
Quantitative Determination of Fluorine Content in Blends of Polylactide (PLA)–Talc Using Near Infrared Spectroscopy

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
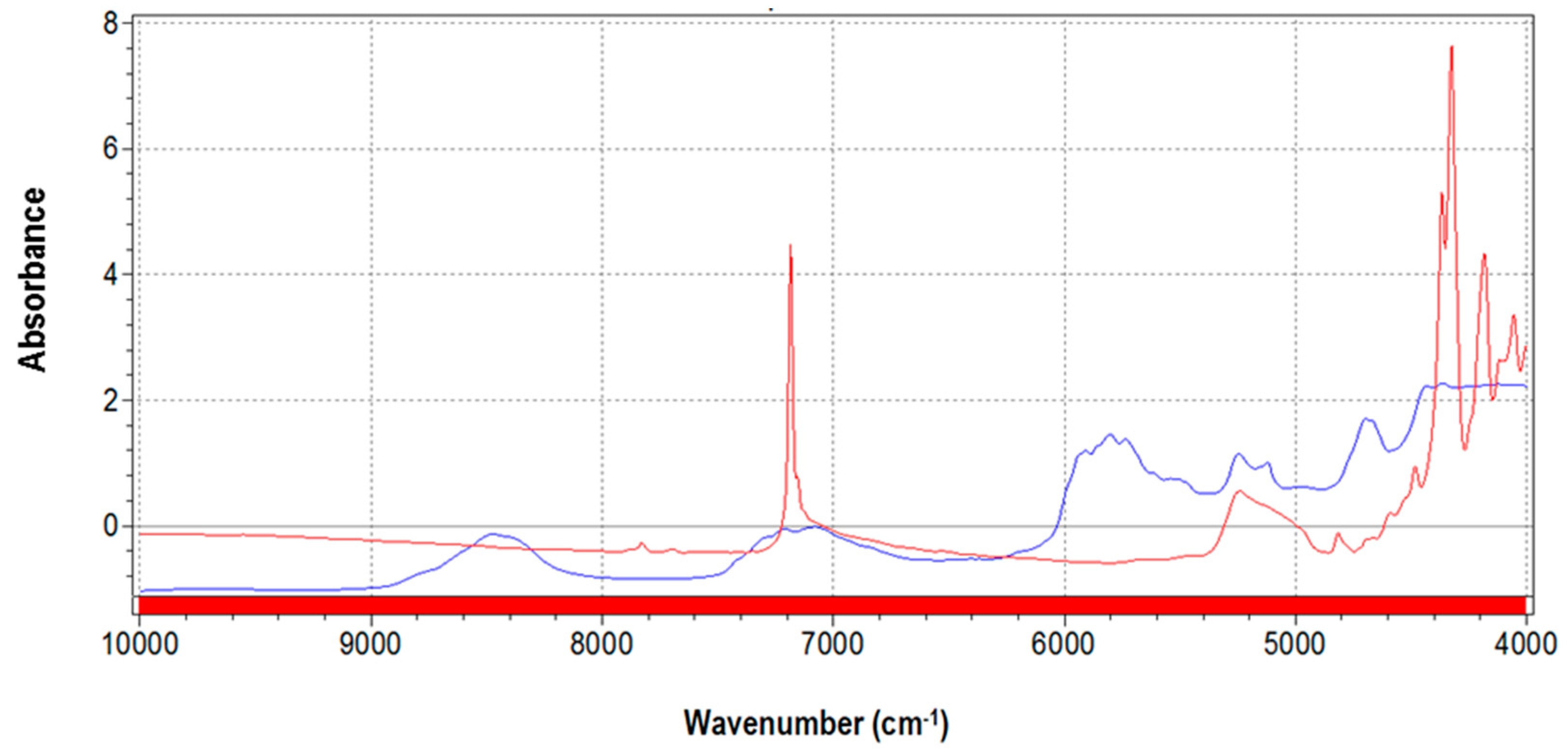
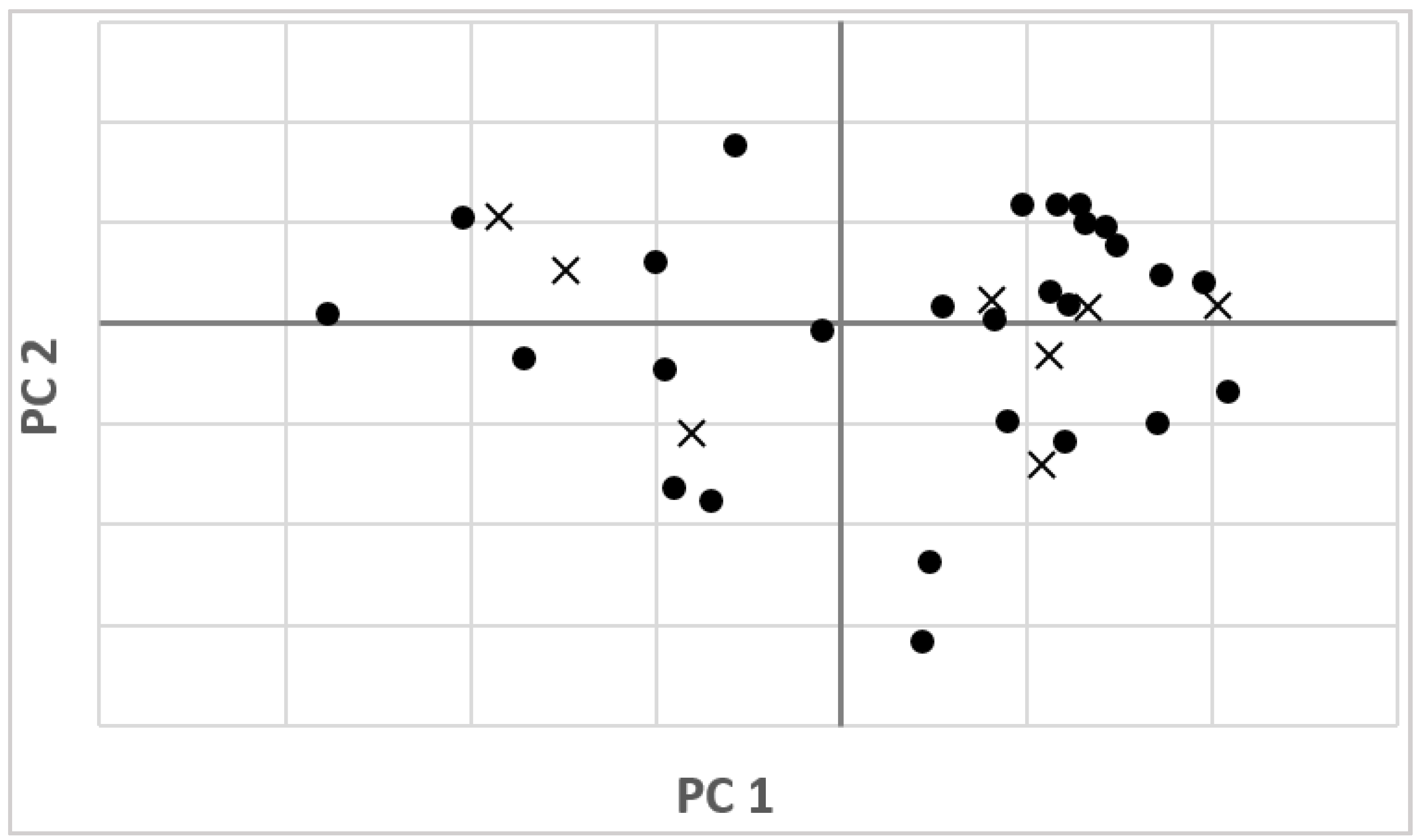
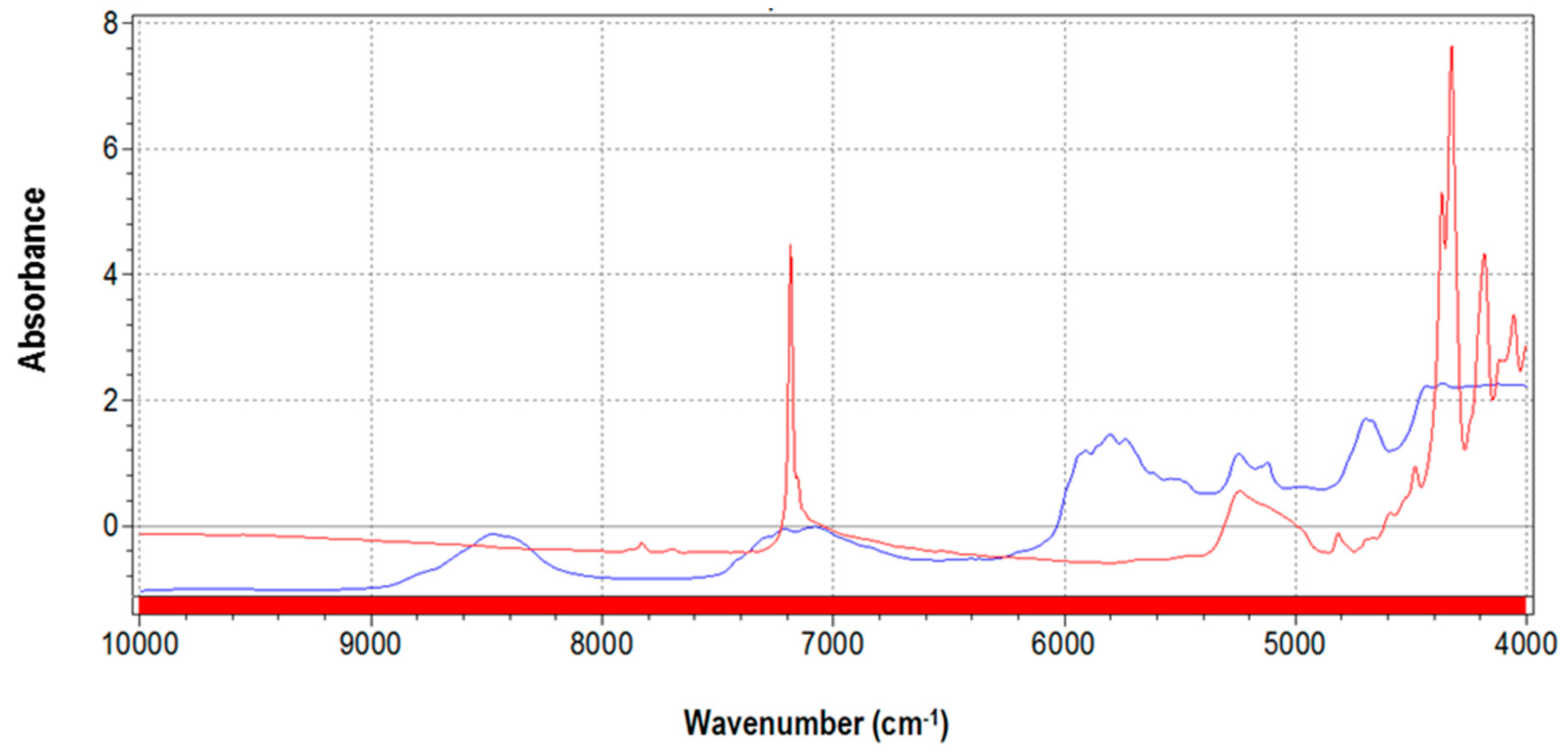
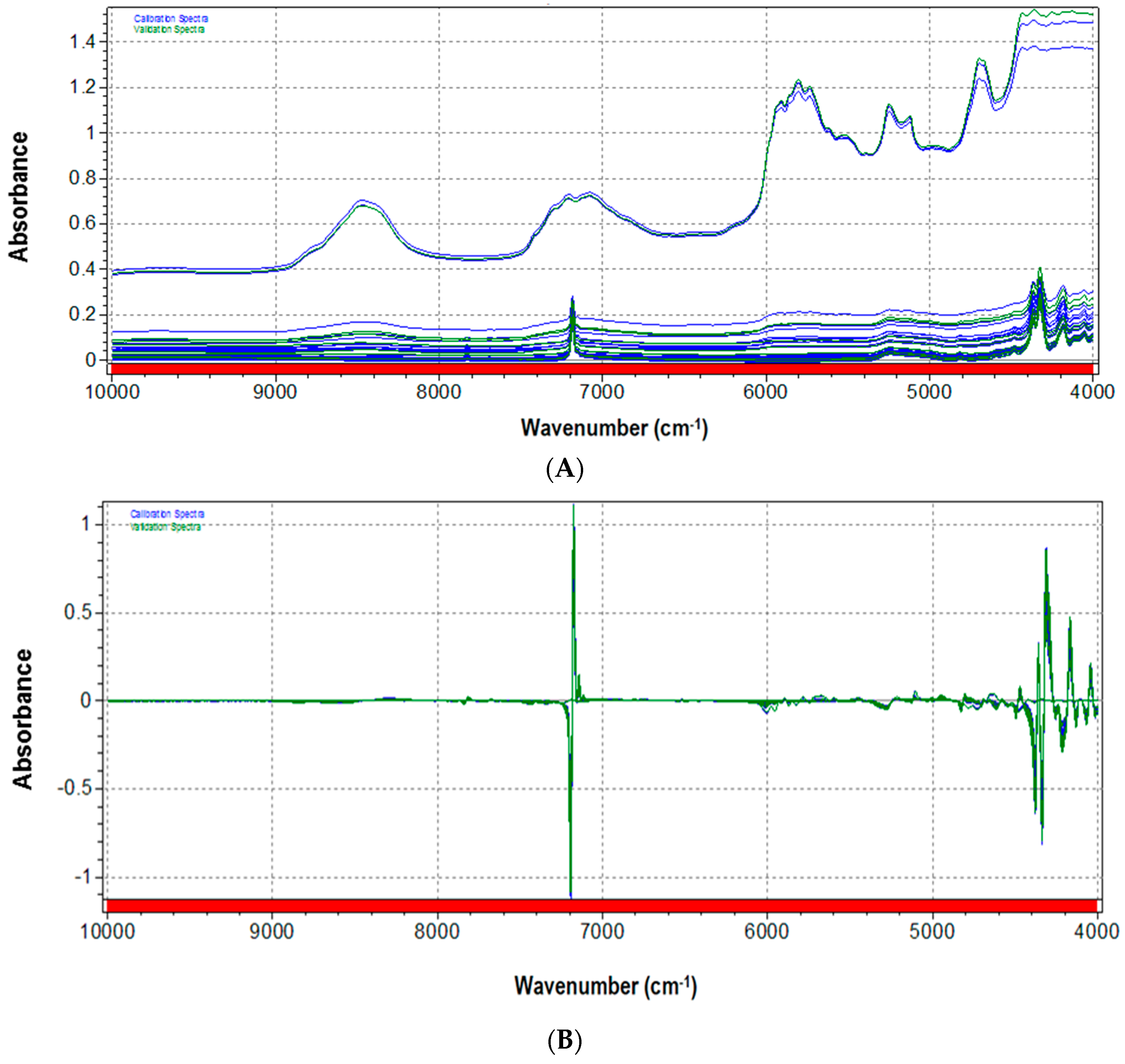
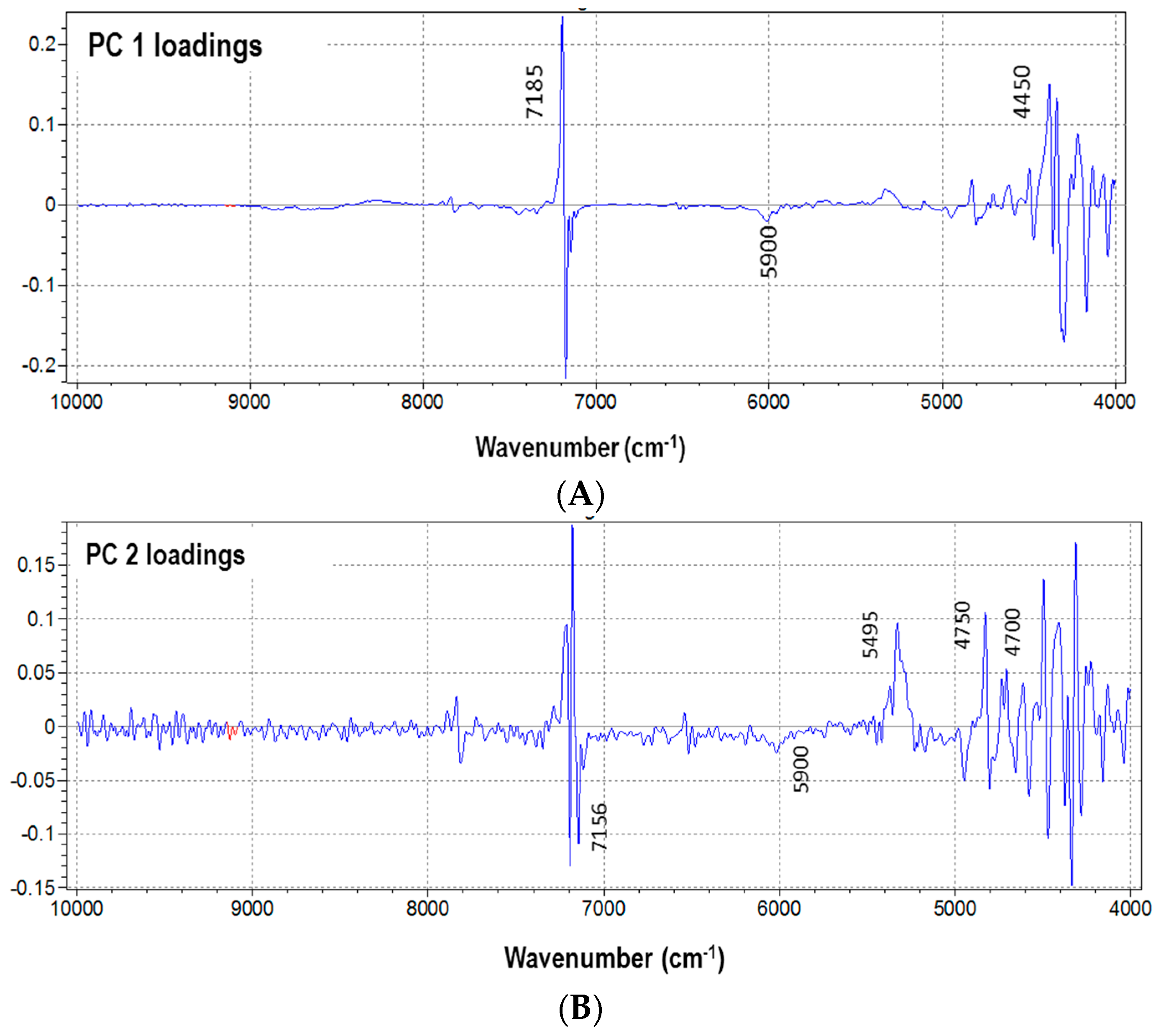
3.1. NIR Spectra and Data Pretreatments
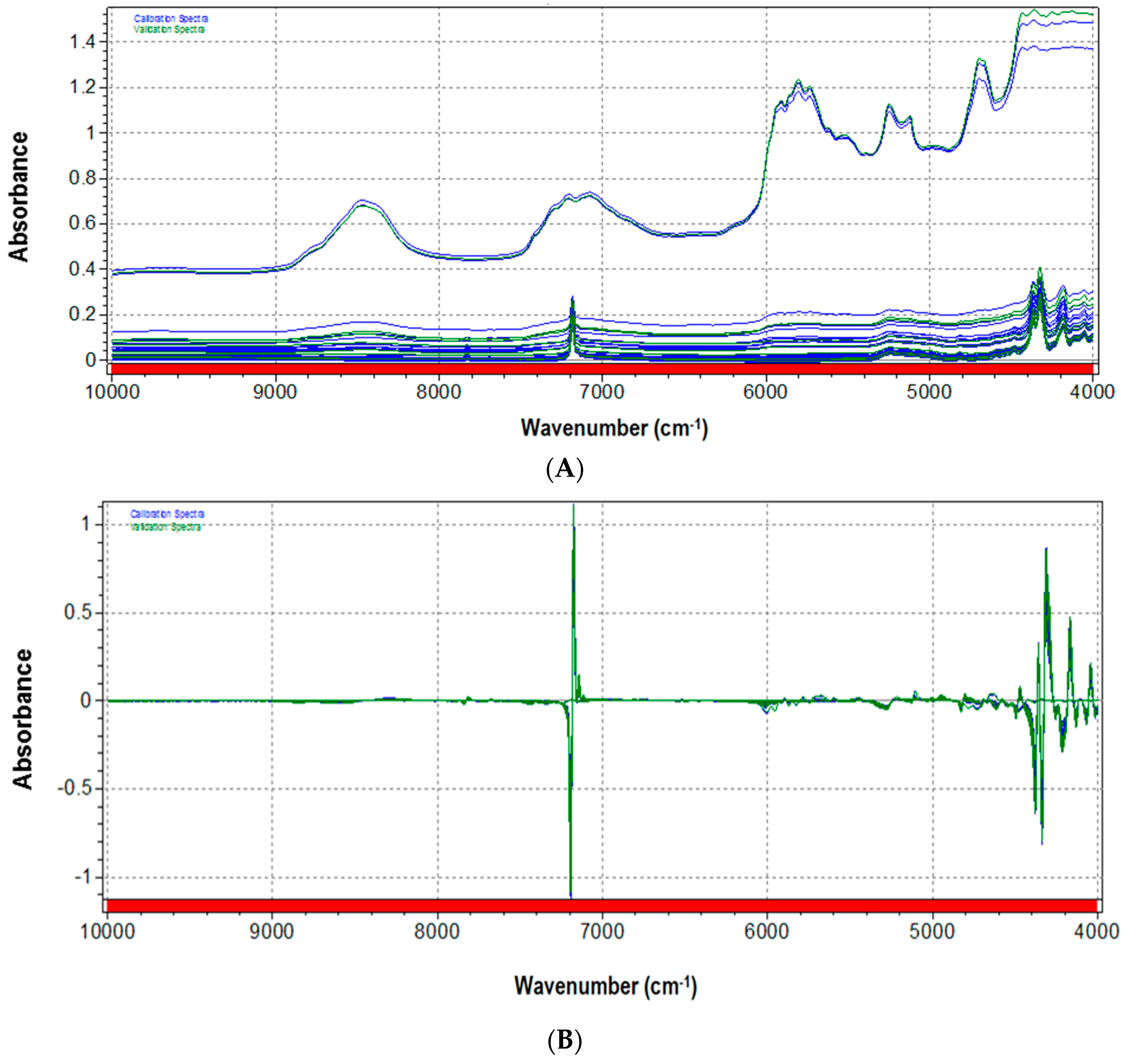
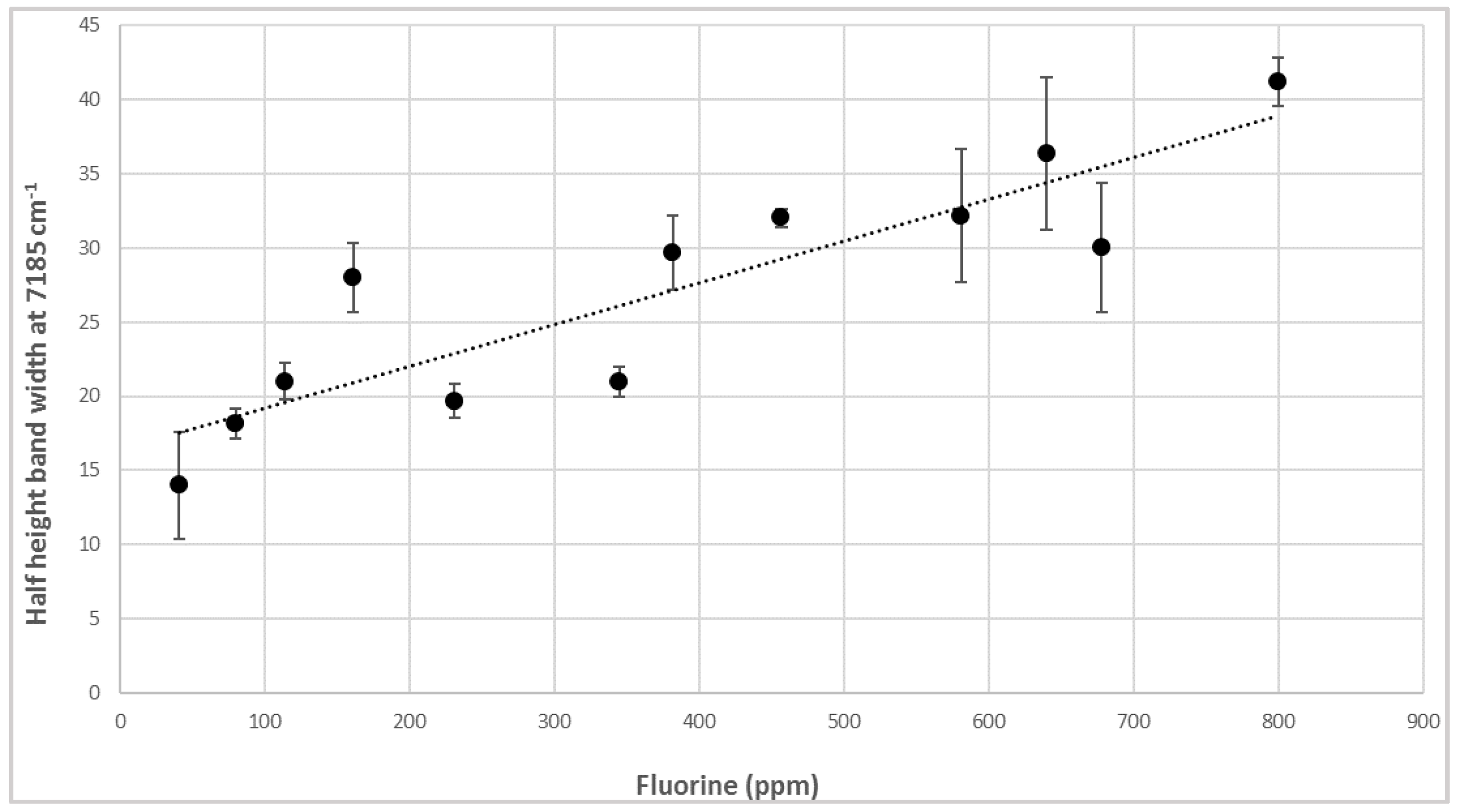
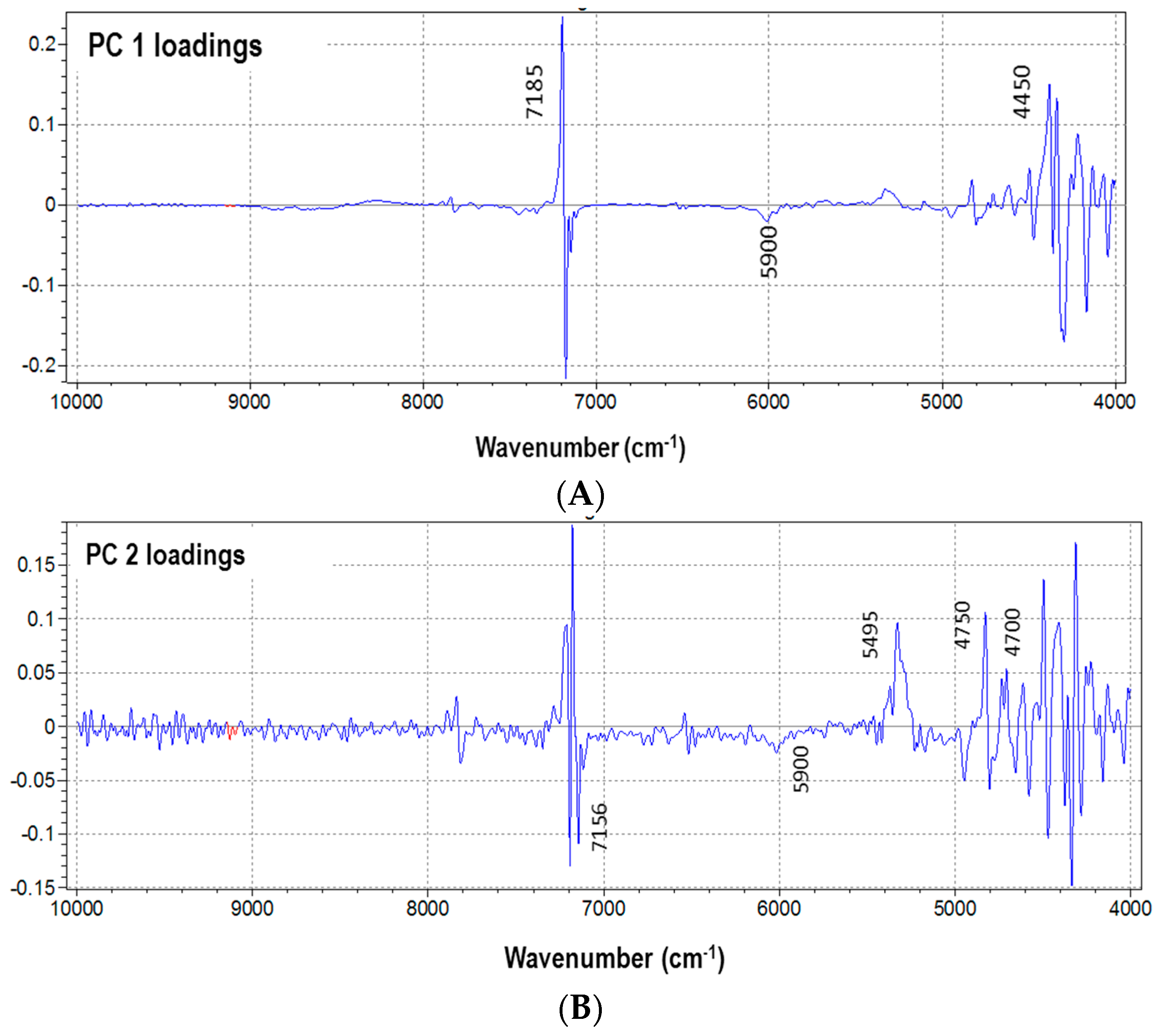
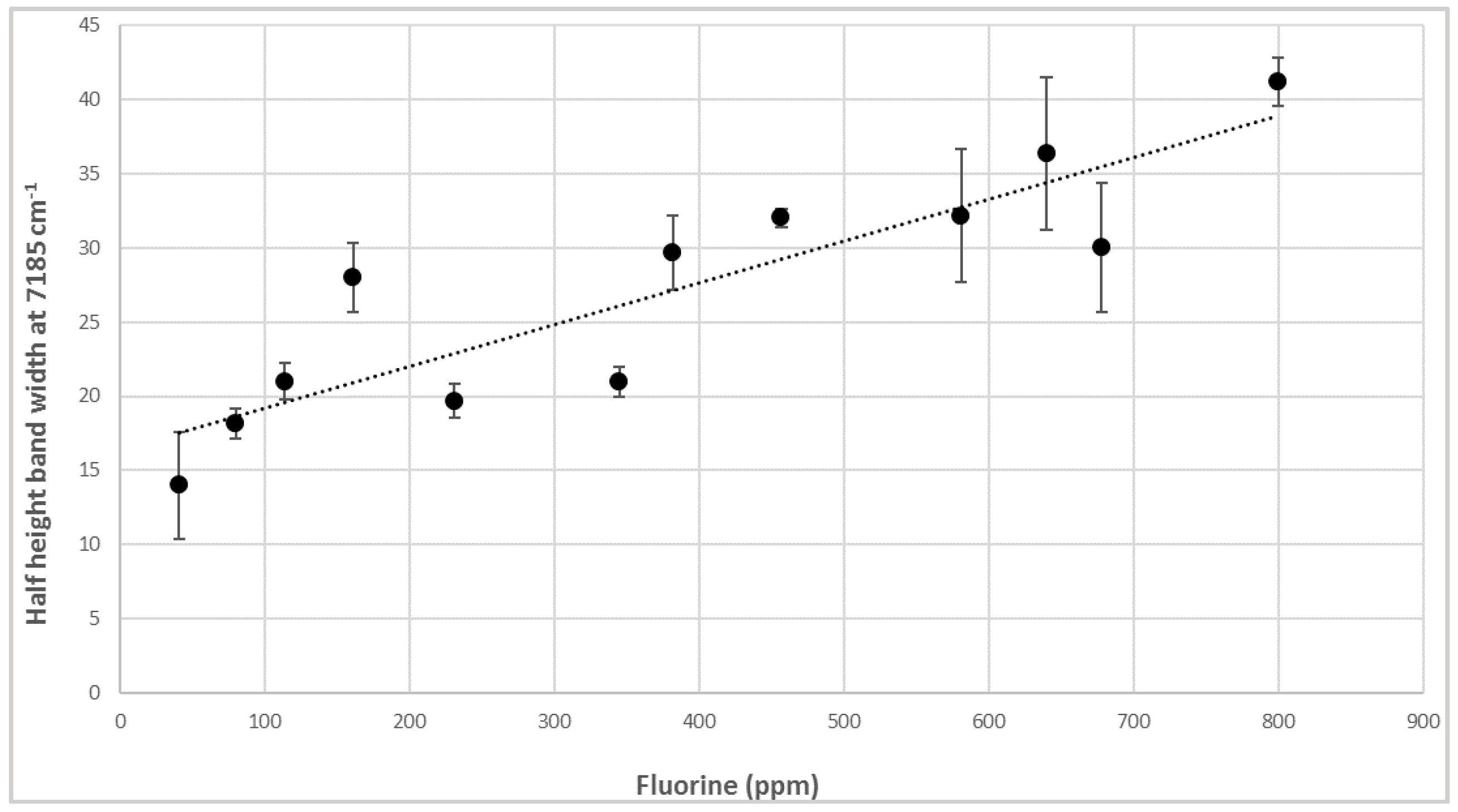
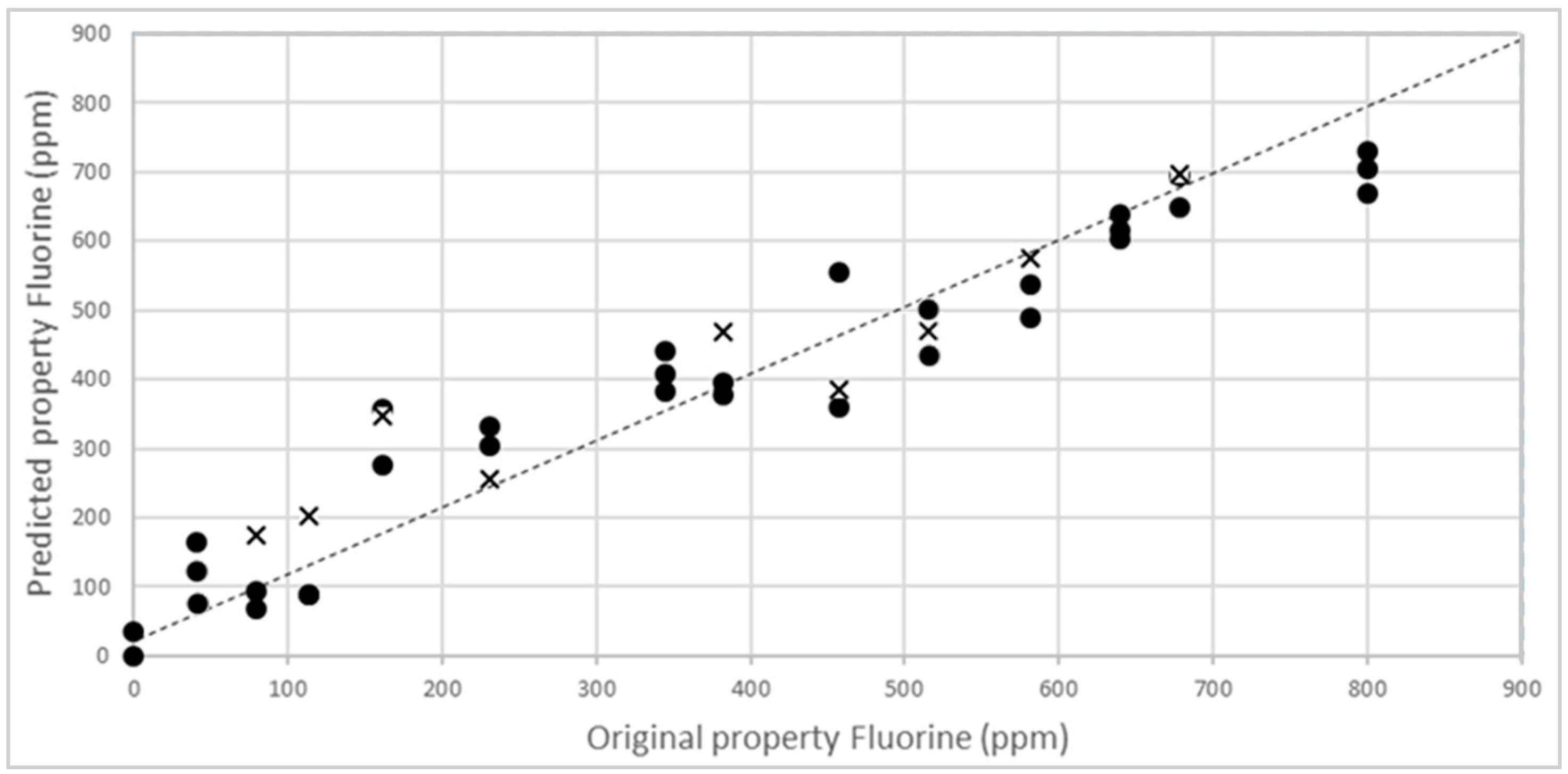
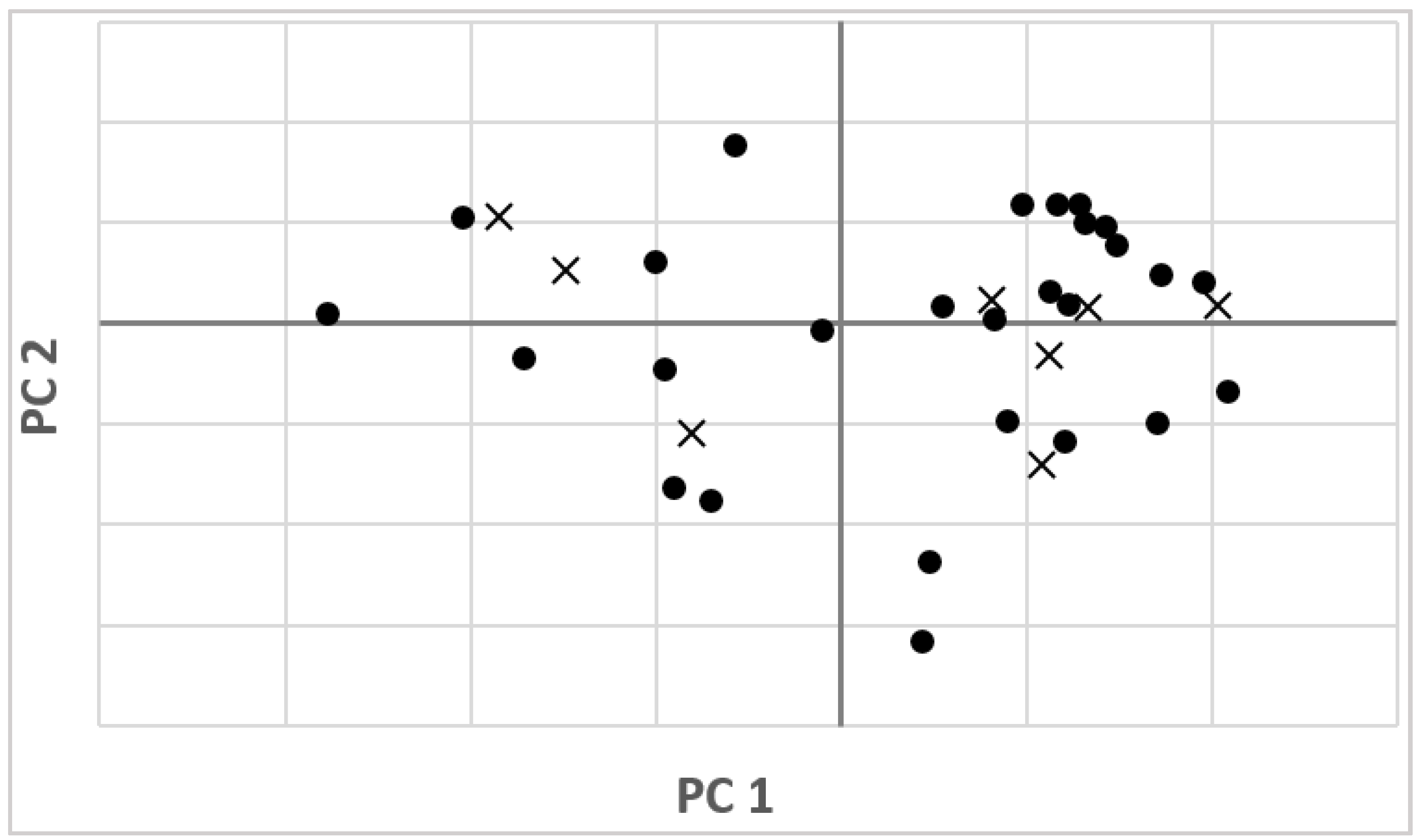
3.2. NIR Calibration and Validation
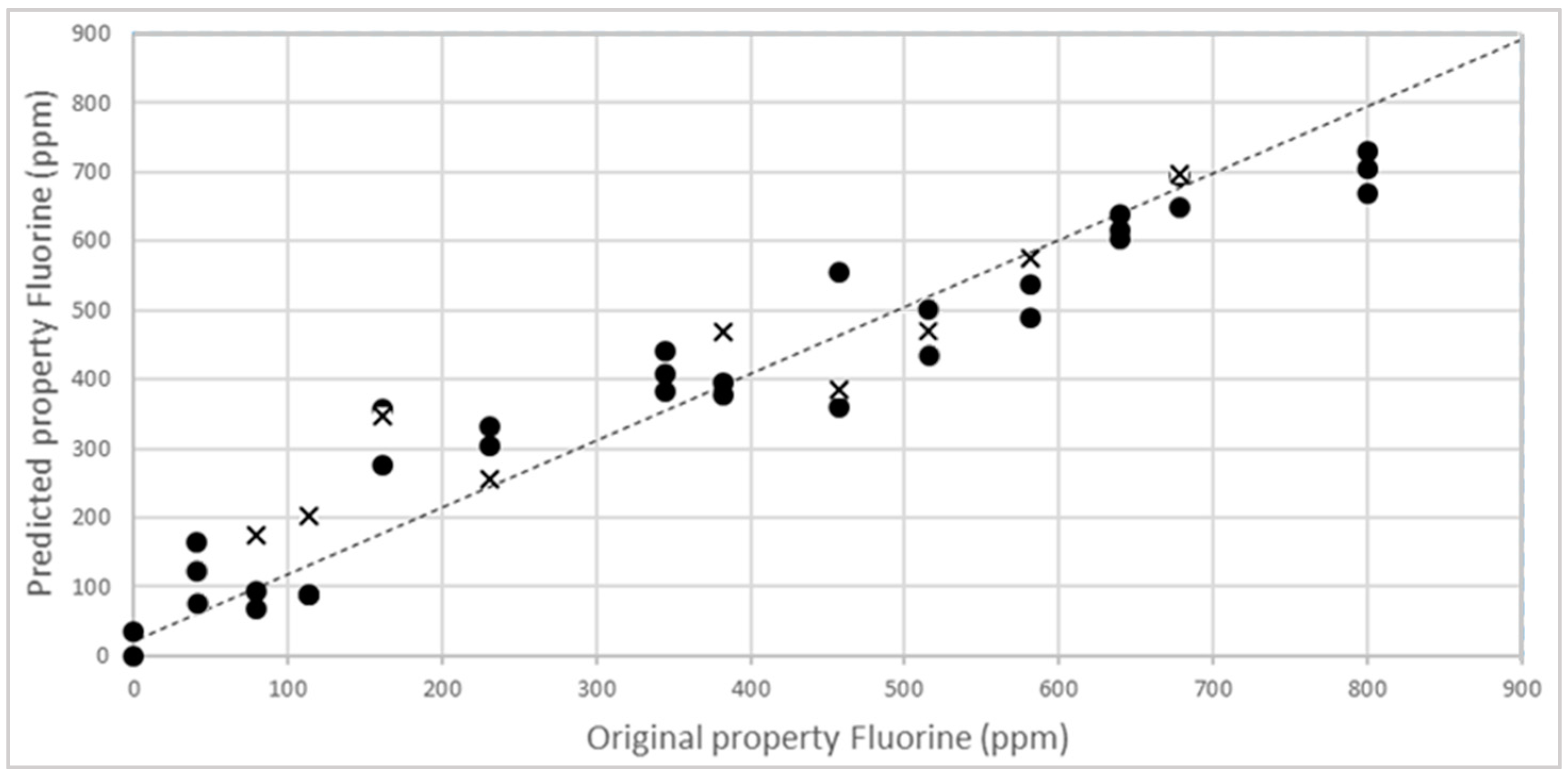
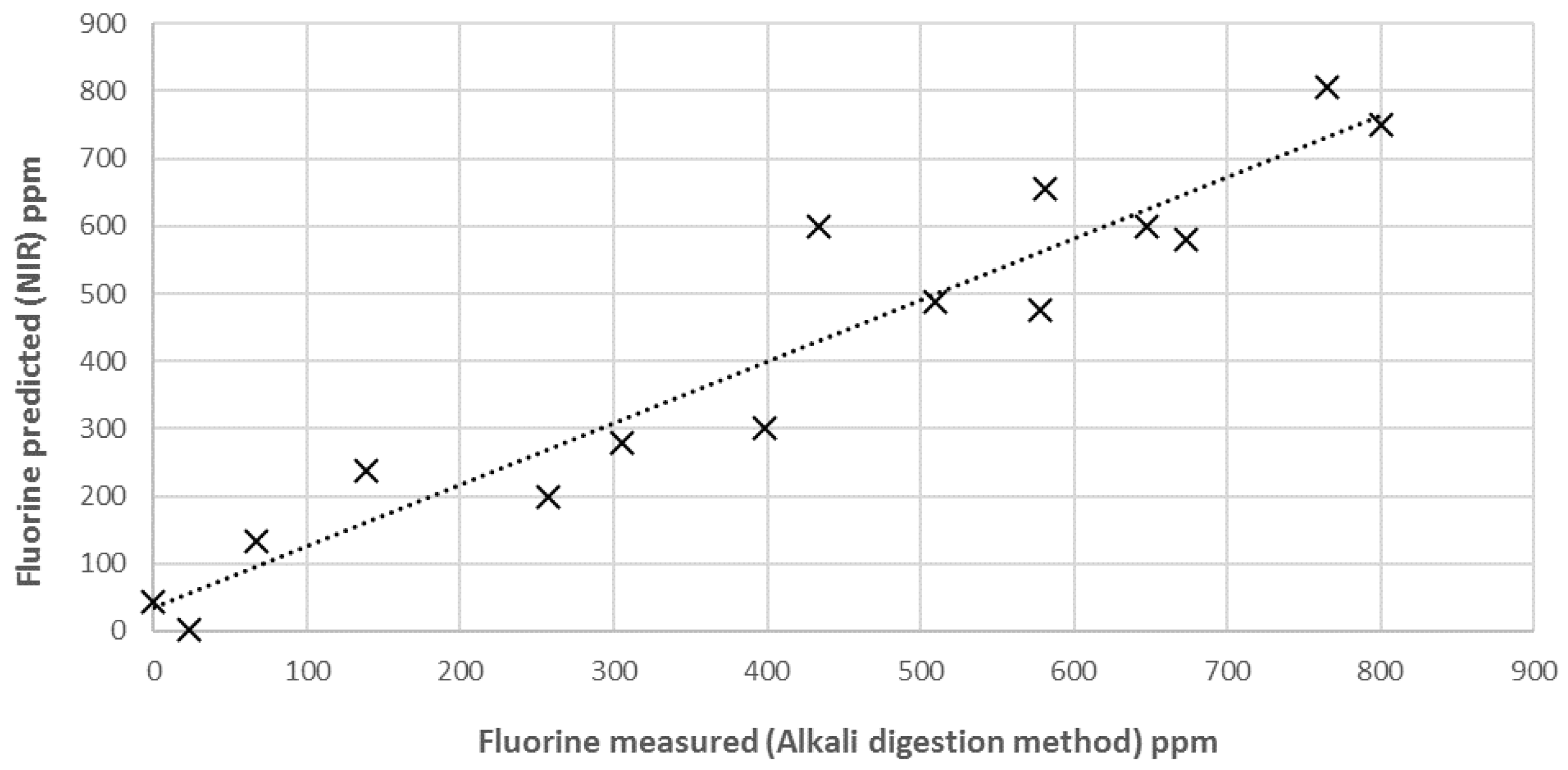
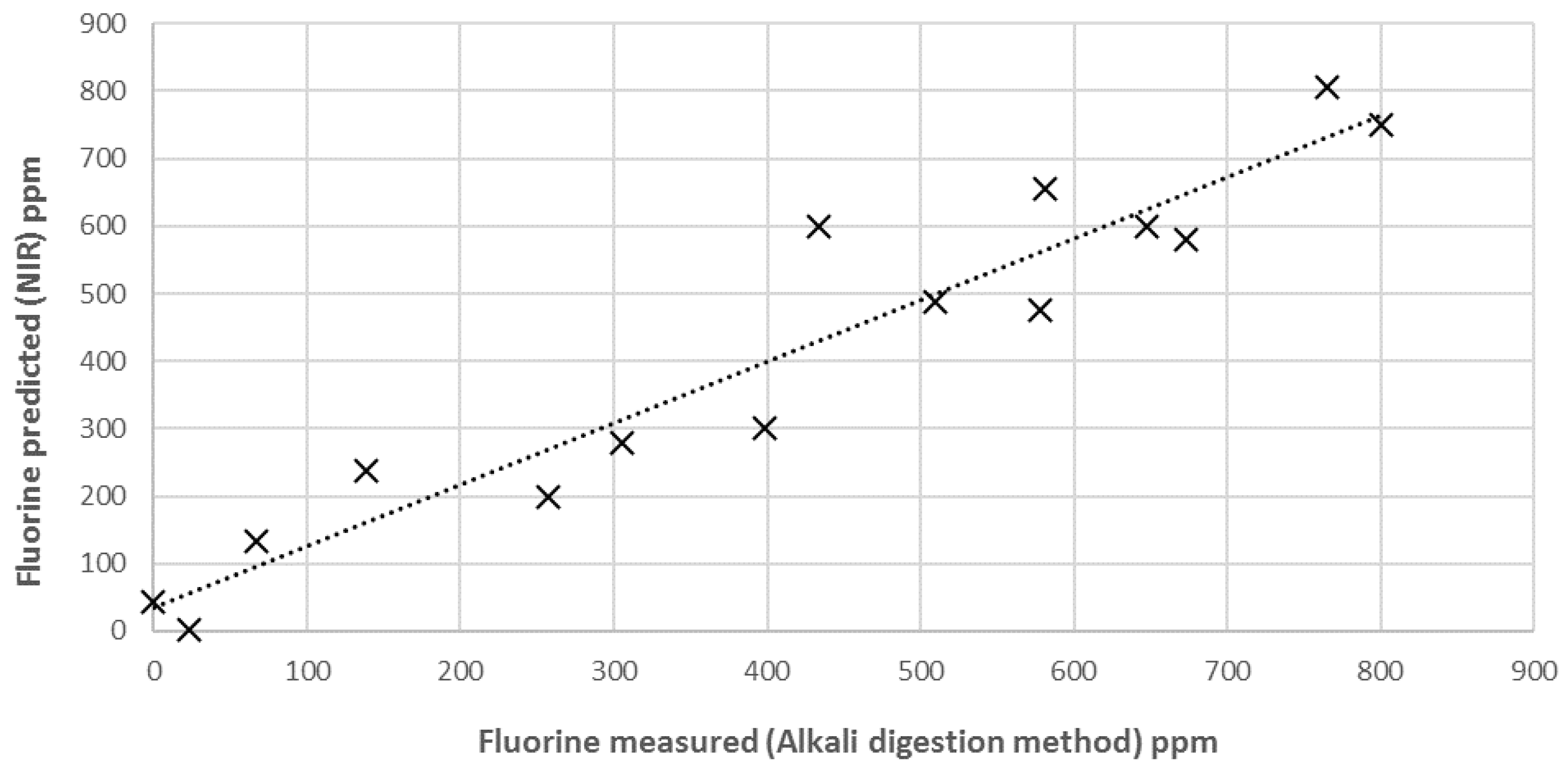
3.3. External Validation of the Model
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Lim, L.T.; Auras, R.; Rubino, M. Processing technologies for poly (lactic acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Conn, R.E.; Kolstad, J.J.; Borzelleca, J.F.; Dixler, D.S.; Filer, L.J., Jr.; LaDu, B.N. Safety assessment of polylactide (PLA) for use as a food-contact polymer. Food Chem. Toxicol. 2011, 33, 273–283. [Google Scholar] [CrossRef]
