Electrochemical Biosensors - Sensor Principles and Architectures

1
Laboratory of Biosensors and Bioelectronics, Institute for Biomedical Engineering, ETH Zurich, Gloriastrasse 35, 8092 Zurich, Switzerland
2
Laboratory for Surface Science and Technology, Department of Materials, ETH Zurich, Wolfgang-Pauli-Strasse 10, 8093 Zurich, Switzerland
*
Author to whom correspondence should be addressed.
Sensors 2008, 8(3), 1400-1458; https://doi.org/10.3390/s80314000
Submission received: 23 January 2008
/
Accepted: 28 January 2008
/
Published: 7 March 2008
(This article belongs to the Special Issue Utilization of Electrochemical Sensors and Biosensors in Biochemistry and Molecular Biology)
Abstract
:Quantification of biological or biochemical processes are of utmost importance for medical, biological and biotechnological applications. However, converting the biological information to an easily processed electronic signal is challenging due to the complexity of connecting an electronic device directly to a biological environment. Electrochemical biosensors provide an attractive means to analyze the content of a biological sample due to the direct conversion of a biological event to an electronic signal. Over the past decades several sensing concepts and related devices have been developed. In this review, the most common traditional techniques, such as cyclic voltammetry, chronoamperometry, chronopotentiometry, impedance spectroscopy, and various field-effect transistor based methods are presented along with selected promising novel approaches, such as nanowire or magnetic nanoparticle-based biosensing. Additional measurement techniques, which have been shown useful in combination with electrochemical detection, are also summarized, such as the electrochemical versions of surface plasmon resonance, optical waveguide lightmode spectroscopy, ellipsometry, quartz crystal microbalance, and scanning probe microscopy.
The signal transduction and the general performance of electrochemical sensors are often determined by the surface architectures that connect the sensing element to the biological sample at the nanometer scale. The most common surface modification techniques, the various electrochemical transduction mechanisms, and the choice of the recognition receptor molecules all influence the ultimate sensitivity of the sensor. New nanotechnology-based approaches, such as the use of engineered ion-channels in lipid bilayers, the encapsulation of enzymes into vesicles, polymersomes, or polyelectrolyte capsules provide additional possibilities for signal amplification.
In particular, this review highlights the importance of the precise control over the delicate interplay between surface nano-architectures, surface functionalization and the chosen sensor transducer principle, as well as the usefulness of complementary characterization tools to interpret and to optimize the sensor response.
1. Introduction
Biosensor-related research has experienced explosive growth over the last two decades. A biosensor is generally defined as an analytical device which converts a biological response into a quantifiable and processable signal [1]. Figure 1 shows schematically the parts comprising a typical biosensor: a) bioreceptors that specifically bind to the analyte; b) an interface architecture where a specific biological event takes place and gives rise to a signal picked up by c) the transducer element; the transducer signal (which could be anything from the in-coupling angle of a laser beam to the current produced at an electrode) is converted to an electronic signal and amplified by a detector circuit using the appropriate reference and sent for processing by, e.g., d) computer software to be converted to a meaningful physical parameter describing the process being investigated; finally, the resulting quantity has to be presented through e) an interface to the human operator. Biosensors can be applied to a large variety of samples including body fluids, food samples, cell cultures and be used to analyze environmental samples.
In order to construct a successful biosensor for the non-specialist market a number of conditions must be met:
- The biocatalyst must be highly specific for the purpose of the analysis, be stable under normal storage conditions and show a low variation between assays.
- The reaction should be as independent as manageable of such physical parameters as stirring, pH and temperature. This will allow analysis of samples with minimal pre-treatment. If the reaction involves cofactors or coenzymes these should, preferably, also be co-immobilized with the enzyme.
- The response should be accurate, precise, reproducible and linear over the concentration range of interest, without dilution or concentration. It should also be free from electrical or other transducer induced noise.
- If the biosensor is to be used for invasive monitoring in clinical situations, the probe must be tiny and biocompatible, having no toxic or antigenic effects. Furthermore, the biosensor should not be prone to inactivation or proteolysis.
- For rapid measurements of analytes from human samples it is desirable that the biosensor can provide real-time analysis.
- The complete biosensor should be cheap, small, portable and capable of being used by semi-skilled operators.
Designed for the purpose, biosensors are generally highly selective due to the possibility to tailor the specific interaction of compounds by immobilizing biological recognition elements on the sensor substrate that have a specific binding affinity to the desired molecule [4]. Typical recognition elements used in biosensors are: enzymes, nucleic acids, antibodies, whole cells, and receptors. Of these, enzymes are among the most common [3]. To fully exploit the specific interaction through biorecognition, the surface architecture of the sensor also must suppress any non-specific interaction. A tremendous research effort has been invested to find surface modifications with specific interaction capabilities over prolonged periods of time in biological fluids [5].
Today, a multitude of instruments referred to as biosensors can be found in labs around the world and there is a growing number of biosensors being used as diagnostic tools in point-of-care testing, but the realization of cheap handheld devices is almost limited to one well-known example: the glucose sensor [6]. In many cases the main limitation in realizing point-of-care testing/sensing devices is the ability to miniaturize the transduction principle and the lack of a cost-effective production method. Thus, they have to be confined to expert users of high-cost equipment in a lab environment and cannot be used e.g. by patients themselves or doctors in the field.
The whole area of biosensors started with the introduction of the first generation glucose oxidase (GOx) biosensor in 1962 [7]. The GOx sensor is still the most widely used, although many improvements (generations) have been added since the 1960's [8]. As exemplified by the glucose sensor, electrochemical biosensors do not suffer the drawback of high sensor setup complexity and cost. This is due to their close link to developments in low-cost production of microelectronic circuits and their easy interface with normal electronic read-out and processing. Other inherent advantages of electrochemical biosensors are their robustness, easy miniaturization, excellent detection limits, also with small analyte volumes, and ability to be used in turbid biofluids with optically absorbing and fluorescing compounds [9, 10]. However, several aspects could be considered to have held back the emergence of additional breakthrough applications built on electrochemical biosensing.
Electrochemical biosensors have suffered from a lack of surface architectures allowing high enough sensitivity and unique identification of the response with the desired biochemical event. For example, pH and ionic strength in biofluids can differ significantly, which affects the response of important classes of biosensors such as immunosensors [10]. Thus, there has recently been an increased emphasis on using nanotechnology to shrink the dimensions of electrochemical sensor elements to sizes which can increase the signal-to-noise ratio for processes designed to occur at the interface of the device and to find ways of using, e.g., multiple enzymatic labels to increase the signal per event. The combination of knowledge in bio- and electrochemistry, solid-state and surface physics, bioengineering, integrated circuit silicon technology and data processing offers the possibility of a new generation of highly specific, sensitive, selective and reliable micro (bio-)chemical sensors and sensor arrays addressing these remaining issues [11]. It is thus timely to summarize recent progress in this diverse field and to discuss its future prospects for development.
After introducing the many incarnations of electrochemical biosensors this review will discuss how electrochemistry has been and can be combined with complementary sensor techniques to enhance data interpretation. The latter we believe to be very important to optimize given biosensor designs and also for the increased use of electrochemical sensors to characterize biointerfaces. Emerging devices for electrochemical biosensors inspired by advances in microelectronics and nanotechnology like the biofield effect transistors, nanowires and other “near-molecular scale devices” will be introduced. The last part of the review will address surface architectures and modifications used in electrochemical biosensors to improve on sensitivity and biospecificity, as well as discuss the emergence of new devices from the multi-disciplinary field where nanotechnology, material science and biology converge.
2. Devices
Biosensor-related publications were sparse in the early 20th century. The early era of biosensing research and development was first sparked with the defining paper by Clark [12, 13] and his invention of the oxygen electrode in 1955/56. The subsequent modification of the oxygen electrode led up to another publication in 1962 [7], which reported the development of the first glucose sensor and the enhancement of electrochemical sensors (e.g. polarographic, potentiometric and conductometric) with enzyme-based transducers. Clark's work and the subsequent transfer of his technology to Yellow Spring Instrument Company led to the successful commercial launch of the first dedicated glucose biosensor in 1975 [14].
Since then, various forms of glucose biosensors have been developed, as well as many other sensing technologies and biosensing devices. This section attempts to describe operating principles of electrochemically-based biosensors by reviewing representative devices and their techniques from the aforementioned categories. Although the general topic of this review is electrochemical biosensing devices, a detailed overview is given in this section of various combinations of electrochemical sensing with other well-established sensing techniques and biosensor devices. Therefore, special attention is given to aspects of complementarity techniques and their advantage of independent, simultaneous measurements with (bio-)electrochemistry. A selection of specific biorecognition elements, structural components and various forms of surface architectures will be reviewed in the following section.
2.1. Electrochemical Detection Techniques
In biosensing the measurement of electrical properties for extracting information from biological systems is normally electrochemical in nature, whereby a bioelectrochemical component serves as the main transduction element. Although biosensing devices employ a variety of recognition elements, electrochemical detection techniques use predominantly enzymes. This is mostly due to their specific binding capabilities and biocatalytic activity [3, 10, 11]. Other biorecognition elements are e.g. antibodies, nucleic acids, cells and micro-organisms [3, 4]. An immunosensor uses antibodies, antibody fragments or antigens to monitor binding events in bioelectrochemical reactions. Detailed information about surface architectures and biorecognition elements is provided in Section 3.
Typically in (bio-)electrochemistry, the reaction under investigation would either generate a measurable current (amperometric), a measurable potential or charge accumulation (potentiometric) or measurably alter the conductive properties of a medium (conductometric) between electrodes [4]. References are also made to other types of electrochemical detection techniques, such as impedimetric, which measures impedance (both resistance and reactance) [15, 16], and field-effect, which uses transistor technology to measure current as a result of a potentiometric effect at a gate electrode [2]. All of these measurement techniques will be introduced here, as well as some devices that employ variations of these techniques.
Since reactions are generally detected only in close proximity to the electrode surface, the electrodes themselves play a crucial role in the performance of electrochemical biosensors. Based on the chosen function of a specific electrode, the electrode material, its surface modification or its dimensions greatly influence its detection ability. Electrochemical sensing usually requires a reference electrode, a counter or auxiliary electrode and a working electrode, also known as the sensing or redox electrode. The reference electrode, commonly made from Ag/AgCl, is kept at a distance from the reaction site in order to maintain a known and stable potential. The working electrode serves as the transduction element in the biochemical reaction, while the counter electrode establishes a connection to the electrolytic solution so that a current can be applied to the working electrode. These electrodes should be both conductive and chemically stable. Therefore, platinum, gold, carbon (e.g. graphite) and silicon compounds are commonly used, depending on the analyte [4, 17]. An excellent summary of the characteristics, such as the varying detection limits, of electrochemical biosensors is provided in a review by Mehrvar et al. [18].
Synergies in nanotechnology and bioelectronics have revealed new possibilities to miniaturize and to optimize existing microscale devices at the nanoscale. It is becoming possible to more accurately measure specific electrical properties in combination with various electrochemical transducers. The higher surface-to-volume ratio of nano-objects makes their electrical properties increasingly susceptible to external influences, especially as these structures continue to shrink toward the atomic limit. Since the nanometer dimensions of these objects are comparable to the size of the target biomolecules, higher measurement sensitivity may result [19], and sensitivity may also increase due to higher capture efficiency [20]. Nanostructures already represent important new components in recently developed electrochemical biosensors, such as the use of nanoparticles as electrochemical labels for DNA sensing [21, 22]. Nanowires, carbon nanotubes, nanoparticles and nanorods are merely some of the familiar objects that are emerging as candidates to become crucial elements of future bioelectronic devices and biosensors [23, 24]. The use of nanowires in biosensing is reviewed in Section 2.2.
Amperometric devices are a type of electrochemical sensor, since they continuously measure current resulting from the oxidation or reduction of an electroactive species in a biochemical reaction [3, 6]. Clark oxygen electrodes perhaps represent the basis for the simplest forms of amperometric biosensors, where a current is produced in proportion to the oxygen concentration. This is measured by the reduction of oxygen at a platinum working electrode in reference to a Ag/AgCl reference electrode at a given potential [4]. Typically, the current is measured at a constant potential and this is referred to as amperometry. If a current is measured during controlled variations of the potential, this is referred to as voltammetry. Furthermore, the peak value of the current measured over a linear potential range is directly proportional to the bulk concentration of the analyte, i.e. the electroactive species [2, 3, 4]. Since not all protein analytes are intrinsically capable to serve as redox partners in electrochemical reactions, these devices use mostly mediated electrochemistry for the electrochemical reaction of the analyte at the working electrode [4, 6]. The role and use of mediators for direct and indirect transduction is reviewed in Section 3.2. Despite the disadvantage of this often indirect sensing system, it is claimed that amperometric devices maintain a sensitivity superior to potentiometric devices [6, 25]. An example of an amperometric device is the aforementioned glucose biosensor, which is based on the amperometric detection of hydrogen peroxide. A very tangible application of amperometry is used in combination with immunosensing techniques to measure levels of the human chorionic gonadotropin β-subunit (β-HCG) in advanced pregnancy testing [26]. Yu et al. have used magnetic microparticles as a means to increase the variety of biocomponent attachment and surface reactions in their amperometric biosensor [27]. Using amperometric methods Kueng et al. developed a biosensing technique for the detection of adenosine-5′-triphosphate (ATP) at physiological pH values with a detection limit of 10 nmol/l [28]. First they co-immobilized glucose oxidase (GOD) and hexokinase (HEX) at electrode surfaces. Upon introducing the electrode to a system containing ATP, glucose was consumed by the enzymatic reaction catalyzed by HEX. This resulted in a decrease in signal in proportion to the ATP concentration. A sample curve for their amperometric measurements is shown in Figure 2.
Potentiometric devices measure the accumulation of a charge potential at the working electrode compared to the reference electrode in an electrochemical cell when zero or no significant current flows between them [3, 4, 10]. In other words, potentiometry provides information about the ion activity in an electrochemical reaction [29]. For potentiometric measurements, the relationship between the concentration and the potential is governed by the Nernst equation, where Ecell represents the observed cell potential at zero current. This is sometimes also refereed to as the electromotive force or EMF
is a constant potential contribution to the cell, R the universal gas constant, T the absolute temperature in degrees Kelvin, n is the charge number of the electrode reaction, F is the Faraday constant and Q is the ratio of ion concentration at the anode to ion concentration at the cathode [30].
The direct determination of the analyte ion concentration with the Nernst equation is referred to as direct potentiometry [3]. The lowest detection limits for potentiometric devices are currently often achieved with ion-selective electrodes (ISE). Therefore, by definition the detection limit is analyte specific and current devices have limits of detection in ranges between 10−8 to 10−11 M. Potentiometric sensors prove suitable for measuring low concentrations in tiny sample volumes, since they ideally offer the benefit of not chemically influencing a sample. The variety of ions, for which low detection limits are possible, is currently quite limited and missing such important analytes as: nickel, manganese, mercury and arsenate ions. Detailed information about potentiometry and their limit of detection (LOD) is provided in the review by Bakker et al. [29].
Potentiometry is also used as an alternative method to electrically determine the point in a (bio)chemical reaction at which equal quantities of opposing solutions reach a state of equilibrium (e.g. 0.1 mol HCl and 0.1 mol NaOH). This is known as measuring a titration end-point with the technique known as potentiometric titration. By performing a titration at constant or zero current, the end point is identified from the variations in electrode potential, which are caused by changes in solution concentration of the potential-determining ion. Many potentiometric devices are also based on various forms of field-effect transistor (FET) devices to measure pH changes, selective ion concentrations and the kinetics of biocatalytic reactions involving enzymes [31]. Such FET devices are reviewed in greater detail in Section 2.1.4. A further example and novel optical/electrochemical hybrid technique is known as a Light Addressable Potentiometric Sensor (LAPS) [32, 33, 34, 35, 36]. LAPS is a silicon-based detector that takes advantage of the photovoltaic effect to selectively determine the point of measurement. By scanning with a focused light source it is possible to determine the spatially resolved surface potential distribution along the interface of the sample and substrate surfaces [37]. A highlighted example of a potentiometric biosensor from Shishkanova et al. is illustrated in Figure 3. The detection principle relies on the hybridization process of single-stranded oligonucleotides close to a PVC membrane, which induces a measurable redistribution of the ion concentration within intermolecular regions.
Conductometric devices measure the ability of an analyte (e.g. electrolyte solutions) or a medium (e.g. nanowires) to conduct an electrical current between electrodes or reference nodes. Although conductometric devices can be considered as a subset of impedimetric devices, techniques for measuring capacitance changes is reviewed later in combination with electrochemical impedance spectroscopy in Section 2.1.3. In most cases conductometric devices have been strongly associated with enzymes, where the ionic strength, and thus the conductivity, of a solution between two electrodes changes as a result of an enzymatic reaction. Thus, conductometric devices can be used to study enzymatic reactions that produce changes in the concentration of charged species in a solution [10]. The variable ionic background of clinical samples and the requirement to measure small conductivity changes in media of high ionic strength limit the applicability of such enzyme-based conductometric devices for biosensing [2]. Another approach is to directly monitor the changes in conductance of an electrode as a result of the immobilization of e.g. enzymes, complementary antibody-antigen pairs, etc. onto the electrode surface.
The construction of multi-analyte conductance biosensors and conductive polymer-based devices has been made possible by the rapid development of semiconductor technology and sensor integration with microelectronic devices, such as FET devices [39, 40]. Now there is an increased interest in conductometric immunosensors in combination with nanostructures, and especially nanowires, for biosensing [19, 24]. Although conductometric sensing has not been as extensively implemented as it could be [3], there are examples of successful development of these devices for practical application, such as drug detection in human urine and pollutant detection in environmental testing [41]. Whole cells have also been used as a biorecognition element in conductometric biosensors for toxicity analysis by immobilizing the cells to a transducer of interdigitated electrodes [42].
2.1.1. Cyclic Voltammetry (CV)
Voltammetry belongs to a category of electro-analytical methods, through which information about an analyte is obtained by varying a potential and then measuring the resulting current. It is, therefore, an amperometric technique. Since there are many ways to vary a potential, there are also many forms of voltammetry, such as: polarography (DC Voltage) [43], linear sweep, differential staircase, normal pulse, reverse pulse, differential pulse and more [3, 44]. Cyclic voltammetry is one of the most widely used forms and it is useful to obtain information about the redox potential and electrochemical reaction rates (e.g. the chemical rate constant) of analyte solutions. In this case, the voltage is swept between two values at a fixed rate, however, when the voltage reaches V2 the scan is reversed and the voltage is swept back to V1, as is illustrated in Figure 4i). The scan rate, (V2 − V1)/(t2 − t1), is a critical factor, since the duration of a scan must provide sufficient time to allow for a meaningful chemical reaction to occur. Varying the scan rate, therefore, yields correspondingly varied results [3, 45].
The voltage is measured between the reference electrode and the working electrode, while the current is measured between the working electrode and the counter electrode. The obtained measurements are plotted as current vs. voltage, also known as a voltammogram. As the voltage is increased toward the electrochemical reduction potential of the analyte, the current will also increase. With increasing voltage toward V2 past this reduction potential, the current decreases, having formed a peak as the analyte concentration near the electrode surface diminishes, since the oxidation potential has been exceeded. As the voltage is reversed to complete the scan toward V1, the reaction will begin to reoxidize the product from the initial reaction. This produces an increase in current of opposite polarity as compared to the forward scan, but again decreases, having formed a second peak as the voltage scan continues toward V1. The reverse scan also provides information about the reversibility of a reaction at a given scan rate [46].
The shape of the voltammogram for a given compound depends not only on the scan rate and the electrode surface, which is different after each adsorption step, but can also depend on the catalyst concentration. For example, increasing the concentration of reaction specific enzymes at a given scan rate will result in a higher current compared to the non-catalyzed reaction [45, 48, 49]. Figure 4 provides an overview of selected cyclic voltammograms from the work of Li et al. [47]. They describe a cholesterol biosensor based on the entrapment of cholesterol oxidase in a silicic sol-gel matrix (please refer to Section 3.5.5.) at a Prussian Blue-modified electrode. The electrode modification is aimed to improve the sensitivity and the selectivity of the biosensor. By means of CV they were able to determine a 3σ detection limit of nearly 1.2×10−7 M compared to other reported detection limits on the order of 10−5 M from literature at the time. They further characterized their system using chronoamperometry (see Section 2.1.2.) and obtained a 3σ detection limit of 5.4×10−7 M. CV is not only useful for sensing purposes, but also as a tool to characterize the processes that take place on the sensing electrode. Liu et al. have for example studied the electrochemical characteristics and characterized the modified process of a biosensor for the determination of the H2O2 concentration with reportedly high sensitivity. Horseradish peroxidase (HRP) was immobilized on a platinum disk electrode modified with a gold colloid-cysteine-nafion film in an approach to enhance the direct electron transfer between enzymes and electrodes without chemical mediators [48].
2.1.2. Chronoamperometry and Chronopotentiometry
Another amperometric technique is known as chronoamperometry, where a square-wave potential is applied to the working electrode and a steady state current is measured as a function of time [3, 46]. Alterations in the current arise from the expansion or reduction of the diffusion layer at the electrode. The concept of a diffusion layer was introduced by Nernst and states that there is a stationary thin layer of solution in contact with the electrode surface. The local analyte concentration drops to zero at the electrode surface and diffusion controls the transfer of analyte from the bulk solution of higher concentration to the electrode. This results in a concentration gradient away from electrode surface. In the bulk solution the concentration of analyte is maintained at a value of c0 by convective transfer.
This technique of chronoamperometry is, therefore, closely related to the Cottrell equation [3, 50], which is shown in Equation 2. It defines the current-time dependence for linear diffusion control at a planar electrode. In the Cottrell equation, the current I is dependent on F Faraday's constant, n the number of transferred electrons per molecule, A the electrode area, c0 the analyte concentration, D the diffusion coefficient and time t. From the Cottrell equation the current depends on the rate at which the analyte diffuses to the electrode.
Figure 5 shows complementary chronoamperometric data on the same system analyzed by CV in Figure 4 [47]. Often used in addition to other techniques, such as cyclic voltammetry, for time-dependent system characterization, chronoamperometry is a sensitive technique which does not require the labelling of reactants. One interesting application of chronoamperometry is the real-time monitoring of electrochemical neurotransmission by Michael et al. [51]. They provide a technical description and comparison of the temporal and chemical resolution of amperometry, fast-scan cyclic voltammetry and high-speed chronoamperometry in studies with single cells, brain slices and intact animals. In chronopotentiometry the potential is measured as a function of time in response to a constant or square-wave current. This technique was used by Martins et al. to investigate the adsorption of human serum albumin (HSA) to SAMs [52].
2.1.3. Electrochemical Impedance Spectroscopy (EIS)
The first publication of electrochemical impedance spectroscopy dates to 1975 [53]. Through the application of a small sinusoidally varying potential U, one measures the resulting current response I [44, 54]. By varying the excitation frequency f of the applied potential over a range of frequencies, one can calculate the complex impedance, sum of the real and imaginary impedance components, of the system as a function of the frequency (i.e. angular frequency w). Therefore, EIS combines the analysis of both real and imaginary components of impedance, namely the electrical resistance and reactance, as shown in Equation 3 [49, 55].
EIS possesses the ability to study any intrinsic material property or specific processes that could influence the conductivity/resistivity or capacitivity of an electrochemical system. Therefore, EIS is a useful tool in the development and analysis of materials for biosensor transduction, such as the study of polymer degradation. For example, by coating conductive electrodes with insulating polymers and then introducing this to an enzymatic reaction, the direct or indirect degradation of the polymer coating can be studied. If during the reaction existing free ions are able to penetrate into the polymer, the insulating nature of the polymer would be compromised and result in modified impedance characteristics of the transducing element [56, 57]. A recent example of electrochemical EIS was its use to characterize the fabrication process of a hydrogen peroxide (HRP) biosensor [58].
For electrochemical sensing, impedance techniques are useful to monitor changes in electrical properties arising from biorecognition events at the surfaces of modified electrodes. For example, changes in the conductance of the electrode can be measured as a result of protein immobilization and antibody-antigen reactions on the electrode surface [44, 45, 49, 59]. To additionally measure changes in capacitance with EIS, sometimes referred to as Faradaic impedance spectroscopy, one scheme as proposed by Katz and Willner is to construct an array of interdigitated electrodes and to monitor antibody-antigen reactions in the gaps between the electrodes, as illustrated in Figure 6a. Binding events of complementary antibody-antigen components alter the electrical properties in the gap between two electrodes, where changes in gap conductivity correspond to changes in the real impedance component Zr(w) and changes in the gap capacitance correspond to changes in the imaginary impedance component Zi(w) [44].
Interesting is also the use of impedimetric sensing to detect the binding state of DNA. In an experiment reported by Hang et al. a microarray configuration of interdigitated electrodes was used [60]. This is illustrated in Figure 6b and shows their intent to demonstrate the potential of EIS for low-density DNA microarrays. Single-stranded DNA (ssDNA) was immobilized to a modified surface between the electrodes to act as the biorecognition element. The immobilized ssDNA is associated with counter cations, which initially support ionic conductivity. However, upon hybridization with the complementary DNA strand to the ssDNA there is a reduction in the density of these cations. This reduction further inhibits the free displacement of ions near the surface and leads to a corresponding increase in the overall electrical impedance between the interdigitated electrodes [16, 44, 60, 61, 62, 63].
Finally as a side note, Electrochemical Impedance Spectroscopy also shares the acronym EIS with the sensing system known as an Electrolyte-Insulator Semiconductor [64]. Interestingly and in relation to the topic of DNA biosensing, electrolyte-insulator-semiconductor systems have been used to measure changes in capacitive impedance as a result of DNA hybridization [65]. Detection occurs when the thickness of the dielectric layer increases as a result of target DNA hybridization at the electrode interface. Subsequently, the measured capacitance decreases according to the following equation, where C is the capacitance, ε is the dielectric constant in vacuum, ε0 is the dielectric constant of the layer, A is the active area and d is the thickness of the dielectric layer:
2.1.4. Field-Effect Transistor (FET)
The FET is a type of transistor that uses an electric field to control the conductivity of a channel (i.e. a region depleted of charge carriers) between two electrodes (i.e. the source and drain) in a semiconducting material. Control of the conductivity is achieved by varying the electric field potential, relative to the source and drain electrode, at a third electrode, known as the gate. Depending on the configuration and doping of the semiconducting material, the presence of a sufficient positive or negative potential at the gate electrode would either attract charge carriers (e.g. electrons) or repel charge carriers in the conduction channel. This would either fill or empty the depletion region of charge carriers and thus form or deform the effective electrical dimensions of the conducting channel. This controls the conductance between the source and drain electrodes. In linear mode, when drain-to-source voltage is much less than the gate-to-source voltage, an FET operates much like a variable resistor to switch between conductive and non-conductive states. Alternatively in saturation mode, an FET operates as a constant-current source and is often used as a voltage amplifier. In this mode the level of constant current is determined by the gate-to-source voltage. FET devices are preferred for weak-signal and/or high impedance applications, hence their widespread use in the growing field of electrochemical biosensing [6, 66].
Although there are many different types of FET devices: MOSFET, JFET, MESFET, CHEMFET, etc., the current focus in biosensing applications is on ISFET (ion-selective field-effect transistor) and EnFET (enzyme field-effect transistor) devices [3, 6, 31, 67, 68]. The conversion of an FET into a sensing device normally involves the replacement of the metal gate electrode by a biochemically sensitive surface (e.g. an analyte-selective membrane or an ion-conductive solution), which is brought into contact with the analyte solution [69]. Also present in the analyte solution is a reference electrode, which completes the circuit via the gate-voltage bias [3, 11].
One of the most popular methods for the construction of FET-based biosensing devices is the immobilization of enzymes at the gate surface of pH-sensitive ISFET devices, creating an EnFET. The method of enzyme-immobilization is a critical aspect of the device's performance and sensitivity [11, 70]. Biocatalytic reactions influence the presence of accumulated charge carriers at the gate surface in proportion to the original analyte concentration. This produces an electrical signal in the form of a measurable drain current [67, 71]. The majority of current EnFET applications involve the analysis of penicillin, glucose and urea [3], but more applications are emerging for the analysis of an ever-expanding variety of analytes [68]. Figure 7 illustrates the sensing principle of a penicillin-sensitive EnFET.
Other than a few ISFET-based devices, mainly for pH testing, FET-based biosensors have not yet been successfully commercialized [72, 73, 74, 75]. Some of the typical problems include: the dependence of the resulting signal on the method of enzyme immobilization, the speed of response and the recovery times between measurements, the difficulty of establishing standard methods of enzyme-layer deposition and surface patterning techniques compatible with silicon-based device fabrication techniques [68]. However, research is continuing to overcome these problems. Many designs have been suggested for EnFET biosensors [76, 77, 78, 79, 80, 81, 82, 83], as well as other forms of FET-based biosensors (e.g. ImmunoFET, GenFET, cell-based FET, etc.), which have already been generally labelled as BioFET devices, or simply BioFETs [68]. Despite the mentioned problems, they remain strong candidates for reusable multiple-analyte sensing arrays or disposable electronic biosensors due to such factors as the ever-shrinking size of FET devices, their low-power operation, their high-density fabrication methods, and their interconnectivity with micro/nanoelectronic elements [34].
Another alternative in FET-based biosensing, which has generated an enormous amount of interest in the past years, is the use of nano-objects, such as nanowires or carbon nanotubes, in combination with FET technology. The use of nanowires in sensing applications is reviewed in detail in Section 2.2. Perhaps the most well-known and controversial work on the subject of nano-object based FET biosensing was presented by Cui et al. [85, 86]. In such sensing devices, the source and drain of a planar FET device is bridged by a nano-object. Binding to the surface of these nano-objects alters their ability to conduct, which serves as the detection mechanism. Although not functionalized with a biorecognition element, Figure 8 provides an illustration of a silicon nanowire FET as presented by Koo et al. [84]. Recently Gao et al. have built silicon nanowire arrays for the label-free detection of DNA [87]. A summarized schematic representation of their sensing principle and their measurements are shown in Figure 9.
2.2. Nanowires
Nanowires belong to a growing family of nano-objects, which also includes e.g. nanotubes, nanoparticles, nanorods, nanobelts, nanosprings, thin films and more [21, 22, 23, 88, 89, 90]. Nanowires are reviewed here in depth for their versatile roles, not only in electrochemical biosensing, but also in bioelectronic and nanoelectronic applications. Nanowires are increasingly being used as building blocks for biosensing techniques. Their implementation as highly-sensitive electrodes is one obvious example, such as the platinum electrode network proposed by Wang et al. for glucose detection [91]. As immediately suggested by their name, nanowires have diameters in the nanometer range. Thus, their diameters have a length scale comparable to the atoms of which they are comprised and are sometimes referred to as quantum wires. Additionally, nanowires are referred to as one-dimensional structures, since their lengths are orders of magnitude larger than their diameters. Nonetheless, one might still pose the question why wires with nanometer dimensions are more suitable or more sensitive than larger wires for sensing applications.
As a wire decreases in diameter to the nanometer regime, the ratio of surface atoms compared to interior atoms, i.e. the surface-to-volume ratio, drastically increases. Therefore, external influences by charged particles or biological species increasingly influence the conduction both on the wire surface and in the wire interior. The penetration into the wire interior is typically determined by the Debye screening length. Furthermore, quantum mechanical models of wave-like electron transport through the nanowire, rather than classical mechanical models of ballistic electron transport, become necessary as the diameter of nanowires approaches the order of the material-dependant Fermi wavelength [92, 93]. This implies that conductivity in nanowires is subject to quantum confinement into separate quantized energy levels. In other words, conductance in a nanowire of a sufficiently small diameter is related to the sum of electron transport in the separate conduction channels of the nanowire defined by their quantization energy. The thinner the wire, the smaller the available unaffected volume, and hence the smaller the probability of uninterrupted conduction. Figure 10 illustrates this concept, but uses the term ‘conduction channel’ in a more classical sense as a mental construct to aid in visualization. Following this methodology, Elfstrom et al. have demonstrated the size-dependent surface charge sensitivity of silicon nanowires [94]. In semiconducting materials the presence of surface charges induces a field effect, which alters the local carrier concentration and, therefore, the conducting ability of the material. Their experiments and simulations confirm that nanowires exhibit large changes in conductance when exposed to acidic buffer solutions, but larger wires exhibit no change.
