Spectrometric and Voltammetric Analysis of Urease – Nickel Nanoelectrode as an Electrochemical Sensor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1 Chemicals
2.2 Enzyme
| 10 | 20 | 30 | 40 | 50 | 60 |
| MNHFNRRQVL | PAVPHLLNII | QVEATLPNGT | KLVTVHDPIA | NENGDLEEAL | YGSFLPVPSL |
| 70 | 80 | 90 | 100 | 110 | 120 |
| DKFAESKEEH | KIPGEIICAD | GRLTLNPGRK | AVFLKVVNHG | DRPIQVGSHY | HFIEVNPYLT |
| 130 | 140 | 150 | 160 | 170 | 180 |
| FDRRKAYGMR | LNIAAGDSVR | FEPGDHKTVN | LVSIGGNKII | RGGNAIADGP | VNEANCKAAM |
| 190 | 200 | 210 | 220 | 230 | 240 |
| EIVCRREFGH | KEEEEASEGV | TTGDPDCPFT | KAIPREEYAN | KYGPTIGDKI | RLGDTDLIAE |
| 250 | 260 | 270 | 280 | 290 | 300 |
| IEKDFALYGD | ESVFGGGKVI | RDGMGQSSGH | PPAMSLDTVI | TSAVIIDYTG | IIKADIGIKD |
| 310 | 320 | 330 | 340 | 350 | 360 |
| GLIASIGKAG | NPDIMNGVFP | NMIIGVNTEV | ICGEGLIVTA | GGIDCHVHYI | CPQSLDEAIS |
| 370 | 380 | 390 | 400 | 410 | 420 |
| SGITTVVGGG | TGPTDGSRAT | TCTPAPTQMK | LMLQSTDDIP | LNFGFTGKGS | GSHPDELHEI |
| 430 | 440 | 450 | 460 | 470 | 480 |
| IKAGAMGLKL | HEDWGCTPAA | IDNCLAVAEQ | HDIQVNIHTD | TVNESGFVEH | TIAAFNGRTI |
| 490 | 500 | 510 | 520 | 530 | 540 |
| HTYHSEGAGG | GHAPDIIKVC | SMKNVLPSST | NTTRPLTSNT | VDEHLDMLMV | CHKLNREIPE |
| 550 | 560 | 570 | 580 | 590 | 600 |
| DLAFASSRVR | EQTIAAEDIL | HHIGGISIIS | SDAQAVGRIG | EVISCTWQTA | DKMKAERGPL |
| 610 | 620 | 630 | 640 | 650 | 660 |
| QPDGSDNDNF | RIKRYIAKYT | INPAIVNGIS | QYVGSVEVGK | LADLVIWKPS | FFGAKPDIVI |
| 670 | 680 | 690 | 700 | 710 | 720 |
| KGGSIAWADM | GDPNGSIPTP | EPVLMRPMYG | TLGKAGSALS | IAFVSKAALD | LGVKVLYGLN |
| 725 | |||||
| KGWNP |
2.3 Electrochemical measurements
Adsorptive transfer stripping differential pulse voltammetric analysis of urease
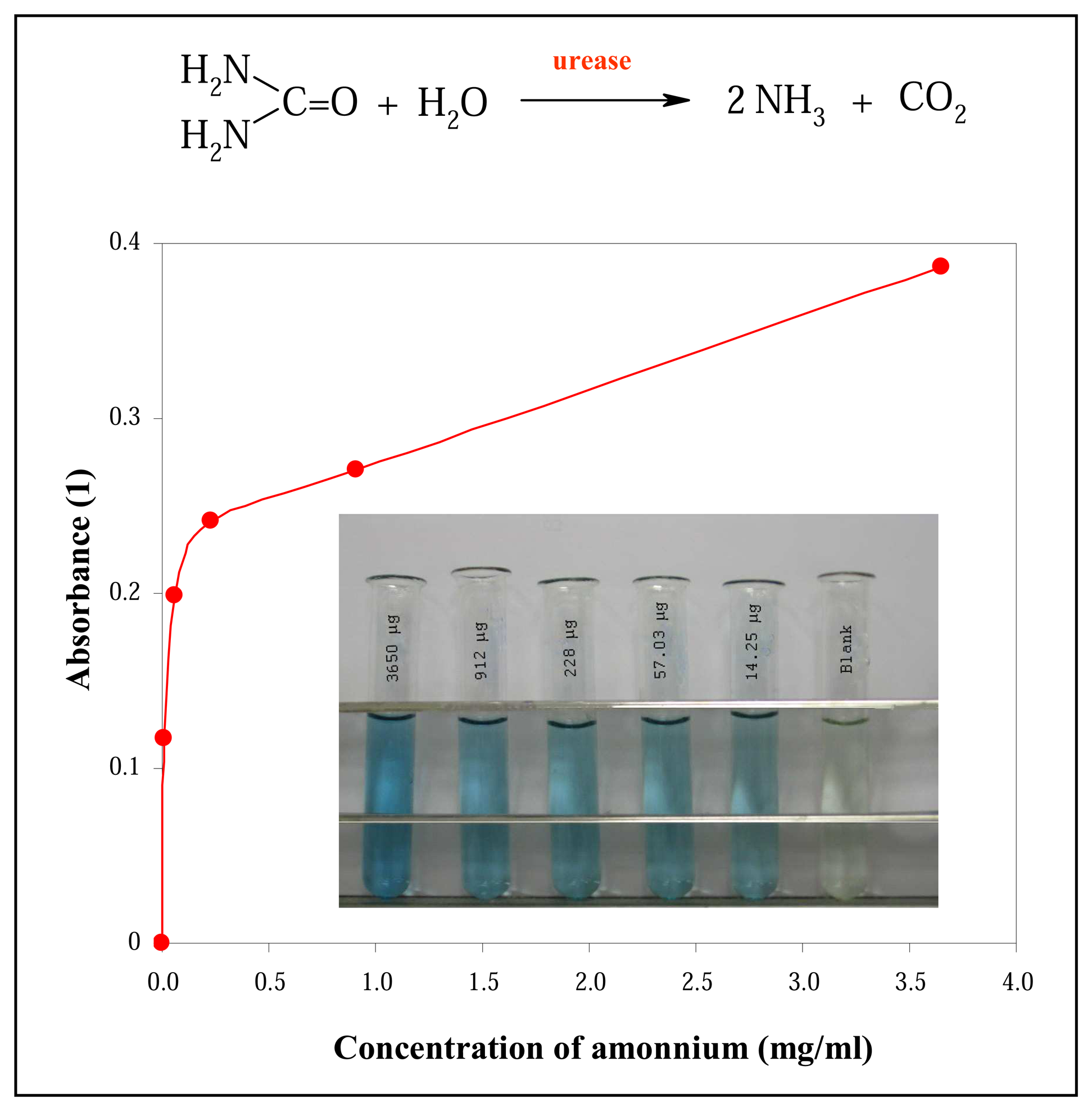
2.4 An indophenol assay for the detection of ammonium – Berthelot method
2.5 pH measurement
2.6 Scanning electron microscope
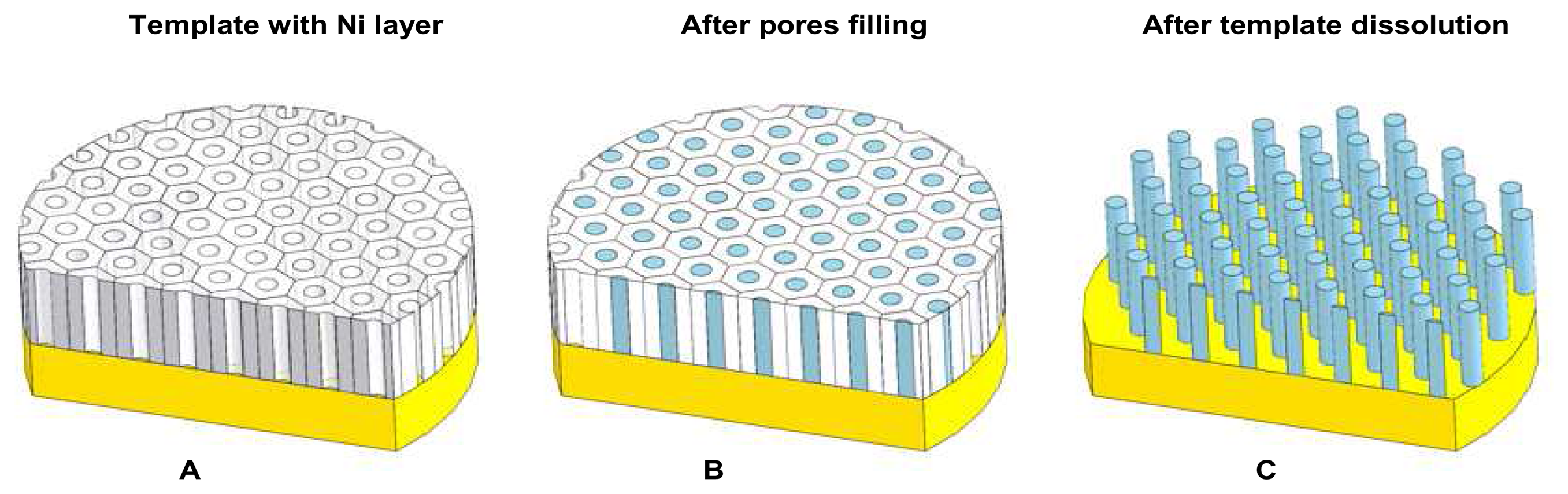
2.7 Ni nanopillars creation
2.8 Ni nanoelectrode preparation for experimental measurements
3. Results and Discussion
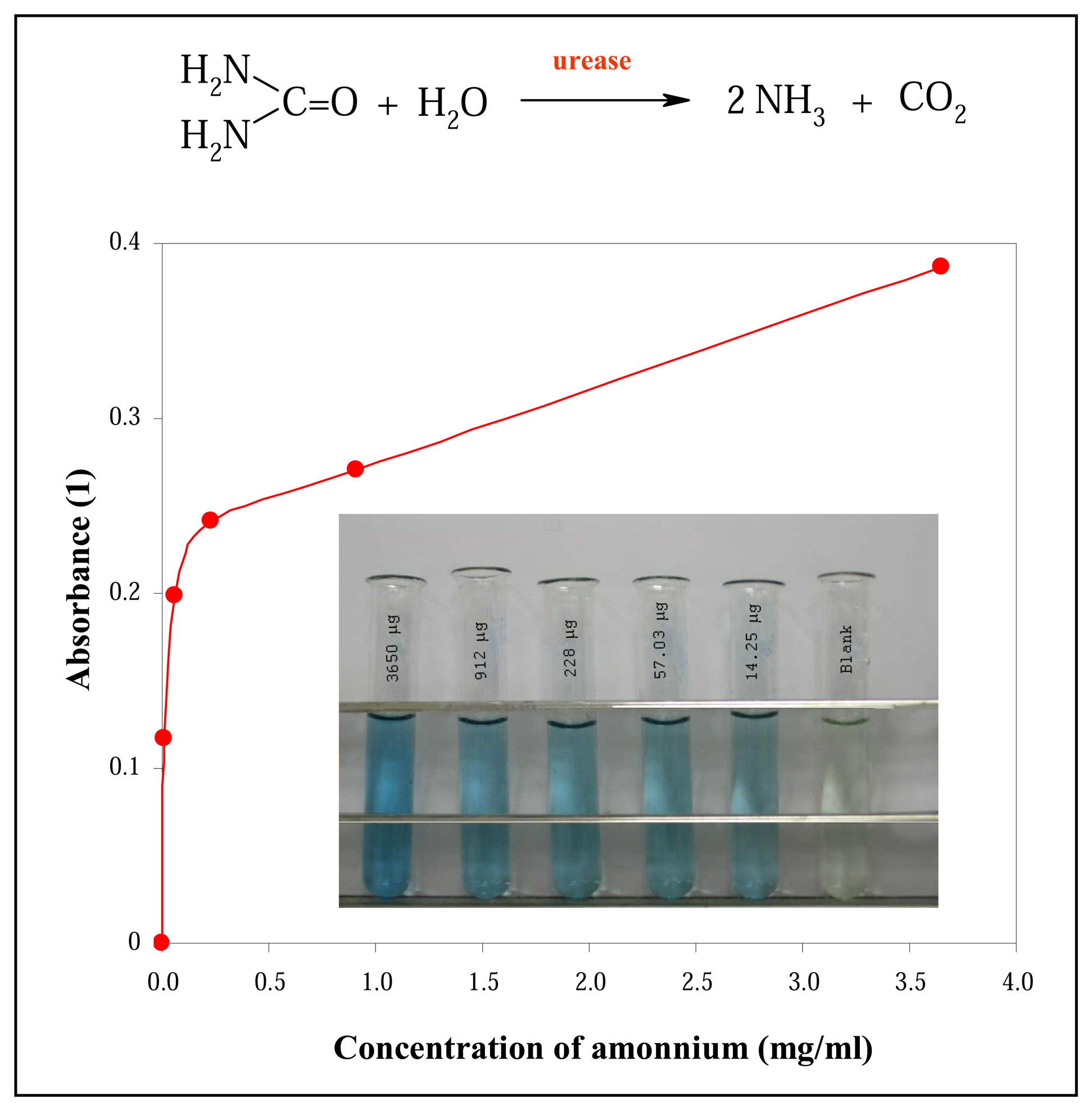
3.1 Spectrophotometric determination of urease activity
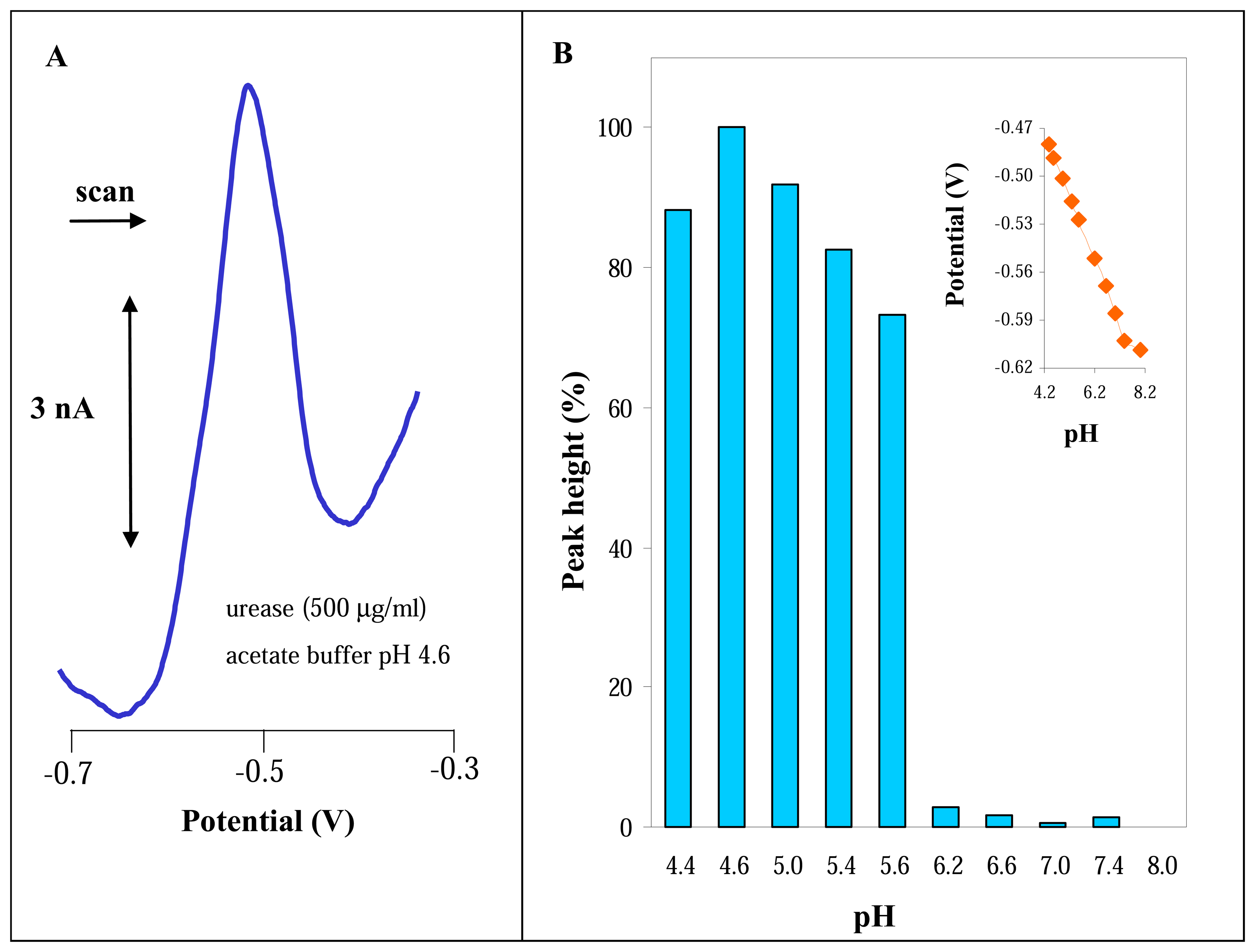
3.2 Electrochemical study of urease
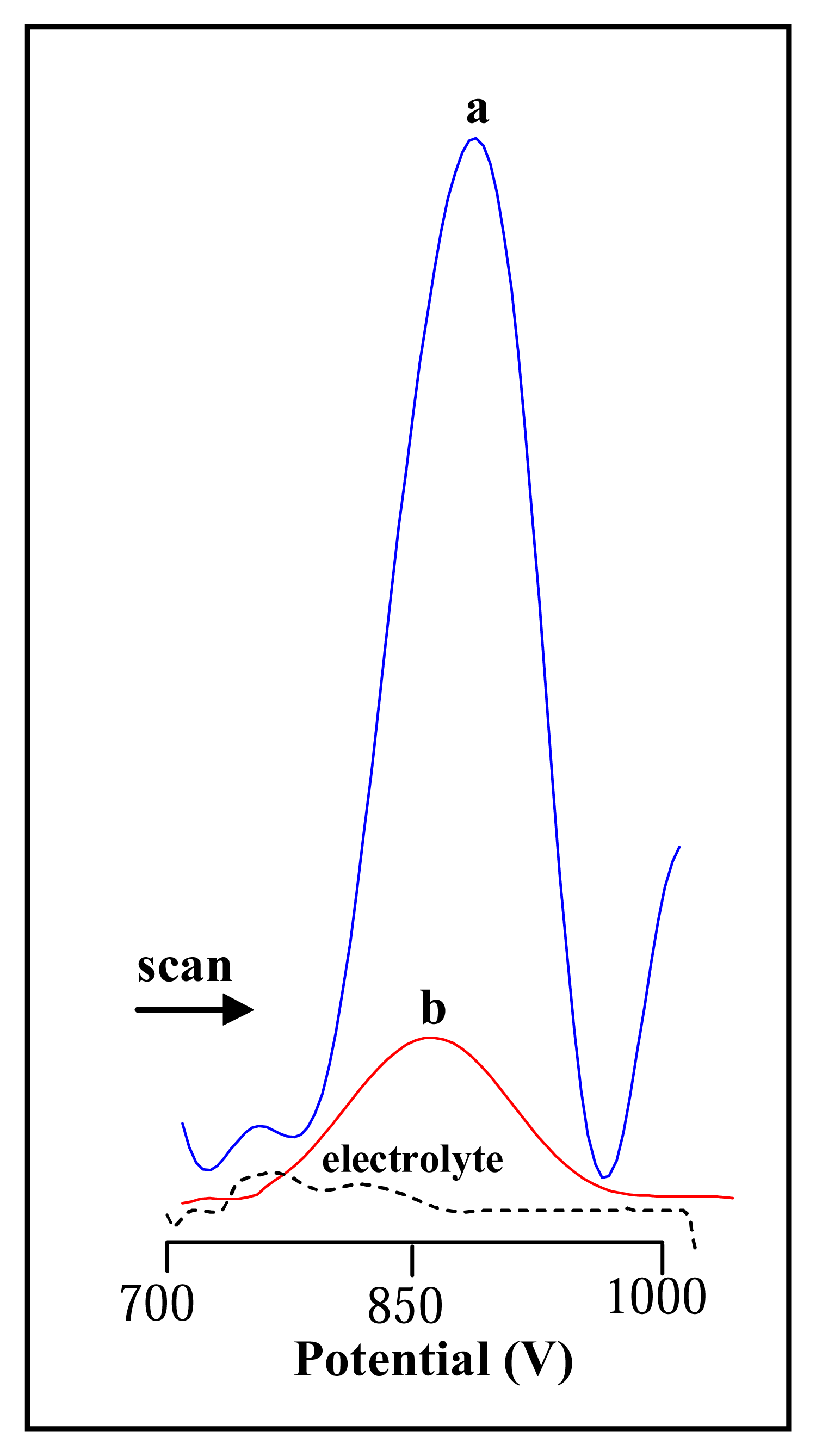
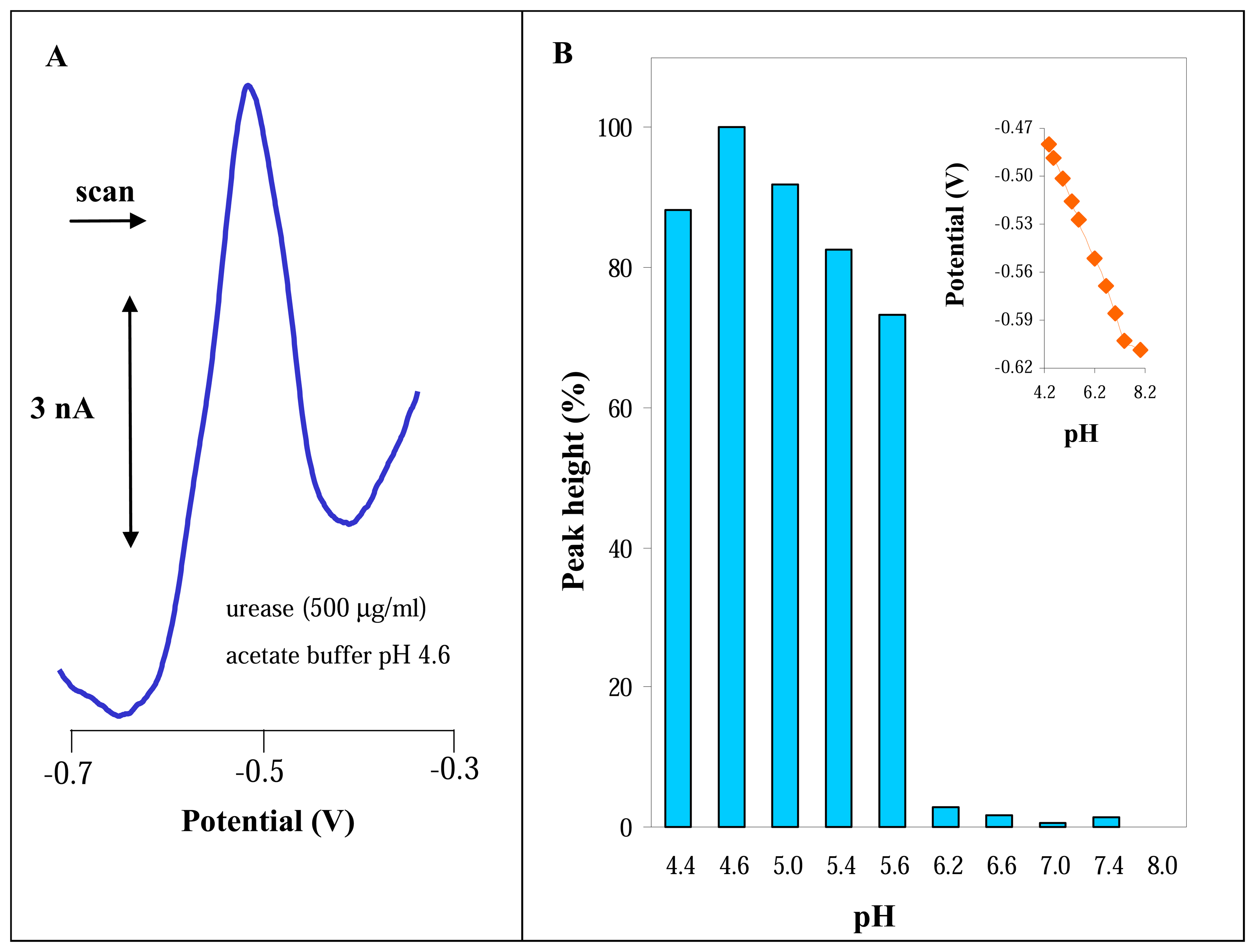
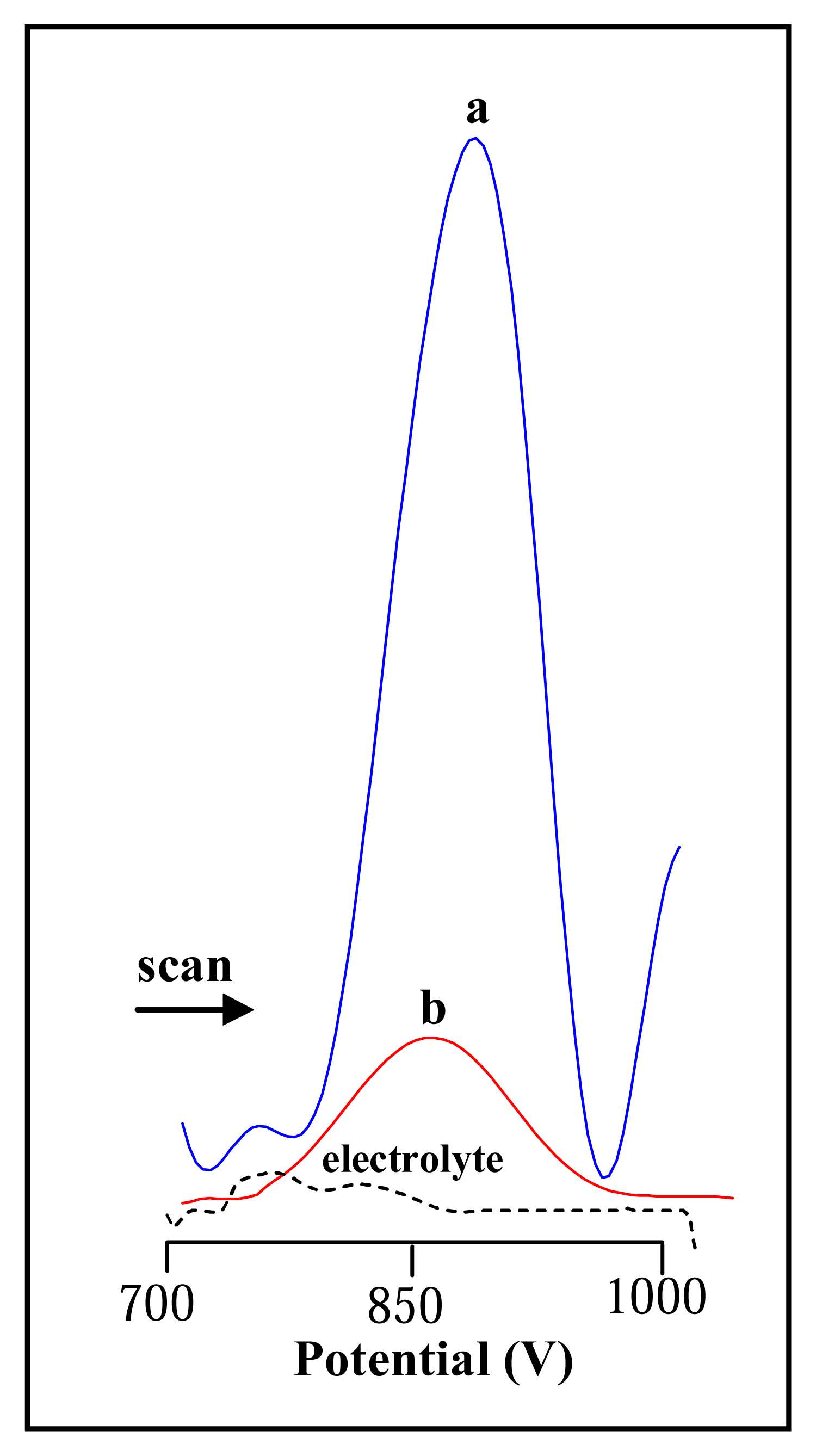
3.2.1 Differential pulse voltammetric analysis of urease
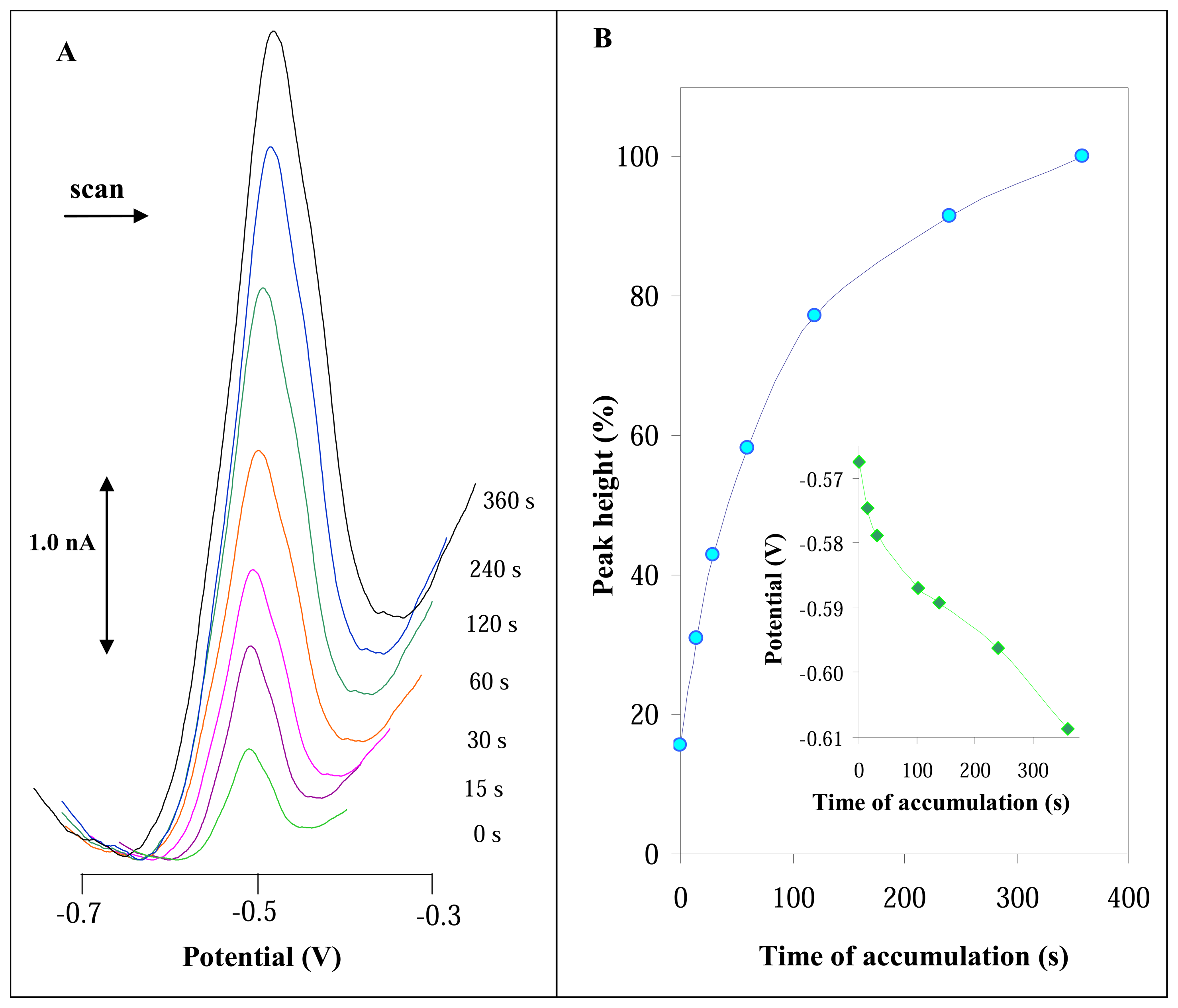
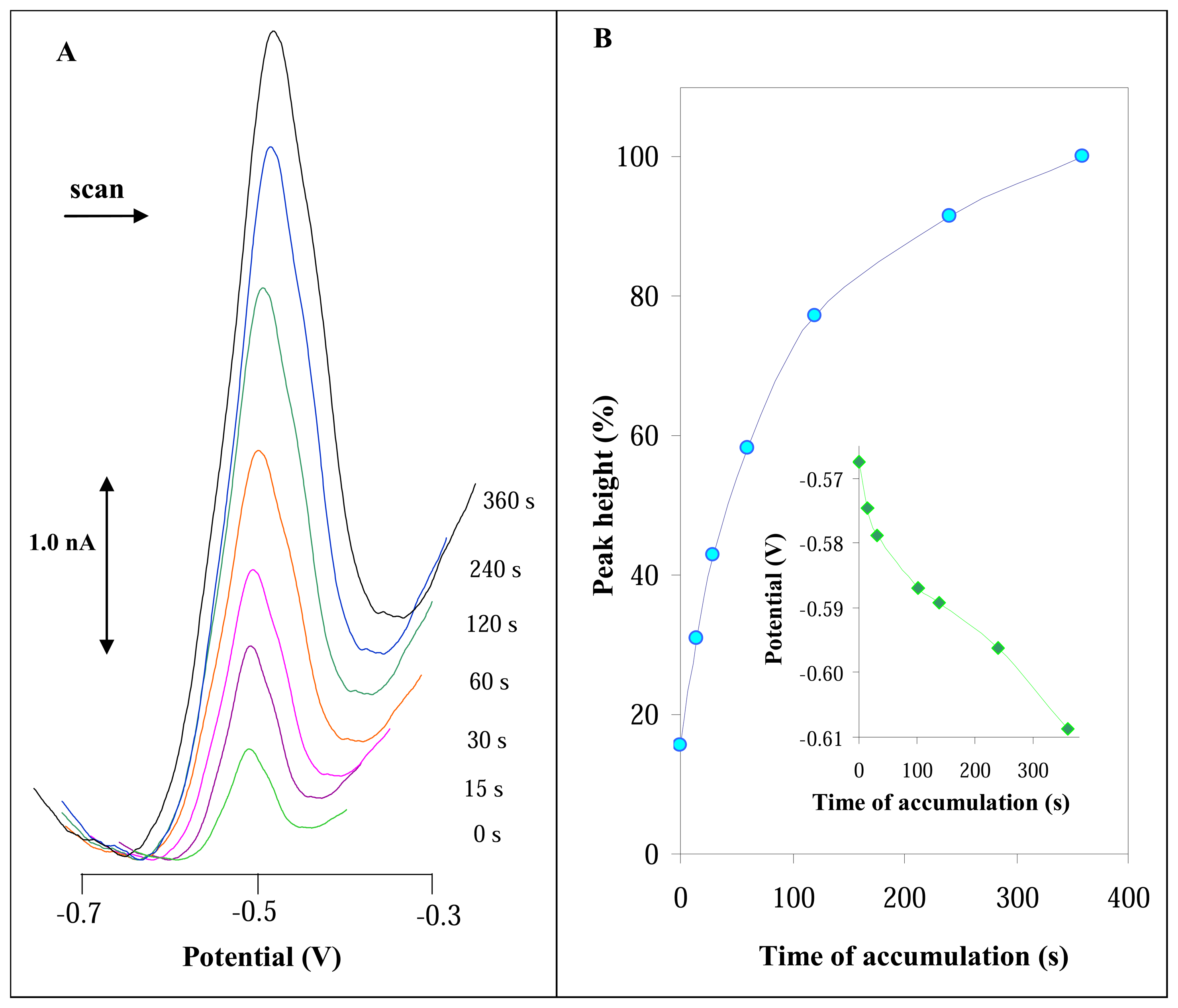
3.2.2 Influence of accumulation time on DPV signal of urease
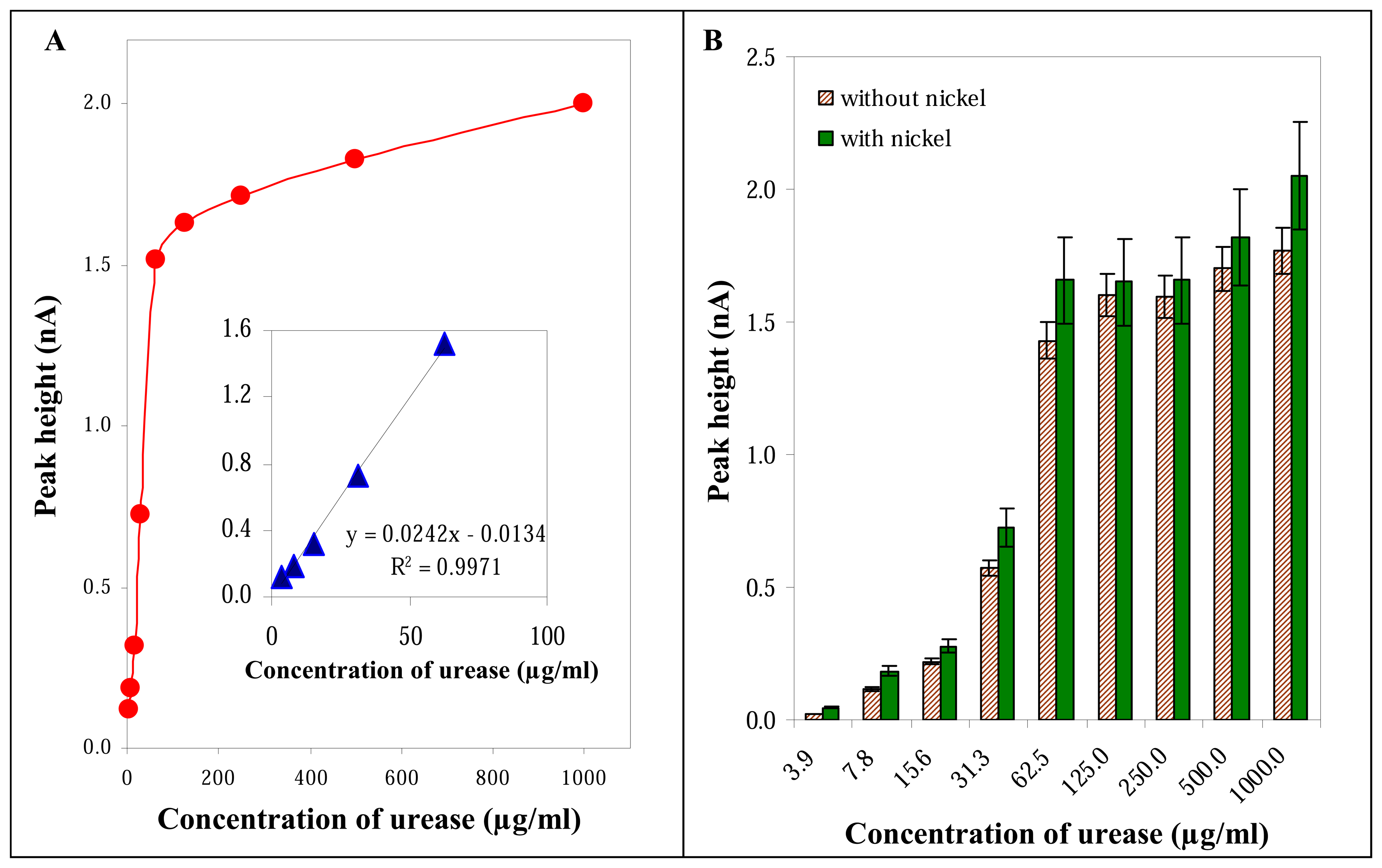
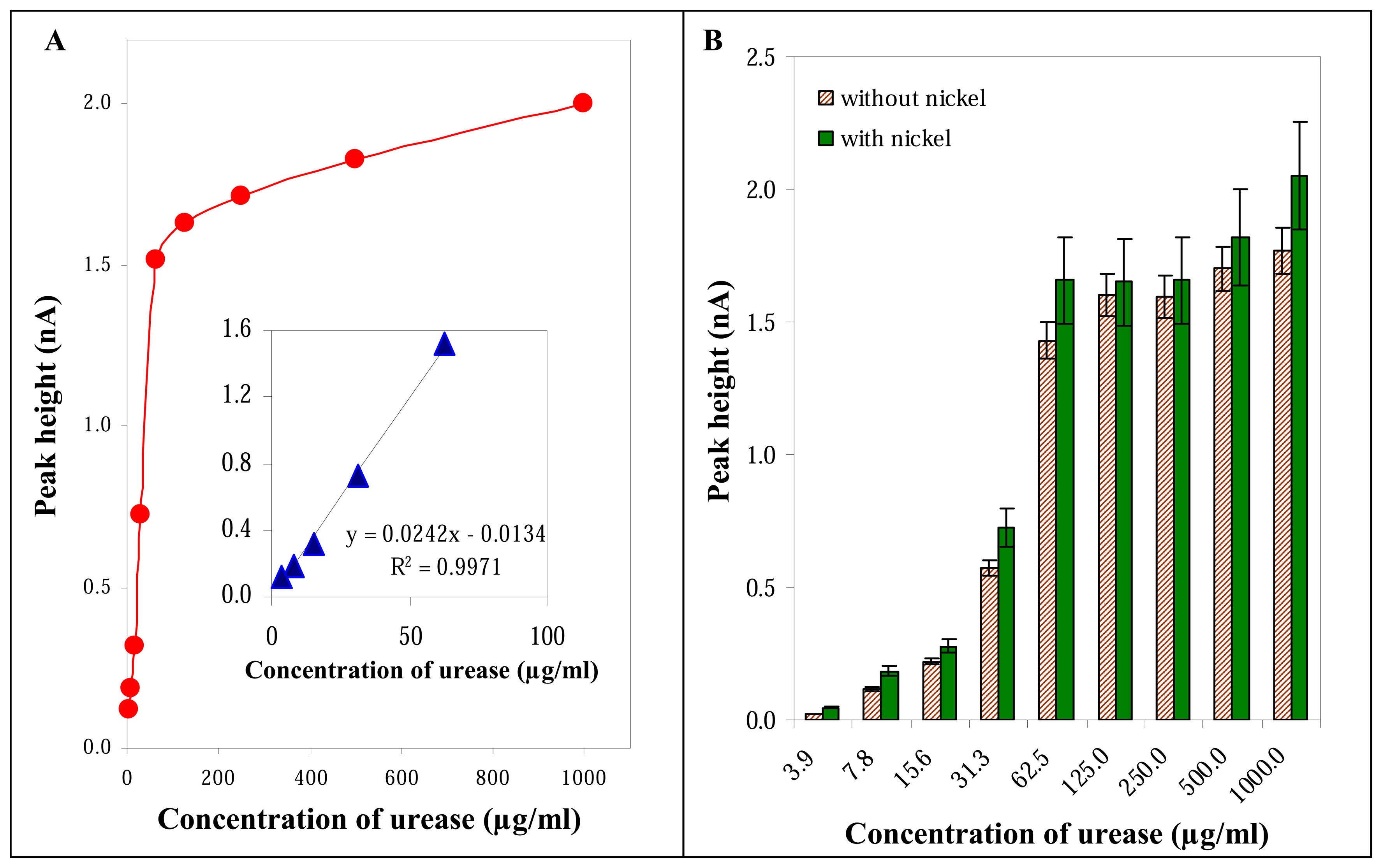
3.2.3 Influence of urease concentration
3.3 Influence of nickel on the electrochemical signal of urease
3.4 SEM analysis of Ni nanoelectrode surface
3.5 Detection of urease by Ni nanoelectrodes
4. Conclusion