- Garlotta, D.A. Literature review of poly (lactic acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S.; Hernandez, R. Mechanical, physical, and barrier properties of poly (lactide) films. J. Plast. Film Sheeting 2003, 19, 123–135. [Google Scholar] [CrossRef]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic acid technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Lasprilla, A.J.; Martinez, G.A.; Lunelli, B.H.; Jardini, A.L.; Maciel Filho, R. Poly-lactic acid synthesis for application in biomedical devices—A review. Biotechnol. Adv. 2012, 30, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Chieng, B.W.; Ibrahim, N.A.; Yunus, W.M.Z.W.; Hussein, M.Z. Poly(lacticacid)/Poly(ethyleneglycol) Polymer Nanocomposites: Effects of Graphene Nanoplatelets. Polymers 2014, 6, 93–104. [Google Scholar] [CrossRef]
- Aldana, D.S.; Villa, E.D.; de dios Hernández, M.; Sánchez, G.G.; Cruz, Q.R.; Gallardo, S.F.; Castillo, H.P.; Casarrubias, L.B. Barrier Properties of Polylactic Acid in Cellulose Based Packages Using Montmorillonite as Filler. Polymers 2014, 6, 2386–2403. [Google Scholar] [CrossRef]
- Botta, L.; Fiore, V.; Scalici, T.; Valenza, A.; Scaffaro, R. New Polylactic Acid Composites Reinforced with Artichoke Fibers. Materials 2015, 8, 7770–7779. [Google Scholar] [CrossRef] [Green Version]
