1. Introduction
Lead is a naturally-occurring heavy metal constituting 10–30 mg·kg
−1 of Earth’s crust [
1]. Due to its unique characteristics it has been used extensively in the manufacturing of a wide range of products including: lead-acid batteries, paint, plumbing pipes, leaded gasoline, and ammunition. It is soft, highly malleable, dense, ductile, resistant to corrosion, non-degradable, and able to react with organic chemicals. Lead has only started being phased out of use in manufacturing over the last few decades even though its toxicity has been documented for hundreds of years [
2]. Lead poisoning can occur as a consequence of ingesting, inhaling, or via direct contact with lead-containing compounds [
1]. Once it enters the body it is able to cross the blood-brain barrier and damage the central and peripheral nervous systems [
3]. This is through the mimicking of other essential metals, such as calcium, iron, and zinc, thus interfering with enzymes’ abilities to catalyze reactions [
4,
5]. Damage to the central nervous system is particularly prevalent in children as lead interferes with synapse formation, neurochemical development, and the organization of ion channels, leading to reduced cognitive ability [
6]. The World Health Organization (WHO) 2011 guidelines recommend a provisional guideline value of 10 µg·L
−1 for lead in drinking water [
7].
Detection of heavy metals in the environment requires use of analytical techniques, including atomic absorption spectrometry and inductively-coupled plasma-atomic emission spectrometry [
8]. These methods of analysis require transport of the potentially contaminated sample to a laboratory and specialist knowledge, thus resulting in a time delay between sample collection and contaminant identification. There is an increasing demand for complementary and alternative in situ detection methods, which can also determine the amount of bioavailable heavy metal [
9]. The bioavailability of lead depends on multiple factors; the physicochemical properties of the lead-containing compound, the environmental matrix, as well as the biological recipient and its physiology [
10].
Bacteria respond rapidly to low concentrations of heavy metals in comparison to other members of soil biota; therefore, they are of increasing importance for use in toxicity tests of groundwater, soil, and sediments [
11,
12]. Lead resistance has been reported in both Gram-negative and Gram-positive bacteria [
13]. These mechanisms are often coupled with reduction and efflux systems of other heavy metals. For example, lead resistance has been reported to be coupled with cadmium [
14] and mercury [
15] resistance. The first lead-specific resistance locus (
pbrTRABCD) was identified on a mobile genetic element, the pMOL30 plasmid of
Ralstonia metallidurans CH34 (later re-named
Cupriavidus metallidurans CH34) [
16]. The locus encodes proteins that are involved in lead uptake (PbrT), efflux (PbrA; a P-type ATPase, PbrB; a phosphatase; and PbrC, a prolipoproprotein signal peptidase), and sequestration (PbrD). The
pbrABCD operon is regulated by a transcriptional regulator, PbrR, belonging to the MerR family of metal ion-sensing regulatory proteins. Similar lead loci (
pbrRABC) have also been identified on other plasmids, such as the pLVPK virulence plasmid from
Klebsiella pneumoniae [
17] and the pEC-IMPQ plasmid from
Enterobacter cloacae [
18].
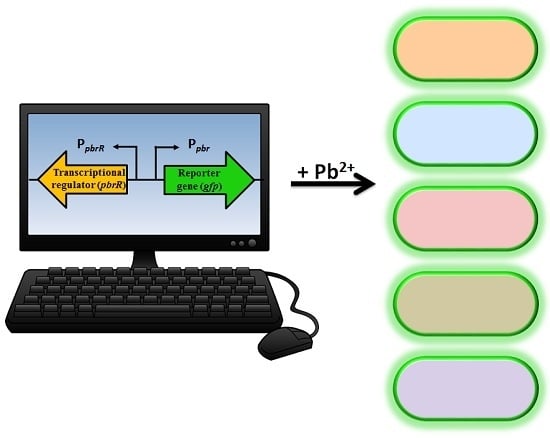
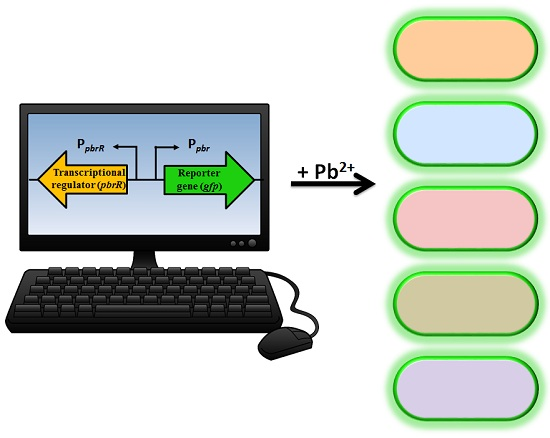
The understanding of resistance mechanisms has enabled synthetic biologists to re-design the system to enable expression of a detectable and measurable output signal, which is directly proportional to the concentration of a specific analyte [
9]. A biosensor genetic element containing a regulatory gene, together with its promoter region and a reporter gene, can be introduced into microbial species either on a plasmid or directly into the chromosome producing a microbial biosensor. Previous plasmid-based lead microbial biosensors have utilized the genetic element from the cadmium resistance mechanism with a luminescent output [
19,
20]. Not surprisingly, these lead biosensors also responded to low levels of cadmium (and zinc) due to the genetic element used. A chromosomal-based lead microbial biosensor utilized
pbrR and its native divergent promoter region, P
pbr, to allow expression of the luciferase operon (
luxCDABE). However, in addition to lead, the biosensor was also responsive to mercury, cadmium, and zinc [
21]. A range of microorganisms have been utilized as lead microbial biosensors, including Gram-negative
Alcaligenes eutrophus CH34 [
22],
Escherichia coli and
Pseudomonas fluorescens [
21], and Gram-positive
Staphylococcus aureus and
Bacillus subtilis [
19,
20].
