
Investigation of Two Novel Approaches for Detection of Sulfate Ion and Methane Dissolved in Sediment Pore Water Using Raman Spectroscopy


Abstract
:1. Introduction
2. Methods
2.1. The Principle of Raman Signal Enhancement
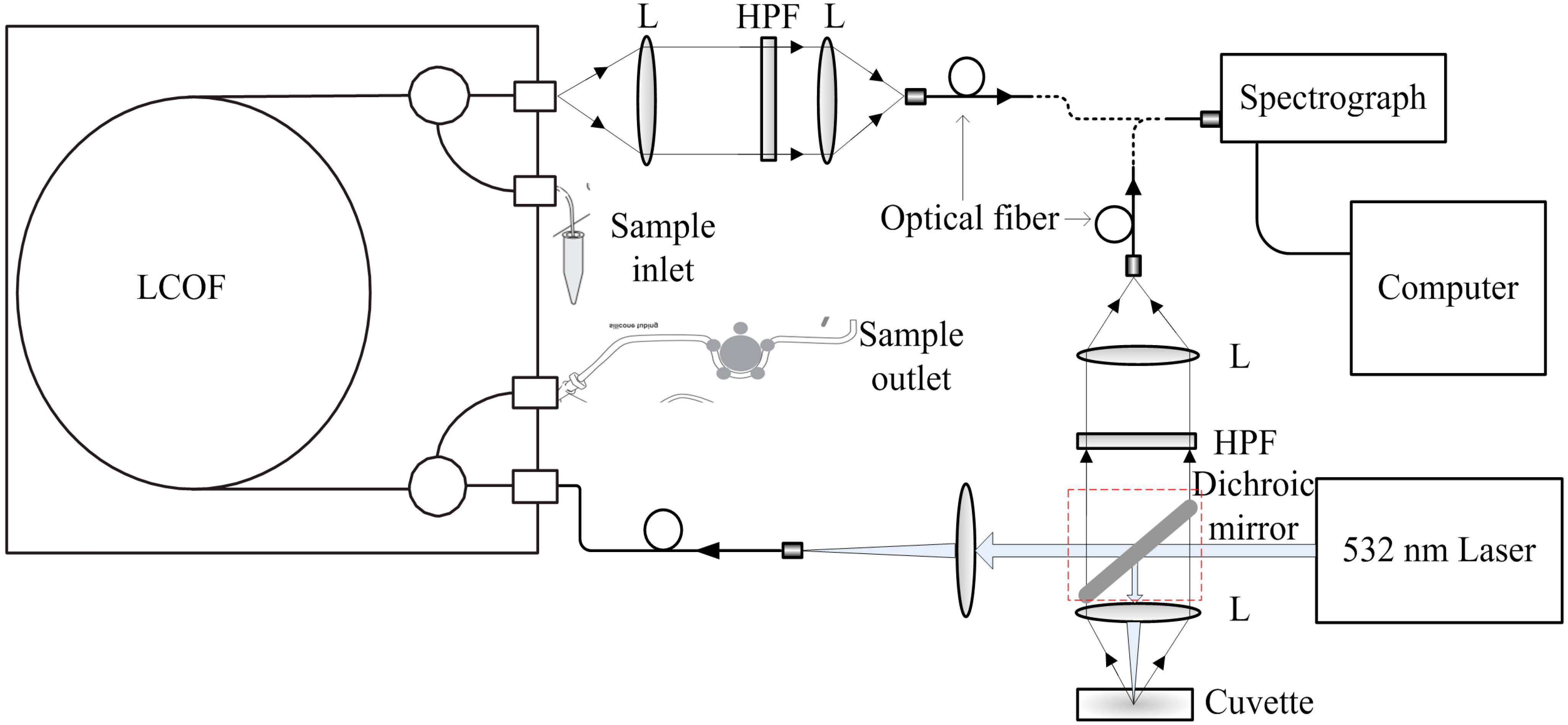
2.2. Instrumental Setup
2.3. Sampling
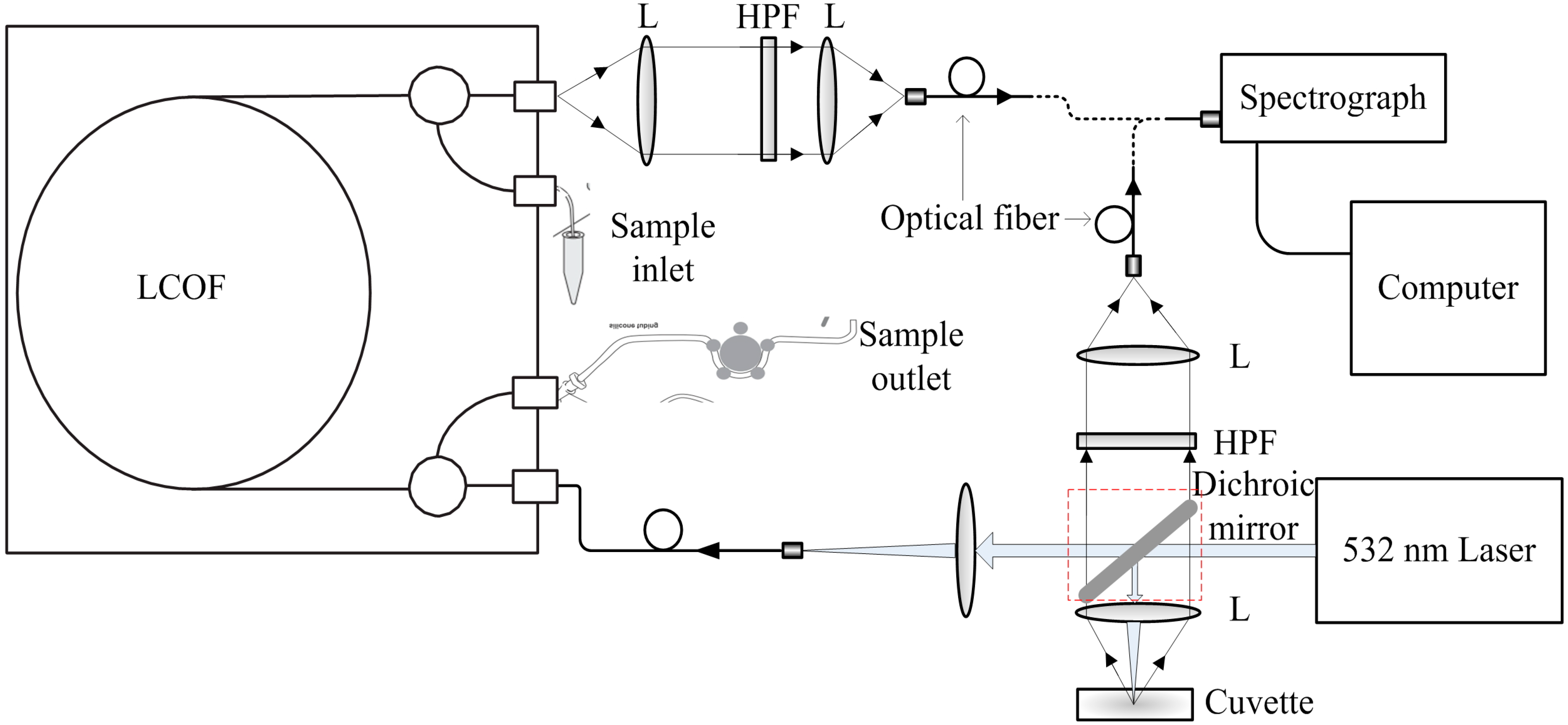