Assuming that the wire is free of impurities, the conductance of each channel is described by G = 2e2/h, where e is the elemental charge of the electron and h is Planck's constant [96, 97, 98]. A simple experiment by Foley et al. demonstrates this stepwise decrease in conductance at the atomic level by pulling gold wires [95], as shown in Figure 11.
Nanowires, therefore, represent very attractive bioelectrochemical transducer components since their diameters are comparable to the size of the biochemical analytes under analysis and since their conductance is sensitive to surface perturbations. As stated earlier in this review, nanowires have already been incorporated into FET devices for biosensing purposes, such as the detection of pH, protein and DNA binding, viral and cancer markers. Figure 12 illustrates the integration of nanowires in a single FET device, but also within an array configuration to demonstrate the concept of multi-analyte biosensing using nanowires. The Lieber group has also reported attempts to interconnect nanowires with neurons to study their electrical behaviour [19, 99, 100]. Yeh et al. have used a combination of gold nanowires and nanoparticles to increase the electron transfer rates in redox enzymes [101]. To overcome the problems of large-scale nanowire production Stadler et al. have combined Extreme Ultraviolet Light Interference Lithography (EUV-IL) with biological patterning to produce high density line arrays of self-assembling DNA-tagged gold nanowires, as shown in Figure 13 [24]. For additional information Wanekaya et al. provide an informative review of nanowire-based electrochemical biosensors [23].
The successful implementation of nanowire-based biosensing devices depends on overcoming the challenges of their precise and reproducible placement in large-scale manufacturing processes, their specific biological functionalization, and their interconnection with both biological and nano-/microelectronic systems. Whether the nanowires are grown, pulled, tagged and enhanced, or deposited, the accurate characterization and subsequent controlled manipulation of the electrical properties of nanowires is essential to their application-specific performance.
2.3. Electrochemistry in Combination with Complementary Biosensor Techniques
Obviously, all sensing techniques demonstrate specific strengths in different, yet sometimes overlapping, areas of application. For example, both electrochemical and optical sensing techniques, can allow for real-time, in situ, non-destructive and label-free analysis of solutions, biolayers, surfaces, thin films, bulk materials and interfaces [102, 103, 104]. On one hand, electrochemical techniques, such as CV, enable the in situ influence and monitoring of e.g. redox reactions, system electrical response and reaction reversibility. On the other hand, optical techniques are known for their ability to measure mass adsorption kinetics (e.g. optical waveguide lightmode spectroscopy (OWLS), surface plasmon resonance (SPR) and ellipsometry). Gravimetric techniques like quartz crystal microbalance with dissipation monitoring (QCM-D) and imaging techniques like scanning probe microscopies, such as AFM, and fluorescent microscopies, such as confocal laser scanning microscopy (CLSM) can also be successfully combined with electrochemical techniques to enhance understanding of biointerfacial phenomena. These techniques provide high sensitivity close to the surface of the transducing element (electrode, waveguide, tip, etc.). The shared high interfacial sensitivity of electrochemical and other types of biosensor results in the simultaneous extraction of a richer set of initial data in addition to the benefit of increased control over the sensing environment.
The following sections will provide an overview of the combination of electrochemical sensing setups with representative alternative biosensor techniques. We put special emphasis on the complementary data and enhanced interpretations that can be obtained, as well as recent advancements in the area.
2.3.1. Electrochemical Surface-Plasmon Resonance (EC-SPR)
In SPR configurations the surface between two mediums of differing refractive indexes is coated with a thin film of conducting material, which is often a noble metal, such as Ag, Au or Cu. If this thin film is irradiated by light of a certain wavelength at a specific incident angle, termed the SPR angle, the evanescent field wave from Total Internal Reflection (TIR) can excite an electron wave, known as a surface plasmon, which propagates along the metal surface [105, 106]. The coupled electromagnetic evanescent field extends typically ∼200 nm from the conductive surface (e.g. Au excited at red wavelengths) as its intensity decays exponentially. Therefore, the sensing mechanism of SPR spectroscopy is based on the measurement of small changes in the refractive index in the vicinity of the surface of a conductive thin film [105, 107, 108, 109].
For use as a biosensor, the changes in refractive index in the vicinity of the conductive thin film surface (i.e. within the evanescent field) can be induced by the presence of biomolecules. The surface concentration or mass coverage of which can then be calculated using the de Feijter formula [111, 112]. SPR biosensors can be used in the determination of a number of surface binding interactions, such as: small molecule adsorption [109], protein adsorption on self-assembled monolayers, antibody-antigen binding, DNA and RNA hybridization [113, 114], protein-DNA interactions [106], binding kinetics, affinity constants, equilibrium constants, as well as receptor-ligand interactions in immunosensing [105, 115] and many more.
In EC-SPR, the combination of electrochemistry and SPR, the thin metal film on the substrate serves not only to excite surface plasmons, but also acts as a working electrode for electrochemical detection or control [117]. This important dual role of the thin metal film can be seen in a typical EC-SPR schematic in Figure 14. One advantage of the EC-SPR configuration is the ability to simultaneously obtain information about the electrochemical and optical properties of films with thicknesses in the nanometer range. EC-SPR has often been applied to study the formation and the properties of thin films and mono-/multi-biolayers using, for example, self-assembly or electro-polymerization methods [110, 118, 119]. One can also use an electrochemical configuration in combination with SPR to optically measure e.g. reaction kinetics of biomolecules in the presence of electric fields. Such experiments were performed by Georgiadis et al. and Heaton et al. to monitor the in situ hybridization of DNA in the presence of different electrochemical fields [114, 120, 121]. A further and recent use of EC-SPR was the characterization of multilayer assembly processes on gold nanoparticles [122]. Kang et al. used EC-SPR to develop a biosensor to detect the enzymatic reaction between horseradish peroxidase and a film of polyaniline (PAn) on a gold electrode in the presence of H2O2. The combination of SPR and electrochemical oxidation/reduction enabled them to demonstrate large changes in SPR responses as a result of the transition in conductivity of a PAn film on a gold electrode surface. Their results are summarized in Figure 15.
2.3.2. Waveguide-Based Techniques and Electrochemistry
Waveguide based optical evanescent sensors have equivalent sensitivity to small numbers of adsorbed analytes compared to most other biosensors. However, because of the need for both, an optically transparent substrate and interface (electrode), fewer combinations of waveguide spectroscopy and electrochemistry to SPR and electrochemistry have been made, since optically transparent electrodes are rare. Optical Waveguide Lightmode Spectroscopy(OWLS) is a label-free technique that allows for in situ measuring of adsorption, desorption, adhesion and biospecific binding processes [112, 123]. This technique is based on linearly polarized laser light that is coupled into a waveguide at two well-defined incident angles. These incoupling angles are sensitive to changes in the refractive index within the evanescent field above the surface of the waveguide. Monitoring of the changes in the incoupling angles enables determination of the adsorbed mass and the number of adsorbed molecules [123] using de Feijter's formula [111, 112].
Electrochemical OWLS (EC-OWLS) combines evanescent-field optical sensing with the electrochemical control of surface adsorption processes. It gives the possibility to directly observe mass adsorption as a function of an applied potential [124, 125]. The electrodes are connected according to the scheme shown in Figure 16. A transparent conductive electrode is made by coating the waveguide with a thin film of Indium Tin Oxide (ITO).
An example of such a combined experiment is given in Figure 17. First cyclic voltammetry was performed in order to get stable electrochemical conditions. Then a three step adsorption was performed. Poly(L-lysine)-grafted-poly(ethylene glycol) with 50 % of the side chains conjugated with biotin was adsorbed first, followed by neutravidin and biotin conjugated GOx. Subsequently, a potential of 1 Volt was applied several times in order to measure the current generated by the enzymes at different substrate concentrations.
2.3.3. Ellipsometry and Electrochemistry
Classic ellipsometry is focused on the external reflection of a light beam from a reflective surface, where linearly-polarized light of a known orientation is reflected as elliptically-polarized light. From the changes in the ellipsometric angles (Ψ, Δ), the optical properties, thickness, morphology or roughness of layers or films on the surface can be calculated [6, 102, 103] and used to determine e.g. the amount of adsorbed protein on a surface [103, 104].
Since ellipsometry can be performed on most reflective substrates it can be easily conducted on an electrode surface and combined with electrochemistry. Ying et al. have used ellipsometry and electrochemical methods to study protein adsorption on metal surfaces and specifically human serum albumin on gold surfaces [126]. Chronoamperometry and ellipsometry were combined for the study of immunosensor interfaces based on methods of Immunoglobulin G adsorption onto mixed self-assembled monolayers [127]. Yu et al. have combined imaging ellipsometry with electrochemistry to investigate the influence of electrostatic interaction on fibrinogen adsorption on gold surfaces [128].
2.3.4. Electrochemical Quartz Crystal Microbalance with Dissipation monitoring (EC-QCM-D)
The Quartz Crystal Microbalance with Dissipation monitoring measures changes in the frequency f and dissipation factor D of an oscillating quartz crystal upon adsorption of a viscoelastic layer [129]. The oscillation is based on the piezoelectric effect and the crystals typically have a fundamental resonance frequency of 5 MHz, which decreases upon mass adsorption. Modern versions of QCM, like the QCM-D, are insensitive to potentials applied at the solution interface and to local changes in ionic strength and can thus be readily combined with electrochemical sensing.
The measured mass includes hydrodynamically coupled water such as water associated with the hydration layer of e.g. proteins and/or water moved in cavities or by rough features in the film [130, 131]. The dissipation factor is a measure of energy loss and increases with e.g. higher viscosity of the solution or of adlayers. The main advantages compared to other common methods for measuring biomolecule adsorption, such as ellipsometry or surface plasmon resonance, are the flexibility to use virtually any surface coating and the information about the viscoelastic properties of the adlayer obtained through simultaneous measurements of both f and D. This includes any electrode material of interest. As with e.g. SPR or ellipsometry no labelling is required [129].
By using a conductive surface coating or the working electrode of the crystal itself, QCM allows for simultaneous electrochemical experiments [129, 132]. For electrochemical QCM a specially designed flowcell is required; typically the QCM-crystal acts as the working electrode, Ag/AgCl on the upper side of the flowcell as the reference electrode and a platinum wire in the outlet tube as the counter electrode [129, 133]. The same setup is also used for adsorption steps that are sensitive to an applied potential [129].
2.3.5. Scanning Probe Microscopy (SPM)
The invention of the scanning tunnelling microscope (STM) [136, 137] in the early 1980's has inspired various forms of SPM techniques. Despite the now diverse palette of SPM techniques, of which only the most common will be reviewed here, STM remains a promising technique for experiments combined with electrochemistry, such as Bae et al. [138].
Perhaps the most commonly employed techniques of SPM for imaging and characterization is Atomic Force Microscopy (AFM) [139]. AFM devices have evolved into versatile and powerful tools, which enable the investigation and imaging of surfaces with even molecular resolution. This is accomplished by monitoring the interaction force between the sample surface and a probe (e.g. a sharp tip), which is attached to the end of a force-sensing cantilever. The details and theory of AFM operation are not explained here, but can be read in a number of other sources [140, 141, 142]. Although AFM is often used ex situ in a separate characterization step before and/or after an electrochemical analysis [143, 144, 145, 146], the focus here will be placed on AFM's simultaneous combination with electrochemistry: EC-AFM. In other words, the imaging of the sample is performed simultaneously with the electrochemical modifications.
One area of investigation with generic EC-AFM methods is the study of electrode surface modification or performance under certain electrochemical conditions [110, 147, 148], which is important in application-specific electrode development for biosensors and chemosensors. Although not biosensing, but in the related field of biomechanics, Eliaz et al. have performed ex situ and in situ imaging, potentiostatic and potentiodynamic measurements by means of EC-AFM. Their studies were aimed at contributing to the improved osseointegration of titanium-based alloys in orthopedic devices and implants [149].
The development of ultramicroelectrodes (UMEs) and their combination with the SPM also paved the way for an SPM technique known as scanning electrochemical microscopy (SECM) [150, 151, 152]. SECM is used to locally investigate the electrochemical activity and/or the topography of a surface. Designed for either amperometric or potentiometric measurements, a UME probe, which is normally in the shape of a tip, is scanned along the surface of interest and is used to induce chemical changes and to collect electrochemical information. Two recent reviews on SECM from Sun et al. and Edwards et al. provide a very comprehensive overview of its general application and theory, as well as its use in current research [153, 154]. In short, SECM has proven useful in the detection and imaging of heterogeneous electron transfer reactions at metal/solution interfaces, electron transfer kinetics at solid/liquid interfaces, various electrocatalytic reactions, predominately the hydrogen oxidation reaction (HOR) and the oxygen reduction reaction (ORR), lateral charge and mass transfers and biological cells [153, 155, 156].
Numerous hybrid techniques have evolved around SECM [153], including its combination with surface plasmon resonance (SPR-SECM) [158], electrochemical quartz crystal microbalance (EC-QCM-SECM) [159, 160, 161], fluorescence spectroscopy (FS-SECM) [162], photoelectrochemical microscopy (PEM) or near-field scanning optical microscopy (NSOM-SECM) [163], electrogenerated chemiluminescence (ECL-SECM) [164] and atomic force microscopy (AFM-SECM) [153, 154, 165]. The most widely used of these hybrid techniques would be AFM-SECM. The general concept of the hybrid probe used in AFM-SECM measurements is illustrated in Figure 18.
AFM-SECM is capable of obtaining simultaneous electrochemical and topographical information at high spatial resolution through the integration of an electrode into the AFM probe. In other words, the structural information is simultaneously correlated with chemical activity of the substrate [167]. These hybrid probes are, therefore, quite effective for the identification and chemical mapping of active surface sites [168]. Critical to the resulting synergy of AFM-SECM is the proper modification of the probe. Please refer to the reference literature for information on the fabrication and application of various types of AFM-SECM probes [153, 154, 168]. Gardner et al. have studied the correlation of membrane structure and transport activity across a synthetic polyethylene terephthalate (PET) membrane [169].
Figure 19 shows the topographical imaging of the morphology of model neurons directly in the cell growth media without a redox mediator as performed by Kurulugama et al. [166]. Kreung et al. have mapped glucose oxidase (GOx) activity on a micropatterned surface and through a synthetic membrane [157, 170]. The same group has imaged immobilized horseradish peroxidase with the same techniques [171]. Simultaneous imaging topography and the methodology of using electrochemistry to agitate interfacial systems has been applied to crystal dissolution studies [172].
2.4. Magnetic Biosensors
Despite the almost automatic association of the words “magnetic” and “sensing” with Magnetic Resonance Imaging (MRI), the focus of this section is the use of magnetism in combination with electrochemistry for biosensing. Therefore, some interesting transduction methods in magnetic biosensing use magnetic remanence and/or relaxation [173, 174, 175, 176], mixed excitation frequency response [177, 178], cantilever movements (e.g. force amplified biological sensor, FABS) [179], inductance [180, 181], induced current [182] or magnetoresistance, such as giant magnetoresistance (GMR) [183, 184, 185, 186]. From these, the magnetoresistive techniques will be highlighted here due to the frequent combination of this technique with electrochemistry in biosensing.
Discovered by Thomson in 1856, the magnetoresistive effect is the change of the resistivity of a material in response to a magnetic field. An illustrated overview of the use of the magnetoresistive effect as a general sensing mechanism, as well as a specific sensing scheme for the detection of pre-hybridized biotinylated DNA are provided in Figure 20 [185]. At a fixed sensing current and in the presence of an applied magnetic field, the changes in resistance can be measured as changes in the voltage. The changes in resistance arise from the magnetic fringe field resulting from the magnetic moment of functionalized magnetic label (e.g. a magnetic nanoparticle) upon binding with complementary surface-bound biomolecules.
In combination with electrochemistry Helali et al. used a magnetic monolayer of streptavidin-coated magnetic particles to construct an ‘immunomagnetic electrochemical sensor’ for the detection of atrazine [187]. Tang et al. have demonstrated the use of Fe2O3/Au magnetic nanoparticles for direct electrochemical immunoassay sensing [188]. They have also proposed the extension of their technique for further protein-magnetic nanoparticle based detection, such as with DNA or enzymes. A novel approach to the application of the magnetoresistive effect in biosensing is the Bead Array Counter (BARC) as proposed by Baselt et al. [184, 189]. Designed to be a multi-analyte biosensor, BARC uses GMR and DNA hybridization to magnetic microbeads for analyte detection and identification. One specific application of a BARC biosensing is the detection and identification of biological warfare agents as designed by Edelstein et al. [190].
3. Surface Architecture
In this chapter the different components of an electronic biosensor are discussed. It starts with the sensor surface that needs to be conductive and depending on the detection technique also transparent. A surface coating is to provide inertness and/or functional groups for tethering of the recognition element, which is often an antibody. The recognition elements (antibodies) are surface immobilized to specifically catch biologically relevant molecules (antigens for an antibody based sensor) that need to be detected. Many biosensors with some notable exceptions employ labelling to quantify the binding, e.g., the binding of an additional antibody with a fluorescent label in the case of ELISA. In electrochemical biosensors the label is usually an enzyme that catalyzes certain reactions in cases where the bound molecule in itself does not significantly alter the charge transfer process across the electrode interface. Finally, the electrons generated during the recognition event or usually the enzymatic label reaction with a substrate also need to be detected. If the reaction takes place away from the electrode interface mediators can be used to shuttle the electrons between the reaction site and the surface.
3.1. Surface Materials and Modifications
Nowadays, a wide variety of different materials is used for the preparation of surfaces for biosensing applications. Depending on the measurement technique, they need to fulfill, special requirements, such as electrical conductivity for electrochemical techniques or transparency for optical devices. Most commonly used are glass and other oxide surfaces because of their favourable characteristics, especially their optical characteristics [191]. Widely used are also gold, microporous gold, graphite, glass carbon [192, 193] and Indium Tin Oxide (ITO) [194].
Electrodes are increasingly screen printed. This approach has advantages such as simple and low cost fabrication as well as easy mass production [192].
Another class of materials used for fabricating electrochemical biosensors are various conducting polymers. These include polyaniline, polypyrrole and polystyrene, which can be coated onto other sensor substrates. Typically, the polymers are adsorbed to gold surfaces. This leads to surfaces with good stability, excellent redox recyclability and easy handling [4, 191, 195].
Gold and other metallic surfaces are also coated with self-assembled monolayers (SAMs) of sulfides (thiols) and disulfides. Molecules with a thiol foot can form highly ordered and well-organized structures functionalized with an organic linker [45, 196]. Different end-functionalizations are used to couple biological recognition elements. For example, carboxyl groups often serve for antibody immobilization, esters form amine couplings and biotin coupling can be used to bind streptavidin and further biotin-functionalized biomolecules [45]. In electrochemical applications it is essential that the SAM allows electron or analyte diffusion [196].
Carbon nanotube (CNT) modified electrodes have advantages in terms of their high surface area, mechanical strength, excellent electrical conductivity and good chemical stability. They are especially interesting because enzymes can be entrapped in the inner cavity [197, 198].
In a more unique approach glassy carbon electrodes were modified with gold nanocrystals. Gold nanoparticles were then bound via cysteamine. The whole procedure leads to high efficiency of the enzyme immobilization [199]. Others have used gold nanorods, which can be flat on a surface or perpendicular to it. They have a diameter of about 20 nm and a length of up to 2 μm [200, 201]. Also semiconductors such as ZnO have been used to fabricate nanorods. They have been functionalized with e.g. biotin [202].
3.2. Electrochemical Signal Transduction
Electrochemical biosensors are mostly based on the principle of direct transduction of the reaction rate into a current [4]. The way has been long from the first electrode-based biosensors to today's state of the art. The first generation were basically oxygen based sensors. In the presence of glucose oxidase (GOx) glucose and oxygen undergo a reaction in which gluconic acid and hydrogen peroxide are formed.
The drawback of this electrode setup is the dependence on the oxygen concentration which is difficult to maintain at a constant level. In the second generation, the oxygen was replaced by other, reversible, oxidizing reagents also known as mediators. They are often based on iron, e.g. ferro/ferricyanide. In the third generation, denaturation of the enzymes was taken into account. In order to prevent unfolding and inactivation they were directly coupled to the electrode. In such a configuration no more mediators are required [3].
In general, it is preferable to have the reaction take place as close to the electrode as possible because the products diffuse in all directions, also away from the surface. This results in a decreasing signal with increasing distance to the surface [203].
Depending on whether a mediator is used or not one talks about direct or indirect transduction. These are elaborated in the following paragraphs.
In biosensors that are based on direct electron transduction the redox enzyme acts as an electrocatalyst. They are also known as third generation biosensors. Such systems do not require a mediator. The surface immobilized enzyme selectively catalyzes the transformation of a specific substrate. The electrons are directly transferred from the electrode to the substrate molecule or vice versa as shown in Figure 21a. A high efficiency of the enzymatic reaction is required [4, 204].
The direct electron transfer slows down with increasing distance between the enzyme and the electrode surface. This effect reduces the sensitivity of mediatorless sensors. Kuznetsov et al. estimated the decrease upon increasing distance by having a hemoglobin layer between the electrode surface and the laccase molecule or conjugating the laccase with an antibody. They found an exponential decrease of the signal with increasing distance to the surface. Because of charge accumulation the signal is still detectable also at very low concentrations in the range of 1 nM [203].
In the indirect transduction the electron transfer mediator shuttles the electrons between the redox center of the enzyme and the electrodes (see Figure 21b). Such mediators are defined as artificial electron acceptors with a low oxidation potential. Usually, they can freely diffuse in solution. It is also possible to co-immobilize them with the enzymes [4, 8].
Mediators are small and mobile molecules, which are able to react rapidly with the enzyme. They are oxidized at the electrode and reduced at the reaction site of the enzyme or vice versa. Therefore, they should exhibit reversible heterogeneous kinetics, not react with oxygen and ideally be pH independent. Most commonly used are ferrocene and its derivates, ferrocyanide, methyleneblue, benzoquinone and N-methyl phenazine [4].
In a similar approach the mediator was replaced by carbon nanotubes (CNTs). Patolsky et al. [205] used CNTs to guide electrons between the enzyme and the surface. They immobilized CNTs in an upright position. On the upper end they covalently bound GOx (see Figure 21c). In this setup the spacial separation prevents tunnelling effects.
3.3. Enzymes
Enzymes are proteins that act as powerful catalysts to convert substrates into products [206]. Some enzymes, also known as redox-enzymes, catalyze reactions that produce or consume electrons. Thereby, the substrate is recognized by a binding pocket of the enzyme, similar to the antibody-antigen interaction. Enzymes with such specific binding pockets are used in enzymatic biosensing - where the electrons are detected. Subsequently, out of the current density through the interface the enzyme- or substrate concentration and/or activity can be calculated [54, 207].
The most commonly used enzymes in biosensing are glucose oxidase (GOx) [8] and horseradish peroxidase (HRP) [208]. The corresponding reaction schemes are given in Equation 5 for GOx and Equation 6 for HRP. Donors for the reaction with HRP are molecules such as phenols, aromatic amines, thioaminosoles or iodide [208]. Other, less commonly used enzymes comprise beta-lactamase [209], urea and urease [10].
One of the major issues when working with enzymes is their stability. This includes shelf life as well as operational stability. Enzymes are very sensitive to their environment. Deactivation, inhibition or unfolding upon adsorption and chemical or thermal inactivation are common if no special precautions are applied [210, 211]. The main immobilization strategies that retain the enzymatic activity include encapsulation, covalent immobilization and site-specific mutagenesis. Compared to direct immobilization, encapsulation provides several advantages. This method provides a more natural environment which causes less inactivation. Furthermore, encapsulation opens the possibility to increase the concentration of immobilized enzymes [210, 211, 212, 213]. Due to these advantages and major efforts towards their implementation, strategies for encapsulation of enzymes will be treated in more detail in Section 3.5. Other stabilization techniques include: enzymes bound to nanoparticles/-fibers or single enzyme nanoparticles [213, 214, 215]. Depending on the treatment different strategies are favourable to inhibit enzyme degradation: e.g. salts, such as sodium chloride, are added to protect the enzymes during freezing processes. Large polyelectrolytes and low molecular weight electrolytes improve the operational stability [210]. The lifetime of enzymatic biosensors is limited by the loss of enzyme activity over time; typically it is limited to 2-8 weeks [10].
3.4. Recognition Elements
3.4.1. Antibodies
Among other recognition elements, antibodies are most widely used because of the high specificity of the antibody-antigen binding. However, they are also very sensitive to their environment. In order to detect antigens, antibodies are surface-immobilized, which can cause a severe loss of their biological activity. Since the activity of surface immobilized antibodies depends on their orientation, it is advantageous to assure that they are not randomly oriented. Other reasons for inactivity include steric hindrance in cases where the density is too high as well as denaturation due to non-specific interactions with the surface [216]. Immobilization techniques include microcontact printing [217], biotin-streptavidin binding [218, 219, 220, 221], direct spotting [218], adsorption to a conductive polymer matrix such as polypyrrole or polyaniline [214] as well as covalent binding [222].
Most commonly used is Immunoglobulin G (IgG) which, like typical antibodies, consists of one Fc and tow Fab' binding sites. It becomes inactive when the Fab' fragment binds to the surface [216]. Often antibodies help to discover new disease markers [223]. In typical applications, monoclonal antibodies are used as specific capture antibodies for e.g. prostate cancer marker (PSA). In such, so called sandwich assays, one antibody acts as a capture antibody while the second enzyme-labelled antibody is bound to the PSA [224].
Electrochemical enzymatic biosensors can be built up similar to the enzyme-linked immunosorbent assay (ELISA). After immobilizing antibodies to a surface, an analyte is introduced to which the antibodies bind specifically. In the most common detection scheme a secondary labelled antibody then binds to the analyte in order to detect its concentration (see Figure 21d). The detection antibodies are coupled to an enzyme, which allows quantitative measurements of the amount of bound antigens by monitoring the electrical signal generated by an enzymatic reaction [225].
A wide variety of antigens, usually in diagnostics, can be detected this way. It is e.g. possible to diagnose Hepatitis C already in an early stage [226]. More recently, capture antigens have also been used to detect cancer markers. Among the most important ones are carcinoembryonic antigen (CEA) [193], carbohydrate antigen 19-9 (CA19-9), carcinoma antigen 125 (CA125), alpha-fetoprotein (AFP), CA15-3, human chorionic gonadotropin (hCG) [192] and prostate specific antigen (PSA) [224], which are used to detect various types of cancer.
3.4.2. Antibody Fragments
Nowadays, antibody fragments are emerging as credible alternatives to antibodies. They provide the same specificity as whole antibodies. Furthermore, they are smaller which can be an advantage for biosensor applications. In their main application field, biosensors and therapeutics, these small, highly specific reagents against target antigens are often used as bi- or trimers [227].
Most commonly used are IgG fragments [216, 217]. Such small, recombinant antibody fragments can be linked to other molecules, for example lipids, drugs and proteins [227]. An antibody that is naturally composed of only heavy chains is the camel antibody. Even though the light chains are missing, it binds very specifically [228]. For other applications biotinylated fragments are adsorbed to streptavidin coated surfaces [219]. As already mentioned for antibodies, the orientation is of great importance. Lu et al. [216] reported a threefold increase of activation upon proper/controlled orientation. In a more complex approach Vikholm et al. adsorbed Fab' fragments directly to gold but embedded them in a protein repellant polymer such that only the antigen binding site stuck out of the polymer [229].
3.4.3. Aptamers
Aptamers are folded single stranded DNA or RNA oligonucleotide sequences with the capacity to recognize various target molecules. They are generated in the SELEX (systematic evolution of ligands by exponential enrichment) process which was first reported by Ellington [230] and Tuerk [231] in 1990. In this approach suitable binding sequences are first isolated from large oligonucleotide libraries and subsequently amplified [6, 232].
The main application for aptamers is in biosensors. While antibodies are used in ELISA, the similar process for aptamers is called ELONA (enzyme linked oligonucleotide assay) [233]. They have many advantages over antibodies such as easier deposition on sensing surfaces, higher reproducibility, longer shelf life, easier regeneration and a higher resistance to denaturation. As antibodies, they are characterized by both, their high affinity and specificity to their targets [6, 232].
3.5. Encapsulation of Enzymes
Several different strategies for enzyme encapsulation to increase signal response and specificity have been envisioned and pursued. Some of the most interesting ones are reviewed below.
3.5.1. Polyelectrolyte Multilayer (PEM) Capsules
These microcapsules were first presented by Caruso and Donath in 1998 [234, 235]. They are fabricated by coating a microparticle with alternating layers of oppositely charged polyelectrolytes, such as the negatively charged poly(sodium styrenesulfonate) (PSS) and the positively charged poly(allylamine hydrochloride) (PAH). The shell thickness can be varied by adjusting the number of layers. Subsequently, the core is dissolved and the material diffuses through the shell into the surrounding solution. The capsules usually have a diameter of 1-10 micrometers [235, 236]. Optionally, coating of poly(L-lysine)-grafted-poly(ethylene glycol) can be applied to make them protein resistant [237].
3.5.2. Vesicles
Liposomes or lipid vesicles consist of bilayer forming amphiphilic molecules such as palmitoyloleoylphosphocholine (POPC), called lipids. Liposome sizes vary between 20 nm and several hundred micrometers. Applications are in e.g. drug delivery, cosmetic emulsions, and optical electrochemical biosensing. In the latter case they are loaded with enzymes. Many different encapsulation methods have been developed during the last decade. Among them, the extrusion technique and dehydration-rehydration have been the most successful ones. The entrapped enzymes catalyze the conversion of substrate molecules into products. Having stable vesicles and enzymes, as well as low enzyme permeability and high substrate permeability are prerequisites for their use in biosensing [240]. However, substrate permeability is typically low, which drastically limits the turnover rate of encapsulated enzymes. To overcome this drawback, membrane channels such as the outer membrane protein F (OmpF) from Escherichia coli were incorporated into the membrane [241]. They allow for diffusion of molecules with a molecular weight up to 400 Da [242]. Hill et al. incorporated glucose oxidase, horseradish per-oxidase and the combination of both into vesicles. There was no major loss in enzymatic activity [243]. In another application the enzyme beta-lactamase was incorporated into POPC vesicles [242].
3.5.3. Polymeric Micelles
Polymeric micelles are built from amphiphilic block copolymers. The inner, hydrophobic side usually consists of poly(acrylic acid) (PAA) or polyesters; for the outer, hydrophilic shell poly(ethylene glycol) (PEG) is most widely used. Their size can vary between 10 and 100 nm [244, 245, 246]. Thurmond et al. cross-linked the shells to increase stability [247]. Even though the main application is in drug delivery where drugs are enclosed, enzymes have also successfully been embedded in the core through electrostatic interaction with polyion complex (PIC) block copolymers [248]. Another way is to PEGy-late the enzymes, whereas the PEG is part of the PIC [249]. This allows for potential use in enzymatic biosensing.
3.5.4. Hydrogel
Since encapsulated enzymes do not need to be mobile, hydrogels offer a suitable immobilization technique. Thus, cross-linked polymers form a biocompatible hydrogel that entraps the enzymes. The water soluble redox polymer should not cross-link too much in order to allow swelling in water. At the same time it mediates electron transfer between the enzymes and the electrode via electron diffusion through electron exchange between mobile polymer segments. In applications, such as electronic biosensors, it is important that the entrapped enzymes are not denaturated and remain in their active state. Otherwise, the sensitivity and selectivity of the biosensor is decreased drastically [250, 251, 252, 253]. Mao et al. introduced a new redox hydrogel, consisting of a polymer with 13-atom-long tethers between the redox centers and the polymer backbone. This led to an electron diffusion coefficient one order of magnitude higher than for previously known systems [254].