The aim of this study was to develop lead microbial biosensors that could be used for ecotoxicological assessment of lead-polluted environmental samples. Furthermore, we aimed to increase knowledge of the use of
pbrR and its divergent promoter region in lead biosensor constructs, and their potential for differential response in various Gram-negative bacteria. A plasmid-based, lead biosensor construct was developed and transferred into
Pseudomonas aeruginosa PAO1,
Shewanella oneidensis MR-1 and two wild-type
Enterobacter sp. isolated from heavy metal contaminated soil. The latter two were selected because of their environmental relevance and bioremediation abilities [
23]. The biosensor construct contained the
pbrR gene and its native divergent promoter region (P
pbr) from the pLVPK plasmid of
K. pneumoniae CG43, in addition to a promoterless
gfp gene. Quantitative analyses were utilized to determine the limit of detection, the response time and the specificity to lead nitrate (Pb(II)) of the five microbial biosensors. To determine the feasibility of the microbial biosensors for in situ studies, two spiked environmental samples, tap water and groundwater, were tested. Previously, lead biosensors have only been tested in response to lead in soil samples [
20], therefore, to the best of the authors’ knowledge this is the first study to develop lead-specific biosensors with a fluorescent output for testing of lead-contaminated water samples.
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
The bacterial strains and plasmids used in this study are listed in
Table 1.
E. coli DH5α and
P. aeruginosa PAO1 were grown at 37 °C, while
S. oneidensis MR-1,
Enterobacter sp. NCR3 and
Enterobacter sp. LCR17 were grown at 28 °C. All strains were cultured on nutrient agar (NA) and in nutrient yeast broth (NYB) supplemented with antimicrobial agents, as necessary. NA contains 3.5% blood agar base (
w/
v) (1.5% (
w/
v) agar, 1% (
w/
v) ‘Lab-Lemco’ powder, 1% (
w/
v) peptone, 0.5% (
w/
v) NaCl) and 0.5% (
w/
v) yeast extract. NYB contains 2.5% (
w/
v) nutrient broth and 0.5% (
w/
v) yeast extract.
2.2. Antimicrobial Agents
Stocks of antibiotics and heavy metals were prepared with sterile distilled water (dH2O) at 104 µg·mL−1. Antimicrobial agents were freshly diluted in dH2O to appropriate concentrations as necessary. Selection for the transfer of pCR2.1®-TOPO® TA cloning vector (InvitrogenTM, Life Technologies, Carlsbad, CA, USA) and the broad-host range plasmid, pBBR1MCS-5, was achieved on NA containing ampicillin (100 µg·mL−1) and gentamicin sulfate (10 µg·mL−1), respectively. A nalidixic acid resistant (7 µg·mL−1) laboratory strain E. coli DH5α was used for construction of the lead biosensor construct. Heavy metals used for testing of the biosensors included: sodium arsenite (As(III)), cadmium chloride (Cd(II)), copper chloride (Cu(II)), chromium oxide (Cr(VI)), lead nitrate (Pb(II)), mercury chloride (Hg(II)) and zinc chloride (Zn(II)). Antibiotics (ampicillin, gentamicin sulphate and nalidixic acid) were stored at 4 °C and heavy metal compounds were stored at room temperature. All antimicrobial agents were obtained from Sigma-Aldrich Pty Ltd. (St. Louis, MO, USA).