2.4. Spectra Acquisition
3. Results and Discussion
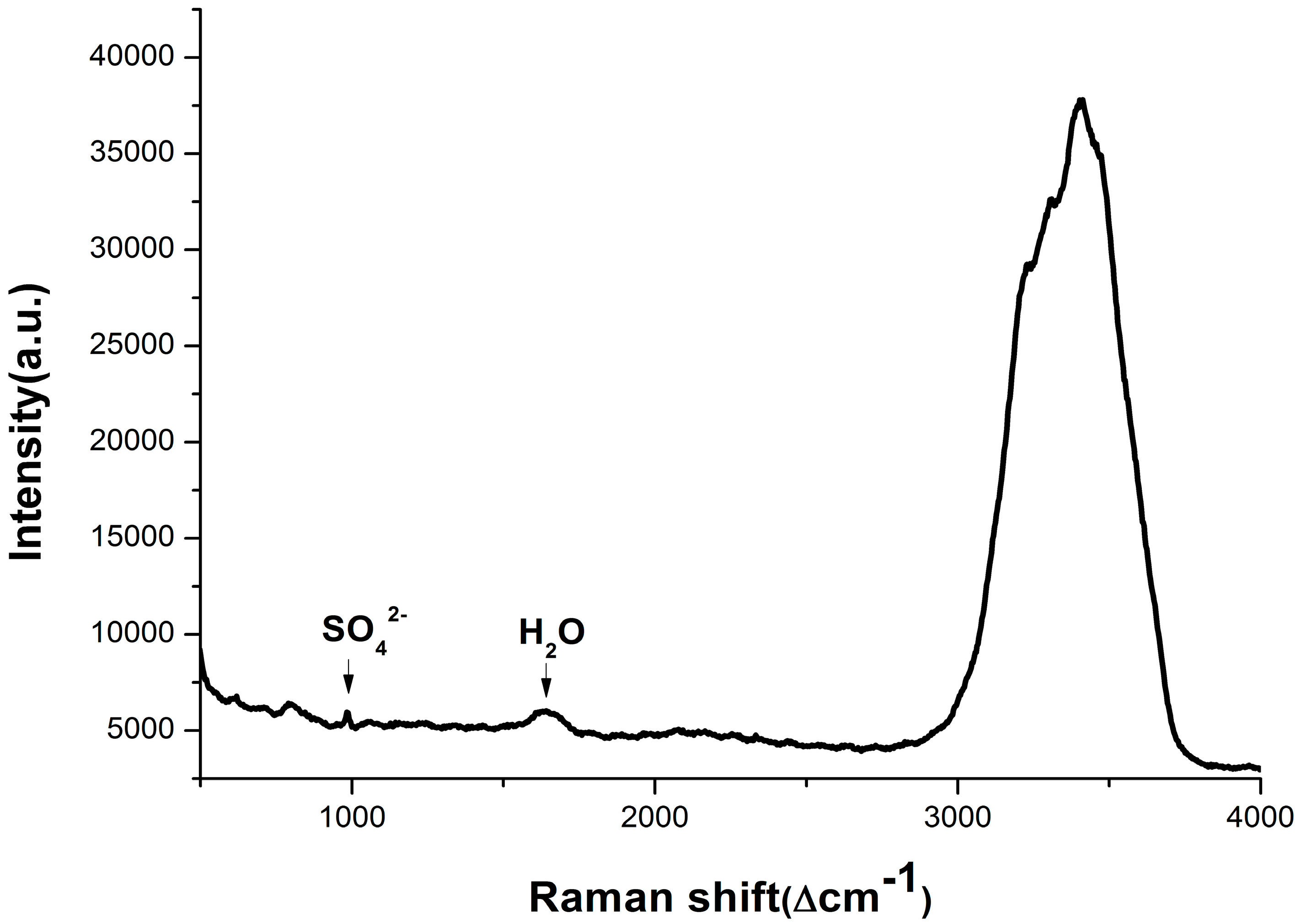
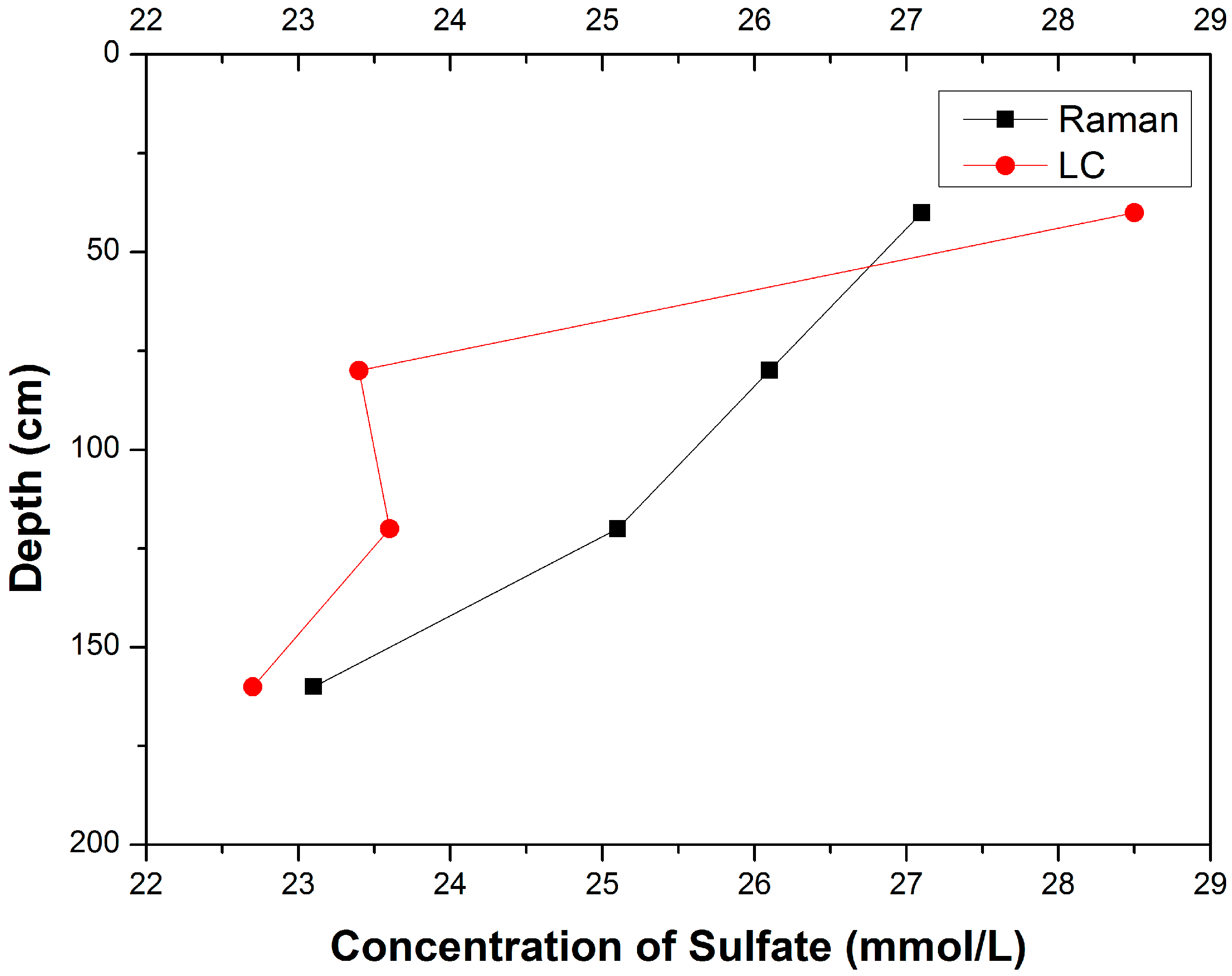
