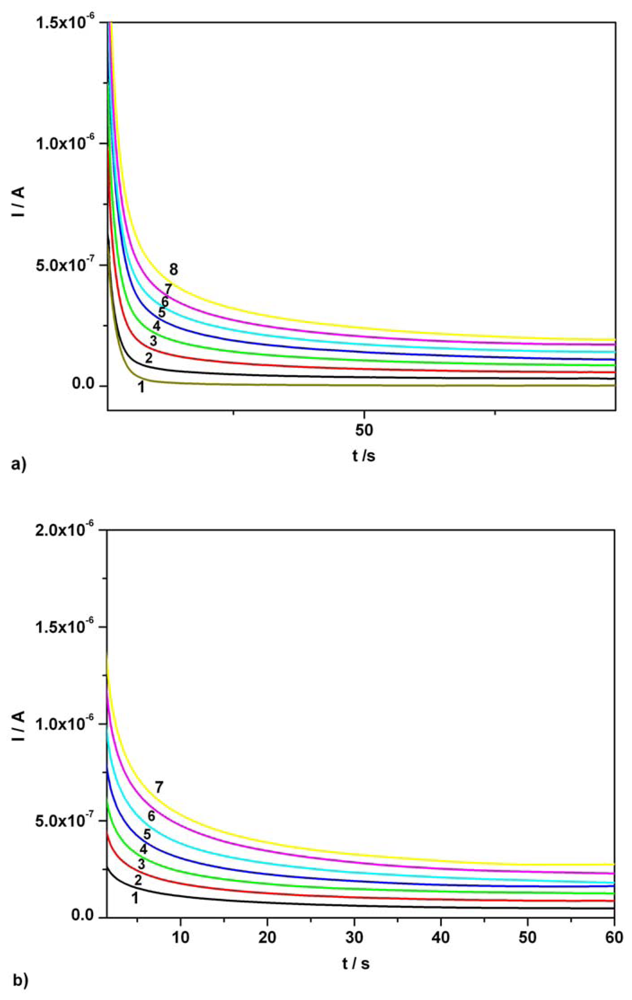
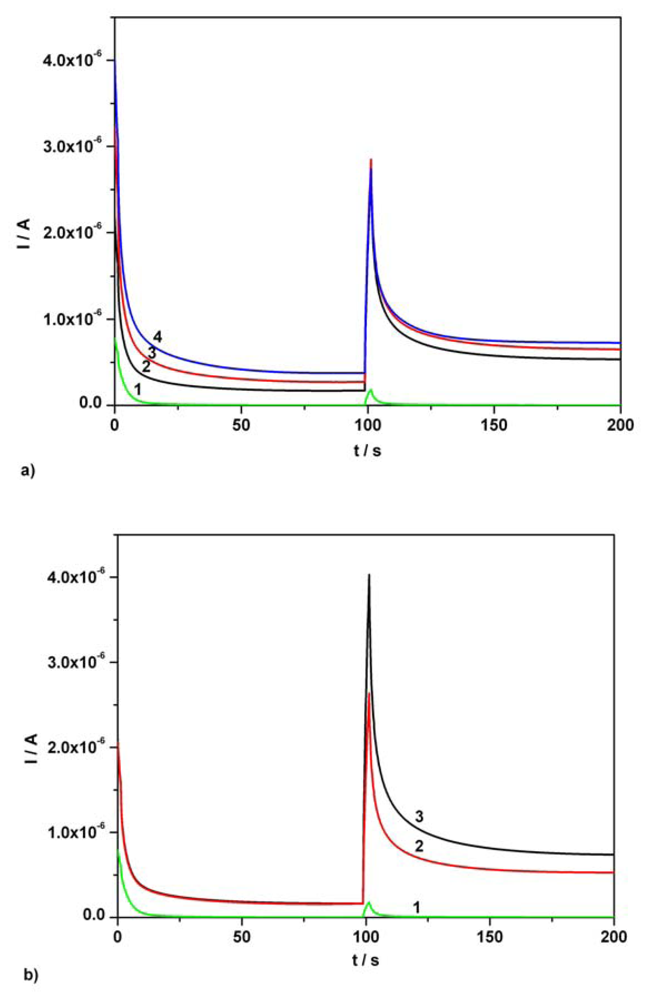
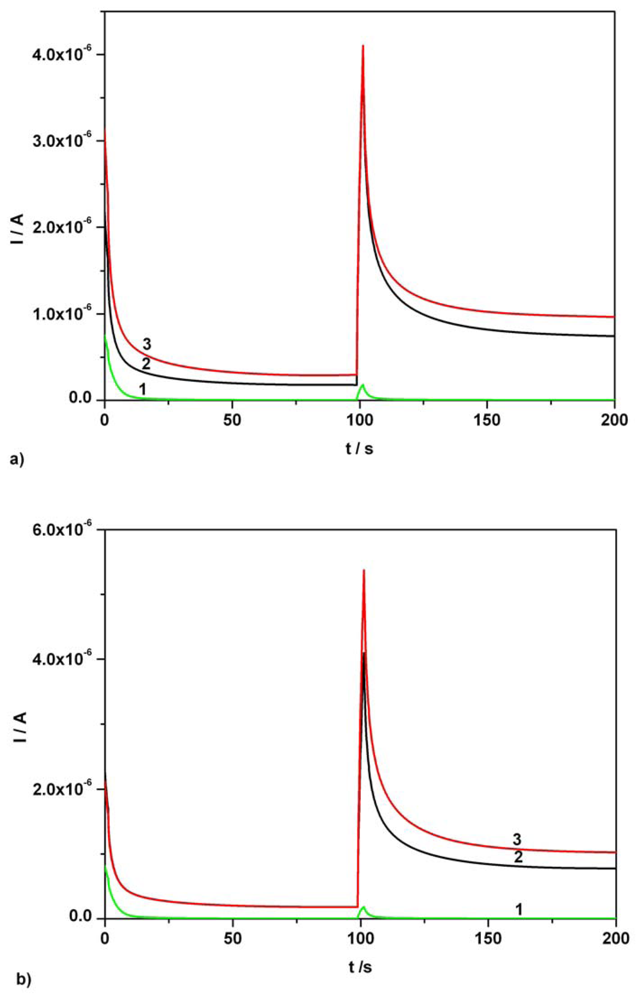
3.2. Chronoamperometry data
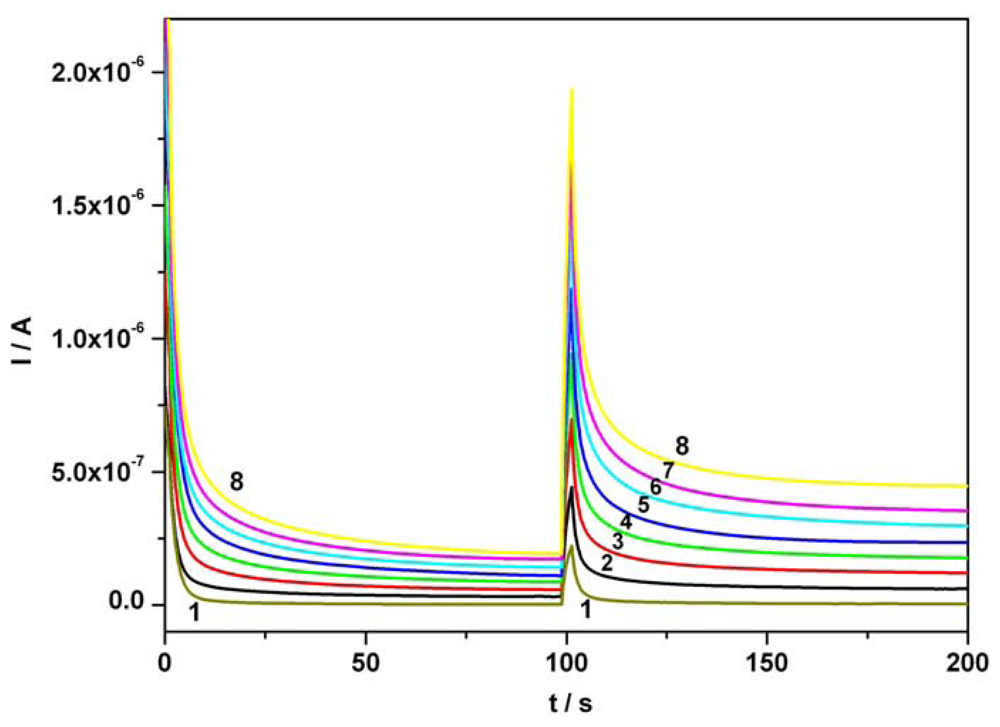
3.3. Chronoamperometric sequential and simultaneous determination of AA and AC from real samples
4. Conclusions
Acknowledgments
References and Notes
- Ojani, R.; Raoof, J.-B.; Zamani, S. Electrochemical Behaviour of Chloranil Chemically Modified Carbon Paste Electrode. Application to the Electrocatalytic Determination of Ascorbic Acid. Electroanalysis 2005, 17, 1740–1745. [Google Scholar]
- Kleszczewski, T.; Kleszczewska, E. Flow Injection Spectrophotometric Determination of L-ascorbic acid in Biological Matters. J. Pharm. Biomed. Anal. 2002, 29, 755–759. [Google Scholar]
- O'Connell, P.J.; Gormally, C.; Pravda, M.; Guilbault, G.G. Development of an Amperometric L-ascorbic acid (Vitamin C) Sensor Based on Electropolymerised Aniline for Pharmaceutical and Food Analysis. Anal. Chim. Acta. 2001, 431, 239–247. [Google Scholar]
- Sabzi, R.E.; Pournaghi-Azar, M.H. Electrocatalytic Determination of Ascorbic Acid on a Glassy Carbon Electrode Chemically Modified with Cobalt Pentacyanonitrosylferrate. Anal. Sci. 2005, 21, 689–692. [Google Scholar]
- Zeng, W.; Martinuzzi, F.; MacGregor, A. Development and Application of a Novel UV Method for the Analysis of Ascorbic Acid. J. Pharm. Biomed. Anal. 2005, 36, 1107–1111. [Google Scholar]
- Săndulescu, R.; Mirel, S.; Oprean, R. The Development of Spectrophotometric and Electroanalytical Methods for Ascorbic Acid and Acetaminophen and Their Applications in the Analysis of Effervescent Dosage Forms. J. Pharm. Biomed. Anal. 2000, 23, 77–87. [Google Scholar]
- Zhang, L.; Sun, Y.; Lin, X. Separation of Anodic Peaks of Ascorbic Acid and Dopamine at an α-alanine Covalently Modified Glassy Carbon Electrode. Analyst 2001, 126, 1760–1763. [Google Scholar]
- Ramesh, P.; Sampath, S. Selective Determination of Uric Acid in Presence of Ascorbic Acid and Dopamine at Neutral pH Using Exfoliated Graphite Electrodes. Electroanalysis 2004, 16, 866–869. [Google Scholar]