3.5.5. Sol-Gel
Like hydrogels, the sol-gel technique is applied directly on surfaces. In the first experiments porous SiO2 glass was used. Braun et al. were the first ones to report about successful enzyme entrapment in a sol-gel matrix [255]. The material is formed via partial or complete hydrolysis of a suitable precursor, usually silanes, mixed with biomolecules. Subsequently, gelation and aging follows. Aging strengthens the network and the drying process simultaneously leads to shrinkage [256, 257]. Tunable pore sizes and pore size distribution, optical transparency, no need of a mediator, mechanical rigidity, chemical inertness, permeability and thermal stability gives them highly desirable properties [257, 258]. Typical applications include selective coatings for optical and electrochemical biosensors [257], for example carbon ceramic composite electrodes (CCE) for amperometric biosensors [258].
3.6. Supported Lipid Bilayer Sensor Architectures
Lipid bilayers can be used to build biosensors that make use of the transport process across the membrane. Functional lipid membranes have not yet found as extensive use in biosensor applications [259] as the enzyme or antibody systems described above and are yet not competitive with other screening methods [260]. However, there is a strong desire to use them in particular to probe function of membrane proteins, e.g. in drug screening applications. Lipid bilayers provide the only alternative for sensors aiming to probe or to make use of membrane proteins, which currently comprise > 50% of all drug targets, and many of these proteins are directly involved in charge transfer processes across the membrane [261]. A direct electrical readout of ionic currents generated by ion channels, for example, bears the advantage of an amplification of the readout signal of membrane functions without need for labelling compared to current state-of-the-art techniques for membrane protein screening.
While most electrochemical measurements utilizing lipid membranes have been conducted using, so called, black lipid membranes (BLMs), which are formed across apertures separating two aqueous compartments, we will focus on lipid bilayer architectures stabilized by a solid support. Only these can be formed free from solvents and detergents, in a miniaturized format and with sufficient stability to be viable for commercial biosensor applications. There are recent examples of advances in combining nanostructures with lipid bilayers which effectively fuse the approaches of aperture spanning membranes with solid supported lipid bilayers [262, 263]. This combination exploits the advantages of the two approaches. For a further overview of recent advances see Reimhult et al. [264]. The main techniques to form lipid bilayers on various, also porous, surfaces include vesicle fusion, painting, detergent dilution and Langmuir-Blodgett deposition [264, 265]. The membranes can be directly adsorbed or tethered to the surface, but also separated from the sensor substrate by a hydrogel [265, 266, 267]. When a lipid membrane is adsorbed in close proximity to a solid substrate like an electrode it is generally referred to as a supported lipid bilayer (SLB), but several different classes can be identified in terms of the surface architecture. They can be divided into hybrid lipid layers (Figure 22a), solid SLB (Figure 22b), tethered lipid bilayers (tSLB, Figure 22c), polymer-supported lipid bilayers (pSLB, Figure 22d) and pore-spanning lipid membranes (nano- or micro-BLM, Figure 22e). Of these the hybrid lipid bilayers have been the ones used the most for electrochemical measurements, because of the ease with which they can be prepared on electrode materials like Au [268]. However, in recent years they have been replaced by the more advanced tSLBs [269, 270], which have two leaflets of lipids and a hydrophilic spacer group between the electrode and the lipid bilayer. This allows incorporation of smaller transmembrane proteins and produces membranes with high resistance.
Using SLBs to study transmembrane protein function or to construct the ion-channel sensors discussed below requires a lipid bilayer of high integrity to provide a good electrochemical seal. Without a good seal the current to the underlying electrode (or the impedance) is dominated by conduction through defects in the membrane instead of charge transport through the integrated proteins. Incorporation of membrane proteins like ion channels and pumps, which enable the diffusion of charged molecules, allows study of their transport properties through electrochemical measurements in configurations ranging from cyclic voltammetry to impedance spectroscopy and membrane-FET. Among these, impedance spectroscopy has been the most common characterization method due to the possibility to model the membrane from the impedance characteristics. To date, only a few membrane proteins have been successfully incorporated into supported lipid bilayers for electrochemical measurements, and they are mainly limited to small ion channels that insert easily in native conformation into lipid bilayers also when there is only a small aqueous reservoir between the lipid bilayer and the support. Examples of such peptides are gramicidin, melittin, alamethicin, and valinomycin [271, 272, 273, 274, 275]. Larger proteins forming ion channels, like Outer membrane protein F (OmpF) from E. Coli, and other peptides have also been investigated [274, 276, 277, 278, 279]. Incorporation is mostly achieved by direct insertion of the small hydrophobic channel proteins directly from solution. Furthermore, Neumann and coworkers have pioneered a method for tethering proteins to the surface and then form a lipid bilayer around it by detergent-lipid micelle dilution [280]. As demonstrated in the work by Ataka et al. also large membrane proteins and proteins giving rise only to low charge transport across the lipid membrane can now under certain circumstances be used for biosensing, although the technique needs to be developed further to be generally applicable. There have been similar demonstrations for other complex proteolipid systems, also with direct biosensor applications [281, 282, 283].
Ion-channels in SLBs have also been exploited as a means of amplifying biorecognition events for electrochemical sensing [267]. Ion channels are membrane proteins with highly selective pores that regulate the transport of inorganic ions, for example K+, Na+ or Ca2+, and/or charged molecules creating electrochemical gradients of cell membranes [285]. Independent of the triggering mechanism, Figure 23 illustrates the simplified structure of an ion channel and the transport of cations through the channel. When the ion channel is triggered to open, the selective pore allows the transport of only the channel-specific ion through the channel. Dependent on the trigger mechanism, this process continues until the potential difference across the membrane (voltage and/or ion concentration) is sufficiently equalized or until the pore closes in response to another stimulus [284]. The most well-known biosensor of this type is the ion channel switch (ICS™) biosensor, developed by Cornell et al. The principle of the biosensor is illustrated and briefly described in Figure 24 [286, 287]. The ion channel switch uses a tSLB and an underlying gold substrate as the working electrode to measure the change in admittance when dimerization occurs for anchored and gated gramicidin ion channels in a two-dimensional sandwich assay. This clever construction utilizes the fluidity of the lipid bilayer to achieve a 103 times higher sensitivity to a capture event than if direct binding to the antibody would be used for gating. Since each active gramicidin ion channel allows very high flux of cations (∼107 ions per second), the amplification per recognition event by the tethered Fab' fragments is amplified by many orders of magnitude. Gramicidin has also been used to construct biosensors by, e.g., Nikolelis et al. [288] and Blake et al. [289]. Other channel peptides and proteins have also been used to construct biosensors, e.g., OmpF by Stora et al. [290].
4. Conclusions & Outlook
The development and research of (bio)sensors is becoming one of the most popular scientific areas at the intersection of the biological and the engineering sciences. Semiconducting technology has matured so much that we see now a rapid infiltration of new nanotechnology-based approaches in the field of sensors. The resulting new discipline of nano-bio-sensing is a good example of how engineering sciences, physics, chemistry, and biology coincide at the nanometer scale: The traditional separation between transducers and bioreceptors is being replaced by an integrative approach as the nano-transducers start to take part in the recognition event and as the receptors start to become active transducing elements of the sensors (Figure 1).
Another explanation to why (nano)biosensing cuts out a major piece of the nano-bio cake is the need for it. Early, low-cost point-of-care detection of markers for diseases is crucial to diagnose and to manage health problems world wide, especially in the developing world. This has been evident already from the inception of the field when the need for distributed monitoring of diabetes mellitus was - and is - driving both academic and commercial efforts towards developing glucose biosensors. Nanotechnology now promises a solution for integration and high throughput for a vast range of electrochemical sensor applications. Miniaturization, functional sensitivity, simplified read-out and multiplexing are the answers for many current challenges of drug discovery and personalized medical care. In addition, nanotechnology provides new physical phenomena that can be utilized for sensing with a potential to reach the ultimate, single molecule sensitivity. For example, nanowires directly convert a binding event to an electrical signal (Figure 12).
Recently, gene-chip technology has become another prospering industry, which is growing at an impressive rate and opening new areas of application ranging from systems biology, drug discovery and forensics, to diagnostics [10]. In addition, the first nanobiosensors are also already on the market illustrating that the advancement of nanotechnology also brought a paradigm change into the already mature field of biosensing. It is easy to imagine how related technologies will have a further impact on formats for electrochemical biosensing. The fusion of electrochemical biosensing with nanotechnology and the growing need for inexpensive, mass production of single-use biosensors promises to change the unfortunate fact that glucose test strips are the sole product of the electrochemical biosensor industry to have achieved commercial success.
In this review we focused on the interplay between the sensors and the corresponding transducer surface nano-architectures with special focus on electronic sensing. In the first section we introduced traditional electrical measuring methods that have largely contributed to the current advanced understanding of the existing transduction mechanisms. After this we discussed the use of field-effect transistor technology for biosensing with special focus on the efforts being invested into the combination of existing FET technology and nanotechnology, such as nanowires and carbon nanotubes [19, 23].
As we approach the nanoscale, the characterization and optimization of the devices becomes more and more difficult. Novel tools that combine different sensing methods can provide the necessary complementary information that is needed to understand the limitations and to optimize the performance of the new techniques. Therefore, we introduced existing methods (e.g. electrochemical SPR, QCM, optical waveguides and AFM) which allow parallel complementary investigations of the biochemical processes that take place at the interface between the devices and the biological sample. Besides their importance in characterization, these techniques also provide unique possibilities for research. It is now possible to manipulate interfaces with electric fields, to study the effect of applied fields onto binding affinities of charged molecules and to control the release of drugs from polyelectrolyte layers or other embedded systems by electronic means [114, 291].
The enormous interest in microarray-based assays originally arose from work using DNA chips [292]. On the other hand protein microarrays are of general interest for all diagnostic applications where the parallel analysis of several parameters is required [292]. They are mainly used to measure abundances of multiple proteins, usually to discover associations with disease or new biomarkers or to improve the selectivity and specificity of tests [225]. In general, arrays allow for the detection of whole cells and enzyme activity and allow for measuring changes in modification or expression levels of cancer-related proteins [293]. In addition to the nanowire arrays, we described an emerging microarray technology based on magnetic particles that has potential in high-sensitivity diagnostics and defense applications in the final part of the device section.
The various generations of enzymatic glucose sensors beautifully demonstrate the integration of transducers, biochemistry, and nanotechnology. In order to eliminate dependence on oxygen levels the mediator concept was introduced; later nanotechnology-based approaches help to overcome problems with low efficiency of the electron transfer between the enzyme and the sensor electrode (Figure 21).
Furthermore, supramolecular architectures enabled unprecedented signal amplification schemes, e.g., incorporating the enzymes into vesicles, polyelectrolyte capsules, sol-gel films or particles. This not only enhanced the stability and improved the shelf-life of sensors, but also improved the sensitivity. However, further new ideas are needed if we want to address new target molecules in the future. While glucose is present in mM concentrations in blood, the diagnosing of cancer markers requires the detection of analytes at pM concentrations. Nanotechnological approaches, both artificial and biologically inspired, which have been demonstrated down to single-molecule sensitivities, are likely to provide the detection schemes that will make this possible.
Natural receptor schemes that operate at such low concentrations (e.g. taste sensing or hormone action) usually involve membrane-based amplification schemes. Ion-channels and G-protein coupled receptors are highly sophisticated nanomachines that successfully solve the problem of selective and efficient amplification of a binding event. Such concepts can be borrowed from nature and used for engineered purposes. Engineered ion-channels embedded in a supported lipid bilayer can be used to amplify the signal of a biosensor as depicted in Figure 24. Future approaches include the use of nanoporous substrates to increase the stability of membranes by spanning apertures only 10-100 nm in diameter [264, 294, 295]. These can be miniaturized in a chip format and also allow very sensitive recording of single protein activity by e.g. voltage-clamp electrochemical setups. Such systems are promising for drug discovery since they directly measure membrane protein functionality when they are e.g. exposed to drug candidates.
At present, biosensor research is not only driving the ever-accelerating race to construct smaller, faster, cheaper and more efficient devices, but may also ultimately result in the successful integration of electronic and biological systems. Thus, the future development of highly sensitive, highly specific, multi-analysis, nanoscale biosensors and bioelectronics will require the combination of much interdisciplinary knowledge from areas such as: quantum, solid-state and surface physics, biology and bioengineering, surface biochemistry, medicine and electrical engineering. Any advancement in this field will have an effect on the future of diagnostics and health care. Personalized and preventive medicine, bedside diagnostics, and drug discovery will all benefit from the novel electronic sensing technologies that were summarized in this review article.
Acknowledgments
The authors would like to acknowledge CCMX-MMNS-Nanowires project, CCMX “Platform for high-density parallel screening of membrane receptor function” project and the ETH Zurich for funding.
References
- Lowe, C.R. Biosensors. Trends in Biotechnology 1984, 2(3), 59–65. [Google Scholar]
- Thevenot, D.R.; Toth, K.; Durst, R. A.; Wilson, G. S. Electrochemical biosensors: recommended definitions and classification. Biosensors & Bioelectronics 2001, 16(1-2), 121–131. [Google Scholar]
- Eggins, B. Chemical sensors and biosensors.; Analytical Techniques in the Sciences. John Wiley & Sons: West Sussex, 2002. [Google Scholar]
- Chaubey, A.; Malhotra, B.D. Mediated biosensors. Biosensors & Bioelectronics 2002, 17(6-7), 441–456. [Google Scholar]
- Kasemo, B. Biological surface science. Surface Science 2002, 500(1-3), 656–677. [Google Scholar]
- Luppa, P.B.; Sokoll, L. J.; Chan, D. W. Immunosensors - principles and applications to clinical chemistry. Clinica Chimica Acta 2001, 314(1-2), 1–26. [Google Scholar]
- Clark, L.C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Annals of the New York Academy of Sciences 1962, 102(1), 29, –&. [Google Scholar]
- Fang, A.P.; Ng, H. T.; Li, S. F.Y. A high-performance glucose biosensor based on monomolecular layer of glucose oxidase covalently immobilised on indium-tin oxide surface. Biosensors & Bioelectronics 2003, 19(1), 43–49. [Google Scholar]
- Wilson, M.S. Electrochemical immunosensors for the simultaneous detection of two tumor markers. Analytical Chemistry 2005, 77(5), 1496–1502. [Google Scholar]
- D'Orazio, P. Biosensors in clinical chemistry. Clinica Chimica Acta 2003, 334(1-2), 41–69. [Google Scholar]
- Schoning, M.J.; Poghossian, A. Recent advances in biologically sensitive field-effect transistors (biofets). Analyst 2002, 127(9), 1137–1151. [Google Scholar]
- Clark, L.C. Monitor and control of blood and tissue oxygen tensions. Transactions American Society for Artificial Internal Organs 1956, 2, 41, –&. [Google Scholar]
- Clark, L.C.; Clark, E. W. A personalized history of the clark oxygen-electrode. International Anesthesiology Clinics 1987, 25(3), 1–29. [Google Scholar]
- Wang, J. Glucose biosensors: 40 years of advances and challenges. Electroanalysis 2001, 13(12), 983–988. [Google Scholar]
- Mirsky, V.M.; Riepl, M.; Wolfbeis, O. S. Capacitive monitoring of protein immobilization and antigen-antibody reactions on monomolecular alkylthiol films on gold electrodes. Biosensors & Bioelectronics 1997, 12(9-10), 977–989. [Google Scholar]
- Guiseppi-Elie, A.; Lingerfelt, L. Impedimetric detection of dna hybridization: Towards near-patient dna diagnostics. In Immobilisation of DNA on Chips I, volume 260 of Topics in Current Chemistry; pp. 161–186. Springer-Verlag Berlin: Berlin, 2005. [Google Scholar]
- Ben, A.M.; Korpan, Y.; Gonchar, M.; El'skaya, A.; Maaref, M. A.; Jaffrezic-Renault, N.; Martelet, C. Formaldehyde assay by capacitance versus voltage and impedance measurements using bi-layer bio-recognition membrane. Biosensors & Bioelectronics 2006, 22(5), 575–581. [Google Scholar]
- Mehrvar, M.; Abdi, M. Recent developments, characteristics, and potential applications of electrochemical biosensors. Analytical Sciences 2004, 20(8), 1113–1126. [Google Scholar]
- Patolsky, F.; Zheng, G.F.; Lieber, C. M. Nanowire-based biosensors. Analytical Chemistry 2006, 78(13), 4260–4269. [Google Scholar]
- Nair, P.; Alam, M. Dimensionally frustrated diffusion towards fractal adsorbers. Physical Review Letters 2007, 99(25), 256101. [Google Scholar]
- Merkoci, A. Electrochemical biosensing with nanoparticles. Febs Journal 2007, 274(2), 310–316. [Google Scholar]
- Park, S.J.; Taton, T. A.; Mirkin, C. A. Array-based electrical detection of dna with nanoparticle probes. Science 2002, 295(5559), 1503–1506. [Google Scholar]
- Wanekaya, A.K.; Chen, W.; Myung, N. V.; Mulchandani, A. Nanowire-based electrochemical biosensors. Electroanalysis 2006, 18(6), 533–550. [Google Scholar]
- Stadler, B.; Solak, H.H.; Frerker, S.; Bonroy, K.; Frederix, F.; Voros, J.; Grandin, H. M. Nanopatterning of gold colloids for label-free biosensing. Nanotechnology 2007, 18(15), 6. [Google Scholar]
- Wang, J. Electrochemical biosensors: Towards point-of-care cancer diagnostics. Biosensors & Bioelectronics 2006, 21(10), 1887–1892. [Google Scholar]
- Santandreu, M.; Alegret, S.; Fabregas, E. Determination of beta-hcg using amperometric immunosensors based on a conducting immunocomposite. Analytica Chimica Acta 1999, 396(2-3), 181–188. [Google Scholar]
- Yu, D.H.; Blankert, B.; Bodoki, E.; Bollo, S.; Vire, J. C.; Sandulescu, R.; Nomura, A.; Kauffmann, J. M. Amperometric biosensor based on horseradish peroxidase-immobilised magnetic microparticles. Sensors and Actuators B-Chemical 2006, 113(2), 749–754. [Google Scholar]
- Kueng, A.; Kranz, C.; Mizaikoff, B. Amperometric atp biosensor based on polymer entrapped enzymes. Biosensors & Bioelectronics 2004, 19(10), 1301–1307. [Google Scholar]
- Bakker, E.; Pretsch, E. Potentiometric sensors for trace-level analysis. Trac-Trends in Analytical Chemistry 2005, 24(3), 199–207. [Google Scholar]
- Buerk, D. Biosensors. Theory and Applications; Technomic Publishing: Lancaster, 1993. [Google Scholar]
- Caras, S.; Janata, J. Field-effect transistor sensitive to penicillin. Analytical Chemistry 1980, 52(12), 1935–1937. [Google Scholar]
- Hafeman, D.G.; Parce, J. W.; McConnell, H. M. Light-addressable potentiometric sensor for biochemical systems. Science 1988, 240(4856), 1182–1185. [Google Scholar]
- Mourzina, Y.; Yoshinobu, T.; Schubert, J.; Luth, H.; Iwasaki, H.; Schoning, M.J. Ion-selective light-addressable potentiometric sensor (laps) with chalcogenide thin film prepared by pulsed laser deposition. Sensors and Actuators B-Chemical 2001, 80(2), 136–140. [Google Scholar]
- Poghossian, A.; Yoshinobu, T.; Simonis, A.; Ecken, H.; Luth, H.; Schoning, M.J. Penicillin detection by means of field-effect based sensors: Enfet, capacitive eis sensor or laps? Sensors and Actuators B-Chemical 2001, 78(1-3), 237–242. [Google Scholar]
- Xu, G.; Ye, X.; Qin, L.; Xu, Y.; Li, Y.; Li, R.; Wang, P. Cell-based biosensors based on light-addressable potentiometric sensors for single cell monitoring. Biosensors & Bioelectronics 2005, 20, 17571763. [Google Scholar]
- Kloock, J.P.; Moreno, L.; Bratov, A.; Huachupoma, S.; Xu, J.; Wagner, T.; Yoshinobu, T.; Ermolenko, Y.; Vlasov, Y. G.; Schoning, M. J. Pld-prepared cadmium sensors based on chalcogenide glasses- isfet, laps and mu ise semiconductor structures. Sensors and Actuators B-Chemical 2006, 118(1-2), 149–155. [Google Scholar]
- Stein, B.; George, M.; Gaub, H.E.; Parak, W. J. Extracellular measurements of averaged ionic currents with the light-addressable potentiometric sensor (laps). Sensors and Actuators B-Chemical 2004, 98(2-3), 299–304. [Google Scholar]
- Shishkanova, T.V.; Volf, R.; Krondak, M.; Kral, V. Functionalization of pvc membrane with ss oligonucleotides for a potentiometric biosensor. Biosensors & Bioelectronics 2007, 22(11), 2712–2717. [Google Scholar]
- Cullen, D.C.; Sethi, R. S.; Lowe, C. R. Multianalyte miniature conductance biosensor. Analytica Chimica Acta 1990, 231(1), 33–40. [Google Scholar]
- Contractor, A.Q.; Sureshkumar, T. N.; Narayanan, R.; Sukeerthi, S.; Lal, R.; Srinivasa, R. S. Conducting polymer-based biosensors. Electrochimica Acta 1994, 39(8-9), 1321–1324. [Google Scholar]
- Yagiuda, K.; Hemmi, A.; Ito, S.; Asano, Y.; Fushinuki, Y.; Chen, C.Y.; Karube, I. Development of a conductivity-based immunosensor for sensitive detection of methamphetamine (stimulant drug) in human urine. Biosensors & Bioelectronics 1996, 11(8), 703–707. [Google Scholar]
- Chouteau, C.; Dzyadevych, S.; Chovelon, J.M.; Durrieu, C. Development of novel conductometric biosensors based on immobilised whole cell chlorella vulgaris microalgae. Biosensors & Bioelectronics 2004, 19(9), 1089–1096. [Google Scholar]
- Heyrovsky, J. The development of polarographic analysis. Analyst 1956, 81(961), 189, –&. [Google Scholar]
- Katz, E.; Willner, I. Probing biomolecular interactions at conductive and semiconductive surfaces by impedance spectroscopy: Routes to impedimetric immunosensors, dna-sensors, and enzyme biosensors. Electroanalysis 2003, 15(11), 913–947. [Google Scholar]
- Pei, R.J.; Cheng, Z. L.; Wang, E. K.; Yang, X. R. Amplification of antigen-antibody interactions based on biotin labeled protein-streptavidin network complex using impedance spectroscopy. Biosensors & Bioelectronics 2001, 16(6), 355–361. [Google Scholar]
- Gosser, D. K. Cyclic Voltammetry, Simulation and Analysis of Reaction Mechanisms.; VHC Publishers, Inc., 1994. [Google Scholar]
- Li, J.P.; Peng, T. Z.; Peng, Y. Q. A cholesterol biosensor based on entrapment of cholesterol oxidase in a silicic sol-gel matrix at a prussian blue modified electrode. Electroanalysis 2003, 15(12), 1031–1037. [Google Scholar]
- Liu, Y.; Yuan, R.; Chai, Y.Q.; Tang, D. P.; Dai, J. Y.; Zhong, X. Direct electrochemistry of horseradish peroxidase immobilized on gold colloid/cysteine/nafion-modified platinum disk electrode. Sensors and Actuators B-Chemical 2006, 115(1), 109–115. [Google Scholar]
- Patolsky, F.; Zayats, M.; Katz, E.; Willner, I. Precipitation of an insoluble product on enzyme monolayer electrodes for biosensor applications: Characterization by faradaic impedance spectroscopy, cyclic voltammetry, and microgravimetric quartz crystal microbalance analyses. Analytical Chemistry 1999, 71(15), 3171–3180. [Google Scholar]
- Nahir, T.M.; Buck, R. P. Modified cottrell behavior in thin-layers - applied voltage steps under diffusion control for constant-resistance systems. Journal of Electroanalytical Chemistry 1992, 341(1-2), 1–14. [Google Scholar]
- Michael, D.J.; Wightman, R. M. Electrochemical monitoring of biogenic amine neurotransmission in real time. Journal of Pharmaceutical and Biomedical Analysis 1999, 19(1-2), 33–46. [Google Scholar]
- Martins, M. C. L.; Fonseca, C.; Barbosa, M. A.; Ratner, B. D. Albumin adsorption on alkanethiols self-assembled monolayers on gold electrodes studied by chronopotentiometry. Biomaterials 2003, 24(21), 3697–3706. [Google Scholar]
- Lorenz, W.; Schulze, K.D. Application of transform-impedance spectrometry. Journal of Electro-analytical Chemistry 1975, 65(1), 141–153. [Google Scholar]
- Willner, I.; Katz, E. e. Bioelectronics.Wiley-VCH, 1st edition; 2005. [Google Scholar]
- Tlili, A.; Abdelghani, A.; Ameur, S.; Jaffrezic-Renault, N. Impedance spectroscopy and affinity measurement of specific antibody-antigen interaction. Materials Science & Engineering C-Biomimetic and Supramolecular Systems 2006, 26(2-3), 546–550. [Google Scholar]
- Fernandez-Sanchez, C.; McNeil, C.J.; Rawson, K. Electrochemical impedance spectroscopy studies of polymer degradation: application to biosensor development. Trac-Trends in Analytical Chemistry 2005, 24(1), 37–48. [Google Scholar]
- Sumner, C.; Sabot, A.; Turner, K.; Krause, S. A transducer based on enzyme-induced degradation of thin polymer films monitored by surface plasmon resonance. Analytical Chemistry 2000, 72(21), 5225–5232. [Google Scholar]
- Tong, Z.Q.; Yuan, R.; Chai, Y. Q.; Xie, Y.; Chen, S. H. A novel and simple biomolecules immobilization method: Electro-deposition zro2 doped with hrp for fabrication of hydrogen peroxide biosensor. Journal of Biotechnology 2007, 128(3), 567–575. [Google Scholar]
- Bakker, E. Electrochemical sensors. Analytical Chemistry 2004, 76(12), 3285–3298. [Google Scholar]
- Hang, T.C.; Guiseppi-Elie, A. Frequency dependent and surface characterization of dna immobilization and hybridization. Biosensors & Bioelectronics 2004, 19(11), 1537–1548. [Google Scholar]
- Patolsky, F.; Katz, E.; Bardea, A.; Willner, I. Enzyme-linked amplified electrochemical sensing of oligonucleotide-dna interactions by means of the precipitation of an insoluble product and using impedance spectroscopy. Langmuir 1999, 15(11), 3703–3706. [Google Scholar]
- Vagin, M.Y.; Karyakin, A. A.; Hianik, T. Surfactant bilayers for the direct electrochemical detection of affinity interactions. Bioelectrochemistry 2002, 56(1-2), 91–93. [Google Scholar]
- Ma, K.S.; Zhou, H.; Zoval, J.; Madou, M. Dna hybridization detection by label free versus impedance amplifying label with impedance spectroscopy. Sensors and Actuators B-Chemical 2006, 114(1), 58–64. [Google Scholar]
- Vlasov, Y.G.; Tarantov, Y. A.; Bobrov, P. V. Analytical characteristics and sensitivity mechanisms of electrolyte-insulator-semiconductor system-based chemical sensors - a critical review. Analytical and Bioanalytical Chemistry 2003, 376(6), 788–796. [Google Scholar]
- Berney, H.; West, J.; Haefele, E.; Alderman, J.; Lane, W.; Collins, J.K. A dna diagnostic biosensor: development, characterisation and performance. Sensors and Actuators B-Chemical 2000, 68(1-3), 100–108. [Google Scholar]
- Horenstein, M. Microelectronic Circuits and Devices; Prentice-Hall: Englewood Cliffs, 1990. [Google Scholar]