- Suksut, B.; Deeprasertkul, C. Effect of Nucleating Agents on Physical Properties of Poly(lactic acid) and Its Blend with Natural Rubber. J. Polym. Environ. 2001, 19, 288–296. [Google Scholar] [CrossRef]
- Ke, T.; Sun, X. Melting behavior and crystallization kinetics of starch and poly (lactic acid) composites. J. Appl. Polym. Sci. 2003, 89, 1203–1210. [Google Scholar] [CrossRef]
- Ross, M.; Smith, W.L.; Ashton, W.H. Triclinic Talc and Associated Amphiboles from Gouverneur Mining District, New York. Am. Mineral. 1968, 53, 752–769. [Google Scholar]
- EN 13432:2000. Packaging: Requirements for Packaging Recoverable through Composting and Biodegradation; EU Commission: Brussels, Belgium, 2000. [Google Scholar]
- Leonidov, V.Y.; O’Hare, P.A.G. Fluorine combustion calorimetry: Progress in recent years and possibilities of further development. Pure Appl. Chem. 1992, 64, 103–110. [Google Scholar] [CrossRef]
- Malde, M.K.; Bjorvatn, K.; Julshamn, K. Determination of fluoride in food by the use of alkali fusion and fluoride ion-selective electrode. Food Chem. 2001, 3, 373–379. [Google Scholar] [CrossRef]