Acknowledgments
References
- Sumner, J.B. The isolation and crystallization of the enzyme urease. J. Biol. Chem. 1926, 69, 435–441. [Google Scholar]
- Mobley, M.L.; Hausinger, R.P. Microbial ureases: significance, regulation and molecular characterization. Microbiol. Rev. 1989, 53, 85–108. [Google Scholar]
- Witte, C.P.; Rosso, M.G.; Romeis, T. Identification of three urease accessory proteins that are required for urease activation in Arabidopsis. Plant Physiol. 2005, 139, 1155–1162. [Google Scholar]
- Witte, C.P.; Tiller, S.; Isidore, E.; Davies, H.V.; Taylor, M.A. Analysis of two alleles of the urease gene from potato: polymorphisms, expression, and extensive alternative splicing of the corresponding mRNA. J. Exp. Bot. 2005, 56, 91–99. [Google Scholar]
- Witte, C.P.; Tiller, S.A.; Taylor, M.A.; Davies, H.V. Leaf urea metabolism in potato. Urease activity profile and patterns of recovery and distribution of N-15 after foliar urea application in wild-type and urease-antisense transgenics. Plant Physiol. 2002, 128, 1129–1136. [Google Scholar]
- Polacco, J.C.; Winkler, R.G. Soyaben leaf urease: a seed enzyme. Plant Physiol. 1984, 74, 800–803. [Google Scholar]
- de Melo, W.J.; Aguiar, P.D.; de Melo, G.M.P.; de Melo, V.P. Nickel in a tropical soil treated with sewage sludge and cropped with maize in a long-term field study. Soil Biol. Biochem. 2007, 39, 1341–1347. [Google Scholar]
- Thoren, A.K. Urea transformation of wetland microbial communities. Microb. Ecol. 2007, 53, 221–232. [Google Scholar]
- Kojima, S.; Bohner, A.; von Wiren, N. Molecular mechanisms of urea transport in plants. J. Membr. Biol. 2006, 212, 83–91. [Google Scholar]
- Kohler, J.; Caravaca, F.; Carrasco, L.; Roldan, A. Interactions between a plant growth-promoting rhizobacterium, an AM fungus and a phosphate-solubilising fungus in the rhizosphere of Lactuca sativa. Appl. Soil Ecol. 2007, 35, 480–487. [Google Scholar]
- Varel, V.H.; Wells, J.E.; Miller, D.N. Combination of a urease inhibitor and a plant essential oil to control coliform bacteria, odour production and ammonia loss from cattle waste. J. Appl. Microbiol. 2007, 102, 472–477. [Google Scholar]
- Yang, Z.X.; Liu, S.Q.; Zheng, D.W.; Feng, S.D. Effects of cadium, zinc and lead on soil enzyme activities. J. Environ. Sci. 2006, 18, 1135–1141. [Google Scholar]
- Dixon, N.E.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack-Bean Urease (Ec 3.5.1.5) -Metalloenzyme - Simple Biological Role for Nickel. J. Am. Chem. Soc. 1975, 97, 4131–4133. [Google Scholar]
- Zehnalek, J.; Adam, V.; Kizek, R. Assimilation of nitrate, ammonium and amide nitrogen by agricultural crops. Chem. Listy 2006, 100, 508–514. [Google Scholar]
- Robotis, J. Two rapid urease tests comparison (CLO Vs PRONTO DRY) for the detection of HP infection. Gastroenterology 2003, 124, A176–A177. [Google Scholar]