- Arvand, M.; Sohrabnezhad, Sh.; Mousavi, M.F.; Shamsipur, M.; Zanjanchi, M.A. Electrochemical Study of Methylene Blue Incorporated into Modernite Type Zeolite and Its Application for Amperometric Determination of Ascorbic Acid in Real Samples. Anal. Chim. Acta 2003, 491, 193–201. [Google Scholar]
- Medina, A.R.; De Córdova, M.L.F.; Diaz, A.M. A Rapid and Selective Solid-Phase UV Spectrophotometric Method for Determination of Ascorbic Acid in Pharmaceutical Preparations and Urine. J. Pharm. Biomed. Anal. 1999, 20, 247–254. [Google Scholar]
- Senthil Kumar, S.; Sriman Narayanan, S. Amperometric Sensor for the Determination of Ascorbic Acid Based on Cobalt Hexacyanoferrate Modified Electrode Fabricated Through a New Route. Chem. Pharm. Bull. 2006, 54, 963–967. [Google Scholar]
- Ijeri, V.S.; Algarra, M.; Martins, A. Electrocatalytic Determination of Vitamin C Using Calixarene Modified Carbon Paste Electrodes. Electroanalysis 2004, 16, 2082–2086. [Google Scholar]
- Wantz, F.; Banks, C.E.; Compton, R.G. Direct Oxidation of Ascorbic Acid at an Edge Plane Pyrolitic Graphite Electrode: a Comparaison of the Electroanalytical Response with Other Carbon Electrodes. Electroanalysis 2005, 17, 1529–1533. [Google Scholar]
- Casella, I.G. Electrooxidation of Ascorbic Acid on the Dispersed Platinum Glassy Carbon Electrode and its Amperometric Determination in Flow Injection Analysis. Electroanalysis 1996, 8, 128–134. [Google Scholar]
- Park, S.-G.; Park, J.-E.; Cho, E.-I.; Hwang, J.-H.; Ohsaka, T. Electrochemical Detection of Ascorbic Acid and Serotonin at a Boron-Doped Diamond Electrode Modified with Poly (N, N-dimethylalanine). Res. Chem. Intermed. 2006, 32, 595–601. [Google Scholar]
- Roy, P.R.; Saha, M.S.; Okajima, T.; Park, S.-G.; Fujishima, A.; Ohsaka, T. Selective Detection of Dopamine and Its Metabolite, DOPAC, in the Presence of Ascorbic Acid Using Diamond Electrode Modified by the Polymer Film. Electroanalysis 2004, 16, 1777–1784. [Google Scholar]
- Van Staden, J.F.; Tsanwani, M. Determination of Paracetamol in Pharmaceutical Formulations Using a Sequential Injection System. Talanta 2002, 58, 1095–1101. [Google Scholar]
- Espinosa Bosch, M.; Ruiz Sánchez, A.J.; Sánchez Rojas, F.; Bosch Ojeda, C. Determination of Paracetamol: Historical Evolution. J. Pharm. Biomed. Anal. 2006, 42, 291–321. [Google Scholar]