- Miskolczi, N.; Hall, W.J.; Borsodi, N.; Williams, P.T.; Angyal, A. A comparison of different analytical techniques (energy dispersive X-ray fluorescent spectrometry, bomb calorimetry combined with ion chromatography and inductively coupled plasma-optical emission spectrometry) for the determination of the bromine and antimony content of samples. Microchem. J. 2011, 99, 60–66. [Google Scholar]
- Pasquini, C. Near Infrared Spectroscopy: Fundamentals, Practical Aspects and Analytical Applications. J. Braz. Chem. Soc. 2003, 14, 198–219. [Google Scholar] [CrossRef]
- Siesler, H.W.; Ozaki, Y.; Kawata, S.; Heise, H.M. Near-Infrared Spectroscopy: Principles, Instruments, Applications; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Mulbry, W.; Reeves Miller, P. Use of mid- and near-infrared spectroscopy to track degradation of bio-based eating utensils during composting. Bioresour. Technol. 2012, 109, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Huck, C. The future role of near infrared spectroscopy in polymer and chemical analysis. NIR News 2016, 27, 17–23. [Google Scholar] [CrossRef]
- Santos, A.F.; Lima, E.L.; Pinto, J.C. In-line evaluation of average particle size in styrene suspension polymerizations using near-infrared spectroscopy. J. Appl. Polym. Sci. 1998, 70, 1737–1745. [Google Scholar] [CrossRef]