- Vilaichone, R.; Mahachai, V.; Nunthapisud, P.; Thong-Gyam, D.; Kullavanijaya, P. Gastric juice urease test and brushing urease test for Helicobacter pylori detection. Gut 2002, 51, A111–A112. [Google Scholar]
- Wisniewska, M.; Nilsson, H.O.; Bak-Romaniszyn, L.; Rechcinski, T.; Bielanski, W.; Planeta-Malecka, I.; Plonka, M.; Konturek, S.; Wadstrom, T.; Rudnicka, W.; Chmiela, M. Detection of specific Helicobacter pylori DNA and antigens in stool samples in dyspeptic patients and healthy subjects. Microbiol. Immunol. 2004, 46, 657–665. [Google Scholar]
- Xia, H.H.X.; Wong, B.C.Y. Gastric biopsy-based rapid urease tests for the detection of Helicobacter pylori: Progress, advantages and limitations. J. Gastroenterol. Hepatol. 2002, 17, 629–632. [Google Scholar]
- Yee, Y.K.; Yip, K.T.; Que, T.L.; Chang, K.K.; Li, K.F.; Lee, C.K.; Wong, S.W.; Lau, S.F.; Szeto, M.L. Efficacy of enzyme immunoassay for the detection of Helicobacter pylori antigens in frozen stool specimens: local validation. Aliment. Pharmacol. Ther. 2002, 16, 1739–1742. [Google Scholar]
- Berdoz, J.; Corthesy, B. Human polymeric IgA is superior to IgG and single-chain Fv of the same monoclonal specificity to inhibit urease activity associated with Helicobacter pylori. Mol. Immunol. 2004, 41, 1013–1022. [Google Scholar]
- Tripathi, R.N.; Nath, N.; Gandhi, A.P. Studies on the quality of canned baked soybeans. J. Food Sci. Technol. -Mysore 2004, 41, 131–134. [Google Scholar]
- Kizek, R.; Trnková, L.; Palecek, E. Determination of metallothionein at the femtomole level by constant current stripping chronopotentiomentry. Anal. Chem. 2001, 73, 4801–4807. [Google Scholar]
- Kizek, R.; Vacek, J.; Trnkova, L.; Klejdus, B.; Havel, L. Application of catalytic reactions on a mercury electrode for electrochemical detection of metallothioneins. Chem. Listy 2004, 98, 166–173. [Google Scholar]
- Masarik, M.; Stobiecka, A.; Kizek, R.; Jelen, F.; Pechan, Z.; Hoyer, W.; Jovin, T.M.; Subramaniam, V.; Palecek, E. Sensitive electrochemical detection of native and aggregated alpha-synuclein protein involved in Parkinson's disease. Electroanalysis 2004, 16, 1172–1181. [Google Scholar]
- Palecek, E.; Masarik, M.; Kizek, R.; Kuhlmeier, D.; Hassmann, J.; Schulein, J. Sensitive electrochemical determination of unlabeled MutS protein and detection of point mutations in DNA. Anal. Chem. 2004, 76, 5930–5936. [Google Scholar]
- Prusa, R.; Potesil, D.; Masarik, M.; Adam, V.; Kizek, R.; Jelen, F. Fast and sensitive electrochemical detection of native, denatured, and aggregated forms of tumor suppressor protein p53. Mol. Biol. Cell 2004, 15, 249A–249A. [Google Scholar]
- Kukacka, J.; Zitka, O.; Horna, A.; Stejskal, K.; Zehnalek, J.; Adam, V.; Havel, L.; Zeman, L.; Prusa, R.; Trnkova, L.; Kizek, R. A new tool for distinguishing of different structural forms of lactoferrin. Faseb J. 2007, 21, A635–A635. [Google Scholar]