- Luo, X.L.; Xu, J. J.; Zhao, W.; Chen, H. Y. Glucose biosensor based on enfet doped with sio2 nanoparticles. Sensors and Actuators B-Chemical 2004, 97(2-3), 249–255. [Google Scholar]
- Schoning, M.J.; Poghossian, A. Bio feds (field-effect devices): State-of-the-art and new directions. Electroanalysis 2006, 18(19-20), 1893–1900. [Google Scholar]
- Errachid, A.; Zine, N.; Samitier, J.; Bausells, J. Fet-based chemical sensor systems fabricated with standard technologies. Electroanalysis 2004, 16(22), 1843–1851. [Google Scholar]
- Poghossian, A.S. Method of fabrication of isfet-based biosensors on an si-sio2-si structure. Sensors and Actuators B-Chemical 1997, 44(1-3), 361–364. [Google Scholar]
- Estrela, P.; Stewart, A.G.; Yan, F.; Migliorato, P. Field effect detection of biomolecular interactions. Electrochimica Acta 2005, 50(25-26), 4995–5000. [Google Scholar]
- Oelssner, W.; Zosel, J.; Guth, U.; Pechstein, T.; Babel, W.; Connery, J.G.; Demuth, C.; Ganseve, M. G.; Verburg, J. B. Encapsulation of isfet sensor chips. Sensors and Actuators B-Chemical 2005, 105(1), 104–117. [Google Scholar]
- Seo, H.I.; Kim, C. S.; Sohn, B. K.; Yeow, T.; Son, M. T.; Haskard, M. Isfet glucose sensor based on a new principle using the electrolysis of hydrogen peroxide. Sensors and Actuators B-Chemical 1997, 40(1), 1–5. [Google Scholar]
- Sohn, B.K.; Cho, B. W.; Kim, C. S.; Kwon, D. H. Isfet glucose and sucrose sensors by using platinum electrode and photo-crosslinkable polymers. Sensors and Actuators B-Chemical 1997, 41(1-3), 7–11. [Google Scholar]
- Poghossian, A.; Luth, H.; Schultze, J.W.; Schoning, M. J. (bio-)chemical and physical microsensor arrays using an identical transducer principle. Electrochimica Acta 2001, 47(1-2), 243–249. [Google Scholar]
- Yeow, T. C. W.; Haskard, M. R.; Mulcahy, D. E.; Seo, H. I.; Kwon, D. H. A very large integrated ph-isfet sensor array chip compatible with standard cmos processes. Sensors and Actuators B-Chemical 1997, 44(1-3), 434–440. [Google Scholar]
- Volotovsky, V.; Kim, N. Cyanide determination by an isfet-based peroxidase biosensor. Biosensors & Bioelectronics 1998, 13(9), 1029–1033. [Google Scholar]
- Kharitonov, A.B.; Zayats, M.; Lichtenstein, A.; Katz, E.; Willner, I. Enzyme monolayer-functionalized field-effect transistors for biosensor applications. Sensors and Actuators B-Chemical 2000, 70(1-3), 222–231. [Google Scholar]
- Zayats, M.; Kharitonov, A.B.; Katz, E.; Buckmann, A. F.; Willner, I. An integrated nad(+)-dependent enzyme-functionalized field-effect transistor (enfet) system: development of a lactate biosensor. Biosensors & Bioelectronics 2000, 15(11-12), 671–680. [Google Scholar]
- Yin, L.T.; Chou, J. C.; Chung, W. Y.; Sun, T. P.; Hsiung, K. P.; Hsiung, S. K. Glucose enfet doped with mno2 powder. Sensors and Actuators B-Chemical 2001, 76(1-3), 187–192. [Google Scholar]
- Park, K.Y.; Choi, S. B.; Lee, M.; Sohn, B. K.; Choi, S. Y. Isfet glucose sensor system with fast recovery characteristics by employing electrolysis. Sensors and Actuators B-Chemical 2002, 83(1-3), 90–97. [Google Scholar]
- Poghossian, A.; Schoning, M.I. Detecting both physical and (bio-)chemical parameters by means of isfet devices. Electroanalysis 2004, 16(22), 1863–1872. [Google Scholar]
- Poghossian, A.; Cherstvy, A.; Ingebrandt, S.; Offenhausser, A.; Schoning, M.J. Possibilities and limitations of label-free detection of dna hybridization with field-effect-based devices. Sensors and Actuators B-Chemical 2005, 111, 470–480. [Google Scholar]
- Koo, S.M.; Edelstein, M. D.; Li, Q. L.; Richter, C. A.; Vogel, E. M. Silicon nanowires as enhancement-mode schottky barrier field-effect transistors. Nanotechnology 2005, 16(9), 1482–1485. [Google Scholar]
- Cui, Y.; Wei, Q.Q.; Park, H. K.; Lieber, C. M. Nanowire nanosensors for highly sensitive and selective detection of biological and chemical species. Science 2001, 293(5533), 1289–1292. [Google Scholar]
- Cui, Y.; Zhong, Z.H.; Wang, D. L.; Wang, W. U.; Lieber, C. M. High performance silicon nanowire field effect transistors. Nano Letters 2003, 3(2), 149–152. [Google Scholar]
- Gao, Z.Q.; Agarwal, A.; Trigg, A. D.; Singh, N.; Fang, C.; Tung, C. H.; Fan, Y.; Buddharaju, K. D.; Kong, J. M. Silicon nanowire arrays for label-free detection of dna. Analytical Chemistry 2007, 79(9), 3291–3297. [Google Scholar]
- Lieber, C.M.; Wang, Z. L. Functional nanowires. Mrs Bulletin 2007, 32(2), 99–108. [Google Scholar]
- Tansil, N.C.; Gao, Z. Q. Nanoparticles in biomolecular detection. Nano Today 2006, 1(1), 28–37. [Google Scholar]
- Xiao, Y.; Patolsky, F.; Katz, E.; Hainfeld, J.F.; Willner, I. Plugging into enzymes: Nanowiring of redox enzymes by a gold nanoparticle. Science 2003, 299(5614), 1877–1881. [Google Scholar]
- Wang, D.H.; Kou, R.; Gil, M. P.; Jakobson, H. P.; Tang, J.; Yu, D. H.; Lu, Y. F. Templated synthesis, characterization, and sensing application of macroscopic platinum nanowire network electrodes. Journal of Nanoscience and Nanotechnology 2005, 5(11), 1904–1909. [Google Scholar]
- Tilke, A.T.; Simmel, F. C.; Lorenz, H.; Blick, R. H.; Kotthaus, J. P. Quantum interference in a one-dimensional silicon nanowire. Physical Review B 2003, 68(7), 6. [Google Scholar]
- Khanal, D. R.; Yim, J. W. L.; Walukiewicz, W.; Wu, J. Effects of quantum confinement on the doping limit of semiconductor nanowires. Nano Letters 2007, 0(0). [Google Scholar]
- Elfstrom, N.; Juhasz, R.; Sychugov, I.; Engfeldt, T.; Karlstrom, A. E.; Linnros, J. Surface charge sensitivity of silicon nanowires: Size dependence. Nano Letters 2007. [Google Scholar]
- Foley, E.L.; Candela, D.; Martini, K. M.; Tuominen, M. T. An undergraduate laboratory experiment on quantized conductance in nanocontacts. American Journal of Physics 1999, 67(5), 389–393. [Google Scholar]
- Landauer, R. Conductance determined by transmission - probes and quantized constriction resistance. Journal of Physics-Condensed Matter 1989, 1(43), 8099–8110. [Google Scholar]
- Agarwal, A.; Sen, D. Conductance of quantum wires: A numerical study of effects of an impurity and interactions. Physical Review B 2006, 73(4), 14. [Google Scholar]
- Das, M.P.; Green, F. Landauer formula without landauer's assumptions. Journal of Physics-Condensed Matter 2003, 15(45), L687–L693. [Google Scholar]
- Patolsky, F.; Timko, B.P.; Zheng, G. F.; Lieber, C. M. Nanowire-based nanoelectronic devices in the life sciences. MRS Bulletin 2007, 32(2), 142–149, Figures 1a & 1c. [Google Scholar]
- Zheng, G.F.; Patolsky, F.; Cui, Y.; Wang, W. U.; Lieber, C. M. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nature Biotechnology 2005, 23(10), 1294–1301. [Google Scholar]
- Yeh, J.I.; Zimmt, M. B.; Zimmerman, A. L. Nanowiring of a redox enzyme by metallized peptides. Biosensors & Bioelectronics 2005, 21(6), 973–978. [Google Scholar]
- Arwin, H.; Poksinski, M.; Johansen, K. Total internal reflection ellipsometry: principles and applications. Applied Optics 2004, 43(15), 3028–3036. [Google Scholar]
- Elwing, H. Protein absorption and ellipsometry in biomaterial research. Biomaterials 1998, 19(4-5), 397–406. [Google Scholar]
- Goodall, D.G.; Stevens, G. W.; Beaglehole, D.; Gee, M. L. Imaging ellipsometry reflectometry for profiling the shape of a deformable droplet as it approaches an interface. Langmuir 1999, 15(13), 4579–4583. [Google Scholar]
- Pattnaik, P. Surface plasmon resonance - applications in understanding receptor-ligand interaction. Applied Biochemistry and Biotechnology 2005, 126(2), 79–92. [Google Scholar]
- McDonnell, J.M. Surface plasmon resonance: towards an understanding of the mechanisms of biological molecular recognition. Current Opinion in Chemical Biology 2001, 5(5), 572–577. [Google Scholar]
- Liedberg, B.; Nylander, C.; Lundstrom, I. Biosensing with surface-plasmon resonance - how it all started. Biosensors & Bioelectronics 1995, 10(8), R1–R9. [Google Scholar]
- Campagnolo, C.; Meyers, K.J.; Ryan, T.; Atkinson, R. C.; Chen, Y. T.; Scanlan, M. J.; Ritter, G.; Old, L. J.; Batt, C. A. Real-time, label-free monitorine, of tumor antigen and serum antibody interactions. Journal of Biochemical and Biophysical Methods 2004, 61(3), 283–298. [Google Scholar]
- Jung, L.S.; Campbell, C. T.; Chinowsky, T. M.; Mar, M. N.; Yee, S. S. Quantitative interpretation of the response of surface plasmon resonance sensors to adsorbed films. Langmuir 1998, 14(19), 5636–5648. [Google Scholar]
- Badia, A.; Arnold, S.; Scheumann, V.; Zizlsperger, M.; Mack, J.; Jung, G.; Knoll, W. Probing the electrochemical deposition and/or desorption of self-assembled and electropolymerizable organic thin films by surface plasmon spectroscopy and atomic force microscopy. Sensors and Actuators B-Chemical 1999, 54(1-2), 145–165. [Google Scholar]
- De Feijter, J.; Benjamins, J.; Veer, V. Ellipsometry as a tool to study the adsorption behaviour of synthetic and biopolymers at the air-water interface. Biopolymers 1978, 17(7), 1759–1772. [Google Scholar]
- Voros, J.; Ramsden, J.J.; Csucs, G.; Szendro, I.; De Paul, S. M.; Textor, M.; Spencer, N. D. Optical grating coupler biosensors. Biomaterials 2002, 23(17), 3699–3710. [Google Scholar]
- Georgiadis, R.; Peterlinz, K.P.; Peterson, A. W. Quantitative measurements and modeling of kinetics in nucleic acid monolayer films using spr spectroscopy. Journal of the American Chemical Society 2000, 122(13), 3166–3173. [Google Scholar]
- Heaton, R.J.; Peterson, A. W.; Georgiadis, R. M. Electrostatic surface plasmon resonance: Direct electric field-induced hybridization and denaturation in monolayer nucleic acid films and label-free discrimination of base mismatches. Proceedings of the National Academy of Sciences of the United States of America 2001, 98(7), 3701–3704. [Google Scholar]
- Mullett, W.M.; Lai, E. P.C.; Yeung, J. M. Surface plasmon resonance-based immunoassays. Methods 2000, 22(1), 77–91. [Google Scholar]
- Kang, X.F.; Cheng, G. J.; Dong, S. J. A novel electrochemical spr biosensor. Electrochemistry Communications 2001, 3(9), 489–493. [Google Scholar]
- Lavers, C.R.; Harris, R. D.; Hao, S.; Wilkinson, J. S.; Odwyer, K.; Brust, M.; Schiffrin, D. J. Electrochemically-controlled wave-guide-coupled surface-plasmon sensing. Journal of Electroanalytical Chemistry 1995, 387(1-2), 11–22. [Google Scholar]
- Baba, A.; Park, M.K.; Advincula, R. C.; Knoll, W. Simultaneous surface plasmon optical and electrochemical investigation of layer-by-layer self-assembled conducting ultrathin polymer films. Langmuir 2002, 18(12), 4648–4652. [Google Scholar]
- Peterlinz, K.A.; Georgiadis, R. In situ kinetics of self-assembly by surface plasmon resonance spectroscopy. Langmuir 1996, 12(20), 4731–4740. [Google Scholar]
- Georgiadis, R.; Peterlinz, K.A.; Rahn, J. R.; Peterson, A. W.; Grassi, J. H. Surface plasmon resonance spectroscopy as a probe of in-plane polymerization in monolayer organic conducting films. Langmuir 2000, 16(17), 6759–6762. [Google Scholar]
- Peterson, A.W.; Heaton, R. J.; Georgiadis, R. M. The effect of surface probe density on dna hybridization. Nucleic Acids Research 2001, 29(24), 5163–5168. [Google Scholar]
- Wang, F.; Wang, J.L.; Chen, H. J.; Dong, S. J. Assembly process of cuhcf/mpa multilayers on gold nanoparticles modified electrode and characterization by electrochemical spr. Journal of Electroanalytical Chemistry 2007, 600(2), 265–274. [Google Scholar]
- Kurrat, R.; Textor, M.; Ramsden, J.J.; Boni, P.; Spencer, N. D. Instrumental improvements in optical waveguide light mode spectroscopy for the study of biomolecule adsorption. Review of Scientific Instruments 1997, 68(5), 2172–2176. [Google Scholar]
- Bearinger, J.P.; Voros, J.; Hubbell, J. A.; Textor, M. Electrochemical optical waveguide lightmode spectroscopy (ec-owls): A pilot study using evanescent-field optical sensing under voltage control to monitor polycationic polymer adsorption onto indium tin oxide (ito)-coated waveguide chips. Biotechnology and Bioengineering 2003, 82(4), 465–473. [Google Scholar]
- Brusatori, M.A.; Van Tassel, P. R. Biosensing under an applied voltage using optical waveguide lightmode spectroscopy. Biosensors & Bioelectronics 2003, 18(10), 1269–1277. [Google Scholar]
- Ying, P.Q.; Viana, A. S.; Abrantes, L. M.; Jin, G. Adsorption of human serum albumin onto gold: a combined electrochemical and ellipsometric study. Journal of Colloid and Interface Science 2004, 279(1), 95–99. [Google Scholar]
- Wang, Z.H.; Viana, A. S.; Jin, G.; Abrantes, L. M. Immunosensor interface based on physical and chemical immunogylobulin g adsorption onto mixed self-assembled monolayers. Bioelectrochemistry 2006, 69(2), 180–186. [Google Scholar]
- Yu, Y.; Jin, G. Influence of electrostatic interaction on fibrinogen adsorption on gold studied by imaging ellipsometry combined with electrochemical methods. Journal of Colloid and Interface Science 2005, 283(2), 477–481. [Google Scholar]
- Marx, K. Quartz crystal microbalance: A useful tool for studying thin polymer films and complexes biomolecular systems at the solution-surface interface. Bio Macromolecules 2002, 4(5), 1099–1120. [Google Scholar]
- Hook, F.; Rodahl, M.; Brzezinski, P.; Kasemo, B. Measurements using the quartz crystal microbalance technique of ferritin monolayers on methyl-thiolated gold: Dependence of energy dissipation and saturation coverage on salt concentration. Journal of Colloid and Interface Science 1998, 208(1), 63–67. [Google Scholar]
- Hook, F.; Voros, J.; Rodahl, M.; Kurrat, R.; Boni, P.; Ramsden, J.J.; Textor, M.; Spencer, N. D.; Tengvall, P.; Gold, J.; Kasemo, B. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids and Surfaces B-Biointerfaces 2002, 24(2), 155–170. [Google Scholar]
- Dong, Y.G. The frequency response of qcm in electrochemically characterizing the immobilization on gold electrode. Sensors and Actuators B-Chemical 2005, 108(1-2), 622–626. [Google Scholar]
- Schumacher, R. The quartz microbalance - a novel-approach to the insitu investigation of interfacial phenomena at the solid liquid junction. Angewandte Chemie-International Edition in English 1990, 29(4), 329–343. [Google Scholar]
- Beissenhirtz, M.K.; Kafka, B.; Schafer, D.; Wolny, M.; Lisdat, F. Electrochemical quartz crystal microbalance studies on cytochrome c/polyelectrolyte multilayer assemblies on gold electrodes. Electroanalysis 2005, 17(21), 1931–1937. [Google Scholar]
- Bott, A. Characterization of films immobilized on an electrode surface using the electrochemical quartz crystal microbalance. Current Separation 1999, 18(3), 79–83. [Google Scholar]
- Binnig, G.; Rohrer, H. Scanning tunneling microscopy. Helvetica Physica Acta 1982, 55(6), 726–735. [Google Scholar]
- Binnig, G.; Rohrer, H. Scanning tunneling microscopy. Ibm Journal of Research and Development 1986, 30(4), 355–369. [Google Scholar]
- Bae, S.E.; Gewirth, A. A. In situ ec-stm studies of mps, sps, and chloride on cu(100): Structural studies of accelerators for dual damascene electrodeposition. Langmuir 2006, 22(25), 10315–10321. [Google Scholar]
- Colton, R.J.; Baselt, D. R.; Dufrene, Y. F.; Green, J. B.D.; Lee, G. U. Scanning probe microscopy. Current Opinion in Chemical Biology 1997, 1(3), 370–377. [Google Scholar]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Physical Review Letters 1986, 56(9), 930–933. [Google Scholar]
- Rugar, D.; Hansma, P. Atomic force microscopy. Physics Today 1990, 43(10), 23–30. [Google Scholar]
- Jalili, N.; Laxminarayana, K. A review of atomic force microscopy imaging systems: application to molecular metrology and biological sciences. Mechatronics 2004, 14(8), 907–945. [Google Scholar]
- Muguruma, H.; Kase, Y. Structure and biosensor characteristics of complex between glucose oxidase and plasma-polymerized nanothin film. Biosensors & Bioelectronics 2006, 22(5), 737–743. [Google Scholar]
- Tominaga, M.; Ohira, A.; Yamaguchi, Y.; Kunitake, M. Electrochemical, afm and qcm studies on ferritin immobilized onto a self-assembled monolayer-modified gold electrode. Journal of Electroanalytical Chemistry 2004, 566(2), 323–329. [Google Scholar]
- Immoos, C.E.; Lee, S. J.; Grinstaff, M. W. Conformationally gated electrochemical gene detection. Chembiochem 2004, 5(8), 1100–1103. [Google Scholar]
- Dong, Y.Z.; Shannon, C. Heterogeneous immunosensing using antigen and antibody monolayers on gold surfaces with electrochemical and scanning probe detection. Analytical Chemistry 2000, 72(11), 2371–2376. [Google Scholar]
- Bergamini, J.F.; Ghilane, J.; Guilloux-Viry, M.; Hapiot, P. In situ ec-afm imaging of cathodic modifications of platinum surfaces performed in dimethylformamide. Electrochemistry Communications 2004, 6(2), 188–192. [Google Scholar]
- Yamaguchi, Y.; Shiota, M.; Nakayama, Y.; Hirai, N.; Hara, S. Combined in situ ec-afm and cv measurement study on lead electrode for lead-acid batteries. Journal of Power Sources 2001, 93(1-2), 104–111. [Google Scholar]
- Eliaz, N.; Eliyahu, M. Electrochemical processes of nucleation and growth of hydroxyapatite on titanium supported by real-time electrochemical atomic force microscopy. Journal of Biomedical Materials Research Part A 2007, 80A(3), 621–634. [Google Scholar]
- Bard, A.J.; Fan, F. R.F.; Kwak, J.; Lev, O. Scanning electrochemical microscopy - introduction and principles. Analytical Chemistry 1989, 61(2), 132–138. [Google Scholar]
- Kwak, J.; Bard, A.J. Scanning electrochemical microscopy - apparatus and two-dimensional scans of conductive and insulating substrates. Analytical Chemistry 1989, 61(17), 1794–1799. [Google Scholar]
- Kwak, J.; Bard, A.J. Scanning electrochemical microscopy - theory of the feedback mode. Analytical Chemistry 1989, 61(11), 1221–1227. [Google Scholar]
- Sun, P.; Laforge, F.O.; Mirkin, M. V. Scanning electrochemical microscopy in the 21st century. Physical Chemistry Chemical Physics 2007, 9(7), 802–823. [Google Scholar]
- Edwards, M.A.; Martin, S.; Whitworth, A. L.; Macpherson, J. V.; Unwin, P. R. Scanning electrochemical microscopy: principles and applications to biophysical systems. Physiological Measurement 2006, 27(12), R63–R108. [Google Scholar]
- Amemiya, S.; Guo, J.D.; Xiong, H.; Gross, D. A. Biological applications of scanning electrochemical microscopy: chemical imaging of single living cells and beyond. Analytical and Bioanalytical Chemistry 2006, 386(3), 458–471. [Google Scholar]
- Kueng, A.; Kranz, C.; Mizaikoff, B. Imaging of atp membrane transport with dual micro-disk electrodes and scanning electrochemical microscopy. Biosensors & Bioelectronics 2005, 21(2), 346–353. [Google Scholar]
- Kueng, A.; Kranz, C.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Afm-tip-integrated amperometric microbiosensors: High-resolution imaging of membrane transport. Angewandte Chemie-International Edition 2005, 44(22), 3419–3422. [Google Scholar]
- Fortin, E.; Defontaine, Y.; Mailley, P.; Livache, T.; Szunerits, S. Micro-imprinting of oligonucleotides and oligonucleotide gradients on gold surfaces: A new approach based on the combination of scanning electrochemical microscopy and surface plasmon resonance imaging (secm/spr-i). Electroanalysis 2005, 17(5-6), 495–503. [Google Scholar]
- Cliffel, D.E.; Bard, A. J. Scanning electrochemical microscopy. 36. a combined scanning electrochemical microscope quartz crystal microbalance instrument for studying thin films. Analytical Chemistry 1998, 70(9), 1993–1998. [Google Scholar]
- Gollas, B.; Bartlett, P.N.; Denuault, G. An instrument for simultaneous eqcm impedance and secm measurements. Analytical Chemistry 2000, 72(2), 349–356. [Google Scholar]
- Xiang, C.H.; Xie, Q. J.; Hu, J. M.; Yao, S. Symmetric current oscillations at tip and substrate electrodes of scanning electrochemical microscope during silver deposition/stripping. Journal of Electroanalytical Chemistry 2005, 584(2), 201–209. [Google Scholar]
- Boldt, F.M.; Heinze, J.; Diez, M.; Petersen, J.; Borsch, M. Real-time ph microscopy down to the molecular level by combined scanning electrochemical microscopy/single-molecule fluorescence spectroscopy. Analytical Chemistry 2004, 76(13), 3473–3481. [Google Scholar]
- Shi, G.D.; Garfias-Mesias, L. F.; Smyrl, W. H. Preparation of a gold-sputtered optical fiber as a microelectrode for electrochemical microscopy. Journal of the Electrochemical Society 1998, 145(6), 2011–2016. [Google Scholar]
- Fan, F.R. F.; Cliffel, D.; Bard, A. J. Scanning electrochemical microscopy. 37. light emission by electrogenerated chemiluminescence at secm tips and their application to scanning optical microscopy. Analytical Chemistry 1998, 70(14), 2941–2948. [Google Scholar]
- Gardner, C.E.; Macpherson, J. V. Atomic force microscopy probes go electrochemical. Analytical Chemistry 2002, 74(21), 576A–584A. [Google Scholar]
- Kurulugama, R.T.; Wipf, D. O.; Takacs, S. A.; Pongmayteegul, S.; Garris, P. A.; Baur, J. E. Scanning electrochemical microscopy of model neurons: Constant distance imaging. Analytical Chemistry 2005, 77(4), 1111–1117. [Google Scholar]
- Macpherson, J.V.; Unwin, P. R. Combined scanning electrochemical-atomic force microscopy. Analytical Chemistry 2000, 72(2), 276–285. [Google Scholar]
- Macpherson, J.V.; Unwin, P. R. Noncontact electrochemical imaging with combined scanning electrochemical atomic force microscopy. Analytical Chemistry 2001, 73(3), 550–557. [Google Scholar]
- Gardner, C.E.; Unwin, P. R.; Macpherson, J. V. Correlation of membrane structure and transport activity using combined scanning electrochemical-atomic force microscopy. Electrochemistry Communications 2005, 7(6), 612–618. [Google Scholar]
- Kueng, A.; Kranz, C.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Integrated afm-secm in tapping mode: Simultaneous topographical and electrochemical imaging of enzyme activity. Angewandte Chemie-International Edition 2003, 42(28), 3238–3240. [Google Scholar]
- Kranz, C.; Kueng, A.; Lugstein, A.; Bertagnolli, E.; Mizaikoff, B. Mapping of enzyme activity by detection of enzymatic products during afm imaging with integrated secm-afm probes. Ultramicroscopy 2004, 100(3-4), 127–134. [Google Scholar]
- Jones, C.E.; Unwin, P. R.; Macpherson, J. V. In situ observation of the surface processes involved in dissolution from the cleavage surface of calcite in aqueous solution using combined scanning electrochemical-atomic force microscopy (secm-afm). Chemphyschem 2003, 4(2), 139–146. [Google Scholar]
- Kotitz, R.; Matz, H.; Trahms, L.; Koch, H.; Weitschies, W.; Rheinlander, T.; Semmler, W.; Bunte, T. Squid based remanence measurements for immunoassays. Ieee Transactions on Applied Superconductivity 1997, 7(2), 3678–3681. [Google Scholar]
- Matz, H.; Drung, D.; Hartwig, S.; Gross, H.; Kotitz, R.; Muller, W.; Vass, A.; Weitschies, W.; Trahms, L. A squid measurement system for immunoassays. Applied Superconductivity 1998, 6(10-12), 577–583. [Google Scholar]
- Kotitz, R.; Weitschies, W.; Trahms, L.; Brewer, W.; Semmler, W. Determination of the binding reaction between avidin and biotin by relaxation measurements of magnetic nanoparticles. Journal of Magnetism and Magnetic Materials 1999, 194(1-3), 62–68. [Google Scholar]
- Lange, J.; Kotitz, R.; Haller, A.; Trahms, L.; Semmler, W.; Weitschies, W. Magnetorelaxometry -a new binding specific detection method based on magnetic nanoparticles. Journal of Magnetism and Magnetic Materials 2002, 252(1-3), 381–383. [Google Scholar]
- Meyer, M.H. F.; Hartmann, M.; Krause, H. J.; Blankenstein, G.; Mueller-Chorus, B.; Oster, J.; Miethe, P.; Keusgen, M. Crp determination based on a novel magnetic biosensor. Biosensors & Bioelectronics 2007, 22(6), 973–979. [Google Scholar]
- Krause, H.-J.; Zhang, Y.; Wolters, N.; Miethe, P.; Plaksin, D. Method and device for selectively detecting ferromagnetic or superparamagnetic particles. 2003. [Google Scholar]
- Baselt, D.R.; Lee, G. U.; Colton, R. J. Biosensor based on force microscope technology. Journal of Vacuum Science & Technology B 1996, 14(2), 789–793. [Google Scholar]
- Larsson, K.; Kriz, K.; Kriz, D. Magnetic transducers in biosensors and bioassays. Analusis 1999, 27(7), 617–621. [Google Scholar]
- Kriz, C.B.; Radevik, K.; Kriz, D. Magnetic permeability measurements in bioanalysis and biosensors. Analytical Chemistry 1996, 68(11), 1966–1970. [Google Scholar]
- Babb, C.W.; Coon, D. R.; Rechnitz, G. A. Biomagnetic neurosensors .3. noninvasive sensors using magnetic stimulation and biomagnetic detection. Analytical Chemistry 1995, 67(4), 763–769. [Google Scholar]
- Baibich, M.N.; Broto, J. M.; Fert, A.; Vandau, F. N.; Petroff, F.; Eitenne, P.; Creuzet, G.; Friederich, A.; Chazelas, J. Giant magnetoresistance of (001)fe/(001) cr magnetic superlattices. Physical Review Letters 1988, 61(21), 2472–2475. [Google Scholar]
- Baselt, D.R.; Lee, G. U.; Natesan, M.; Metzger, S. W.; Sheehan, P. E.; Colton, R. J. A biosensor based on magnetoresistance technology. Biosensors & Bioelectronics 1998, 13(7-8), 731–739. [Google Scholar]
- Graham, D.L.; Ferreira, H. A.; Freitas, P. P. Magnetoresistive-based biosensors and biochips. Trends in Biotechnology 2004, 22(9), 455–462. [Google Scholar]
- Megens, M.; Prins, M. Magnetic biochips: a new option for sensitive diagnostics. Journal of Magnetism and Magnetic Materials 2005, 293(1), 702–708. [Google Scholar]
- Helali, S.; Martelet, C.; Abdelghani, A.; Maaref, M.A.; Jaffrezic-Renault, N. A disposable immunomagnetic electrochemical sensor based on functionalised magnetic beads on gold surface for the detection of atrazine. Electrochimica Acta 2006, 51(24), 5182–5186. [Google Scholar]
- Tang, D.P.; Yuan, R.; Chai, Y. Q. Direct electrochemical immunoassay based on immobilization of protein-magnetic nanoparticle composites on to magnetic electrode surfaces by sterically enhanced magnetic field force. Biotechnology Letters 2006, 28(8), 559–565. [Google Scholar]
- Rife, J.C.; Miller, M. M.; Sheehan, P. E.; Tamanaha, C. R.; Tondra, M.; Whitman, L. J. Design and performance of gmr sensors for the detection of magnetic microbeads in biosensors. Sensors and Actuators a-Physical 2003, 107(3), 209–218. [Google Scholar]
- Edelstein, R.L.; Tamanaha, C. R.; Sheehan, P. E.; Miller, M. M.; Baselt, D. R.; Whitman, L. J.; Colton, R. J. The barc biosensor applied to the detection of biological warfare agents. Biosensors & Bioelectronics 2000, 14(10-11), 805–813. [Google Scholar]
- Pavlickova, P.; Schneider, E.M.; Hug, H. Advances in recombinant antibody microarrays. Clinica Chimica Acta 2004, 343(1-2), 17–35. [Google Scholar]
- Lin, J.H.; Ju, H. X. Electrochemical and chemiluminescent immunosensors for tumor markers. Biosensors & Bioelectronics 2005, 20(8), 1461–1470. [Google Scholar]
- Dai, Z.; Yan, F.; Hua, Y.; Hu, X.Y.; Ju, H. X. Novel amperometric immunosensor for rapid separation-free immunoassay of carcinoembryonic antigen. Journal of Immunological Methods 2004, 287(1-2), 13–20. [Google Scholar]
- Yang, L.J.; Li, Y. B. Afm and impedance spectroscopy characterization of the immobilization of antibodies on indium-tin oxide electrode through self-assembled monolayer of epoxysilane and their capture of escherichia coli o157: H7. Biosensors & Bioelectronics 2005, 20(7), 1407–1416. [Google Scholar]
- Zhang, Y.Y.; Fung, Y. S.; Sun, H.; Zhu, D. R.; Yao, S. Z. Study of protein adsorption on polymer coatings surface by combining quartz crystal microbalance with electrochemical impedance methods. Sensors and Actuators B-Chemical 2005, 108(1-2), 933–942. [Google Scholar]
- Wink, T.; van Zuilen, S.; Bult, A.; van Bennekom, W. Self-assembled monolayers for biosensors. Analyst 1997, 122. [Google Scholar]
- Pan, D.W.; Chen, J. H.; Yao, S. Z.; Tao, W. Y.; Nie, L. H. An amperometric glucose biosensor based on glucose oxidase immobilized in electropolymerized poly(o-aminophenol) and carbon nanotubes composite film on a gold electrode. Analytical Sciences 2005, 21(4), 367–371. [Google Scholar]
- Sotiropoulou, S.; Gavalas, V.; Vamvakaki, V.; Chaniotakis, N.A. Novel carbon materials in biosensor systems. Biosensors & Bioelectronics 2003, 18(2-3), 211–215. [Google Scholar]
- Mena, M.L.; Yanez-Sedeno, P.; Pingarron, J. M. A comparison of different strategies for the construction of amperometric enzyme biosensors using gold nanoparticle-modified electrodes. Analytical Biochemistry 2005, 336(1), 20–27. [Google Scholar]
- Bauer, L.A.; Birenbaum, N. S.; Meyer, G. J. Biological applications of high aspect ratio nanoparticles. Journal of Materials Chemistry 2004, 14(4), 517–526. [Google Scholar]
- Yu, Y.Y.; Chang, S. S.; Lee, C. L.; Wang, C. R.C. Gold nanorods: Electrochemical synthesis and optical properties. Journal of Physical Chemistry B 1997, 101(34), 6661–6664. [Google Scholar]
- Yi, G.C.; Wang, C. R.; Park, W. I. Zno nanorods: synthesis, characterization and applications. Semiconductor Science and Technology 2005, 20(4), S22–S34. [Google Scholar]
- Kuznetsov, B.A.; Shumakovich, G. P.; Koroleva, O. V.; Yaropolov, A. I. On applicability of laccase as label in the mediated and mediatorless electroimmunoassay: effect of distance on the direct electron transfer between laccase and electrode. Biosensors & Bioelectronics 2001, 16(1-2), 73–84. [Google Scholar]
- Ghindilis, A.L.; Atanasov, P.; Wilkins, E. Enzyme-catalyzed direct electron transfer: Fundamentals and analytical applications. Electroanalysis 1997, 9(9), 661–674. [Google Scholar]
- Patolsky, F.; Weizmann, Y.; Willner, I. Long-range electrical contacting of redox enzymes by swcnt connectors. Angewandte Chemie-International Edition 2004, 43(16), 2113–2117. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell. 2002.