- Petit, S.; Martin, F.; Wiewiora, A.; de Parseval, P.; Decarreau, A. Crystal-chemistry of talc: A near infrares (NIR) spectroscopy study. Am. Mineral. 2004, 89, 319–326. [Google Scholar] [CrossRef]
- Wang, Q.; Makishima, A.; Nakamura, E. Determination of fluorine and chlorine by pyrohydrolysis and ion chromatography: comparison with alkaline fusion digestion and ion chromatography. Geostand. Geoanal. Res. 2010, 34, 175–183. [Google Scholar] [CrossRef]
- Barnes, R.; Dhanoa, M.; Lister, J. Standard normal variable transformation and detrending of near infrared diffuse reflectance spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Martens, H.; Naes, T. Multivariate Calibration; J. Wiley and Sons: New York, NY, USA, 1988. [Google Scholar]
- Williams, P.C. Near-Infrared Technology in the Agricultural and Food Industries, 2nd ed.; American Association of Cereal Chemists: Eagan, MN, USA, 2001. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.R-project.org (accessed on 1 August 2016).
- Huck, C.; Ohmacht, R.; Tzabo, S.; Bonn, G.K. Near infrared spectroscopy, cluster and multivariate analysis—Characterization of silica materials for liquid chromatography. J. Near Infrared Spectrosc. 2006, 14, 51–57. [Google Scholar] [CrossRef]
- Rocke, D.M.; Woodruff, D.L. Identification of Outliers in Multivariate Data. J. Am. Stat. Assoc. 1996, 91, 1047–1061. [Google Scholar] [CrossRef]