- Zitka, O.; Horna, A.; Stejskal, K.; Zehnalek, J.; Adam, V.; Havel, L.; Zeman, L.; Kizek, R. Study of structural changes of lactoferrin using flow injection analysis with electrochemical detection on glassy carbon electrode. Acta Chim. Slov. 2007, 54, 68–73. [Google Scholar]
- Zitka, O.; Stejskal, K.; Kleckerova, A.; Adam, V.; Beklova, M.; Horna, A.; Supalkova, V.; Havel, L.; Kizek, R. Utilizing electrochemical techniques for detection of biological samples. Chem. Listy 2007, 101, 225–231. [Google Scholar]
- Adam, V.; Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Trnkova, L.; Jelen, F.; Kizek, R. Study of metallothionein modified electrode surface behavior in the presence of heavy metal ions-biosensor. Electroanalysis 2005, 17, 1649–1657. [Google Scholar]
- Adam, V.; Zehnalek, J.; Petrlova, J.; Potesil, D.; Sures, B.; Trnkova, L.; Jelen, F.; Vitecek, J.; Kizek, R. Phytochelatin modified electrode surface as a sensitive heavy-metal ion biosensor. Sensors 2005, 5, 70–84. [Google Scholar]
- Kizek, R.; Masarik, M.; Kramer, K.J.; Potesil, D.; Bailey, M.; Howard, J.A.; Klejdus, B.; Mikelova, R.; Adam, V.; Trnkova, L.; Jelen, F. An analysis of avidin, biotin and their interaction at attomole levels by voltammetric and chromatographic techniques. Anal. Bioanal. Chem. 2005, 381, 1167–1178. [Google Scholar]
- Huang, X.J.; Choi, Y.K. Chemical sensors based on nanostructured materials. Sens. Actuator B-Chem. 2007, 122, 659–671. [Google Scholar]
- Daniel, S.; Rao, T.P.; Rao, K.S.; Rani, S.U.; Naidu, G.R.K.; Lee, H.Y.; Kawai, T. A review of DNA functionalized/grafted carbon nanotubes and their characterization. Sens. Actuator B-Chem. 2007, 122, 672–682. [Google Scholar]
- Li, L.; Li, G.H.; Fang, X.S. Bi-based nanowire and nanojunction arrays: Fabrication and physical properties. J. Mater. Sci. Technol. 2007, 23, 166–181. [Google Scholar]
- Sque, S.J.; Jones, R.; Oberg, S.; Briddon, P.R. Carbon nanotubes and their interaction with the surface of diamond. Phys. Rev. B 2007, 75, 16. [Google Scholar]
- Billinge, S.J.L.; Levin, I. The problem with determining atomic structure at the nanoscale. Science 2007, 316, 561–565. [Google Scholar]
- Tang, J.; Xing, G.M.; Zhao, F.; Yuan, H.; Zhao, Y.L. Modulation of structural and electronic properties of fullerene and metallofullerenes by surface chemical modifications. J. Nanosci. Nanotechnol. 2007, 7, 1085–1101. [Google Scholar]
- Bandaru, P.R. Electrical properties and applications of carbon nanotube structures. J. Nanosci. Nanotechnol. 2007, 7, 1239–1267. [Google Scholar]
- Chen, Y.; Lin, Y.; Liu, Y.; Doyle, J.; He, N.; Zhuang, X.D.; Bai, J.R.; Blau, W.J. Carbon nanotube-based functional materials for optical limiting. J. Nanosci. Nanotechnol. 2007, 7, 1268–1283. [Google Scholar]