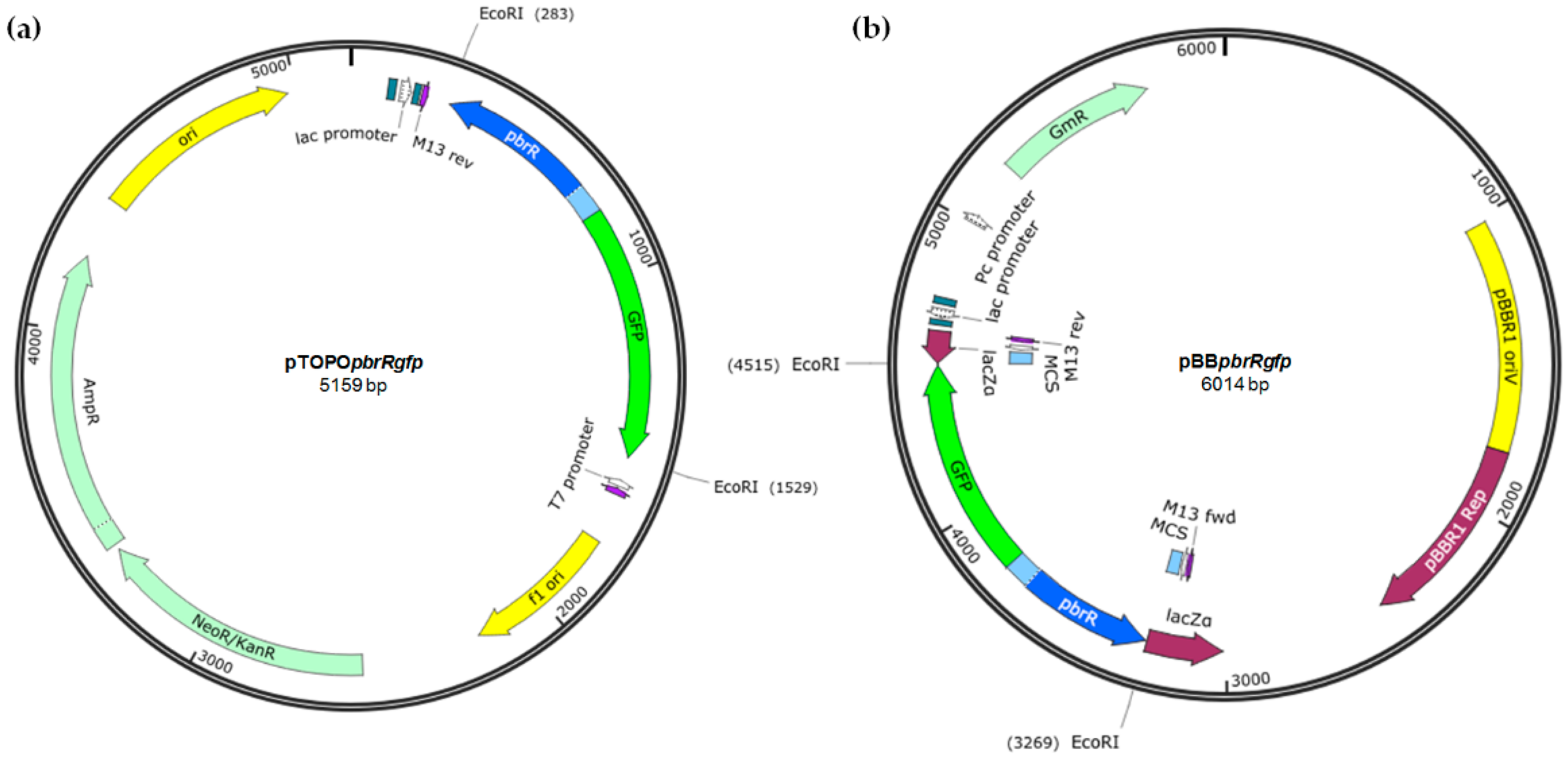
2.3. Construction of the Plasmid-Based Lead Biosensor Construct
The genetic element containing the pbrR gene (435 bp) and the divergent promoter region (Ppbr; 85 bp) that it regulates from K. pneumoniae CG43 plasmid pLVPK (NCBI Accession No. AY378100; coordinates: 155782–56301; 520 bp) and a promoterless gfp gene (gfpmut3b; 720 bp; iGEM biobrick BBa_E0040), was synthesized by Integrated DNA Technologies. The synthesized fragment (1240 bp) was cloned into pCR® 2.1-TOPO® TA (InvitrogenTM, Life Technologies, Carlsbad, CA, USA) (3931 bp) and subsequently subcloned into the EcoRI (Promega (Madison, WI, USA)) site of pBBR1MCS-5 (4768 bp). Blue-white screening was employed to detect the vector containing the recombinant DNA (i.e., pBBpbrRgfp). Isolated plasmid DNA was subjected to EcoRI restriction digestion at 37 °C for 1 h and gel electrophoresis to visualise the release of the correct sized fragment. Both pTOPOpbrRgfp and pBBpbrRgfp were Sanger sequenced at the Australian Genome Research Facility to confirm the genetic element cloned.
2.4. Conjugation Procedure
Aliquots (1 mL) of exponential phase donor (E. coli WM3064(pBBpbrRgfp)) and recipient (either P. aeruginosa PAO1, S. oneidensis MR-1, Enterobacter sp. NCR3 or Enterobacter sp. LCR17) NYB-cultures (~109 cfu·mL−1) were mixed and filtered through a nitrocellulose membrane filter (0.45 µm pore size; 25 mm diametre; Millipore (Cork, County Cork, Ireland). The filter with the bacteria facing uppermost was then placed onto the surface of a pre-warmed NA plate. After incubation (2–3 h for P. aeruginosa PAO1 or 4–5 h for S. oneidensis MR-1 and Enterobacter spp.) the bacteria were resuspended in 2 mL NYB, serially diluted (ten-fold in saline) and aliquots (100 µL) plated onto selective NA containing gentamicin sulfate at 10 µg·mL−1, allowed to dry and incubated under appropriate conditions. Aliquots (100 µL) of donor and recipient cultures were plated separately onto the same selective medium to serve as negative controls.
2.5. Fluorescence Microscopy
Exponential-phase NYB-cultures (OD600 nm = 0.6–0.8) of lead biosensors (E. coli DH5α(pBBpbrRgfp), P. aeruginosa PAO1(pBBpbrRgfp), S. oneidensis MR-1(pBBpbrRgfp), Enterobacter sp. NCR3(pBBpbrRgfp) and Enterobacter sp. LCR17(pBBpbrRgfp)) containing antibiotics were induced with a concentration of 100 µg·mL−1 Pb(II) for 24 h to allow expression of GFP. An identical NYB-culture was left uninduced and also incubated for 24 h to serve as a negative control. After 24 h, a drop of culture (10 µL) was placed onto a glass slide and covered with a cover slip. The cover slip was firmly pressed onto the slide and sealed using nail polish. Samples were viewed using a Nikon Eclipse Ti-E inverted microscope. Phase contrast was used to view the bacterial cells, and the GFP filter (excitation 485 nm and emission 535 nm) was used to visualise the fluorescent cells at ×1000 magnification with oil immersion.