- Goyal, R.N.; Gupta, V.K.; Oyama, M.; Bachheti, N. Differential Pulse Voltammetric Determination of Paracetamol at Nanogold Modified Indium Tin Oxide Electrode. Electrochem. Commun. 2005, 7, 803–807. [Google Scholar]
- Bose, D.; Durgbanshi, A.; Martinavarro-Dominguez, A.; Capella-Peiró, M.E.; Carda-Broch, S.; Esteve-Romero, J.S.; Gil-Agusti, M.T.J. Rapid Determination of Acetaminophen in Physiological Fluids by Liquid Chromatography Using SDS Mobile Phase and ED Detection. J. Chromatogr. Sci. 2005, 43, 313–318. [Google Scholar]
- Li, C.; Zhan, G.; Yang, Q.; Lu, J. Electrochemical Investigation of Acetaminophen with a Carbon Nano-Tube Composite Film Electrode. Bull. Korean Chem. Soc. 2006, 27, 1854–1860. [Google Scholar]
- Guzy, J.; Chovanova, Z.; Marekova, M.; Chavkova, Z.; Tomečkova, V.; Mojžišová, G.; Kušnir, J. Effect of Quercetin on Paracetamol-Induced Rat Liver Mitochondria Dysfunction. Biologia Bratislava 2004, 59, 399–403. [Google Scholar]
- Raghavendran, H.R.B.; Sathivel, A.; Devaki, T. Hepatoprotective Nature of Seaweed Alchoholic Extract on Acetaminophen Induced Hepatic Oxidative Stress. J. Health Sci. 2004, 50, 42–46. [Google Scholar]
- Grundmann, U.; Wörnle, C.; Biedler, A.; Kreuer, S.; Wrobel, M.; Wilhelm, W. The Efficacy of the Non-Opioid Analgesics Parecoxib, Paracetamol and Metamizol for Postoperative Pain Relief after Lumbar Microdiscectomy. Anesth. Analg. 2006, 103, 217–222. [Google Scholar]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S. K.; Levine, M. Vitamin C as an Antioxidant: Evaluation of its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar]
- Brillas, E.; Sirés, I.; Arias, C.; Cabot, P.L.; Centellas, F.; Rodriguez, R.M.; Garrido, J.A. Mineralization of Paracetamol in Aqueous Medium by Anodic Oxidation with a Boron-Doped Diamond Electrode. Chemosphere 2005, 58, 399–406. [Google Scholar]
- De Carvalho, R.M.; Freire, R.S.; Rath, S.; Kubota, L.T. Effects of EDTA on Signal Stability During Electrochemical Detection of Acetaminophen. J. Pharm. Biomed. Anal. 2004, 34, 871–878. [Google Scholar]
- Sena, M.M.; Poppi, R.J. N-way PLS Applied to Simultaneous Spectrophotometric Determination of Acetylsalicylic Acid, Paracetamol and Caffeine. J. Pharm. Biomed. Anal. 2004, 34, 27–34. [Google Scholar]