- Heller, A. Amperometric biosensors. Current Opinion in Biotechnology 1996, 7(1), 50–54. [Google Scholar]
- Azevedo, A.; Martins, V.; Prazeres, D.; Vojinovic, V.; Cabral, J.; Fonseca, L. Horseradish peroxidase: a valuable tool in biotechnology. Biotechnology annual review 2003, 9, 199–247. [Google Scholar]
- Zhen, G.L.; Eggli, V.; Voros, J.; Zammaretti, P.; Textor, M.; Glockshuber, R.; Kuennemann, E. Immobilization of the enzyme beta-lactamase on biotin-derivatized poly(l-lysine)-g-poly(ethylene glycol)-coated sensor chips: A study on oriented attachment and surface activity by enzyme kinetics and in situ optical sensing. Langmuir 2004, 20(24), 10464–10473. [Google Scholar]
- Chaniotakis, N.A. Enzyme stabilization strategies based on electrolytes and polyelectrolytes for biosensor applications. Analytical and Bioanalytical Chemistry 2004, 378(1), 89–95. [Google Scholar]
- O'Fagain, C. Enzyme stabilization - recent experimental progress. Enzyme and Microbial Technology 2003, 33(2-3), 137–149. [Google Scholar]
- Grieshaber, D.; Reimhult, E.; Vrs, J. Enzymatic biosensors towards a multiplexed electronic detection system for early cancer diagnostics. In 2nd IEEE -NEMS; volume 1, pp. 402–405. Bangkok, 2007. [Google Scholar]
- Sotiropoulou, S.; Vamvakaki, V.; Chaniotakis, N.A. Stabilization of enzymes in nanoporous materials for biosensor applications. Biosensors & Bioelectronics 2005, 20(8), 1674–1679. [Google Scholar]
- Sung, W.J.; Bae, Y. H. Glucose oxidase, lactate oxidase, and galactose oxidase enzyme electrode based on polypyrrole with polyanion/peg/enzyme conjugate dopant. Sensors and Actuators B-Chemical 2006, 114(1), 164–169. [Google Scholar]
- Kim, J.; Grate, J.W.; Wang, P. Nanostructures for enzyme stabilization. Chemical Engineering Science 2006, 61(3), 1017–1026. [Google Scholar]
- Lu, B.; Smyth, M.R.; OKennedy, R. Oriented immobilization of antibodies and its applications in immunoassays and immunosensors. Analyst 1996, 121(3), R29–R32. [Google Scholar]
- LaGraff, J.R.; Chu-LaGraff, Q. Scanning force microscopy and fluorescence microscopy of microcontact printed antibodies and antibody fragments. Langmuir 2006, 22(10), 4685–4693. [Google Scholar]
- Wacker, R.; Schroder, H.; Niemeyer, C.M. Performance of antibody microarrays fabricated by either dna-directed immobilization, direct spotting, or streptavidin-biotin attachment: a comparative study. Analytical Biochemistry 2004, 330(2), 281–287. [Google Scholar]
- Peluso, P.; Wilson, D.S.; Do, D.; Tran, H.; Venkatasubbaiah, M.; Quincy, D.; Heidecker, B.; Poindexter, K.; Tolani, N.; Phelan, M.; Witte, K.; Jung, L. S.; Wagner, P.; Nock, S. Optimizing antibody immobilization strategies for the construction of protein microarrays. Analytical Biochemistry 2003, 312(2), 113–124. [Google Scholar]
- Padeste, C.; Grubelnik, A.; Steiger, B.; Hefti, J.; Tiefenauer, L. Molecular architectures for enzyme sensors. PSI annual report 2001 2001. [Google Scholar]
- Zacco, E.; Pividori, M.I.; Alegret, S. Electrochemical biosensing based on universal affinity biocomposite platforms. Biosensors & Bioelectronics 2006, 21(7), 1291–1301. [Google Scholar]
- Danczyk, R.; Krieder, B.; North, A.; Webster, T.; HogenEsch, H.; Rundell, A. Comparison of antibody functionality using different immobilization methods. Biotechnology and Bioengineering 2003, 84(2), 215–223. [Google Scholar]
- Nielsen, U.B.; Geierstanger, B. H. Multiplexed sandwich assays in microarray format. Journal of Immunological Methods 2004, 290(1-2), 107–120. [Google Scholar]
- Sarkar, P.; Pal, P.S.; Ghosh, D.; Setford, S. J.; Tothill, I. E. Amperometric biosensors for detection of the prostate cancer marker (psa). International Journal of Pharmaceutics 2002, 238(1-2), 1–9. [Google Scholar]
- Haab, B.B. Applications of antibody array platforms. Current Opinion in Biotechnology 2006, 17(4), 415–421. [Google Scholar]
- Peterson, J.; Green, G.; Iida, K.; Caldwell, B.; Kerrison, P.; Bernich, S.; Aoyagi, K.; Lee, S.R. Detection of hepatitis c core antigen in the antibody negative ‘window’ phase of hepatitis c infection. Vox Sanguinis 2000, 78(2), 80–85. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nature Biotechnology 2005, 23(9), 1126–1136. [Google Scholar]
- Hamerscasterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally-occurring antibodies devoid of light-chains. Nature 1993, 363(6428), 446–448. [Google Scholar]
- Vikholm, I. Self-assembly of antibody fragments and polymers onto gold for immunosensing. Sensors and Actuators B-Chemical 2005, 106(1), 311–316. [Google Scholar]
- Ellington, A.D.; Szostak, J. W. Invitro selection of rna molecules that bind specific ligands. Nature 1990, 346(6287), 818–822. [Google Scholar]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment - rna ligands to bacteriophage-t4 dna-polymerase. Science 1990, 249(4968), 505–510. [Google Scholar]
- Tombelli, S.; Minunni, A.; Mascini, A. Analytical applications of aptamers. Biosensors & Bioelectronics 2005, 20(12), 2424–2434. [Google Scholar]
- O'Sullivan, C.K. Aptasensors - the future of biosensing. Analytical and Bioanalytical Chemistry 2002, 372(1), 44–48. [Google Scholar]
- Caruso, F.; Caruso, R.A.; Mohwald, H. Nanoengineering of inorganic and hybrid hollow spheres by colloidal templating. Science 1998, 282(5391), 1111–1114. [Google Scholar]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis, S. A.; Mohwald, H. Novel hollow polymer shells by colloid-templated assembly of polyelectrolytes. Angewandte Chemie-International Edition 1998, 37(16), 2202–2205. [Google Scholar]
- Antipov, A.A.; Sukhorukov, G. B. Polyelectrolyte multilayer capsules as vehicles with tunable permeability. Advances in Colloid and Interface Science 2004, 111(1-2), 49–61. [Google Scholar]
- Heuberger, R.; Sukhorukov, G.; Voros, J.; Textor, M.; Mohwald, H. Biofunctional polyelectrolyte multilayers and microcapsules: Control of non-specific and bio-specific protein adsorption. Advanced Functional Materials 2005, 15(3), 357–366. [Google Scholar]
- Gao, C. Y.; Liu, X. Y.; Shen, J. C.; Mohwald, H. Spontaneous deposition of horseradish peroxidase into polyelectrolyte multilayer capsules to improve its activity and stability. Chemical Communications 2002, (17), 1928–1929. [Google Scholar]
- Volodkin, D.V.; Petrov, A. I.; Prevot, M.; Sukhorukov, G. B. Matrix polyelectrolyte microcapsules: New system for macromolecule encapsulation. Langmuir 2004, 20(8), 3398–3406. [Google Scholar]
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity and applications. Biomolecular Engineering 2001, 18(4), 143–177. [Google Scholar]
- Nasseau, M.; Boublik, Y.; Meier, W.; Winterhalter, M.; Fournier, D. Substrate-permeable encapsulation of enzymes maintains effective activity, stabilizes against denaturation, and protects against proteolytic degradation. Biotechnology and Bioengineering 2001, 75(5), 615–618. [Google Scholar]
- Winterhalter, M.; Hilty, C.; Bezrukov, S.M.; Nardin, C.; Meier, W.; Fournier, D. Controlling membrane permeability with bacterial porins: application to encapsulated enzymes. Talanta 2001, 55(5), 965–971. [Google Scholar]
- Hill, K.J.; Kaszuba, M.; Creeth, J. E.; Jones, M. N. Reactive liposomes encapsulating a glucose oxidase-peroxidase system with antibacterial activity. Biochimica Et Biophysica Acta-Biomembranes 1997, 1326(1), 37–46. [Google Scholar]
- Torchilin, V.P. Peg-based micelles as carriers of contrast agents for different imaging modalities. Advanced Drug Delivery Reviews 2002, 54(2), 235–252. [Google Scholar]
- Torchilin, V.P. Targeted polymeric micelles for delivery of poorly soluble drugs. Cellular and Molecular Life Sciences 2004, 61, 2549–2559. [Google Scholar]
- Gaucher, G.; Dufresne, M.H.; Sant, V. P.; Kang, N.; Maysinger, D.; Leroux, J. C. Block copolymer micelles: preparation, characterization and application in drug delivery. Journal of Controlled Release 2005, 109(1-3), 169–188. [Google Scholar]
- Thurmond, K.B.; Huang, H. Y.; Clark, C. G.; Kowalewski, T.; Wooley, K. L. Shell cross-linked polymer micelles: stabilized assemblies with great versatility and potential. Colloids and Surfaces B-Biointerfaces 1999, 16(1-4), 45–54. [Google Scholar]
- Harada, A.; Kataoka, K. Novel polyion complex micelles entrapping enzyme molecules in the core: Preparation of narrowly-distributed micelles from lysozyme and poly(ethylene glycol)-poly(aspartic acid) block copolymer in aqueous medium. Macromolecules 1998, 31(2), 288–294. [Google Scholar]
- Harada, A.; Kataoka, K. Pronounced activity of enzymes through the incorporation into the core of polyion complex micelles made from charged block copolymers. Journal of Controlled Release 2001, 72(1-3), 85–91. [Google Scholar]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. Journal of Physical Chemistry 1992, 96(9), 3579–3587. [Google Scholar]
- Dong, S.J.; Guo, Y. Z. Organic-phase enzyme electrode operated in water-free solvents. Analytical Chemistry 1994, 66(22), 3895–3899. [Google Scholar]
- Mano, N.; Mao, F.; Heller, A. On the parameters affecting the characteristics of the “wired” glucose oxidase anode. Journal of Electroanalytical Chemistry 2005, 574(2), 347–357. [Google Scholar]
- Mitala, J.J.; Michael, A. C. Improving the performance of electrochemical microsensors based on enzymes entrapped in a redox hydrogel. Analytica Chimica Acta 2006, 556(2), 326–332. [Google Scholar]
- Mao, F.; Mano, N.; Heller, A. Long tethers binding redox centers to polymer backbones enhance electron transport in enzyme “wiring” hydrogels. Journal of the American Chemical Society 2003, 125(16), 4951–4957. [Google Scholar]
- Braun, S.; Rappoport, S.; Zusman, R.; Avnir, D.; Ottolenghi, M. Biochemically active sol-gel glasses - the trapping of enzymes. Materials Letters 1990, 10(1-2), 1–5. [Google Scholar]
- Avnir, D.; Coradin, T.; Lev, O.; Livage, J. Recent bio-applications of sol-gel materials. Journal of Materials Chemistry 2006, 16(11), 1013–1030. [Google Scholar]
- Jin, W.; Brennan, J.D. Properties and applications of proteins encapsulated within sol-gel derived materials. Analytica Chimica Acta 2002, 461(1), 1–36. [Google Scholar]
- Lei, C.X.; Hu, S. Q.; Gao, N.; Shen, G. L.; Yu, R. Q. An amperometric hydrogen peroxide biosensor based on immobilizing horseradish peroxidase to a nano-au monolayer supported by sol-gel derived carbon ceramic electrode. Bioelectrochemistry 2004, 65(1), 33–39. [Google Scholar]
- Trojanowicz, M. Miniaturized biochemical sensing devices based on planar bilayer lipid membranes. Fresenius Journal of Analytical Chemistry 2001, 371(2), 246–260. [Google Scholar]
- Xu, J.; Wang, X.B.; Ensign, B.; Li, M.; Wu, L.; Guia, A.; Xu, J. Q. Ion-channel assay technologies: quo vadis? Drug Discovery Today 2001, 6(24), 1278–1287. [Google Scholar]
- Howard, A.D.; McAllister, G.; Feighner, S. D.; Liu, Q. Y.; Nargund, R. P.; Van der Ploeg, L. H.T.; Patchett, A. A. Orphan g-protein-coupled receptors and natural ligand discovery. Trends in Pharmacological Sciences 2001, 22(3), 132–140. [Google Scholar]
- Schmitt, E.; Nurbani, M.; Bushby, R.; Steinem, C. Electrically insulating pore-suspending membranes on highly ordered porous alumina obtained from vesicle spreading. Soft Matter 2008. [Google Scholar]
- Han, X.; DiBernardino, M.; Studer, A.; Sehr, H.; Geissbhler, I.; Winkler, F.; Tiefenauer, L. Nanopore arrays for stable and functional free-standing lipid bilayers. Advanced Materials 2007, 19, 4466–4470. [Google Scholar]