2.6. Fluorescence Assays
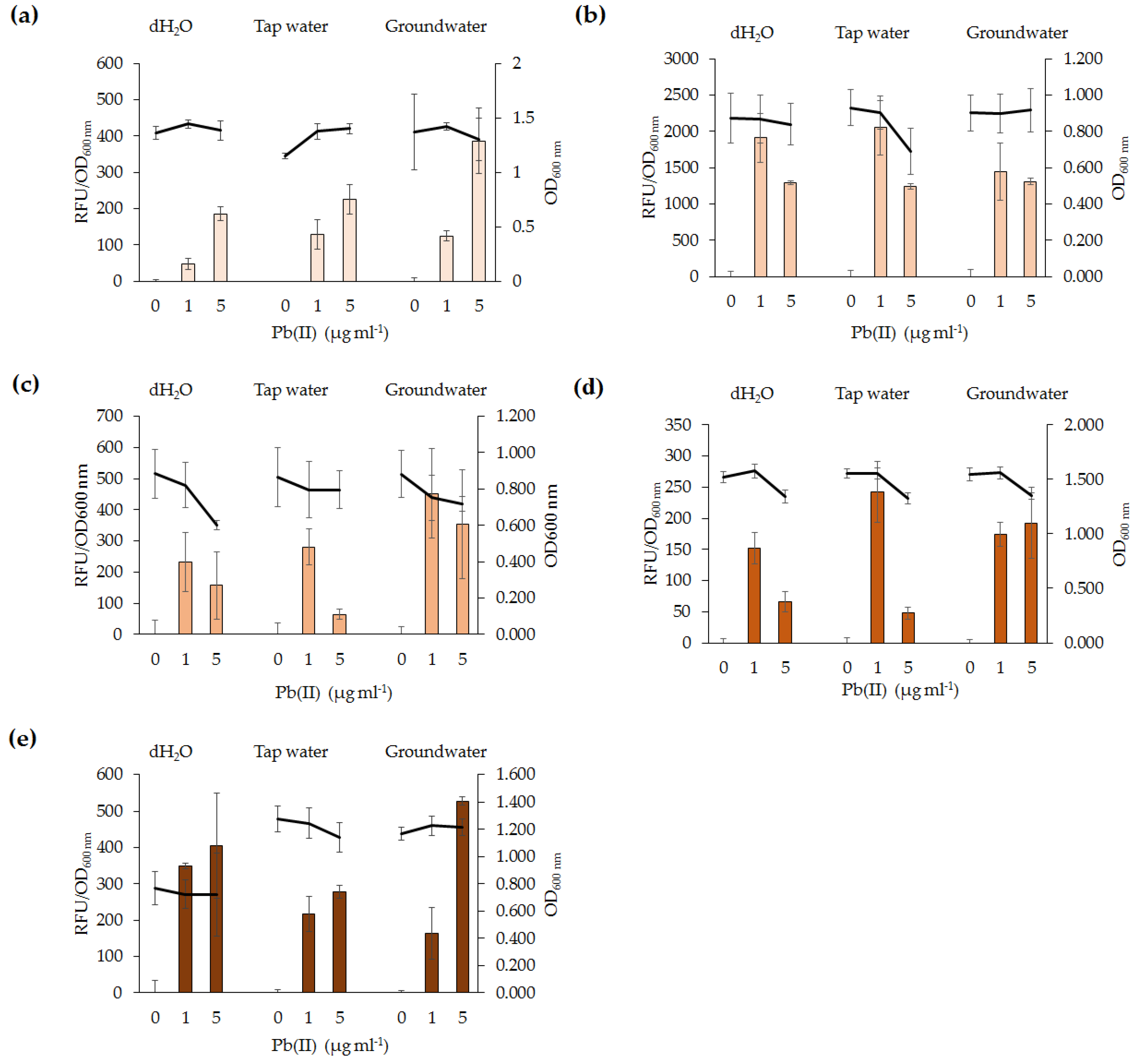
Pb(II) at varying concentrations (or other heavy metals in the case of specificity assays) was added to exponential phase NYB-cultures of lead biosensors and incubated for an appropriate period of time with shaking in the dark. For environmental assays, 900 µL of exponential phase NYB-cultures of lead biosensors were added to either dH2O, tap water or groundwater (in 100 µL aliquots) spiked with either 0, 2, or 5 µg·mL−1 Pb(II). The groundwater and tap water used were collected from a lake system and the laboratory tap in Victoria, Australia. Following up to 4 h of incubation, the cells were pelleted and washed three times in dH2O. For time detection assays cells were pelleted and washed at consecutive time intervals after induction. After washing, cells were aliquoted (200 µL) into black 96-well plates with clear flat bottom which allowed successive measurement of the relative fluorescence units (RFU) and optical density (OD600 nm) of the bacterial cells. A Clariostar plate reader was used for measurements of RFU (with the excitation and emission values set at 485 nm and 535 nm, respectively) and OD600 nm.
For each assay, three independent biological replicates were performed in triplicate, unless otherwise stated. The mean RFU and OD600 nm of triplicate samples was calculated to obtain the mean RFU per OD600 nm of each biological replicate. Standard error was determined through the use of the students t-test (p < 0.05), whereby the mean RFU per OD600 nm of the three biological replicates of the uninduced sample was compared to the other samples being tested. The p value was calculated by comparing the variability between the mean RFU per OD600 nm of the lowest concentration used during the fluorescence assay (i.e., 0 µg·mL−1 Pb(II)) or from the earliest time point during time assays (0 min) to that of other concentrations used or time points, respectively. In each fluorescence assay the mean RFU per OD600 nm of the three biological replicates of the uninduced sample was subtracted from that of the induced samples (i.e., RFU(induced)/OD600 nm(induced) − RFU(uninduced)/OD600 nm(uninduced)).
4. Discussion
In this study, a lead biosensor construct (pBBpbrRgfp) was developed and introduced into several Gram-negative bacterial species, which were then exposed to a range of Pb(II) concentrations for various time periods. Like many other heavy metals, lead can also exist in a number of different forms, oxides and hydroxides, as well as oxyanion complexes that are released into the soil and groundwater. The most common form, Pb(II), was chosen for testing of the biosensors in addition to several other heavy metal ions to investigate the specificity of the developed biosensors.