- Wei, W.; Sethuraman, A.; Jin, C.; Monteiro-Riviere, N.A.; Narayan, R.J. Biological properties of carbon nanotubes. J. Nanosci. Nanotechnol. 2007, 7, 1284–1297. [Google Scholar]
- Wang, J. Carbon-nanotube based electrochemical biosensors: A review. Electroanalysis 2005, 17, 7–14. [Google Scholar]
- Lin, Y.; Taylor, S.; Li, H.P.; Fernando, K.A.S.; Qu, L.W.; Wang, W.; Gu, L.R.; Zhou, B.; Sun, Y.P. Advances toward bioapplications of carbon nanotubes. J. Mater. Chem. 2004, 14, 527–541. [Google Scholar]
- Martin, C.R.; Kohli, P. The emerging field of nanotube biotechnology. Nat. Rev. Drug Discov. 2003, 2, 29–37. [Google Scholar]
- Niyogi, S.; Hamon, M.A.; Hu, H.; Zhao, B.; Bhowmik, P.; Sen, R.; Itkis, M.E.; Haddon, R.C. Chemistry of single-walled carbon nanotubes. Accounts Chem. Res. 2002, 35, 1105–1113. [Google Scholar]
- Baughman, R.H.; Zakhidov, A.A.; de Heer, W.A. Carbon nanotubes - the route toward applications. Science 2002, 297, 787–792. [Google Scholar]
- Thostenson, E.T.; Ren, Z.F.; Chou, T.W. Advances in the science and technology of carbon nanotubes and their composites: a review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar]
- Terrones, M.; Hsu, W.K.; Kroto, H.W.; Walton, D.R.M. Nanotubes: A revolution in materials science and electronics; Springer-Verlag: Berlin, 1999; pp. 189–234. [Google Scholar]
- Subramoney, S. Novel nanocarbons - Structure, properties, and potential applications. Adv. Mater. 1998, 10, 1157, -+. [Google Scholar]
- Ebbesen, T.W. Carbon Nanotubes. Annu. Rev. Mater. Sci. 1994, 24, 235–264. [Google Scholar]
- Leoni, L.; Attiah, D.; Desai, T.A. Nanoporous platforms for cellular sensing and delivery. Sensors 2002, 2, 111–120. [Google Scholar]
- Wang, J. Nanoparticle-based electrochemical bioassays of proteins. Electroanalysis 2007, 19, 769–776. [Google Scholar]
- Cochrane, C.; Koncar, V.; Lewandowski, M.; Dufour, C. Design and development of a flexible strain sensor for textile structures based on a conductive polymer composite. Sensors 2007, 7, 473–492. [Google Scholar]
- Jaffrezic-Renault, N.; Martelet, C.; Chevolot, Y.; Cloarec, J.P. Biosensors and bio-bar code assays based on biofunctionalized magnetic microbeads. Sensors 2007, 7, 589–614. [Google Scholar]
- Lyons, M.E.G.; Keeley, G.P. The redox behaviour of randomly dispersed single walled carbon nanotubes both in the absence and in the presence of adsorbed glucose oxidase. Sensors 2006, 6, 1791–1826. [Google Scholar]
- Lu, J.H.; Cao, X.A.; Pan, C.Y.; Yang, L.F.; Lai, G.B.; Chen, J.L.; Wu, C.Q. Studies of the cataluminescence of benzene homologues on nanosized gamma-Al2O3/Eu2O3 and the development of a gas sensor for benzene homologue vapors. Sensors 2006, 6, 1827–1836. [Google Scholar]
- Liewhiran, C.; Phanichphant, S. Influence of thickness on ethanol sensing characteristics of doctor-bladed thick film from flame-made ZnO nanoparticles. Sensors 2007, 7, 185–201. [Google Scholar]