- Reimhult, E.; Kumar, K. Membrane biosensor platforms using nano- and mocroporous supports. Trends in Biotechnology 2008, 26(2), 82–89. [Google Scholar]
- Castellana, E.T.; Cremer, P. S. Solid supported lipid bilayers: From biophysical studies to sensor design. Surface Science Reports 2006, 61(10), 429–444. [Google Scholar]
- Ide, T.; Ichikawa, T. A novel method for artificial lipid-bilayer formation. Biosensors & Bioelectronics 2005, 21(4), 672–677. [Google Scholar]
- Janshoff, A.; Steinem, C. Transport across artificial membranes - an analytical perspective. Analytical and Bioanalytical Chemistry 2006, 385(3), 433–451. [Google Scholar]
- Sackmann, E. Supported membranes: Scientific and practical applications. Science 1996, 271(5245), 43–48. [Google Scholar]
- Lang, H.; Duschl, C.; Vogel, H. A new class of thiolipids for the attachment of lipid bilayers on gold surfaces. Langmuir 1994, 10(1), 197–210. [Google Scholar]
- Naumann, R.; Schiller, S.M.; Giess, F.; Grohe, B.; Hartman, K. B.; Karcher, I.; Koper, I.; Lubben, J.; Vasilev, K.; Knoll, W. Tethered lipid bilayers on ultraflat gold surfaces. Langmuir 2003, 19(13), 5435–5443. [Google Scholar]
- Naumann, R.; Walz, D.; Schiller, S.M.; Knoll, W. Kinetics of valinomycin-mediated k+ ion transport through tethered bilayer lipid membranes. Journal of Electroanalytical Chemistry 2003, 550, 241–252. [Google Scholar]
- Steinem, C.; Galla, H.J.; Janshoff, A. Interaction of melittin with solid supported membranes. Physical Chemistry Chemical Physics 2000, 2(20), 4580–4585. [Google Scholar]
- Steinem, C.; Janshoff, A.; Galla, H.J.; Sieber, M. Impedance analysis of ion transport through gramicidin channels incorporated in solid supported lipid bilayers. Bioelectrochemistry and Bioenergetics 1997, 42(2), 213–220. [Google Scholar]
- Gritsch, S.; Nollert, P.; Jahnig, F.; Sackmann, E. Impedance spectroscopy of porin and gramicidin pores reconstituted into supported lipid bilayers on indium-tin-oxide electrodes. Langmuir 1998, 14(11), 3118–3125. [Google Scholar]
- Yin, P.; Burns, C.J.; Osman, P. D.J.; Cornell, B. A. A tethered bilayer sensor containing alamethicin channels and its detection of amiloride based inhibitors. Biosensors & Bioelectronics 2003, 18(4), 389–397. [Google Scholar]
- Glazier, S.A.; Vanderah, D. J.; Plant, A. L.; Bayley, H.; Valincius, G.; Kasianowicz, J. J. Reconstitution of the pore-forming toxin alpha-hemolysin in phospholipid/18-octadecyl-1-thiahexa(ethylene oxide) and phospholipid/n-octadecanethiol supported bilayer membranes. Langmuir 2000, 16(26), 10428–10435. [Google Scholar]
- Hirano, A.; Wakabayashi, M.; Matsuno, Y.; Sugawara, M. A single-channel sensor based on gramicidin controlled by molecular recognition at bilayer lipid membranes containing receptor. Biosensors & Bioelectronics 2003, 18(8), 973–983. [Google Scholar]
- Knapp, O.; Benz, R.; Gibert, M.; Marvaud, J.C.; Popoff, M. R. Interaction of clostridium perfringens iota-toxin with lipid bilayer membranes - demonstration of channel formation by the activated binding component ib and channel block by the enzyme component ia. Journal of Biological Chemistry 2002, 277(8), 6143–6152. [Google Scholar]
- Bayley, H.; Jayasinghe, L. Functional engineered channels and pores - (review). Molecular Membrane Biology 2004, 21(4), 209–220. [Google Scholar]
- Ataka, K.; Giess, F.; Knoll, W.; Naumann, R.; Haber-Pohlmeier, S.; Richter, B.; Heberle, J. Oriented attachment and membrane reconstitution of his-tagged cytochrome c oxidase to a gold electrode: In situ monitoring by surface-enhanced infrared absorption spectroscopy. Journal of the American Chemical Society 2004, 126(49), 16199–16206. [Google Scholar]
- Naumann, R.; Baumgart, T.; Graber, P.; Jonczyk, A.; Offenhausser, A.; Knoll, W. Proton transport through a peptide-tethered bilayer lipid membrane by the h+-atp synthase from chloroplasts measured by impedance spectroscopy. Biosensors & Bioelectronics 2002, 17(1-2), 25–34. [Google Scholar]
- Ottenbacher, D.; Kindervater, R.; Gimmel, P.; Klee, B.; Jahnig, F.; Gopel, W. Developing biosensors with ph-isfet transducers utilizing lipid bilayer-membranes with transport proteins. Sensors and Actuators B-Chemical 1992, 6(1-3), 192–196. [Google Scholar]
- Jeuken, L.J. C.; Connell, S. D.; Nurnabi, M.; O'Reilly, J.; Henderson, P. J.F.; Evans, S. D.; Bushby, R. J. Direct electrochemical interaction between a modified gold electrode and a bacterial membrane extract. Langmuir 2005, 21(4), 1481–1488. [Google Scholar]
- Doyle, D.A.; Cabral, J. M.; Pfuetzner, R. A.; Kuo, A. L.; Gulbis, J. M.; Cohen, S. L.; Chait, B. T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of k+ conduction and selectivity. Science 1998, 280(5360), 69–77. [Google Scholar]
- Andersson, M.; Keizer, H.M.; Zhu, C. Y.; Fine, D.; Dodabalapur, A.; Duran, R. S. Detection of single ion channel activity on a chip using tethered bilayer membranes. Langmuir 2007, 23(6), 2924–2927. [Google Scholar]
- Cornell, B.A.; BraachMaksvytis, V. L.B.; King, L. G.; Osman, P. D.J.; Raguse, B.; Wieczorek, L.; Pace, R. J. A biosensor that uses ion-channel switches. Nature 1997, 387(6633), 580–583. [Google Scholar]
- Woodhouse, G.; King, L.; Wieczorek, L.; Osman, P.; Cornell, B. The ion channel switch biosensor. Journal of Molecular Recognition 1999, 12(5), 328–334. [Google Scholar]
- Nikolelis, D.P.; Siontorou, C. G.; Krull, U. J.; Katrivanos, P. L. Ammonium ion minisensors from self-assembled bilayer lipid membranes using gramicidin as an ionophore. modulation of ammonium selectivity by platelet-activating factor. Analytical Chemistry 1996, 68(10), 1735–1741. [Google Scholar]
- Blake, S.; Mayer, T.; Mayer, M.; Yang, J. Monitoring chemical reactions by using ion-channel-forming peptides. Chembiochem 2006, 7(3), 433–435. [Google Scholar]
- Stora, T.; Lakey, J.H.; Vogel, H. Ion-channel gating in transmembrane receptor proteins: Functional activity in tethered lipid membranes. Angewandte Chemie-International Edition 1999, 38(3), 389–392. [Google Scholar]
- Boulmedais, F.; Frisch, B.; Etienne, O.; Lavalle, P.; Picart, C.; Ogier, J.; Voegel, J.C.; Schaaf, P.; Egles, C. Polyelectrolyte multilayer films with pegylated polypeptides as a new type of anti-microbial protection for biomaterials. Biomaterials 2004, 25(11), 2003–2011. [Google Scholar]
- Templin, M.F.; Stoll, D.; Schrenk, M.; Traub, P. C.; Vohringer, C. F.; Joos, T. O. Protein microarray technology. Trends in Biotechnology 2002, 20(4), 160–166. [Google Scholar]
- Haab, B.B. Antibody arrays in cancer research. Molecular & Cellular Proteomics 2005, 4(4), 377–383. [Google Scholar]
- Romer, W.; Lam, Y.H.; Fischer, D.; Watts, A.; Fischer, W. B.; Goring, P.; Wehrspohn, R. B.; Gosele, U.; Steinem, C. Channel activity of a viral transmembrane peptide in micro-blms: Vpu(1-32) from hiv-1. Journal of the American Chemical Society 2004, 126(49), 16267–16274. [Google Scholar]
- Danelon, C.; Perez, J.B.; Santschi, C.; Brugger, J.; Vogel, H. Cell membranes suspended across nanoaperture arrays. Langmuir 2006, 22(1), 22–25. [Google Scholar]
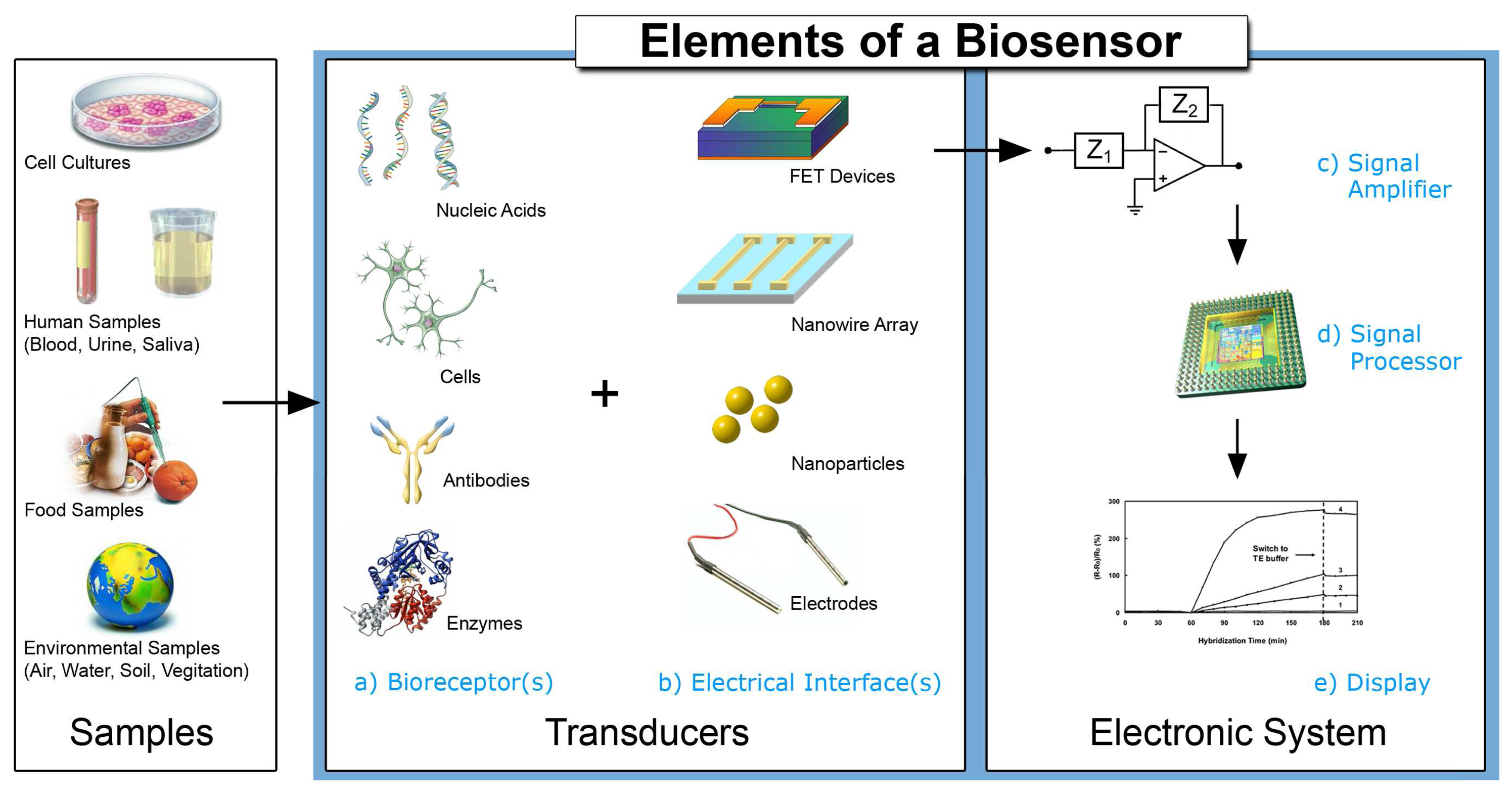
{kind=link}
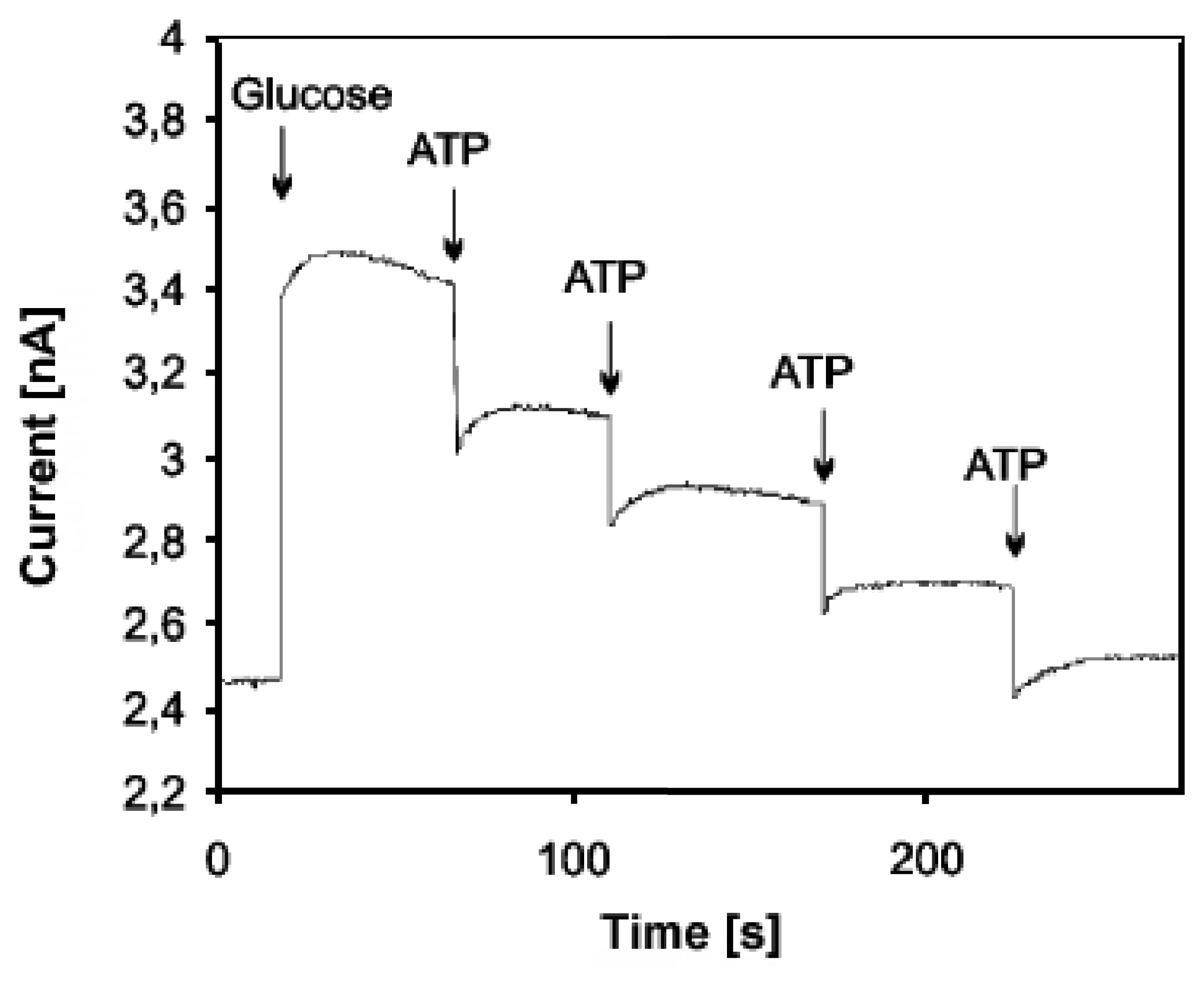{kind=link}
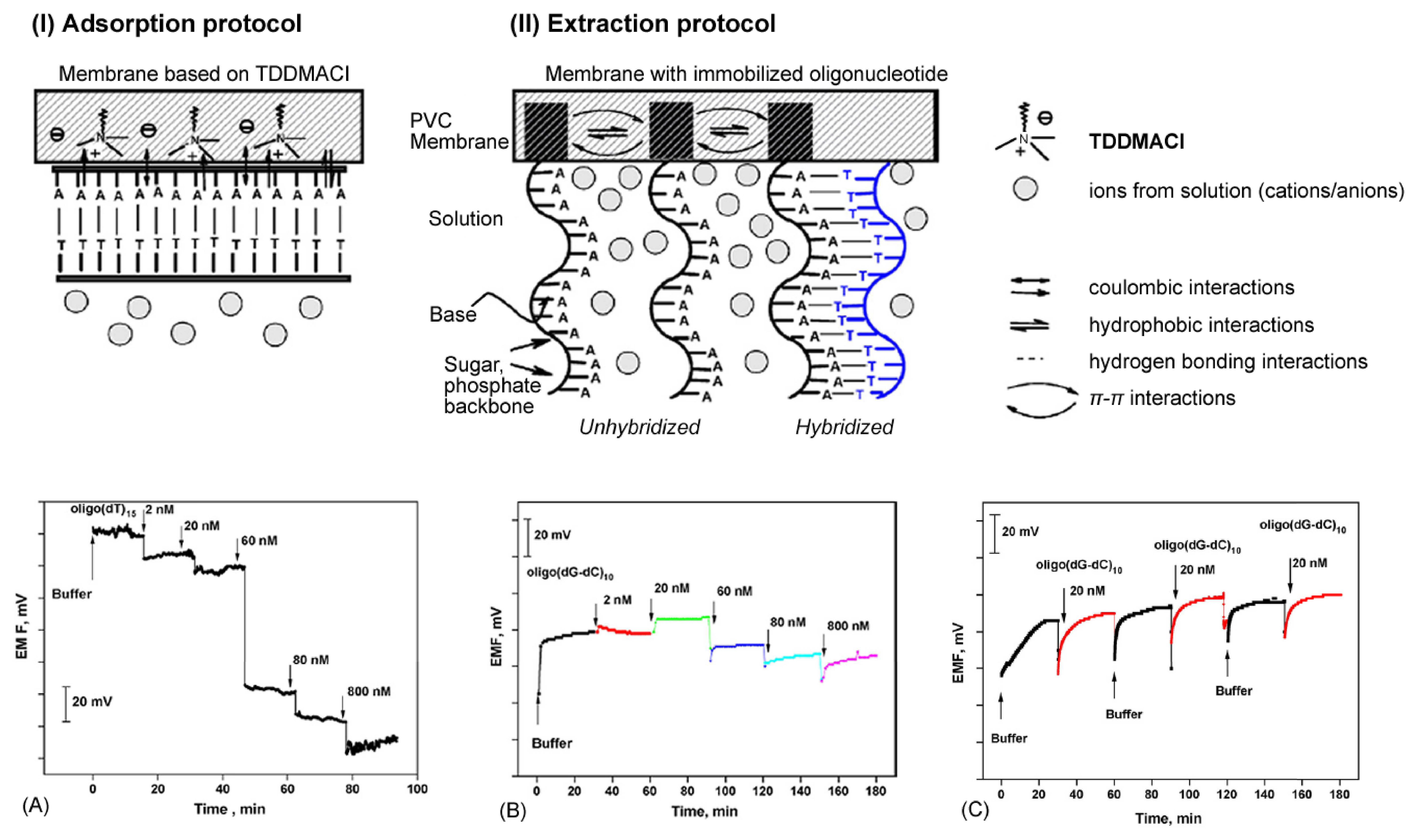{kind=link}
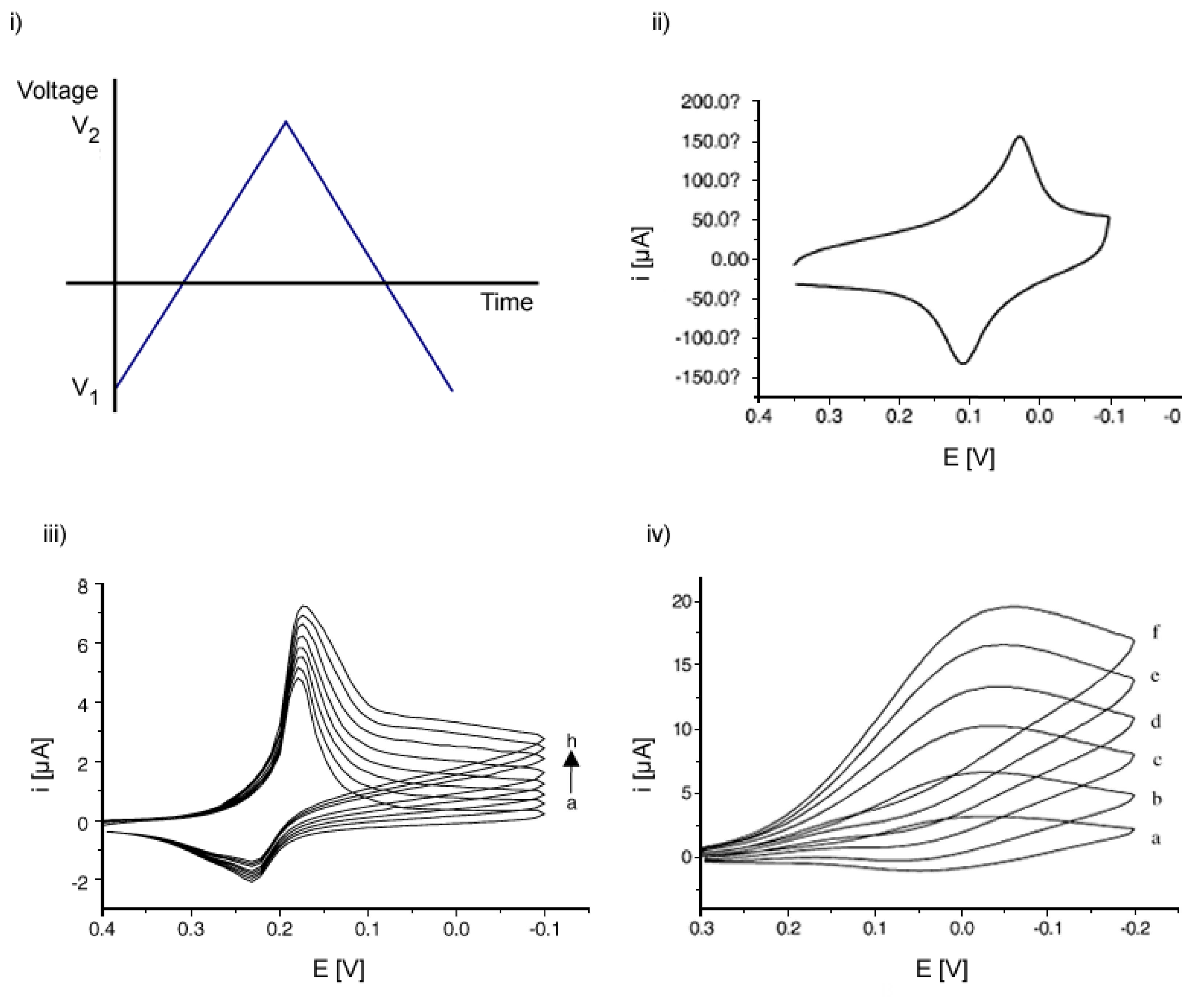{kind=link}
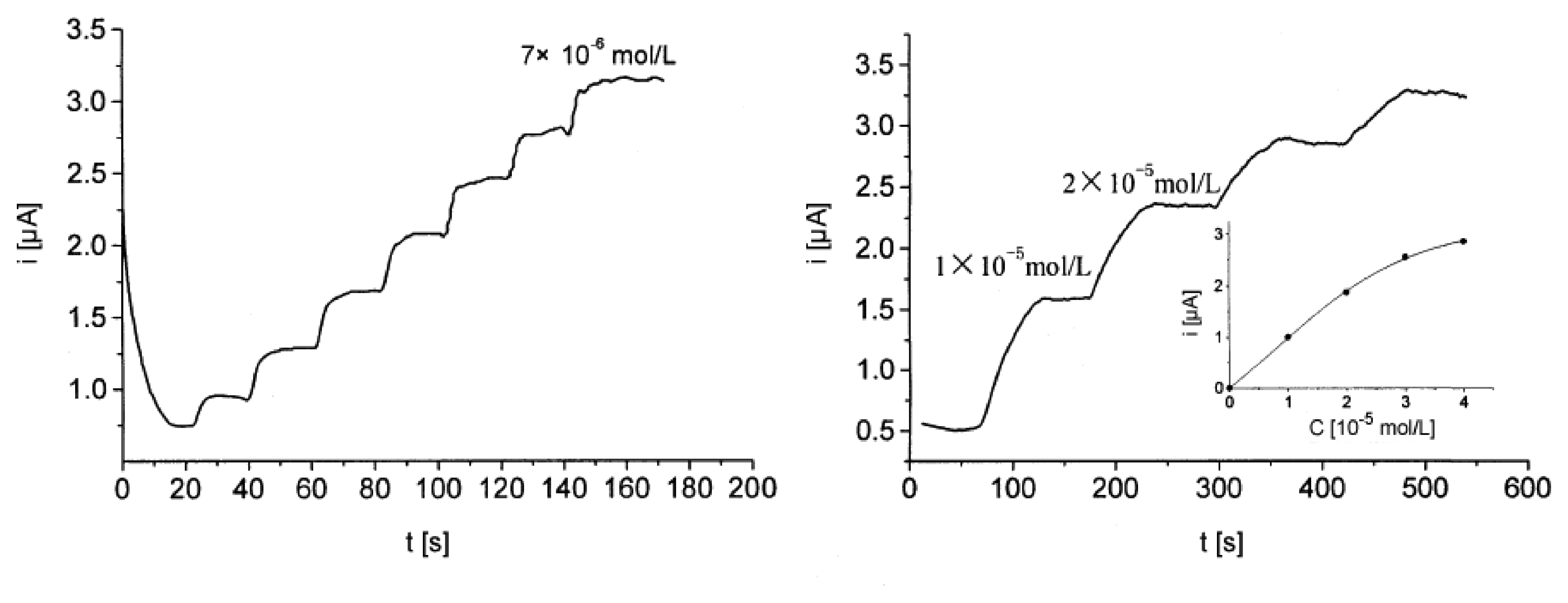{kind=link}
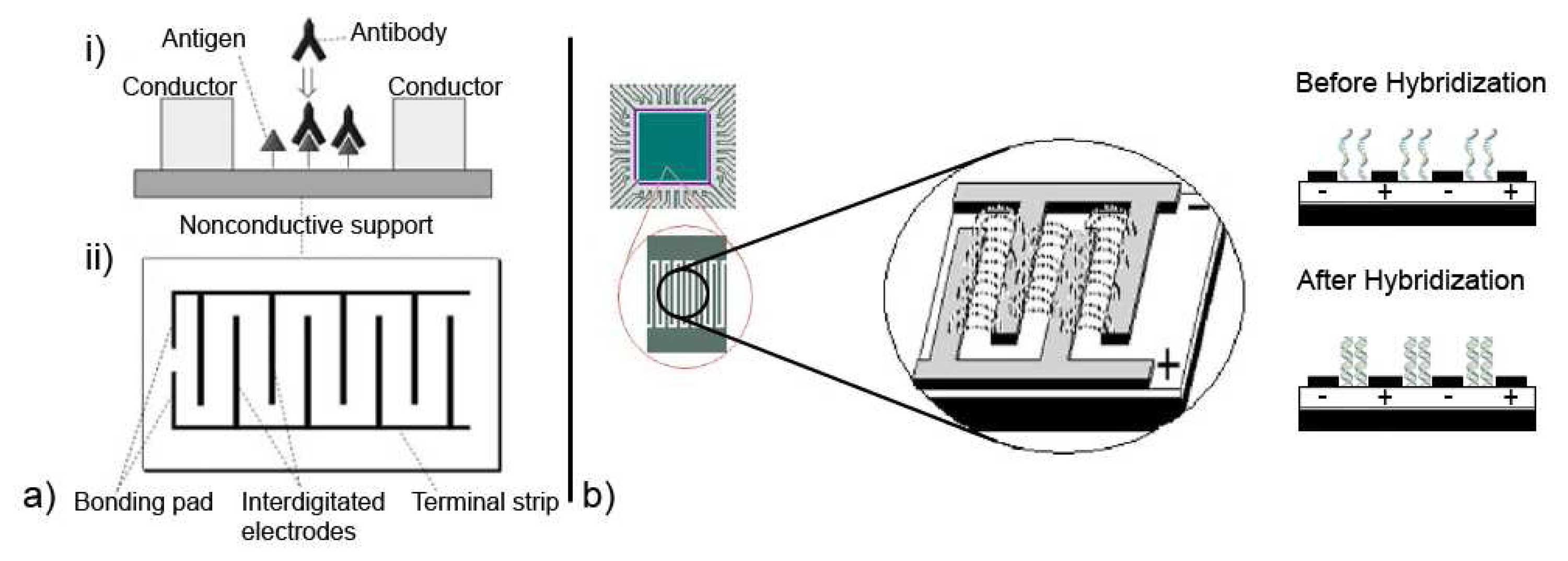{kind=link}
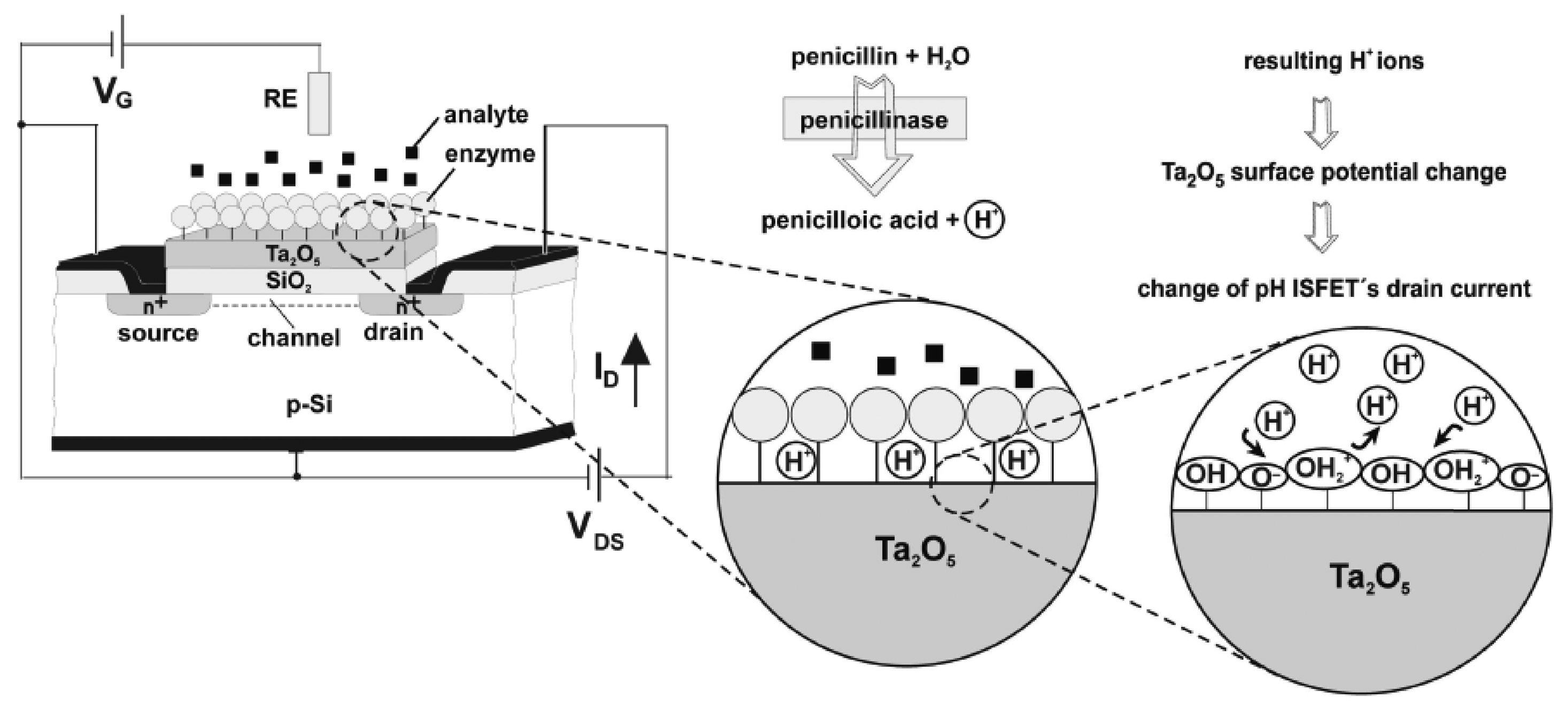{kind=link}
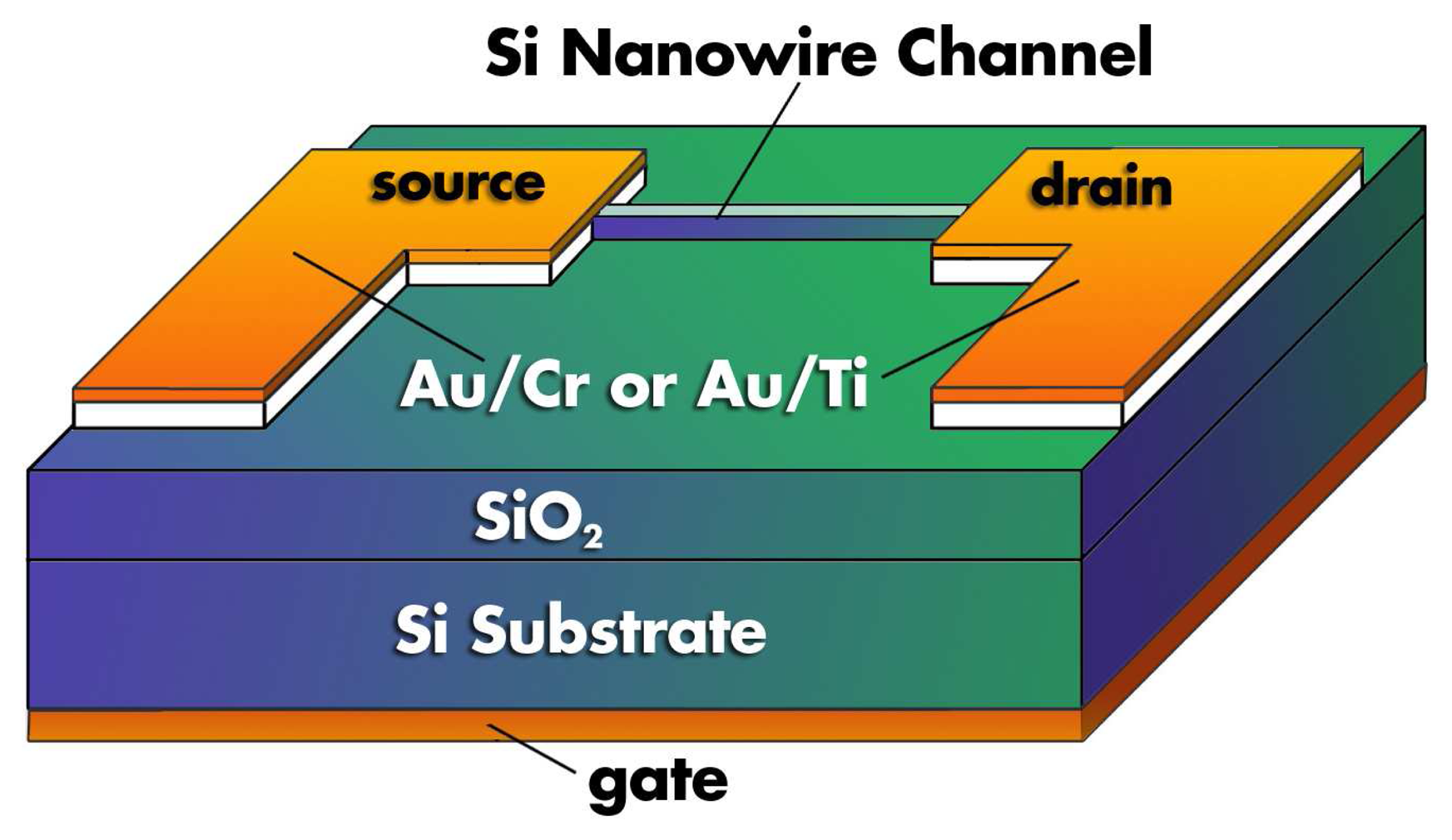{kind=link}
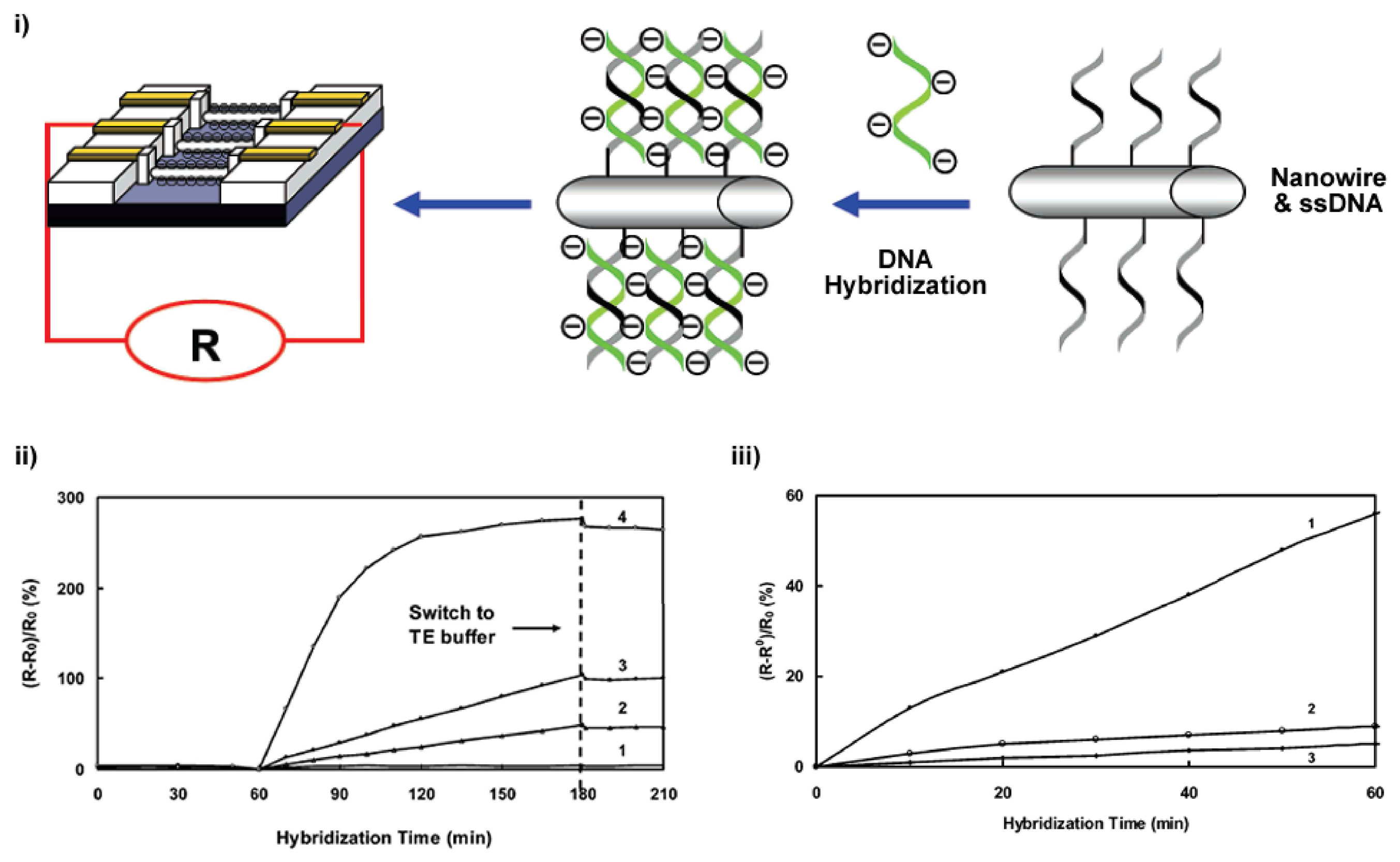{kind=link}
Figure 2.
A sample amperometric measurement: According to Kueng et al. [28] this is a typical hydrodynamic response of their biosensor to glucose followed by several injections of ATP measured in phosphate buffer at 650 mV in reference to Ag/AgCl. The change in current response is proportional to the ATP concentration as glucose is consumed at the glucose oxidase (GOD) and hexokinase (HEX) modified electrode surface.
Figure 2.
A sample amperometric measurement: According to Kueng et al. [28] this is a typical hydrodynamic response of their biosensor to glucose followed by several injections of ATP measured in phosphate buffer at 650 mV in reference to Ag/AgCl. The change in current response is proportional to the ATP concentration as glucose is consumed at the glucose oxidase (GOD) and hexokinase (HEX) modified electrode surface.
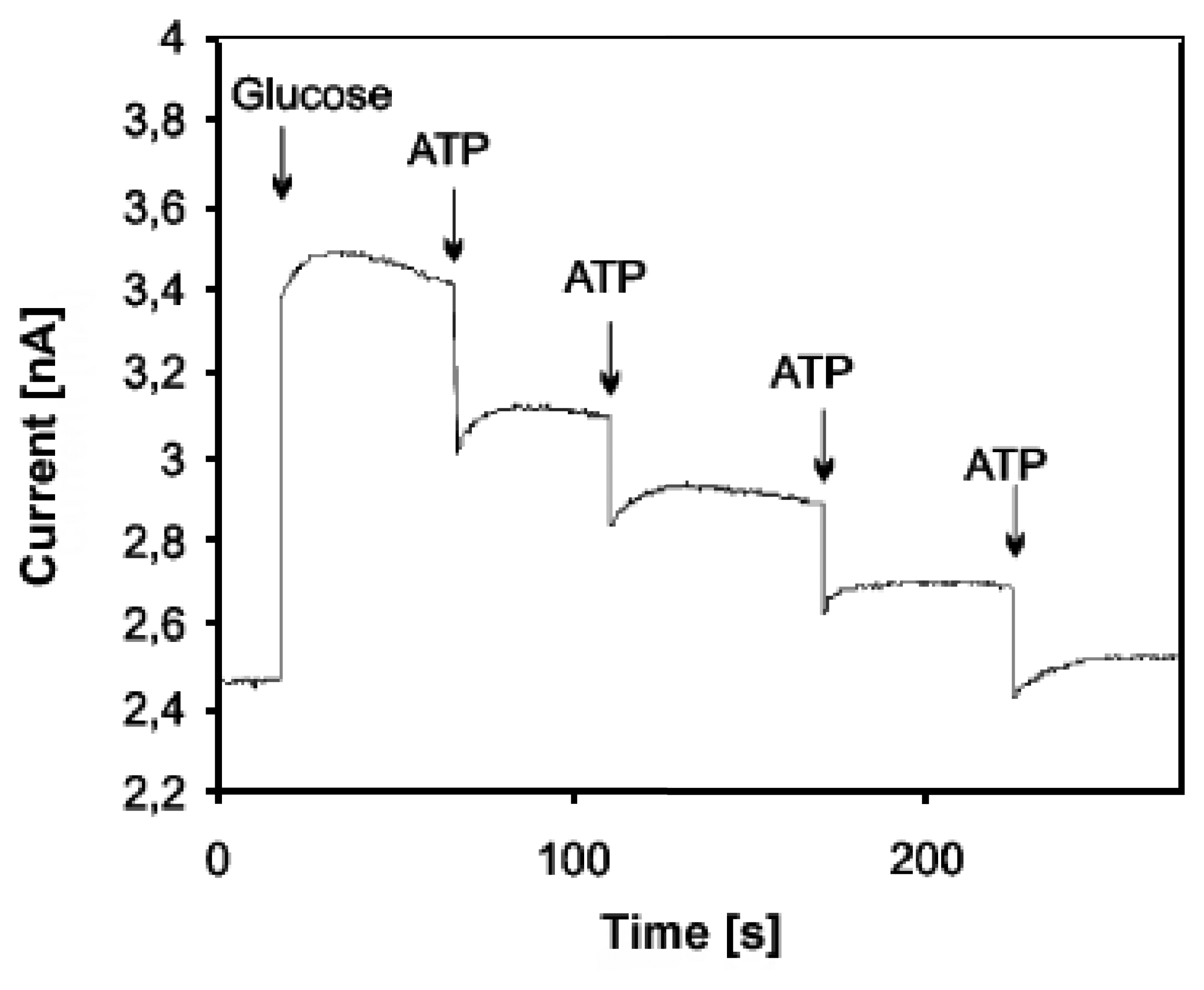
Figure 3.
The schematic and performance results of a potentiometric sensor for the detection of hybridization between complementary single-stranded (ss) oligonucleotides on the phase boundary PVC membrane/solution. The arrangement of the probe is shown in two configurations where the ss oligonucleotide is attached along (I) or parallel (II) to the PVC membrane surface. The potentiometric detection of the hybridization with complimentary strands is shown: (A) with complementary and unspecific interactions with non-complementary oligonucleotides; (B) with complementary oligonucleotides; (C) demonstration of the reproducibility after regeneration in a buffer of 0.01 M NaOH for 15 minutes (upward arrow) and subsequent injections of the corresponding oligonucleotides (downward arrow) [38].
Figure 3.
The schematic and performance results of a potentiometric sensor for the detection of hybridization between complementary single-stranded (ss) oligonucleotides on the phase boundary PVC membrane/solution. The arrangement of the probe is shown in two configurations where the ss oligonucleotide is attached along (I) or parallel (II) to the PVC membrane surface. The potentiometric detection of the hybridization with complimentary strands is shown: (A) with complementary and unspecific interactions with non-complementary oligonucleotides; (B) with complementary oligonucleotides; (C) demonstration of the reproducibility after regeneration in a buffer of 0.01 M NaOH for 15 minutes (upward arrow) and subsequent injections of the corresponding oligonucleotides (downward arrow) [38].
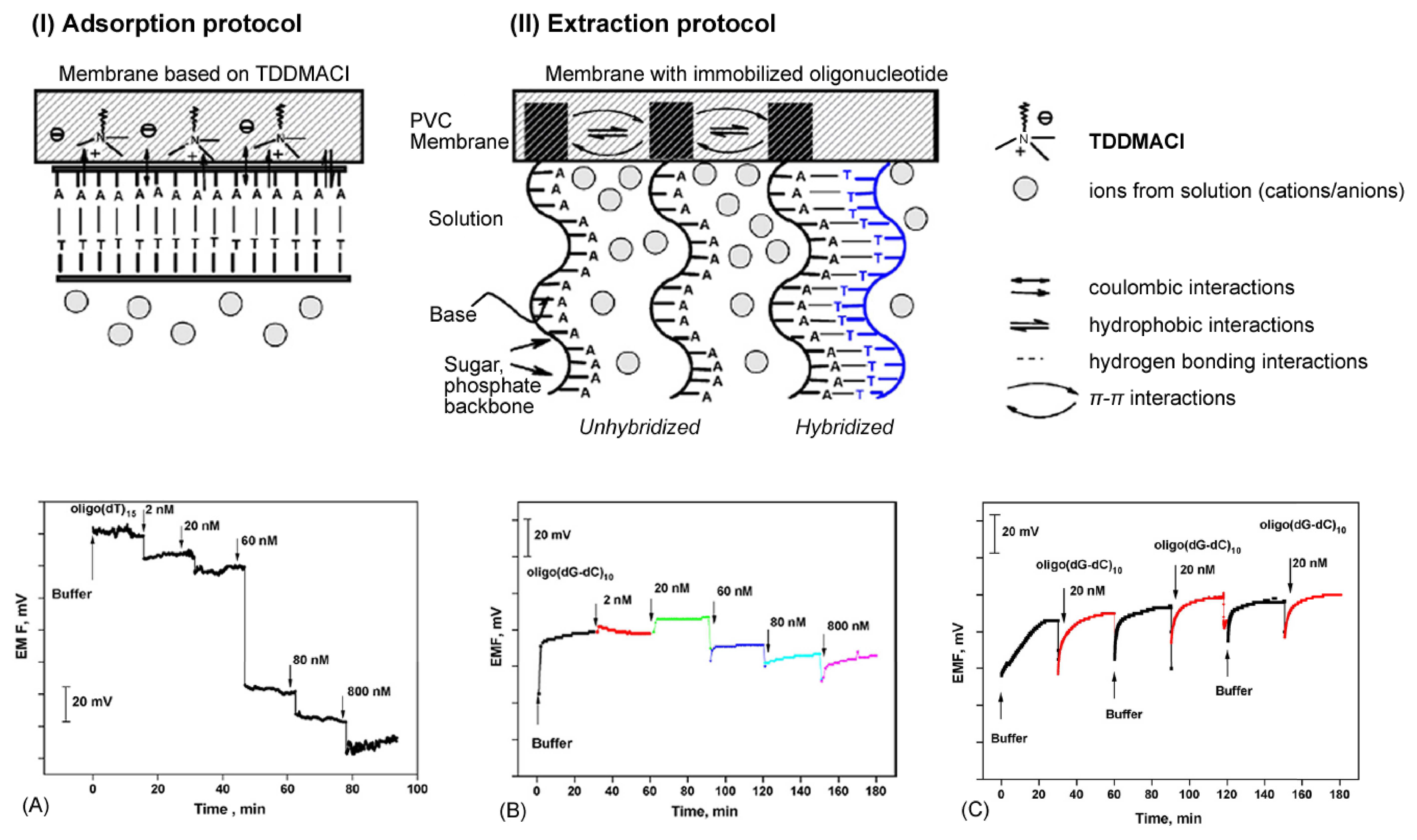
Figure 4.
An overview of selected cyclic voltammograms with reference to the work of Li et al.: i) a single generic linear voltage sweep; ii) a cyclic voltammogram of the Prussian Blue-modified (PB) glassy carbon electrode was prepared in 0.1 M KCl + 0.1 M HCl at a scan rate 50 mV/s; iii) cyclic voltammograms of the PB sol-gel modified glassy carbon electrode and iv) the cholesterol/PB sol-gel modified glassy carbon electrode in a phosphate buffer (pH 6.8) at varying concentrations of the analytic solution: a) blank solution; b-f) blank solution + cholesterol 1×10−5, 2×10−5, 3×10−5, 4×10−5, 5×10−5 mol/L. [47]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Figure 4.
An overview of selected cyclic voltammograms with reference to the work of Li et al.: i) a single generic linear voltage sweep; ii) a cyclic voltammogram of the Prussian Blue-modified (PB) glassy carbon electrode was prepared in 0.1 M KCl + 0.1 M HCl at a scan rate 50 mV/s; iii) cyclic voltammograms of the PB sol-gel modified glassy carbon electrode and iv) the cholesterol/PB sol-gel modified glassy carbon electrode in a phosphate buffer (pH 6.8) at varying concentrations of the analytic solution: a) blank solution; b-f) blank solution + cholesterol 1×10−5, 2×10−5, 3×10−5, 4×10−5, 5×10−5 mol/L. [47]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
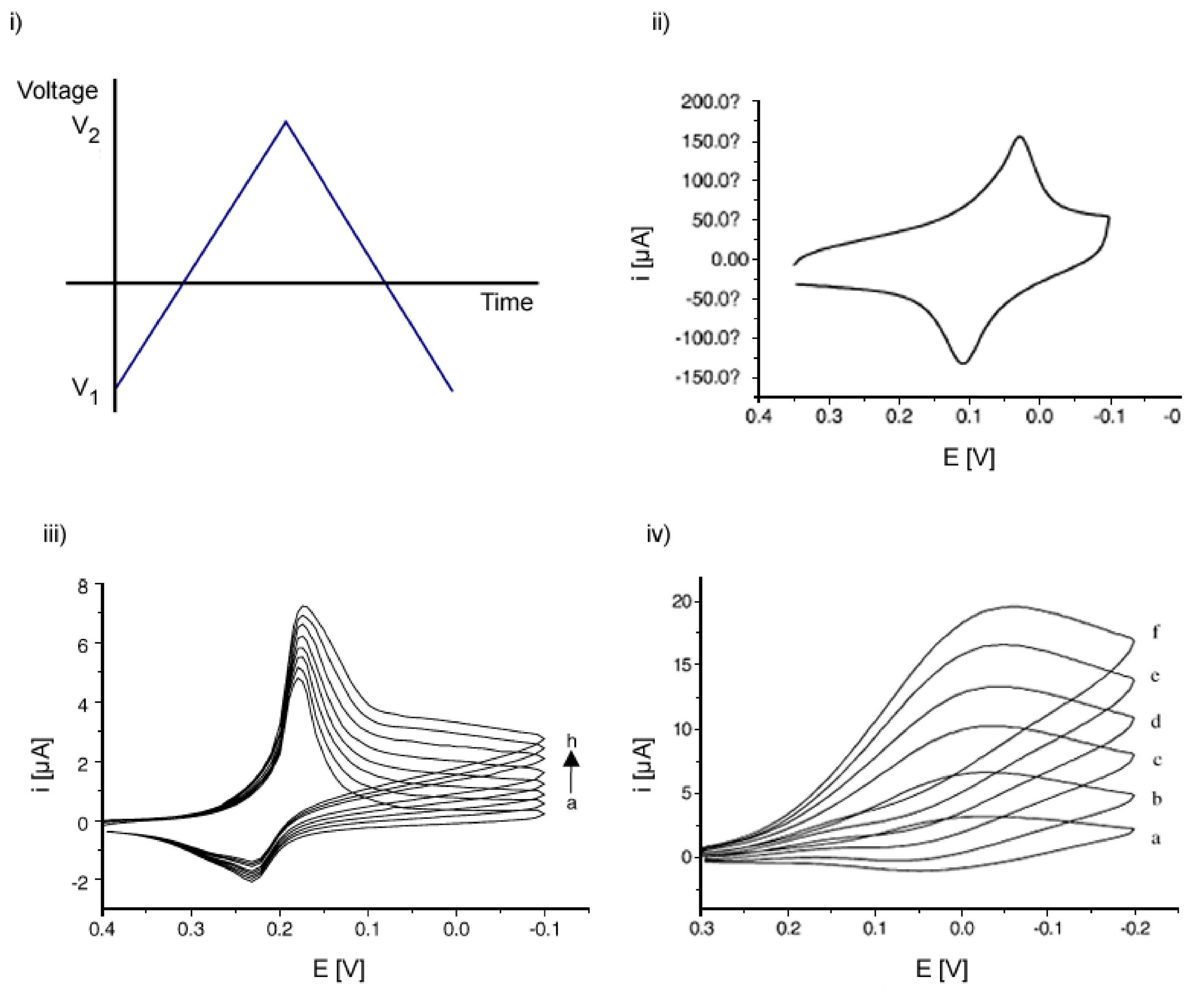
Figure 5.