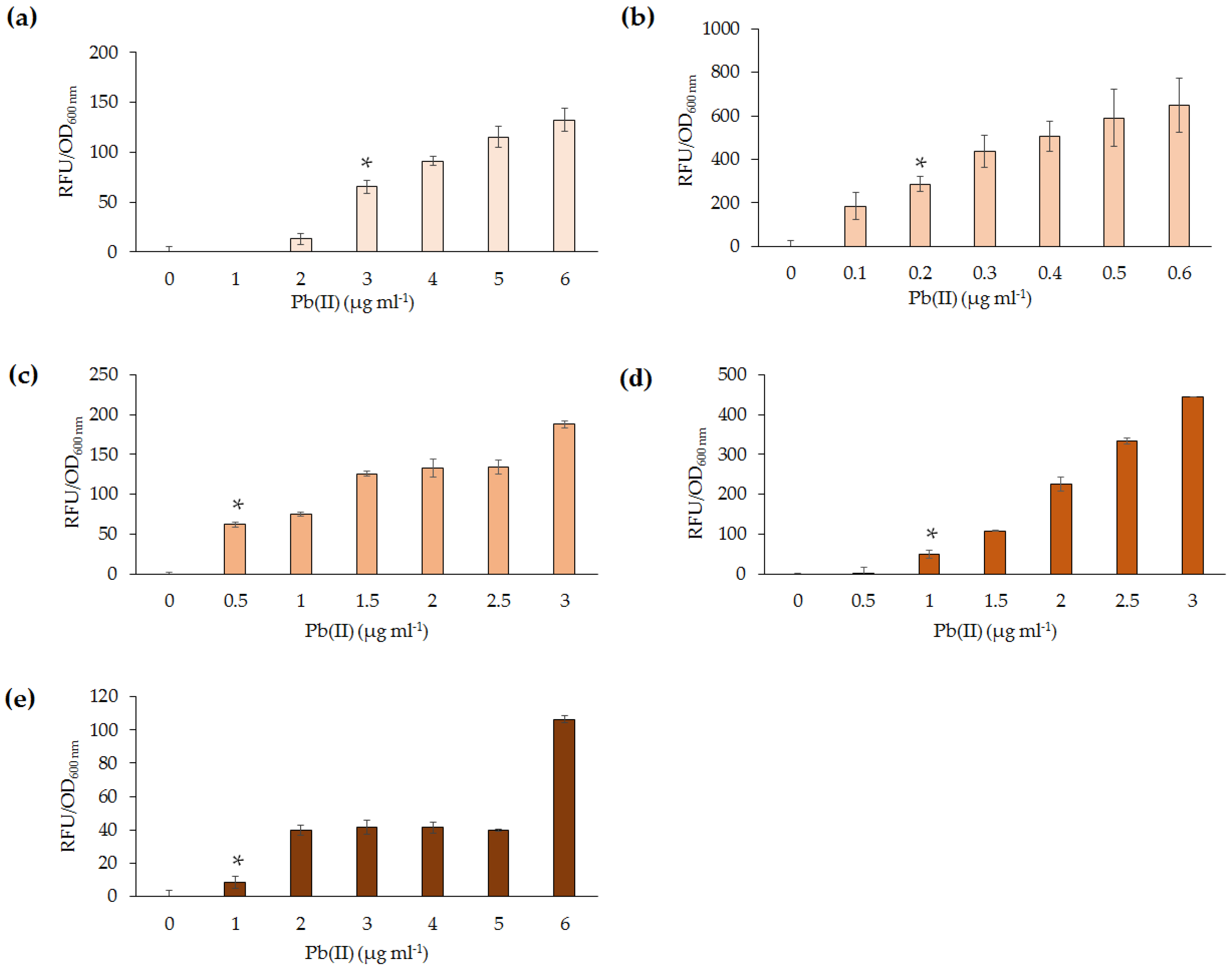
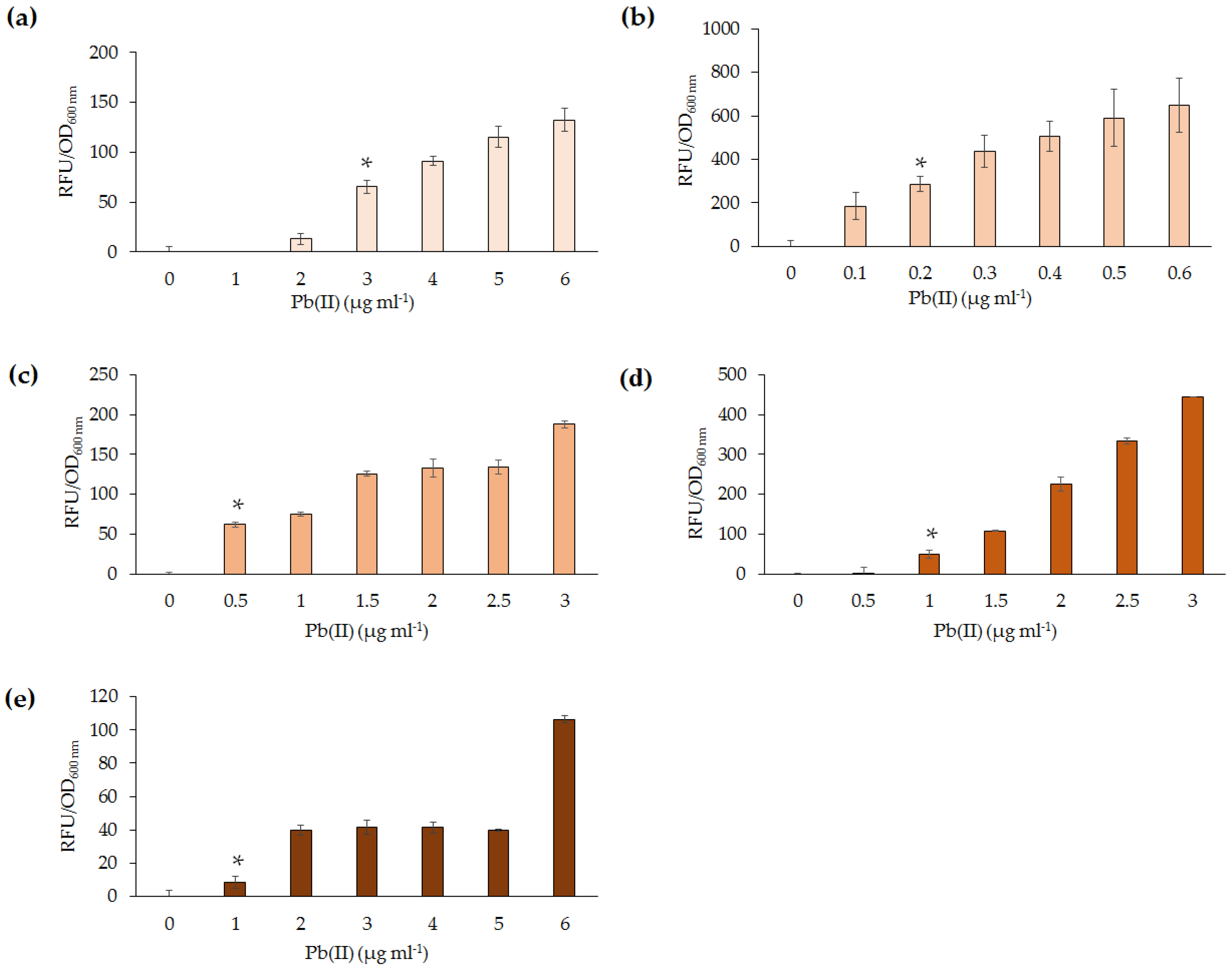
The five lead biosensors developed in this study were able to detect between 0.2 and 1 µg·mL
−1 Pb(II) following 4 h incubation.