- Peng, W.; Li, T.; Li, H.; Wang, E. Direct Injection of Urine and Determination of Acetaminophen by Micellar Liquid Chromatography with Wall-Jet Cell/Carbon Fibre Microelectrode. Anal. Chim. Acta 1994, 298, 415–421. [Google Scholar]
- Navarro, I.; Gonzalez-Arjona, D.; Roldan, E.; Rueda, M. Determination of Paracetamol in Tablets and Blood Plasma by Differential Pulse Voltammetry. J. Pharm. Biomed. Anal. 1988, 6, 969–976. [Google Scholar]
- Lau, O.-W.; Luk, Sh.-F.; Cheung, Y.-M. Simultaneous Determination of Ascorbic Acid, Caffeine and Paracetamol in Drug Formulations by Differential Pulse Voltammetry Using a Glassy Carbon Electrode. Analyst 1989, 114, 1047–1051. [Google Scholar]
- Silva, M.L.S.; Garcia, M.B.Q.; Lima, J.L.F.C.; Barrado, E. Flow System with Electrochemical Detection for Determination of Paracetamol in Pharmaceutical Preparations. Portug. Electrochim. Acta 2006, 24, 261–271. [Google Scholar]
- Quintino, M.S.M.; Araki, K.; Toma, H.E.; Angnes, L. Batch Injection Analysis Utilizing Modified Electrodes with Tetraruthenated Porphyrin Films for Acetaminophen Quantification. Electroanalysis 2002, 14, 1629–1634. [Google Scholar]
- Wang, C.; Hu, X.; Leng, Z.; Yang, G.; Jin, G. Differential Pulse Voltammetry for Determination of Paracetamol at a Pumice Mixed Carbon Paste Electrode. Anal. Lett. 2001, 34, 2747–2759. [Google Scholar]
- Masawat, P.; Liawruangrath, S.; Vaneesorn, Y.; Liawruangrath, B. Design and Fabrication of Low–Cost Flow–Through Cell for the Determination of Acetaminophen in Pharmaceutical Formulation by Flow Injection Cyclic Voltammetry. Talanta 2002, 58, 1221–1234. [Google Scholar]
- Wang, C.; Li, C.; Wang, F.; Wang, Ch. Covalent Modification of Glassy Carbon Electrode with L-cysteine for the Determination of Acetaminophen. Microchim. Acta 2006, 155, 365–371. [Google Scholar]
- Zen, J-M.; Ting, Y-S. Simultaneous Determination of Caffeine and Acetaminophen in Drug Formulations by Square-Wave Voltammetry Using a Chemically Modified Electrode. Anal. Chim. Acta 1997, 342, 175–180. [Google Scholar]