- Durbin, J.; Watson, G.S. Testing for serial correlation in least squares regression. Biometrika 1950, 37, 409–428. [Google Scholar] [PubMed]
- Popo, M.; Romero-Torres, S.; Conde, C.; Romañach, R.J. Blend Uniformity Analysis Using Stream Sampling and Near Infra-Red Spectroscopy. AAPS PharmSciTech 2002, 3, 1–11. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Brereton, R.G. Chemometrics for Pattern Recognition; John Wiley & Sons Ltd.: Bristol, UK, 2009. [Google Scholar]
- Bossart, R.; Keller, H.; Kellerhals, H.; Oelichmann, J. Principal components analysis as a tool for identity control using near-infrared spectroscopy. J. Mol. Struct. 2003, 661, 319–323. [Google Scholar] [CrossRef]
- Lindberg, J.D.; Snyder, D.G. Diffuse reflectance spectra of several several clay minerals. Am. Mineral. 1972, 57, 485–493. [Google Scholar]
- Rywak, A.A.; Burtlitch, J.M. The crystal chemistry and thermal stability of sol-gel prepared fluorine substituted talc. Phys. Chem. Miner. 1996, 23, 418–431. [Google Scholar] [CrossRef]
- Bishop, J.; Madejova, J.; Komadel, P.; Froèschl, H. The influence of structural Fe, Al and Mg on the infrared OH bands in spectra of dioctahedral smectites. Clay Miner. 2002, 37, 607–616. [Google Scholar] [CrossRef]