P. aeruginosa PAO1(pBB
pbrRgfp) was deemed the most sensitive. This may be due to a higher expression of GFP in
Pseudomonas, as a consequence of reduced transcriptional repression by PbrR. Upon exposure to Pb(II) for 10 h, the sensitivity of the
Pseudomonas lead biosensor increased from 0.2 to 0.05 µg·mL
−1 (data not shown). It can, thus, be proposed that, upon further incubation, the
Pseudomonas lead biosensor could detect below 0.01 µg·mL
−1, which is the WHO guideline level for Pb(II) in drinking water [
7]. The limit of detection of previously reported lead biosensors varies significantly. For example,
A. eutrophus luminescent lead biosensor detected ~331 µg·mL
−1 Pb (compound not specified) [
22]. In a separate study,
S. aureus and
B. subtilis luminescent biosensors (p
cadClucFF) detected 0.01 µg·mL
−1 Pb(CH
3COO)
2 after ~2 h exposure [
19]. Some of the factors which can be attributed to differences in detection limits of lead biosensors developed include the genetic element used, the choice of the microorganism, the medium used, as well as the reporter gene. The growth phase of the biosensor culture as well as the concentration of the cells exposed to lead affect the limit of detection. Limitations to the limit of detection may include the culture reaching a stationary phase, which is largely influenced by the availability of nutrients. Nutrient availability influences the growth rate of the organism [
19]. Furthermore, differences in copy number of the plasmid used may contribute to the sensitivity of lead detection and variability observed among different studies which have utilized a range of different plasmids. Therefore, future studies should utilize a high-copy number, broad-host range plasmid.
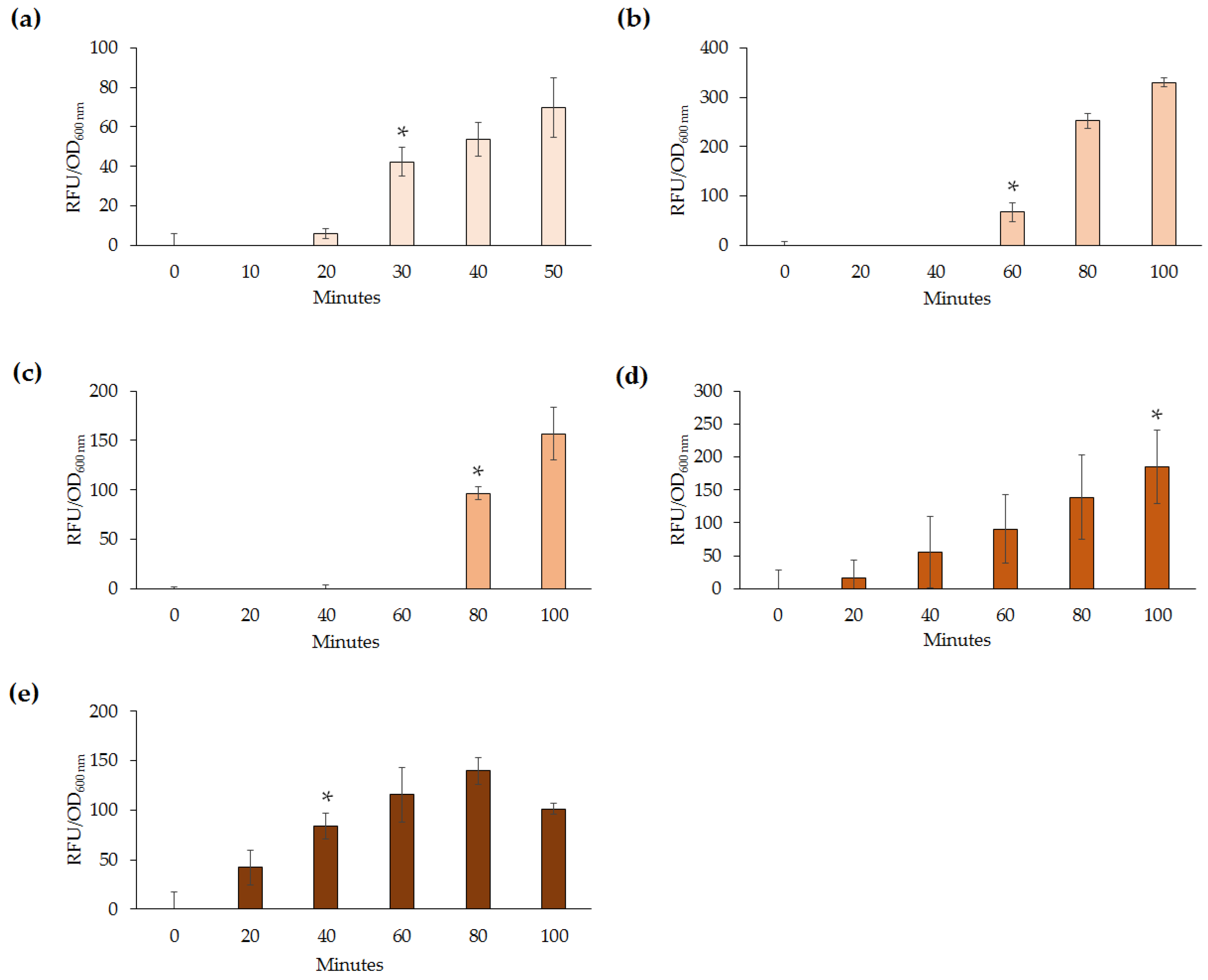
Slight variation of detection abilities (0.2–1 µg·mL
−1) among the lead biosensors were further highlighted upon analysis of response times following exposure to Pb(II). Out of the five biosensors,
E. coli DH5α(pBB
pbrRgfp) and
Enterobacter sp. LCR17(pBB
pbrRgfp) responded quickest at 30 and 40 min, respectively. However, this may be attributed to differences in the growth rate of the microorganisms, as well as the different concentrations of Pb(II) used to test the response times. For example,
E. coli DH5α(pBB
pbrRgfp) was exposed to 5 µg·mL
−1 whereas
P. aeruginosa PAO1(pBB
pbrRgfp) was exposed to 0.2 µg·mL
−1. Response times are highly dependent on the concentration of the heavy metal used and the growth rate of the microorganism. Although a constant concentration could have been used, due to the variability of the lead biosensors’ response to Pb(II), the lowest detectable amount or above was chosen for testing the response times in this study. Previous studies have reported only induction time for lead biosensors tested [
19,
22] and as such, it would be beneficial to determine response times during future analyses.