- Kachoosangi, R.T.; Compton, R.G. A Simple Electroanalytical Methodology for the Simultaneous Determination of Dopamine, Serotonin and Ascorbic Acid Using an Unmodified Edge Plane Pyrolitic Graphite Electrode. Anal. Bioanal. Chem. 2007, 387, 2793–2800. [Google Scholar]
- Felix, F.S.; Brett, C.M.A.; Angnes, L. Carbon Film Resistor Electrode for Amperometric Determination of Acetaminophen in Pharmaceutical Formulations. J. Pharm. Biomed. Anal. 2007, 43, 1622–1627. [Google Scholar]
- Goyal, R.N.; Singh, S.P. Voltammetric Determination of Paracetamol at C60-Modified Carbon Electrode. Electrochim. Acta 2006, 51, 3008–3012. [Google Scholar]
- Wangfuengkanagul, N.; Chailapakul, O. Electrochemical Analysis of Acetaminophen Using a Boron-Doped Diamond Thin Film Electrode Applied to Flow Injection System. J. Pharm. Biomed. Anal. 2002, 28, 841–847. [Google Scholar]
- Coe, S.E.; Sussmann, R.S. Optical, Thermal and Mechanical Properties of CVD Diamond. Diamond Relat. Mater. 2000, 9, 1726–1729. [Google Scholar]
- Witek, M.; Wang, J.; Stotter, J.; Hupert, M.; Haymond, S.; Sonthalia, P.; Swain, G.M.; Zak, J.K.; Chen, Q.; Gruen, D.M.; Butler, J.E.; Kobashi, K.; Tachibana, T. Summary of Recent Progress with Diamond Electrodes in Electroanalysis, Spectroelectrochemistry and Electrocatalysis. J. Wide Bandgap Mater. 2001, 8, 171–188. [Google Scholar]
- Pleskov, Yu.V. Electrochemistry of Diamond. Russ. J. Electrochem. 2002, 38, 1275–1291. [Google Scholar]
- Granger, M.C.; Xu, J.; Strojek, J.W.; Swain, G.M. Polycrystalline Diamond Electrodes: Basic Properties and Applications as Amperometric Detector in Flow Injection Analysis and Liquid. Anal. Chim. Acta 1999, 397, 145–161. [Google Scholar]
- Compton, R.G.; Foord, J.S.; Marken, F. Electroanalysis at Diamond-Like and Doped-Diamond Electrodes. Electroanalysis 2003, 15, 1349–1363. [Google Scholar]
- Kraft, A. Doped Diamond: a Compact Review on a Versatile Electrode Material. Int. J. Electrochem. Sci. 2007, 2, 355–385. [Google Scholar]
- Komatsu, M.; Fujishima, A. Detection of Ascorbic Acid in an Ethanol-Water Mixed Solution on a Conductive Diamond Electrode. Bull. Chem. Soc. Jpn. 2003, 76, 927–933. [Google Scholar]
- Radovan, C.; Manea, F. Determination of Sodium Diethyldithiocarbamate in Water by Anodic Voltammetry Using a Boron-Doped Diamond Electrode. Electroanalysis 2007, 19, 91–95. [Google Scholar]