- Xiao, H.W.; Li, P.; Ren, X.; Jiang, T.; Yeh, J.T. Isothermal crystallization kinetics and crystal structure of poly (lactic acid): Effect of triphenyl phosphate and talc. J. Appl. Polym. Sci. 2010, 118, 3558–3569. [Google Scholar] [CrossRef]
- Shinzawa, H.; Nishida, M.; Tanaka, T.; Kanematsu, W. Crystalline structure and mechanical property of poly (lactic acid) nanocomposite probed by near-infrared (NIR) hyperspectral imaging. Vib. Spectrosc. 2012, 60, 50–53. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wessel, L.F.A.; Reinders, M.J.T.; Shmulevich, I. Fewer permutations, more accurate P-values. Bioinformatics 2009, 25, i161–i168. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, G.E.; Ciurczac, E.W. A Comparison between Validating Laboratory and Process Near-Infrared Spectrophotometers. Spectroscopy 2003, 18, 34–42. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Fluorine |
|---|---|
| Units | ppm |
| SEL-reproducibility | 17.34 |
| #samples | 39 |
| Outliers | 0 |
| Min | 0.00 |
| Mean | 346.92 |
| Max | 800.00 |
| SD | 260.17 |
| WN range/step (cm−1) | 9000–4500/8 |
| Pretreatments | SNV/D1 |
| Regression method | PLS |
| Number of factors | 4 |
| SEC | 86.14 |
| R2cal | 0.9471 |
| SECV | 76.83 |
| R2CV | 0.9464 |
| NIR repeatability | 0.20 |
| DW | 2.29 |