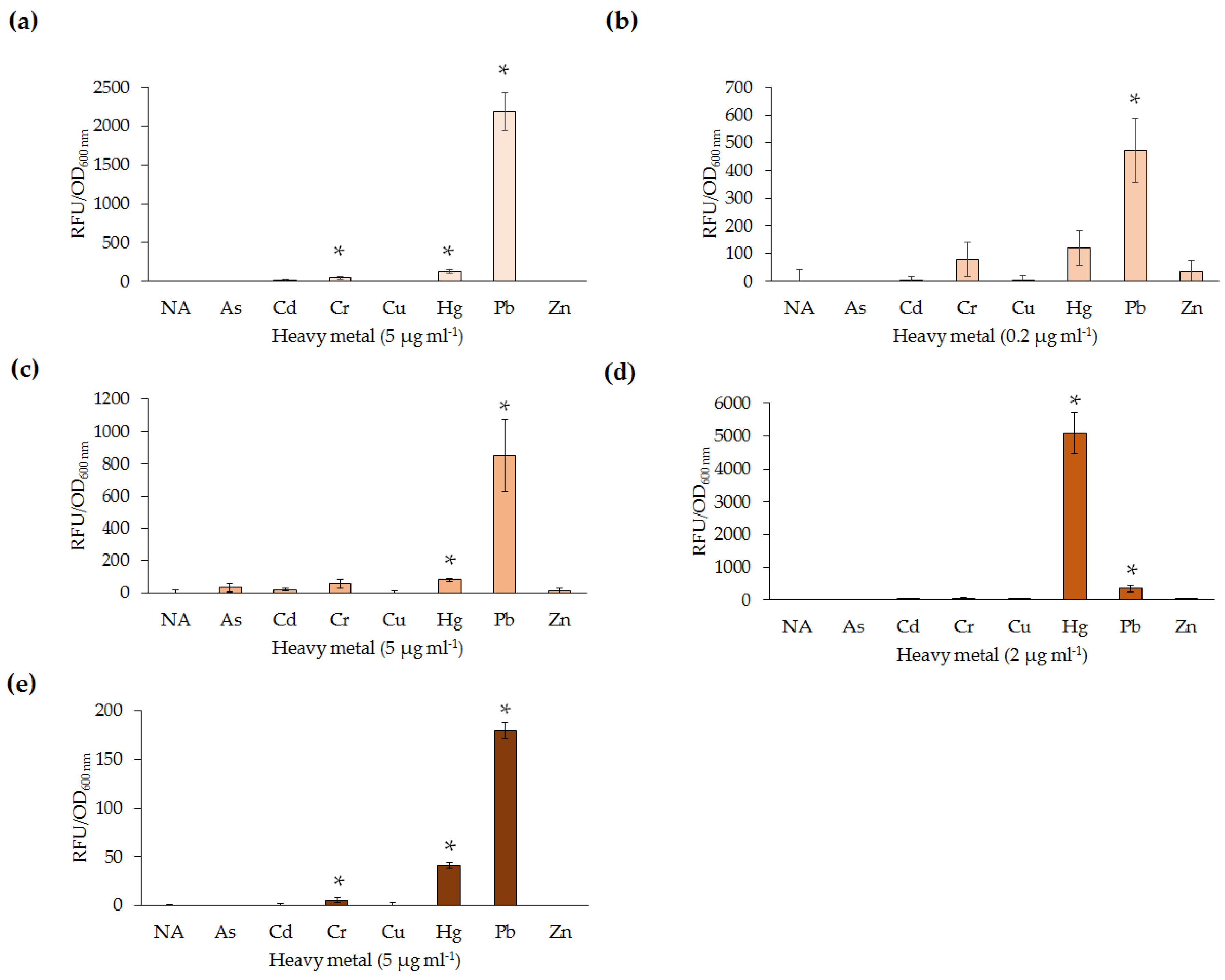
All five lead biosensors developed could detect low levels of Pb(II) (0.2–1 µg·mL
−1) following exposure for 30–100 min. Additionally, the biosensors were able to detect at least one heavy metal ion other than Pb(II), in most instances Hg(II).
E. coli DH5α(pBB
pbrRgfp) and
Enterobacter sp. LCR17(pBB
pbrRgfp) were also responsive to Cr(VI). Interestingly, the response of lead biosensors to Cr(VI) has not previously been reported. Similarly, reported Cr(VI) microbial biosensors were not tested for response to Pb(II) [
29]. Nevertheless, non-specificity of the biosensor constructs was expected due to the similarity of heavy metal ions, as well as previous reports of non-specificity among lead biosensors [
19]. For example, the
S. aureus and
B. subtilis luminescent lead biosensors detected Cd(II), Hg(II), Mn(II), Sb(III), Sn(II), and Zn(II) [
19,
20]. The differences in specificity between various microorganisms may be attributed to different methods of metal uptake into the cell. In contrast to non-specific
S. aureus and
B. subtilis lead biosensors, The
A. eutrophus luminescent lead biosensor only detected Pb (compound not specified) [
22]. This high level of specificity may be due to the medium used or the higher concentration of Pb (331 µg·mL
−1) used for testing. Interestingly,
Enterobacter sp. NCR3(pBB
pbrRgfp) exhibited an elevated response to Hg(II) in comparison to the same concentration of Pb(II). As Hg(II) is generally more toxic than Pb(II), up-regulation of native Hg(II) resistance mechanisms may be responsible for increased uptake and binding of Hg(II) over Pb(II). In nature, the ability to sense a range of heavy metals in the surrounding environment may be hypothesized to provide microorganisms a competitive advantage, resulting in enhanced survival.