- De Stefano, L.; Arcari, P.; Lamberti, A.; Sanges, C.; Rotiroti, L.; Rea, I.; Rendina, I. DNA optical detection based on porous silicon technology: from biosensors to biochips. Sensors 2007, 7, 214–221. [Google Scholar]
- Liu, N.; Hui, J.; Sun, C.Q.; Dong, J.H.; Zhang, L.Z.; Xiao, H. Nanoporous zeolite thin film-based fiber intrinsic Fabry-Perot interferometric sensor for detection of dissolved organics in water. Sensors 2006, 6, 835–847. [Google Scholar]
- Kang, B.S.; Wang, H.T.; Tien, L.C.; Ren, F.; Gila, B.P.; Norton, D.P.; Abernathy, C.R.; Lin, J.S.; Pearton, S.J. Wide bandgap semiconductor nanorod and thin film gas sensors. Sensors 2006, 6, 643–666. [Google Scholar]
- Wang, L.Y.; Kariuki, N.N.; Schadt, M.; Mott, D.; Luo, J.; Zhong, C.J.; Shi, X.J.; Zhang, C.; Hao, W.B.; Lu, S.; Kim, N.; Wang, J.Q. Sensing arrays constructed from nanoparticle thin films and interdigitated microelectrodes. Sensors 2006, 6, 667–679. [Google Scholar]
- Katz, E.; Willner, I. Magneto-controlled quantized electron transfer to surface-confined redox units and metal nanoparticles. Sensors 2006, 6, 420–427. [Google Scholar]
- Zhang, X.L.; Wang, J.X.; Wang, Z.; Wang, S.C. Improvement of amperometric sensor used for determination of nitrate with polypyrrole nanowires modified electrode. Sensors 2005, 5, 580–593. [Google Scholar]
- Bushuev, Y.; Davletbaeva, S.; Muguet, F.F. Hydration simulations of a carbon nanotube, immersed in water, according to the 3-attractor water model. Sensors 2005, 5, 139–147. [Google Scholar]
- Yin, Y.J.; Lu, Y.F.; Wu, P.; Cai, C.X. Direct electrochemistry of redox proteins and enzymes promoted by carbon nanotubes. Sensors 2005, 5, 220–234. [Google Scholar]
- Cheng, F.L.; Zhang, M.L.; Wang, H. Fabrication of polypyrrole nanowire and nanotube arrays. Sensors 2005, 5, 245–249. [Google Scholar]
- Liang, W.; Yuan, Z.B. Direct electrochemistry of glucose oxidase at a gold electrode modified with single-wall carbon nanotubes. Sensors 2003, 3, 544–554. [Google Scholar]
- Witte, C.P.; Medina-Escobar, N. In-gel detection of urease with nitroblue tetrazolium and quantification of the enzyme from different crop plants using the indophenol reaction. Anal. Biochem. 2001, 290, 102–107. [Google Scholar]
- Bollo, S.; Ferreyra, N.F.; Rivas, G.A. Electrooxidation of DNA at glassy carbon electrodes modified with multiwall carbon nanotubes dispersed in chitosan. Electroanalysis 2007, 19, 833–840. [Google Scholar]
- Castaneda, M.T.; Alegret, S.; Merkoci, A. Electrochemical sensing of DNA using gold nanoparticles. Electroanalysis 2007, 19, 743–753. [Google Scholar]
- Erdem, A.; Sayar, F.; Karadeniz, H.; Guven, G.; Ozsoz, M.; Piskin, E. Development of streptavidin carrying magnetic nanoparticles and their applications in electrochemical nucleic acid sensor systems. Electroanalysis 2007, 19, 798–804. [Google Scholar]