Chronoamperometric curves for detecting cholesterol in a phosphate buffer (pH 6.8), using Prussian Blue (PB) modified electrodes and cholesterol oxidase (ChOx)/PB sol-gel electrodes. The working electrode was polarized at -0.05 V (vs. Ag/AgCl) in a stirred solution. A 3σ detection limit of 5.4 × 10−7 M [47]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Figure 5.
Chronoamperometric curves for detecting cholesterol in a phosphate buffer (pH 6.8), using Prussian Blue (PB) modified electrodes and cholesterol oxidase (ChOx)/PB sol-gel electrodes. The working electrode was polarized at -0.05 V (vs. Ag/AgCl) in a stirred solution. A 3σ detection limit of 5.4 × 10−7 M [47]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
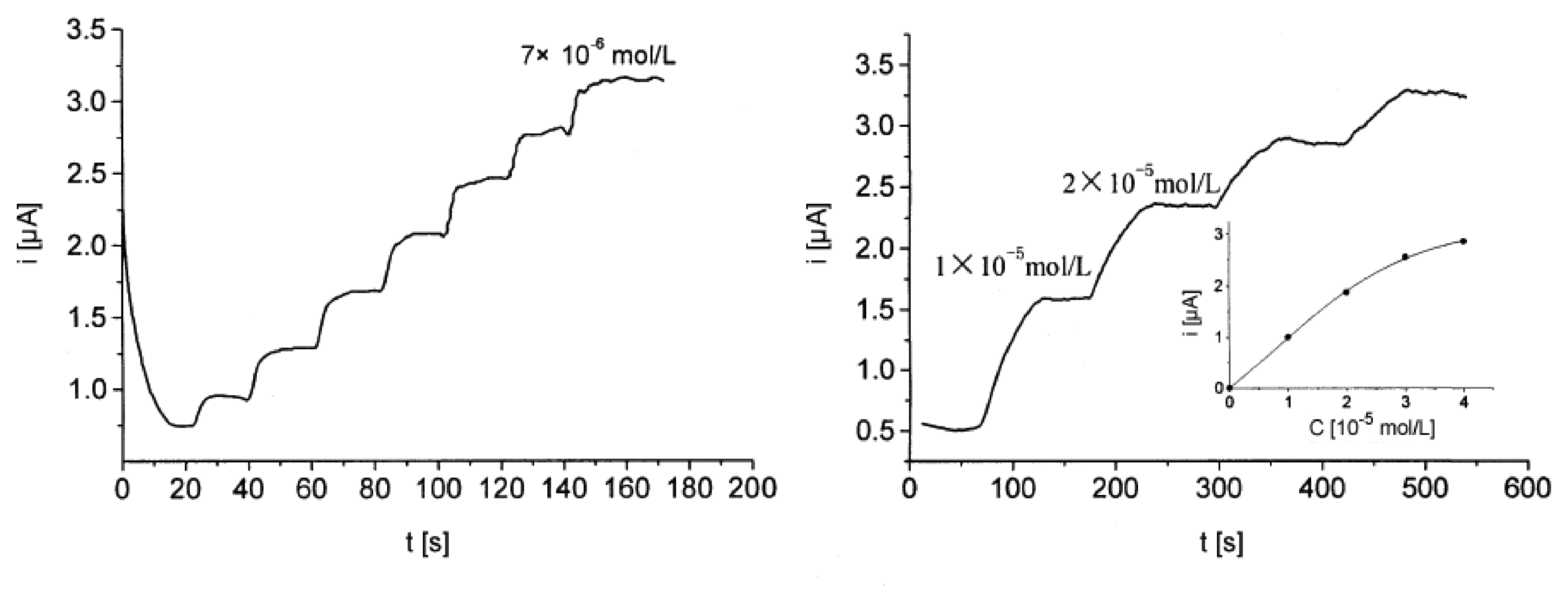
Figure 6.
a) i) Immunosensor for the in-plane impedance measurements between conductive electrodes, ii) interdigitated electrode for the in-plane impedance immunosensing [60]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission; b) illustration of the principle behind the change in network impedance upon hybridization of ssDNA probe oligomers to its complement interdigitated microsensor electrode [16]. With kind permission of Springer Science and Business Media.
Figure 6.
a) i) Immunosensor for the in-plane impedance measurements between conductive electrodes, ii) interdigitated electrode for the in-plane impedance immunosensing [60]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission; b) illustration of the principle behind the change in network impedance upon hybridization of ssDNA probe oligomers to its complement interdigitated microsensor electrode [16]. With kind permission of Springer Science and Business Media.
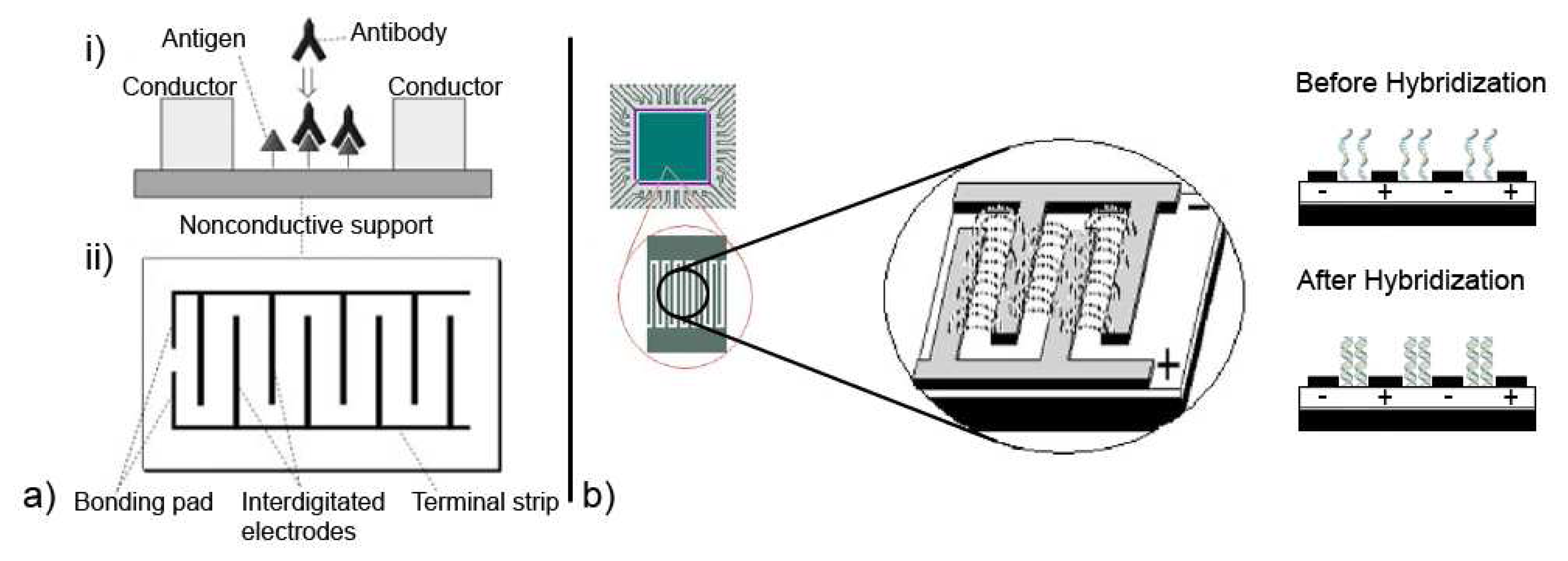
Figure 7.
Structure and principal function of a penicillin-sensitive EnFET. The enzyme penicillinase is immobilized on a pH-sensitive Ta2O5 surface [11]. The reference electrode is labelled as RE. Reproduced by permission of The Royal Society of Chemistry.
Figure 7.
Structure and principal function of a penicillin-sensitive EnFET. The enzyme penicillinase is immobilized on a pH-sensitive Ta2O5 surface [11]. The reference electrode is labelled as RE. Reproduced by permission of The Royal Society of Chemistry.
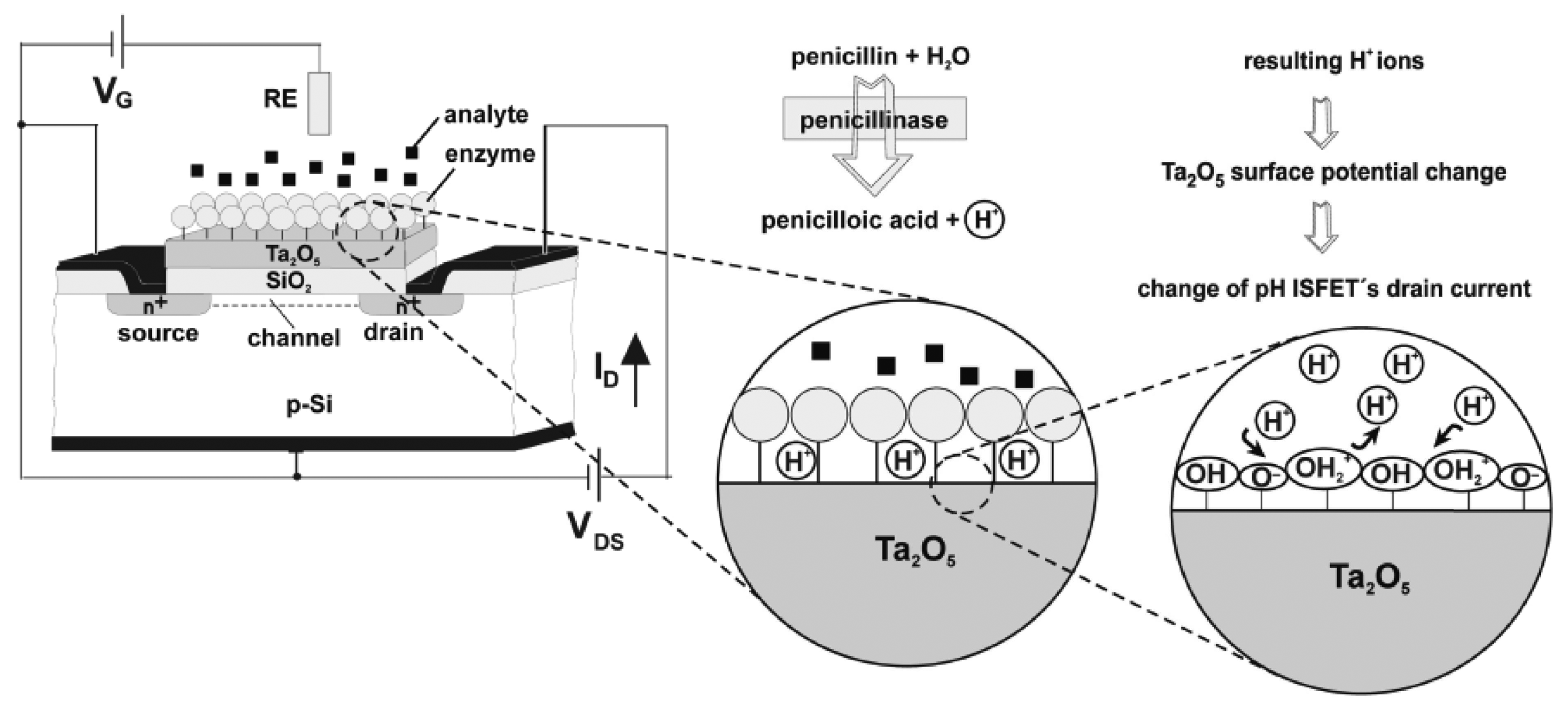
Figure 8.
Schematic diagram of a silicon nanowire FET [84]. The field effect occurs as charged particles or molecules bind to the surface of the nanowire and change its conductive properties, analogous to the gate voltage and channel conduction of a conventional FET.
Figure 8.
Schematic diagram of a silicon nanowire FET [84]. The field effect occurs as charged particles or molecules bind to the surface of the nanowire and change its conductive properties, analogous to the gate voltage and channel conduction of a conventional FET.

Figure 9.
i) A schematic representation of the design to use silicon nanowire FET as a biosensor for the label-free detection of DNA hybridization; ii) the change of resistance of the silicon nanowire array as a function of hybridization time ((1) 1.0 nM non-complementary DNA as control, (2) 25 fM, (3) 100 fM and (4) 1.0 nM target DNA in buffer); iii) the change of resistance of the silicon nanowire array as a function of hybridization time in 25 fM of (1) fully complementary, (2) one-base mismatched, and (3) two-base mismatched target DNA in TE buffer. The TE buffer solution (pH 8.5 10 mM Tris-HCl-1.0 mM EDTA-30 mM NaCl buffer) was used as the hybridization, washing, and working buffer. Reprinted with permission from [87]. Copyright 2007 American Chemical Society.
Figure 9.
i) A schematic representation of the design to use silicon nanowire FET as a biosensor for the label-free detection of DNA hybridization; ii) the change of resistance of the silicon nanowire array as a function of hybridization time ((1) 1.0 nM non-complementary DNA as control, (2) 25 fM, (3) 100 fM and (4) 1.0 nM target DNA in buffer); iii) the change of resistance of the silicon nanowire array as a function of hybridization time in 25 fM of (1) fully complementary, (2) one-base mismatched, and (3) two-base mismatched target DNA in TE buffer. The TE buffer solution (pH 8.5 10 mM Tris-HCl-1.0 mM EDTA-30 mM NaCl buffer) was used as the hybridization, washing, and working buffer. Reprinted with permission from [87]. Copyright 2007 American Chemical Society.
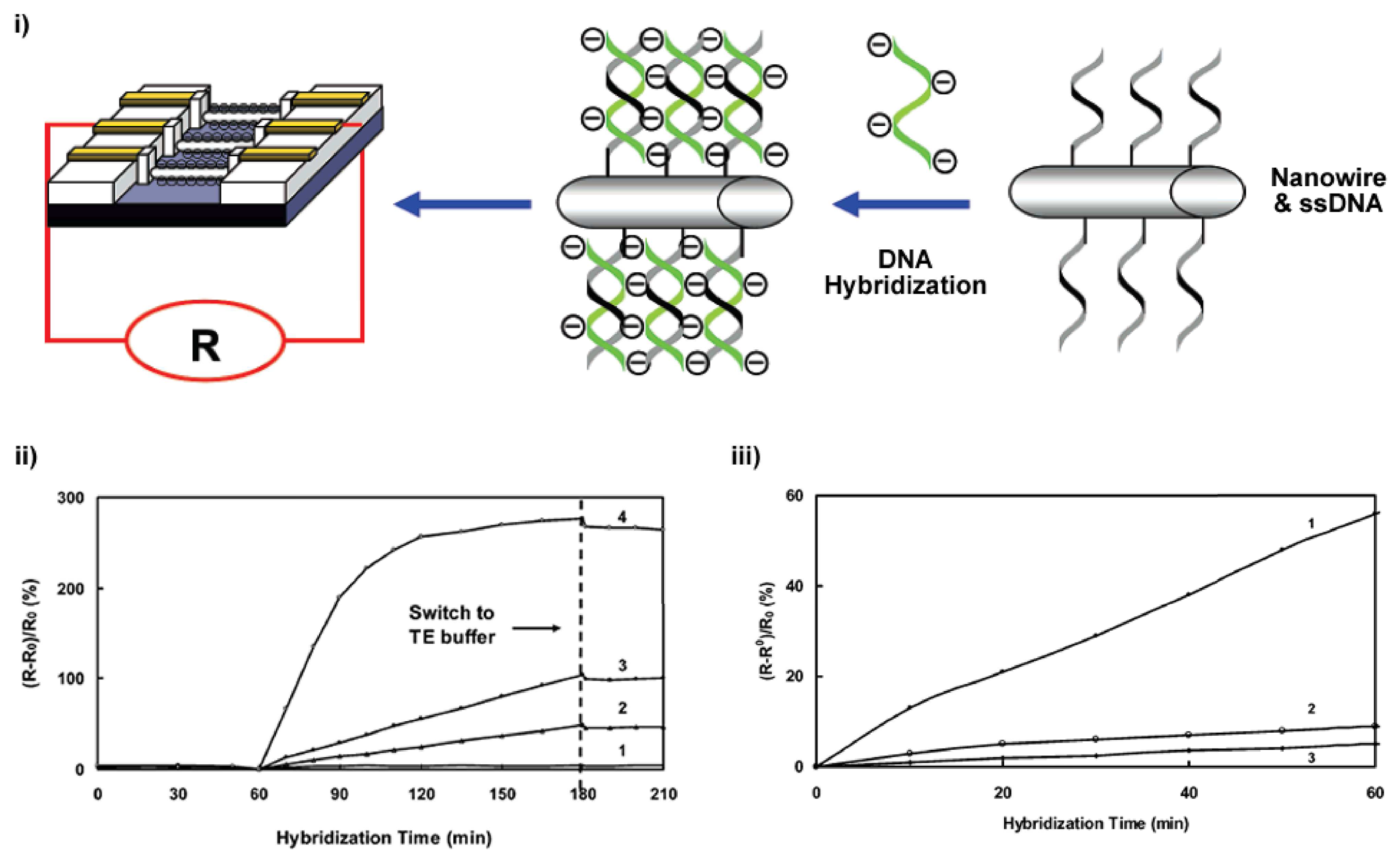
Figure 10.
An illustration to demonstrate the concept of the influence of surface interactions on nanowire conduction. In larger wires, for example with micrometer dimensions, the surface-to-volume ratio is relatively small. Even if surface interactions, such as binding with charge particles or biological species, influence conduction near the surface of the wire, there is still a large portion of the wire's interior available for uninterrupted conduction. To help visualize this, the illustration uses a mental construct of ‘conduction channels’ within the wire. As the dimensions of a wire decrease to the nanometer regime, the surface-to-volume ratio drastically increases. Therefore, the same external influences increasingly influence the conduction both on the wire surface and in the wire interior, thereby decreasing the possibility for uninterrupted conduction.
Figure 10.
An illustration to demonstrate the concept of the influence of surface interactions on nanowire conduction. In larger wires, for example with micrometer dimensions, the surface-to-volume ratio is relatively small. Even if surface interactions, such as binding with charge particles or biological species, influence conduction near the surface of the wire, there is still a large portion of the wire's interior available for uninterrupted conduction. To help visualize this, the illustration uses a mental construct of ‘conduction channels’ within the wire. As the dimensions of a wire decrease to the nanometer regime, the surface-to-volume ratio drastically increases. Therefore, the same external influences increasingly influence the conduction both on the wire surface and in the wire interior, thereby decreasing the possibility for uninterrupted conduction.
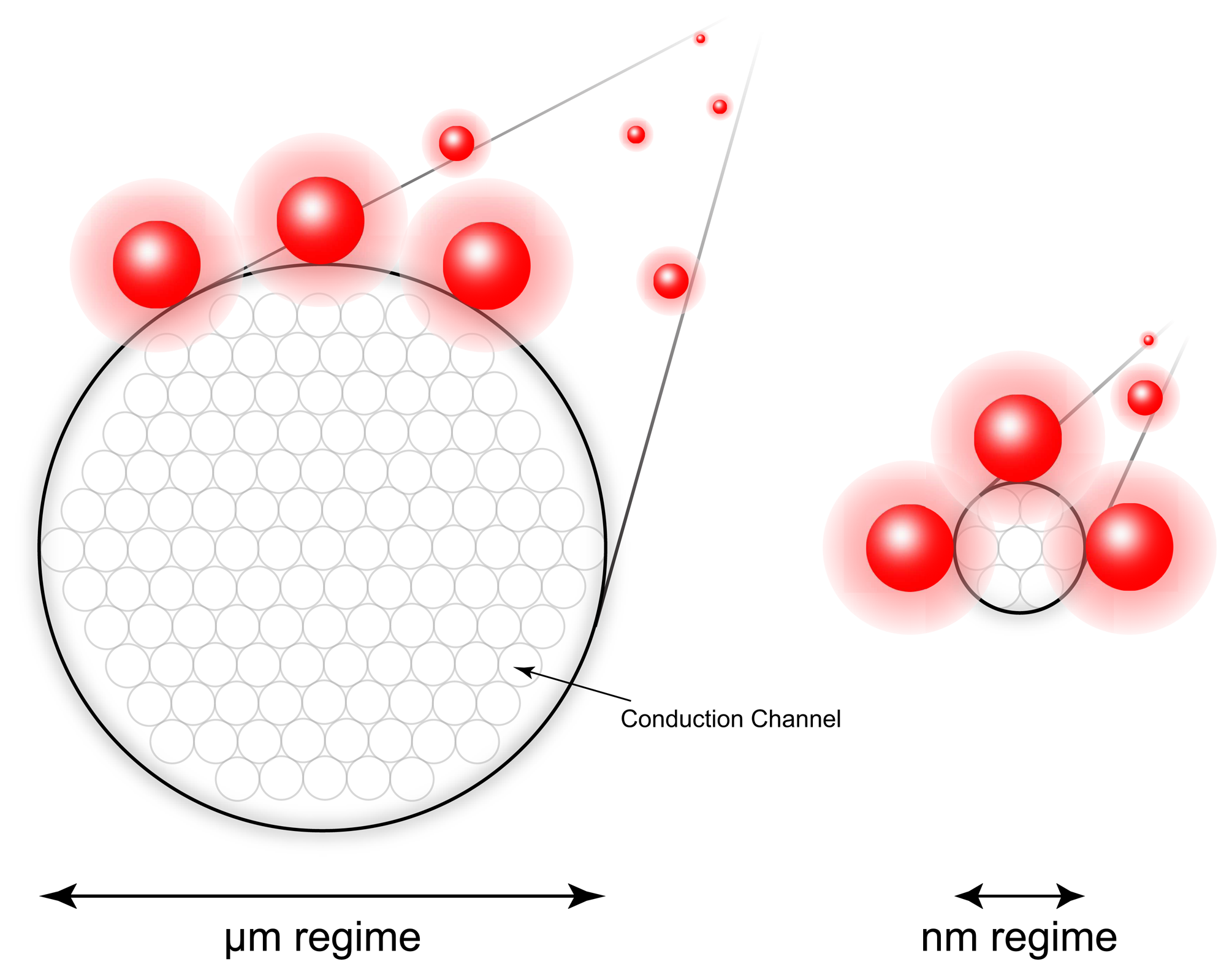
Figure 11.
An overview of the experiment by Foley et al. to measure the step-wise decrease in conductance as a gold nanowire is thinned by pulling. The discrete values of the conductance in units of 2e2/h has been corrected for the impedance of the measurement equipment. Reprinted with permission from [95]. Copyright 1999, American Institute of Physics.
Figure 11.
An overview of the experiment by Foley et al. to measure the step-wise decrease in conductance as a gold nanowire is thinned by pulling. The discrete values of the conductance in units of 2e2/h has been corrected for the impedance of the measurement equipment. Reprinted with permission from [95]. Copyright 1999, American Institute of Physics.

Figure 12.
The image on the left illustrates the sensing principle of nanowire-based biosensing with an FET configuration. The image on the right demonstrates the concept of multi-analyte biosensing with nanowire FET configurations [99]. Reproduced by permission of the MRS Bulletin.
Figure 12.
The image on the left illustrates the sensing principle of nanowire-based biosensing with an FET configuration. The image on the right demonstrates the concept of multi-analyte biosensing with nanowire FET configurations [99]. Reproduced by permission of the MRS Bulletin.
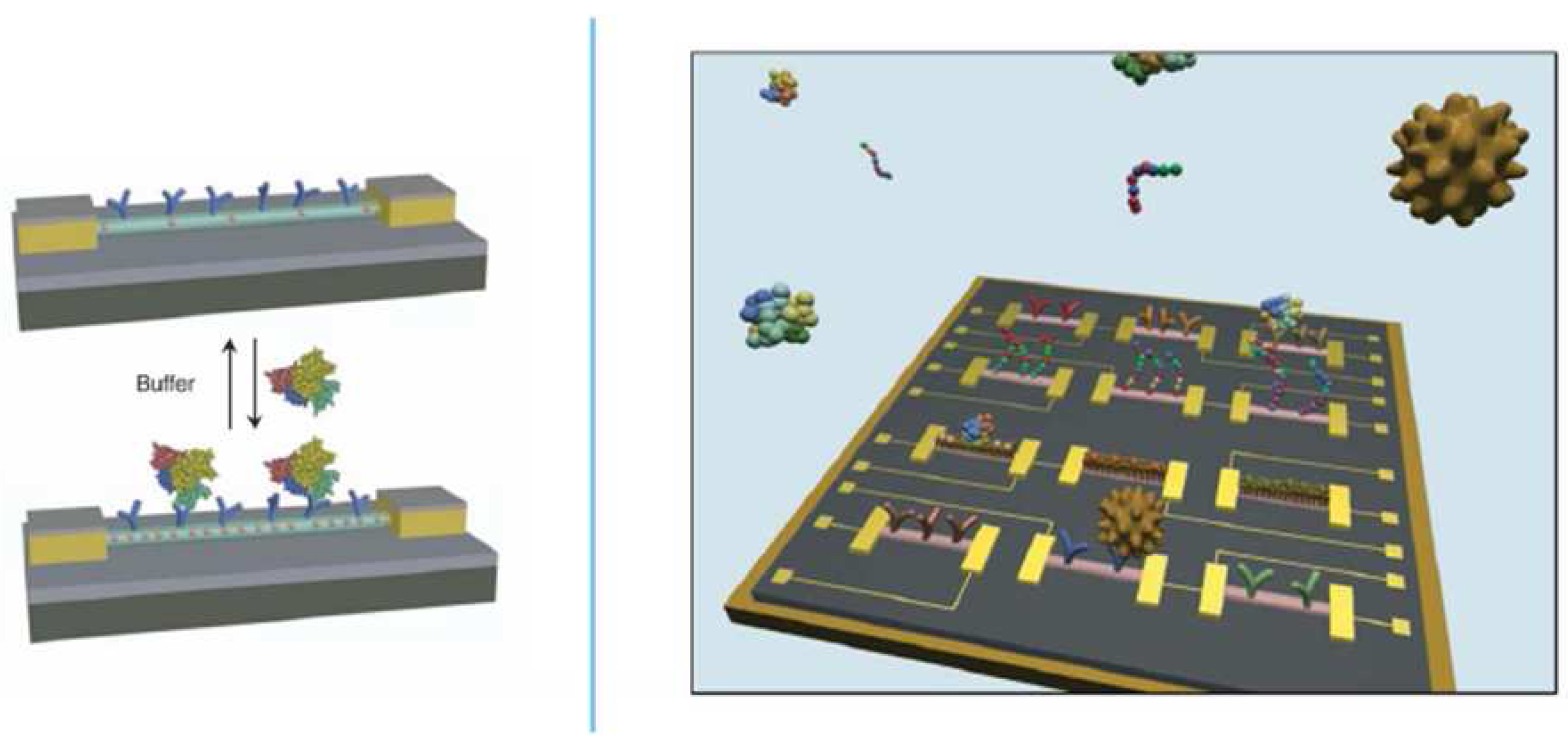
Figure 13.
Array of nanowires made from DNA-tagged Au nanoparticles using line patterns made with EUV-IL [24].
Figure 13.
Array of nanowires made from DNA-tagged Au nanoparticles using line patterns made with EUV-IL [24].
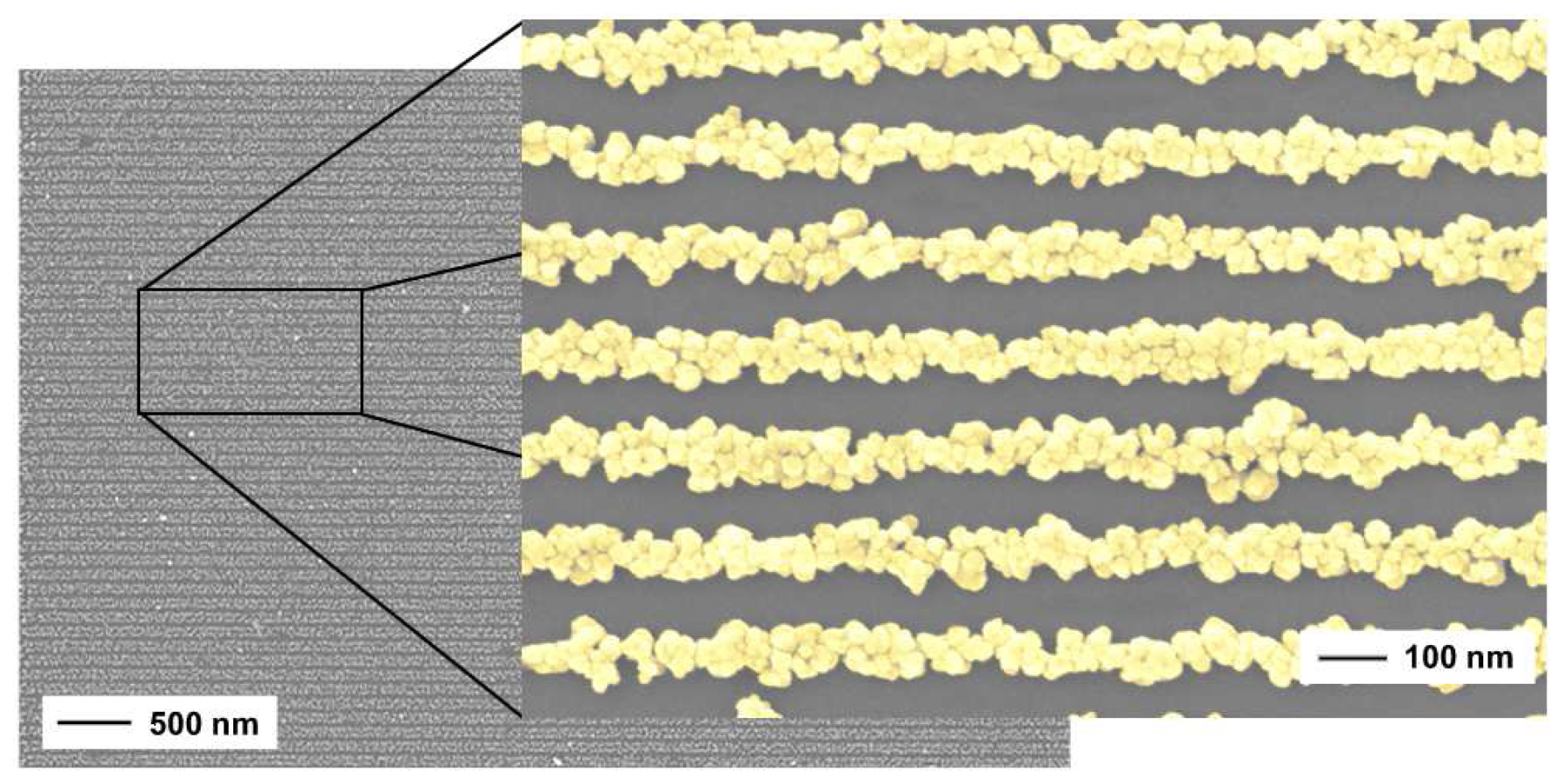
Figure 14.