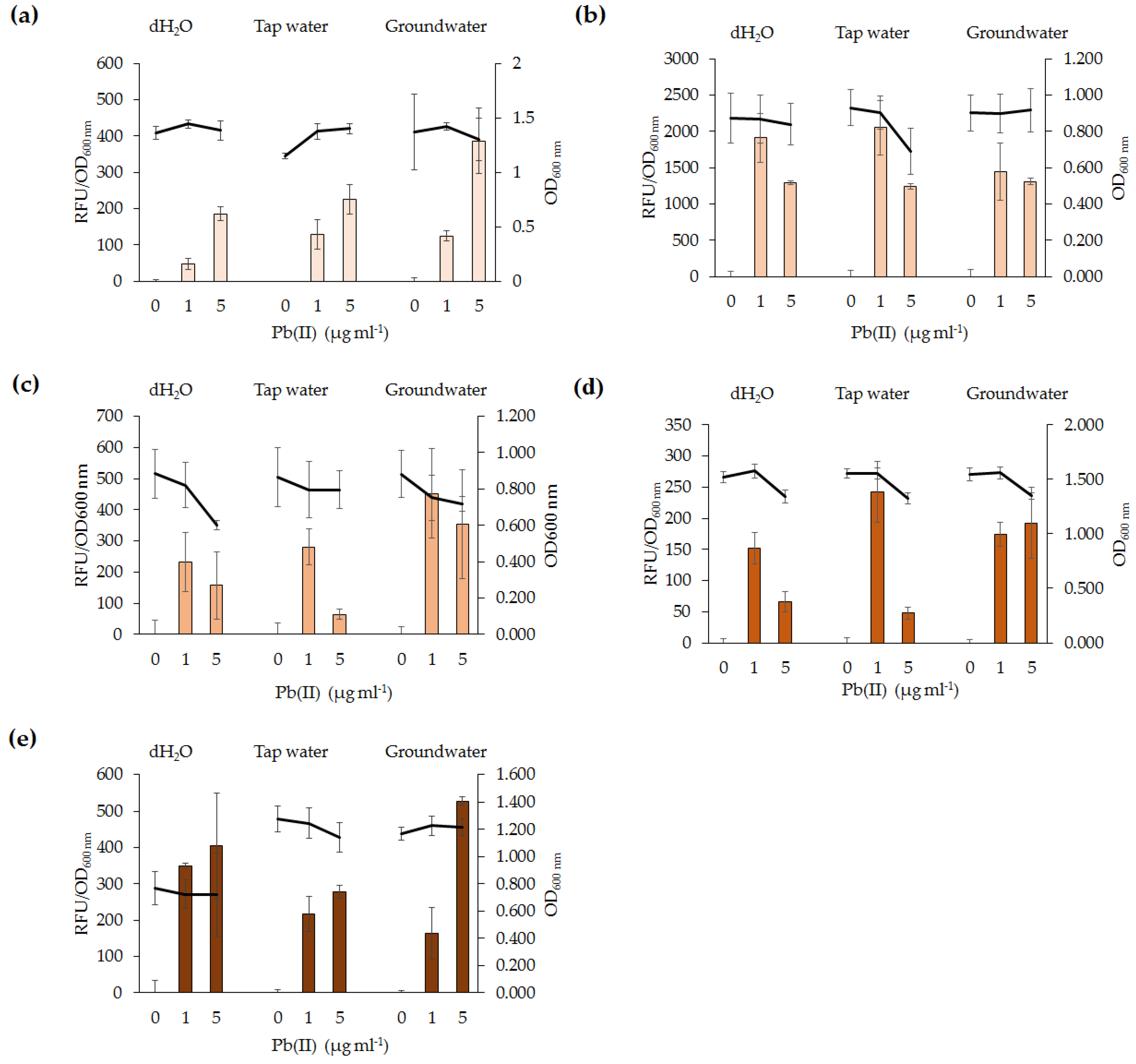
The potential of lead biosensors developed for in situ applications was assessed. All five lead biosensors detected 1 µg·mL−1 Pb(II) in tap water and groundwater. Testing of biosensors in environmental samples which have not been treated via sterilization or filtering is useful to determine the effects of complex environmental samples with resident microbial community. Our findings indicate that short-term exposure of the biosensors to contaminated water samples yield a response to Pb(II). Organic compounds and resident microbial communities in the water samples did not affect the functionality of the lead biosensors. The difference in absolute RFU values between fluorescence assays of the same bacterial species may be explained by the natural variability of the cultures. Overall, the lead biosensors are sensitive, rapid, and generally specific for Pb(II). As such, they could serve as monitoring tools for in situ ecotoxicological assessment of lead-polluted environmental samples. The findings in this study have highlighted variations among biosensors utilizing the same genetic element and, as such, differences between bacterial species should be addressed in future microbial biosensor studies. Future biosensor research should focus on utilizing organisms involved in bioremediation processes, such as immobilization or phytoremediation, and incorporating the biosensor genetic element into their chromosomes. It would also be beneficial for future biosensor studies to develop a more high-throughput method for analysis of microbial biosensors and for detection of Pb(II) in other environmental samples, such as wet weather effluent. This may be performed in parallel to analytical methods for comparison of accuracy and feasibility of the lead biosensors to detect Pb(II) in various environmental samples.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}