- Horibe, T.; Zhang, J.D.; Oyama, M. Effects of capping reagents on the electron transfer reactions on gold nanoparticle-attached indium tin oxide electrodes. Electroanalysis 2007, 19, 847–852. [Google Scholar]
- Hsing, I.M.; Xu, Y.; Zhao, W.T. Micro- and nano-magnetic particles for applications in biosensing. Electroanalysis 2007, 19, 755–768. [Google Scholar]
- Liu, G.D.; Wang, J.; Wu, H.; Lin, Y.Y. Nanovehicles based bioassay labels. Electroanalysis 2007, 19, 777–785. [Google Scholar]
- Merkoci, A. Nanobiomaterials in electroanalysis. Electroanalysis 2007, 19, 739–741. [Google Scholar]
- Rivas, G.A.; Rubianes, M.D.; Pedano, M.L.; Ferreyra, N.F.; Luque, G.L.; Rodriguez, M.C.; Miscoria, S.A. Carbon nanotubes paste electrodes. A new alternative for the development of electrochemical sensors. Electroanalysis 2007, 19, 823–831. [Google Scholar]
- Rajesh; Bisht, V.; Takashima, W.; Kaneto, K. An amperometric urea biosensor based on covalent immobilization of urease onto an electrochemically prepared copolymer poly (N-3-aminopropyl pyrrole-co-pyrrole) film. Biomaterials 2005, 26, 3683–3690. [Google Scholar]
- Gambhir, A.; Gerard, M.; Mulchandani, A.K.; Malhotra, B.D. Coimmobilization of urease and glutamate dehydrogenase in electrochemically prepared polypyrrole-polyvinyl sulfonate films. Appl. Biochem. Biotechnol. 2001, 96, 249–257. [Google Scholar]
- Balugian, R.; Sorochinsky, V.; Seidguseinov, A. Electrochemical Sensor for the Determination of Urea in Blood on the Basis of Immobilized Urease. Artif. Organs 1980, 4, 137–138. [Google Scholar]
- Karube, I.; Suzuki, S. Electrochemical Preparation of Urease-Collagen Membrane. Biochem. Biophys. Res. Commun. 1972, 47, 51, -&. [Google Scholar]
- Palecek, E. Adsorptive transfer stripping voltammetry - determination of nanogram quanitities of DNA immobilized at the electrode surface. Anal. Biochem. 1988, 170, 421–431. [Google Scholar]
- Palecek, E. Adsorptive transfer stripping voltammetry: Effect of electrode potential on the structure of DNA adsorbed at the mercury surface. Bioelectrochem. Bioenerg. 1992, 28, 71–83. [Google Scholar]
- Palecek, E.; Jelen, F.; Postbieglova, I. Adsorptive transfer stripping voltammetry offers new possibilities in DNA research. Studia Biophysica 1989, 130, 51–54. [Google Scholar]
- Palecek, E.; Postbieglova, I. Adsorptive stripping voltammetry of bimacromolecules with transfer of the adsorbed layer. J. Electroanal. Chem. 1986, 214, 359–371. [Google Scholar]
- Adam, V.; Krizkova, S.; Zitka, O.; Trnkova, L.; Petrlova, J.; Beklova, M.; Kizek, R. Determination of apo-metallothionein using adsorptive transfer stripping technique in connection with differential pulse voltammetry. Electroanalysis 2007, 19, 339–347. [Google Scholar]