- Ni, Y.; Wang, Y.; Kokot, S. Differential Pulse Stripping Voltammetric Determination of Paracetamol and Phenobarbital in Pharmaceuticals Assisted by Chemometrics. Anal. Lett. 2004, 37, 3219–3235. [Google Scholar]
- Van Benschoten, J.J.; Lewis, J.Y.; Heineman, W.R.; Roston, D.A.; Kissinger, P.T. Cyclic Voltammetry Experiment. J. Chem. Ed. 1983, 60, 772–776. [Google Scholar]
- Miner, D.J.; Rice, J.R.; Riggin, R.M.; Kissinger, P.T. Voltammetry of Acetaminophen and Its Metabolits. Anal. Chem. 1981, 53, 2258–2263. [Google Scholar]
- Radovan, C.; Cofan, C.; Cinghita, D. Simultaneous Determination of Acetaminophen and Ascorbic Acid at an Unmodified Boron-Doped Diamond Electrode by Differential Pulse Voltammetry in Buffered Media. Electroanalysis 2008. article online in advance of print, published online: Apr 1 2008. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Figure | Concentration range (mM) | Regression equation of linear calibration plot | Sensitivity (μA/mM) | R2 | LOD (μM) |
|---|---|---|---|---|---|---|
| Ascorbic acid | Fig.5a [*]1 | 0.01-0.07 | I = 3.332C + 0.0002 | 3.332 | 0.999 | 0.80 |
| Ascorbic acid | Fig.5a [*]2 | 0.01-0.07 | I = 3.343C + 0.009 | 3.343 | 0.995 | 0.86 |
| Acetaminophen | Fig. 5b [*] | 0.01-0.07 | I = 3.699C + 0.001 | 3.699 | 0.997 | 0.97 |
| Ascorbic acid | Fig. 6 [**] | 0.01-0.07 | I = 3.343C + 0.009 | 3.343 | 0.995 | 0.86 |
| Acetaminophen | Fig. 6 [**] | 0.01-0.07 | I = 3.806C - 0.006 | 3.806 | 0.994 | 1.42 |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cofan, C.; Radovan, C. Simultaneous Chronoamperometric Sensing of Ascorbic Acid and Acetaminophen at a Boron-Doped Diamond Electrode. Sensors 2008, 8, 3952-3969. https://doi.org/10.3390/s8063952
Cofan C, Radovan C. Simultaneous Chronoamperometric Sensing of Ascorbic Acid and Acetaminophen at a Boron-Doped Diamond Electrode. Sensors. 2008; 8(6):3952-3969. https://doi.org/10.3390/s8063952
Chicago/Turabian StyleCofan, Codruţa, and Ciprian Radovan. 2008. "Simultaneous Chronoamperometric Sensing of Ascorbic Acid and Acetaminophen at a Boron-Doped Diamond Electrode" Sensors 8, no. 6: 3952-3969. https://doi.org/10.3390/s8063952