| C-Set Durbin–Watson in range 1.5 to 2.5? | yes |
| Q-value | 0.85 |
| RPDcal | 2.62 |
| RPDCV | 3.60 |
| Parameter | Fluorine |
|---|---|
| Units | ppm |
| #samples | 15 |
| Outliers | 0 |
| Min | 0.00 |
| Mean | 413.20 |
| Max | 800.00 |
| SD | 258.46 |
| RMSEP | 61.08 |
| R2EX.V | 0.8955 |
| NIR repeatability | 0.20 |
| Bias | 0.05 |
| Intercept | 37.46 |
| Slope | 0.85 |
| DW | 1.84 |
| V-Set Durbin-Watson in range 1.5 to 2.5? | yes |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamburini, E.; Tagliati, C.; Bonato, T.; Costa, S.; Scapoli, C.; Pedrini, P. Quantitative Determination of Fluorine Content in Blends of Polylactide (PLA)–Talc Using Near Infrared Spectroscopy. Sensors 2016, 16, 1216. https://doi.org/10.3390/s16081216
Tamburini E, Tagliati C, Bonato T, Costa S, Scapoli C, Pedrini P. Quantitative Determination of Fluorine Content in Blends of Polylactide (PLA)–Talc Using Near Infrared Spectroscopy. Sensors. 2016; 16(8):1216. https://doi.org/10.3390/s16081216
Chicago/Turabian StyleTamburini, Elena, Chiara Tagliati, Tiziano Bonato, Stefania Costa, Chiara Scapoli, and Paola Pedrini. 2016. "Quantitative Determination of Fluorine Content in Blends of Polylactide (PLA)–Talc Using Near Infrared Spectroscopy" Sensors 16, no. 8: 1216. https://doi.org/10.3390/s16081216