A modified schematic illustration of a combined surface-plasmon optical and electrochemical setup from Badia et al. [110].
Figure 14.
A modified schematic illustration of a combined surface-plasmon optical and electrochemical setup from Badia et al. [110].

Figure 15.
Measured results of the biosensor of Kang et al. for the detection of the enzymatic reaction between horseradish peroxidase and a film of polyaniline (PAn) on a gold electrode in the presence of H2O2: i) CV curve (a) and reflectance change (ΔR) (b) as a function of a potential at a fixed incident angle of 62.2°; ii) SPR measurements of reflectivity versus incidence angle at varying electrochemical conditions 1) bare gold electrode 2) reduced PAn film after maintaining a potential of 0 V for 5 minutes 3) oxidized PAn film after maintaining a potential of 0.9 V for 5 min iii) SPR kinetic curves at a fixed angle of 62.8° resulting from the HRP/PAn reaction at various concentrations of H2O2; iv) the relationship between the slope of the ΔR-t curves from iii) and the H2O2 concentration [116].
Figure 15.
Measured results of the biosensor of Kang et al. for the detection of the enzymatic reaction between horseradish peroxidase and a film of polyaniline (PAn) on a gold electrode in the presence of H2O2: i) CV curve (a) and reflectance change (ΔR) (b) as a function of a potential at a fixed incident angle of 62.2°; ii) SPR measurements of reflectivity versus incidence angle at varying electrochemical conditions 1) bare gold electrode 2) reduced PAn film after maintaining a potential of 0 V for 5 minutes 3) oxidized PAn film after maintaining a potential of 0.9 V for 5 min iii) SPR kinetic curves at a fixed angle of 62.8° resulting from the HRP/PAn reaction at various concentrations of H2O2; iv) the relationship between the slope of the ΔR-t curves from iii) and the H2O2 concentration [116].
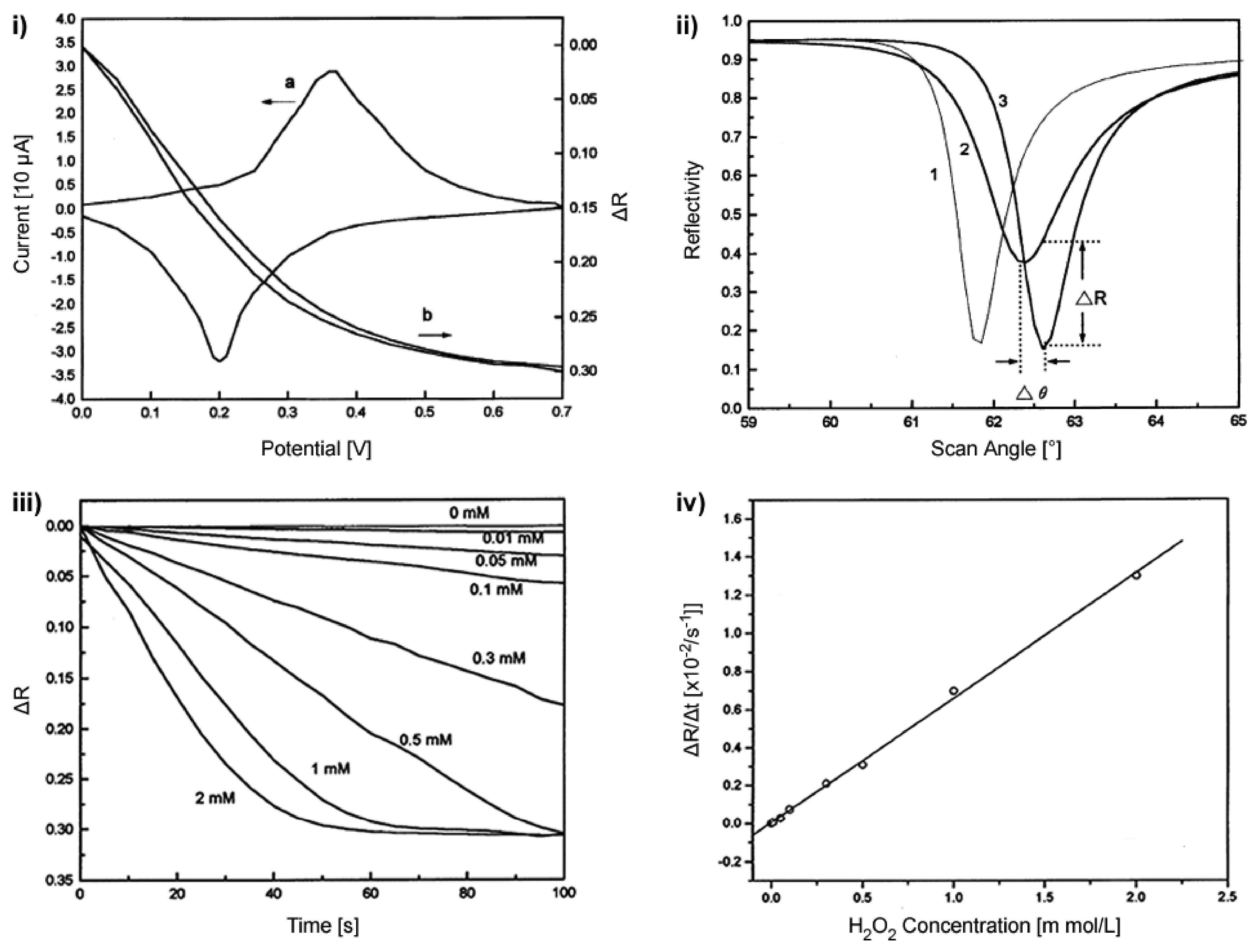
Figure 16.
EC-OWLS flow cell. The laser is coupled into the grating at a certain incoupling angle. The ITO-coated waveguide acts as the working electrode, the reference electrode is placed on the topside of the flowcell and the counter electrode at the outlet.
Figure 16.
EC-OWLS flow cell. The laser is coupled into the grating at a certain incoupling angle. The ITO-coated waveguide acts as the working electrode, the reference electrode is placed on the topside of the flowcell and the counter electrode at the outlet.

Figure 17.
A) EC-OWLS curve showing adsorption as a function of time. Additionally, the signal also reacts to an applied potential. Prior to the adsorption steps the system was electrically cycled to obtain stable conditions. PLL-g-PEG/biotin, neutravidin and biotin conjugated GOx were adsorbed in three steps with buffer rinsing in between. After completing the adsorption, a potential of 1 V was applied several times in order to measure the current generated by the enzymes at different substrate concentrations. B) Current after 10 min as a function of the glucose concentration. The applied potential was 1 V as stated on the left side.
Figure 17.
A) EC-OWLS curve showing adsorption as a function of time. Additionally, the signal also reacts to an applied potential. Prior to the adsorption steps the system was electrically cycled to obtain stable conditions. PLL-g-PEG/biotin, neutravidin and biotin conjugated GOx were adsorbed in three steps with buffer rinsing in between. After completing the adsorption, a potential of 1 V was applied several times in order to measure the current generated by the enzymes at different substrate concentrations. B) Current after 10 min as a function of the glucose concentration. The applied potential was 1 V as stated on the left side.
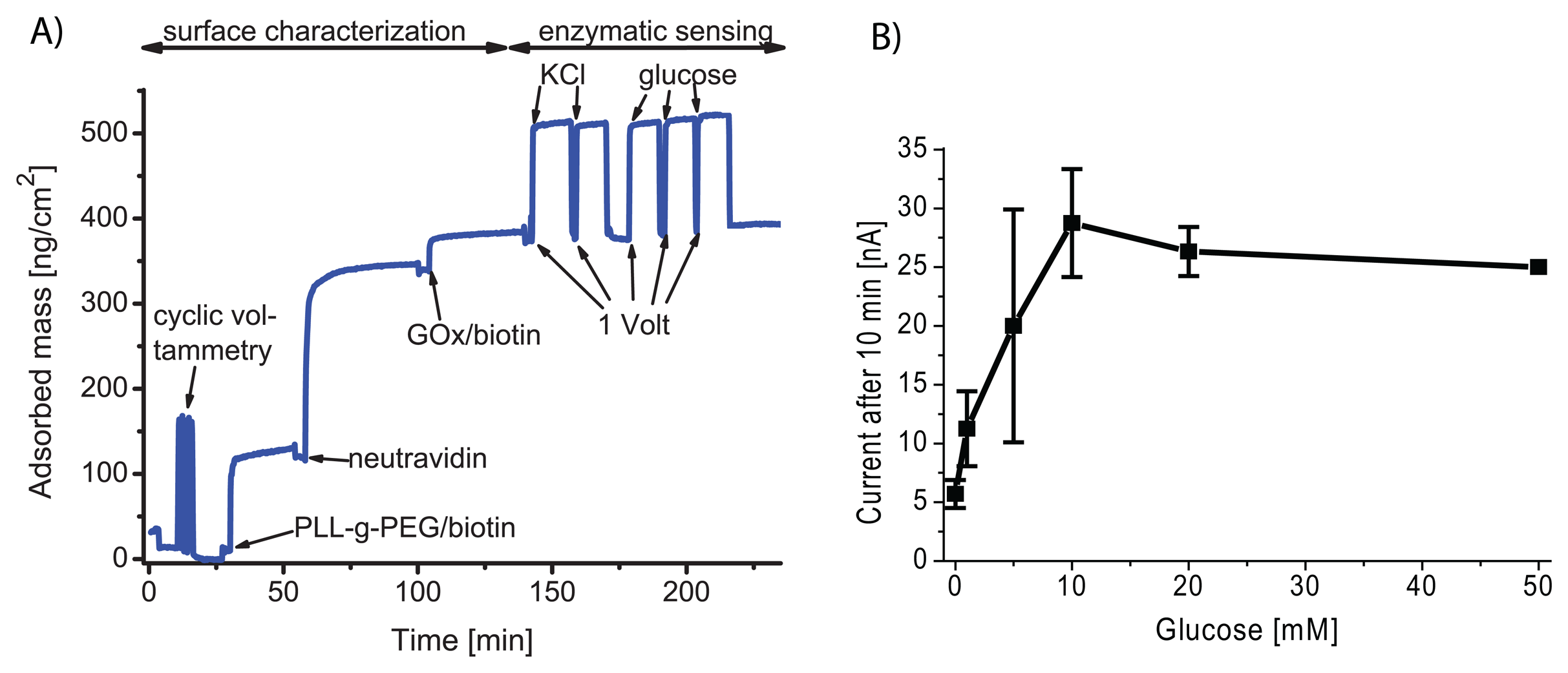
Figure 18.
An overview of an SPM technique combined with electrochemical techniques; a) SEM image and schematic top view of an AFM-tip-integrated frame electrode; b) schematic cross section of an AFM-tip integrated biosensor. Enzymes are shown to be immobilized at the surface of the scanning-probe tip-integrated electrode; c) schematic cross section of the experimental setup for imaging glucose transport through a porous membrane during simultaneous AFM mapping [157]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
Figure 18.
An overview of an SPM technique combined with electrochemical techniques; a) SEM image and schematic top view of an AFM-tip-integrated frame electrode; b) schematic cross section of an AFM-tip integrated biosensor. Enzymes are shown to be immobilized at the surface of the scanning-probe tip-integrated electrode; c) schematic cross section of the experimental setup for imaging glucose transport through a porous membrane during simultaneous AFM mapping [157]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.
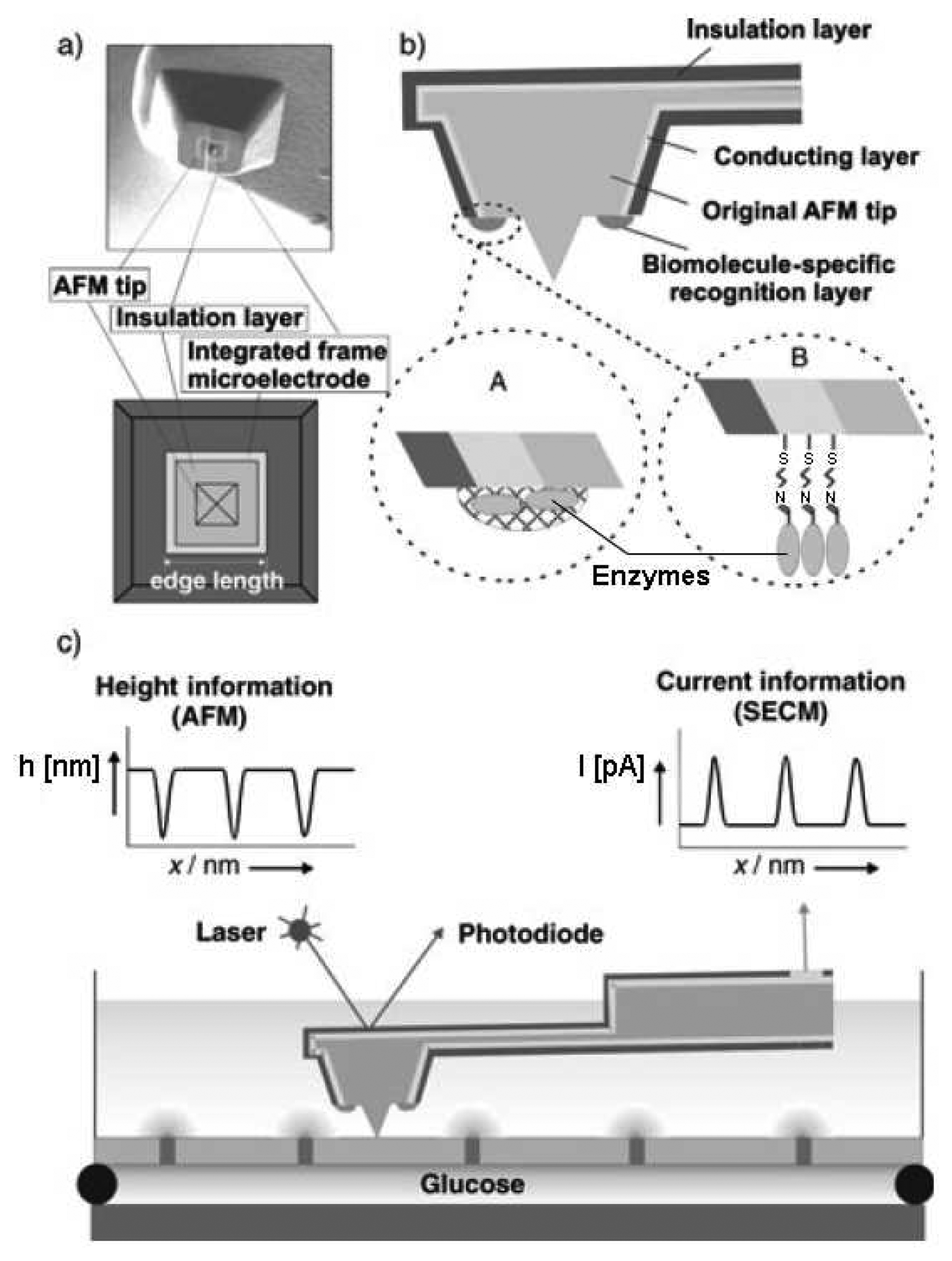
Figure 19.
Topographic images obtained using constant-current mode of SECM imaging as presented by Kurulugama et al. of (A) an undifferentiated PC12 cell, (B) early neurite development and (C) a differentiated PC12 cell. In their work they also provide images using other feedback modes, namely constant-impedance and constant-distance. Reprinted with permission from [166]. Copyright 2005 American Chemical Society.
Figure 19.
Topographic images obtained using constant-current mode of SECM imaging as presented by Kurulugama et al. of (A) an undifferentiated PC12 cell, (B) early neurite development and (C) a differentiated PC12 cell. In their work they also provide images using other feedback modes, namely constant-impedance and constant-distance. Reprinted with permission from [166]. Copyright 2005 American Chemical Society.
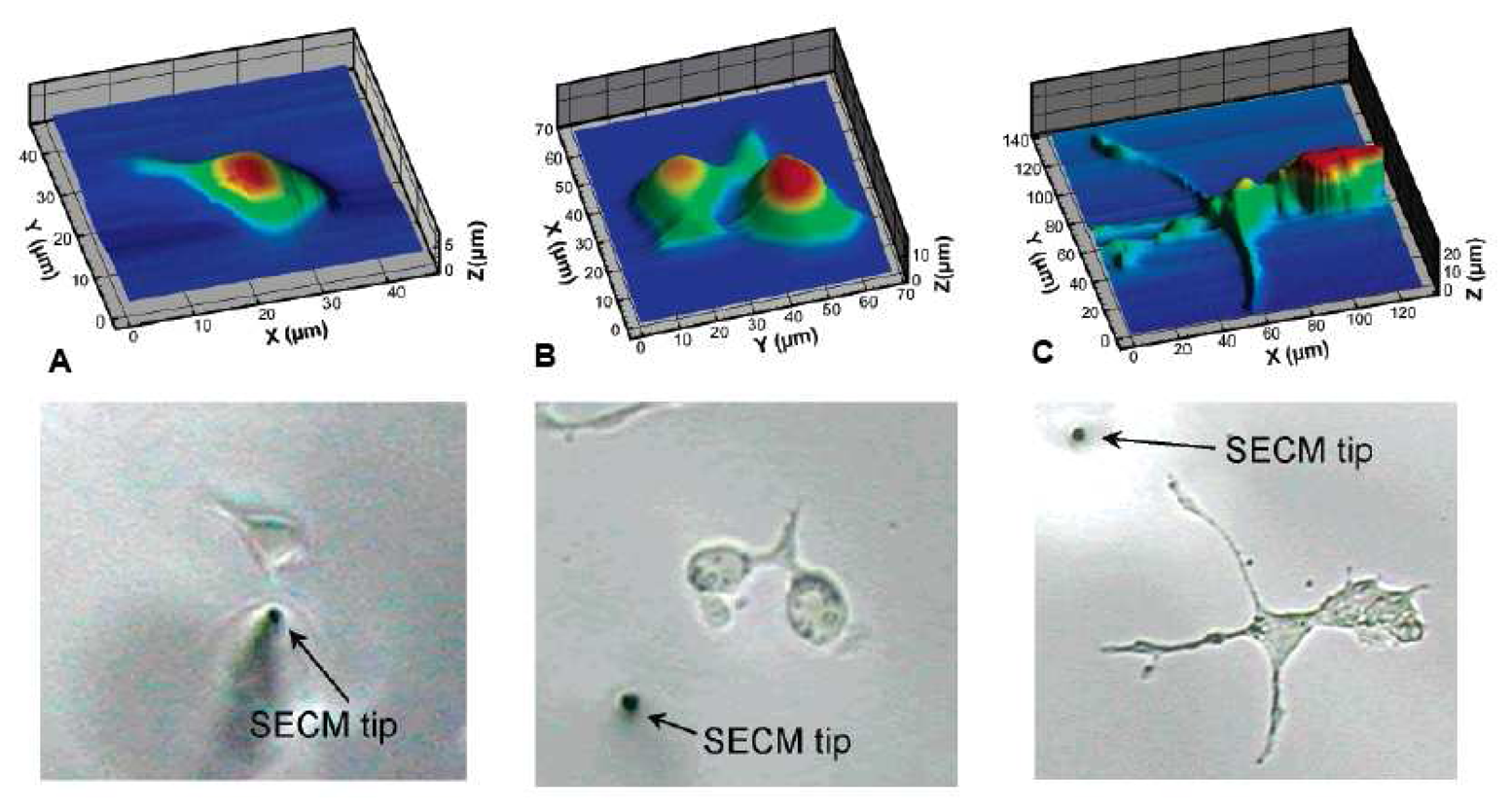
Figure 20.
a) Simplified scheme for magnetically labelled biomolecule detection. The magnetic label, functionalized with biomolecule A, interacts with the complementary biomolecule B at the magnetoresistive sensing surface. The resulting magnetic fringe field changes the resistance of the magnetoresistive sensor, which is measured as a change in voltage (ΔV) at a fixed sensing current (I). b) Simplified scheme for the use of magnetically labelled streptavidin to detect the location of pre-hybridized biotinylated DNA. First, the immobilized DNA on the magnetoresistive sensor surface is hybridized with biotinylated target DNA. Second, the magnetically labelled streptavidin is introduced to detect the hybridized DNA by binding to the biotinylated hybridized target DNA. The magnetic fringe field of the labels is detected by the magnetoresistive sensors as in a) [185].
Figure 20.
a) Simplified scheme for magnetically labelled biomolecule detection. The magnetic label, functionalized with biomolecule A, interacts with the complementary biomolecule B at the magnetoresistive sensing surface. The resulting magnetic fringe field changes the resistance of the magnetoresistive sensor, which is measured as a change in voltage (ΔV) at a fixed sensing current (I). b) Simplified scheme for the use of magnetically labelled streptavidin to detect the location of pre-hybridized biotinylated DNA. First, the immobilized DNA on the magnetoresistive sensor surface is hybridized with biotinylated target DNA. Second, the magnetically labelled streptavidin is introduced to detect the hybridized DNA by binding to the biotinylated hybridized target DNA. The magnetic fringe field of the labels is detected by the magnetoresistive sensors as in a) [185].
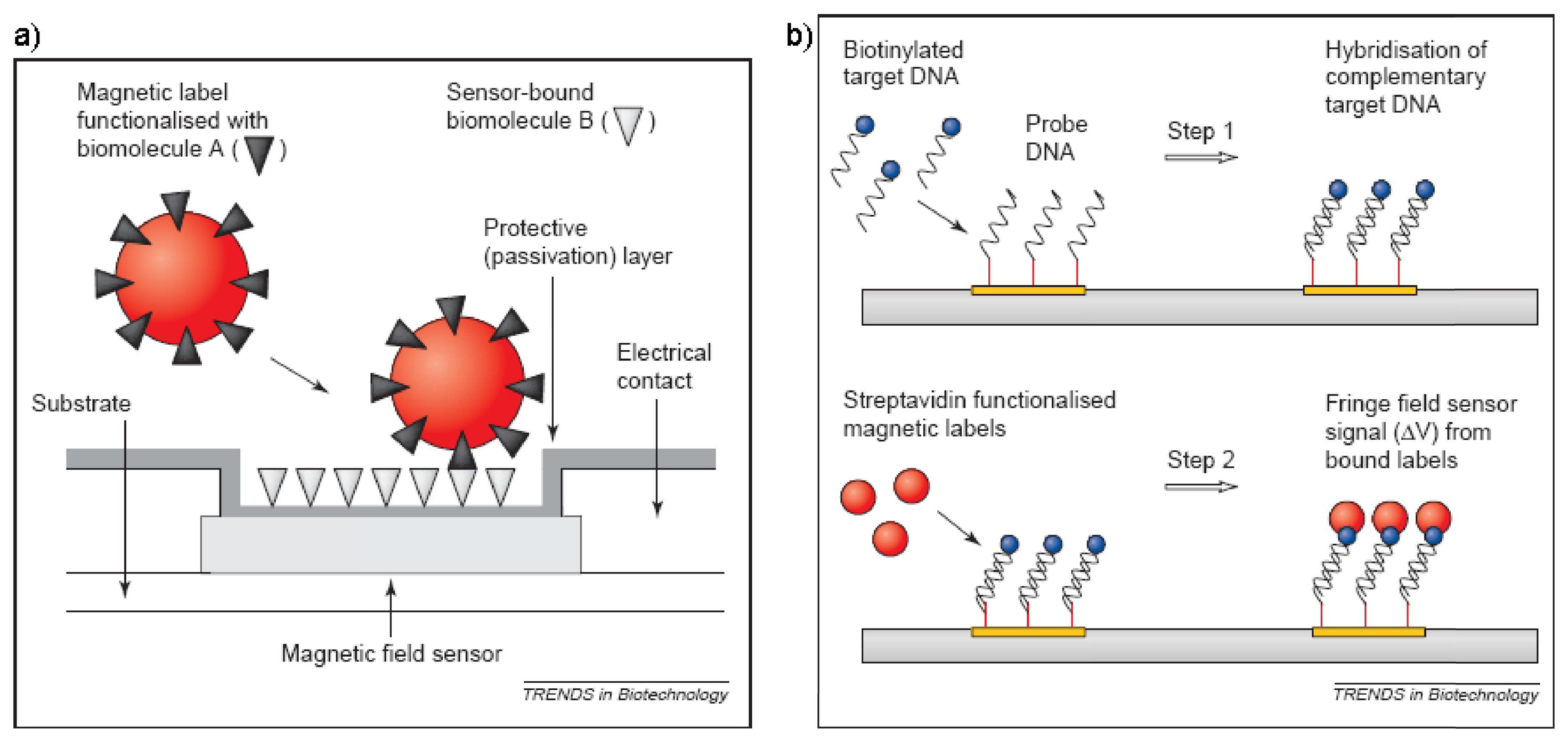
Figure 21.
Scheme of direct and indirect electron transduction. a) Direct transduction: the electrons only generate a measurable current if the reaction takes place close to the surface. b) Indirect transduction: with the help of a mediator, that shuttles the electrons between the reaction site and the surface, much larger distances can be overcome. c) Enzyme bound via conducting CNT. d) ELISA-like sandwich setup, where an enzyme labelled antibody binds to the detected antigen.
Figure 21.
Scheme of direct and indirect electron transduction. a) Direct transduction: the electrons only generate a measurable current if the reaction takes place close to the surface. b) Indirect transduction: with the help of a mediator, that shuttles the electrons between the reaction site and the surface, much larger distances can be overcome. c) Enzyme bound via conducting CNT. d) ELISA-like sandwich setup, where an enzyme labelled antibody binds to the detected antigen.
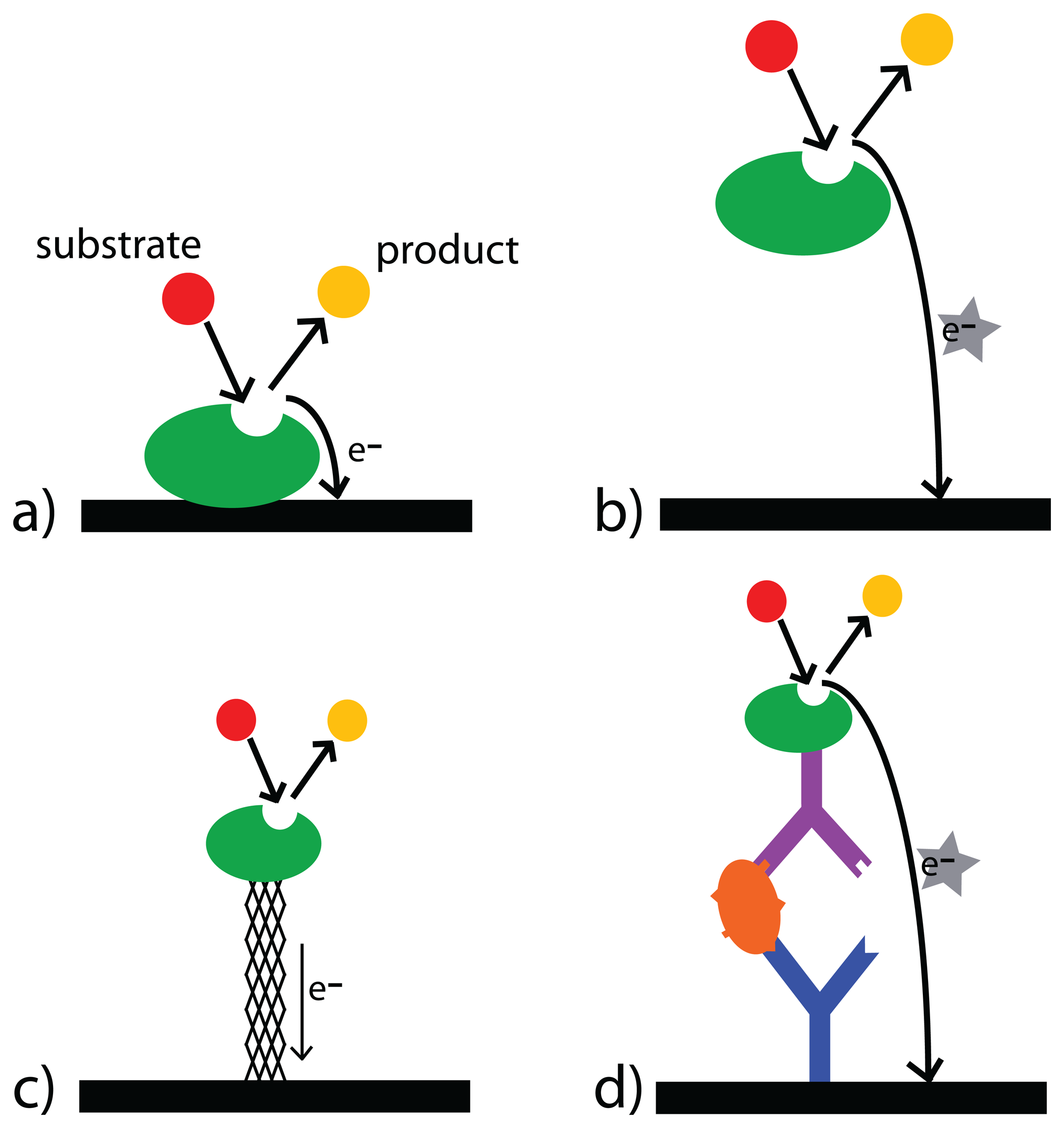
Figure 22.
Schematics of supported lipid bilayer architectures used for sensor applications including electrochemical biosensors: a) hybrid bilayer, b) solid-supported lipid bilayer (SLB), c) tethered lipid bilayer (t-SLB), d) polymer-supported bilayers (pSLB), e) pore-spanning lipid bilayers (nano-BLM) and f) a nano-BLM with an incorporated protein for single channel recordings.
Figure 22.
Schematics of supported lipid bilayer architectures used for sensor applications including electrochemical biosensors: a) hybrid bilayer, b) solid-supported lipid bilayer (SLB), c) tethered lipid bilayer (t-SLB), d) polymer-supported bilayers (pSLB), e) pore-spanning lipid bilayers (nano-BLM) and f) a nano-BLM with an incorporated protein for single channel recordings.
Figure 23.
Upon becoming triggered to the open state, the pore filters and selects only those specific ions or charged molecules that are designed to pass through the channel [284]. From Doyle, D. A. et al., SCIENCE 280:69-77 (April 3, 1998). Reprinted with permission from AAAS.
Figure 23.
Upon becoming triggered to the open state, the pore filters and selects only those specific ions or charged molecules that are designed to pass through the channel [284]. From Doyle, D. A. et al., SCIENCE 280:69-77 (April 3, 1998). Reprinted with permission from AAAS.

Figure 24.
Schematic of an ion-channel switch (ICS) biosensor as created by Cornell et al.: a) the addition of the target analyte inhibits channel formation by anchoring the pore, which lowers the electrochemical conductivity of the membrane, b) an alternative mechanism, where the introduced analyte replaces an existing bond and releases the pore. This then promotes the formation of channels and increases the membrane conductance [286].
Figure 24.
Schematic of an ion-channel switch (ICS) biosensor as created by Cornell et al.: a) the addition of the target analyte inhibits channel formation by anchoring the pore, which lowers the electrochemical conductivity of the membrane, b) an alternative mechanism, where the introduced analyte replaces an existing bond and releases the pore. This then promotes the formation of channels and increases the membrane conductance [286].
© 2008 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Grieshaber, D.; MacKenzie, R.; Vörös, J.; Reimhult, E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors 2008, 8, 1400-1458. https://doi.org/10.3390/s80314000
AMA Style
Grieshaber D, MacKenzie R, Vörös J, Reimhult E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors. 2008; 8(3):1400-1458. https://doi.org/10.3390/s80314000
Chicago/Turabian StyleGrieshaber, Dorothee, Robert MacKenzie, Janos Vörös, and Erik Reimhult. 2008. "Electrochemical Biosensors - Sensor Principles and Architectures" Sensors 8, no. 3: 1400-1458. https://doi.org/10.3390/s80314000