- Petrlova, J.; Potesil, D.; Mikelova, R.; Blastik, O.; Adam, V.; Trnkova, L.; Jelen, F.; Prusa, R.; Kukacka, J.; Kizek, R. Attomole voltammetric determination of metallothionein. Electrochim. Acta 2006, 51, 5112–5119. [Google Scholar]
- Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Adam, V.; Trnkova, L.; Kizek, R. Cisplatin electrochemical biosensor. Electrochim. Acta 2006, 51, 5169–5173. [Google Scholar]
- Kukacka, J.; Krizkova, S.; Zitka, O.; Prusa, R.; Adam, V.; Sures, B.; Beklova, M.; Kizek, R. Study of nucleic acids interactions with platinum based cytostatics using biosensor. Faseb J. 2007, 21, A262–A262. [Google Scholar]
- Krizkova, S.; Adam, V.; Petrlova, J.; Zitka, O.; Stejskal, K.; Zehnalek, J.; Sures, B.; Trnkova, L.; Beklova, M.; Kizek, R. A suggestion of electrochemical biosensor for study of platinum(II)-DNA interactions. Electroanalysis 2007, 19, 331–338. [Google Scholar]
- Adam, V.; Petrlova, J.; Potesil, D.; Lubal, P.; Zehnalek, J.; Sures, B.; Kizek, R. New electrochemical biosensor to determine platinum cytostatics to DNA structure. Chem. Listy 2005, 99, 353–393. [Google Scholar]
- Prusa, R.; Petrlova, J.; Kukacka, J.; Adam, V.; Sures, B.; Beklova, M.; Kizek, R. Study of interaction of glutathiones and metallothionein with cytostatics. Clin. Chem. 2006, 52, A175–A175. [Google Scholar]
- Potesil, D.; Mikelova, R.; Adam, V.; Kizek, R.; Prusa, R. Change of the protein p53 electrochemical signal according to its structural form - Quick and sensitive distinguishing of native, denatured, and aggregated form of the “guardian of the genome”. Protein J. 2006, 25, 23–32. [Google Scholar]
- Xu, Z.; Chen, X.; Qu, X.H.; Jia, J.B.; Dong, S.J. Single-wall carbon nanotube-based voltammetric sensor and biosensor. Biosens. Bioelectron. 2004, 20, 579–584. [Google Scholar]


© 2007 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Hubalek, J.; Hradecky, J.; Adam, V.; Krystofova, O.; Huska, D.; Masarik, M.; Trnkova, L.; Horna, A.; Klosova, K.; Adamek, M.; et al. Spectrometric and Voltammetric Analysis of Urease – Nickel Nanoelectrode as an Electrochemical Sensor. Sensors 2007, 7, 1238-1255. https://doi.org/10.3390/s7071238
Hubalek J, Hradecky J, Adam V, Krystofova O, Huska D, Masarik M, Trnkova L, Horna A, Klosova K, Adamek M, et al. Spectrometric and Voltammetric Analysis of Urease – Nickel Nanoelectrode as an Electrochemical Sensor. Sensors. 2007; 7(7):1238-1255. https://doi.org/10.3390/s7071238
Chicago/Turabian StyleHubalek, Jaromir, Jan Hradecky, Vojtech Adam, Olga Krystofova, Dalibor Huska, Michal Masarik, Libuse Trnkova, Ales Horna, Katerina Klosova, Martin Adamek, and et al. 2007. "Spectrometric and Voltammetric Analysis of Urease – Nickel Nanoelectrode as an Electrochemical Sensor" Sensors 7, no. 7: 1238-1255. https://doi.org/10.3390/s7071238