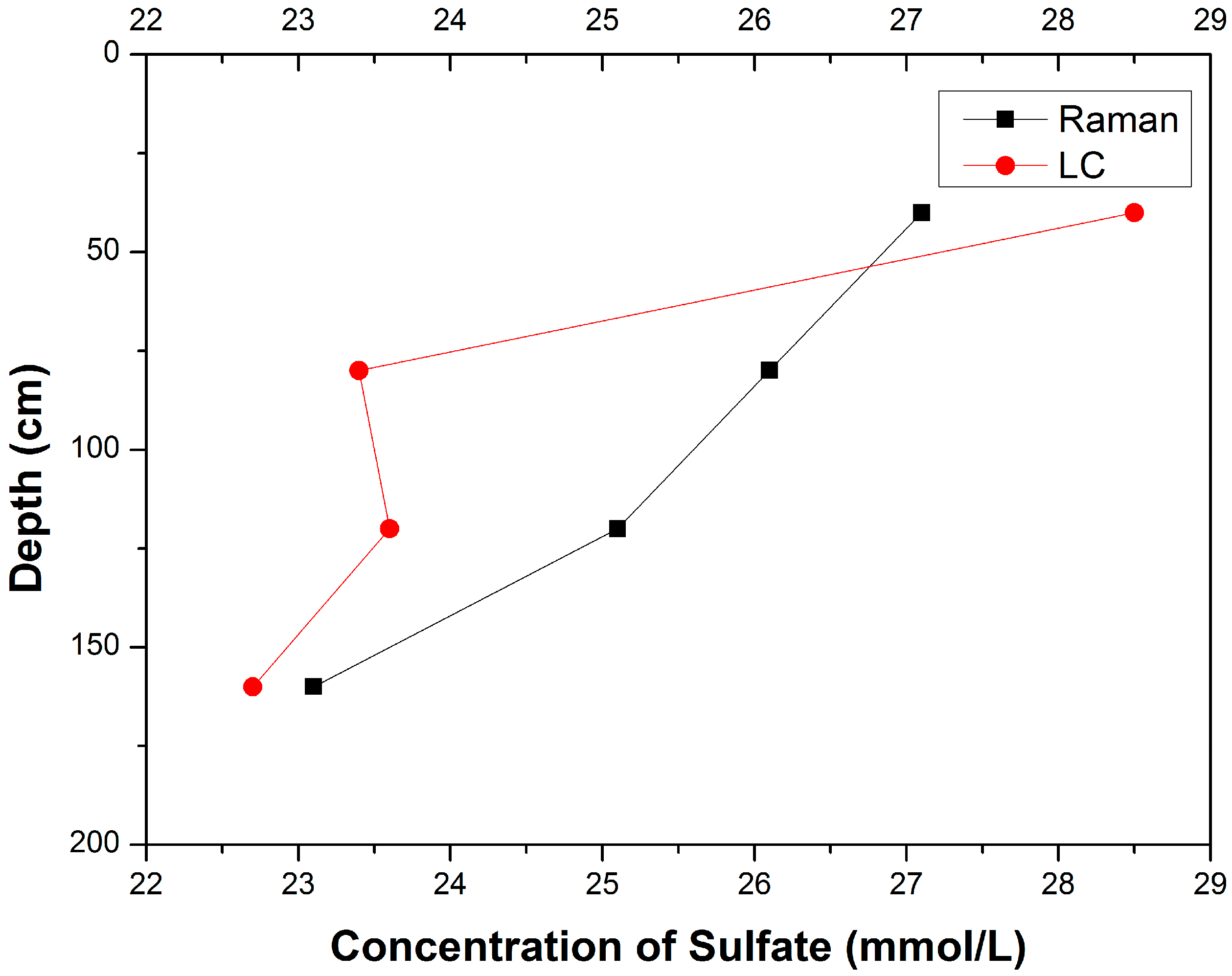
3.1. Composition Analysis of Pore Water Samples with Different Depth
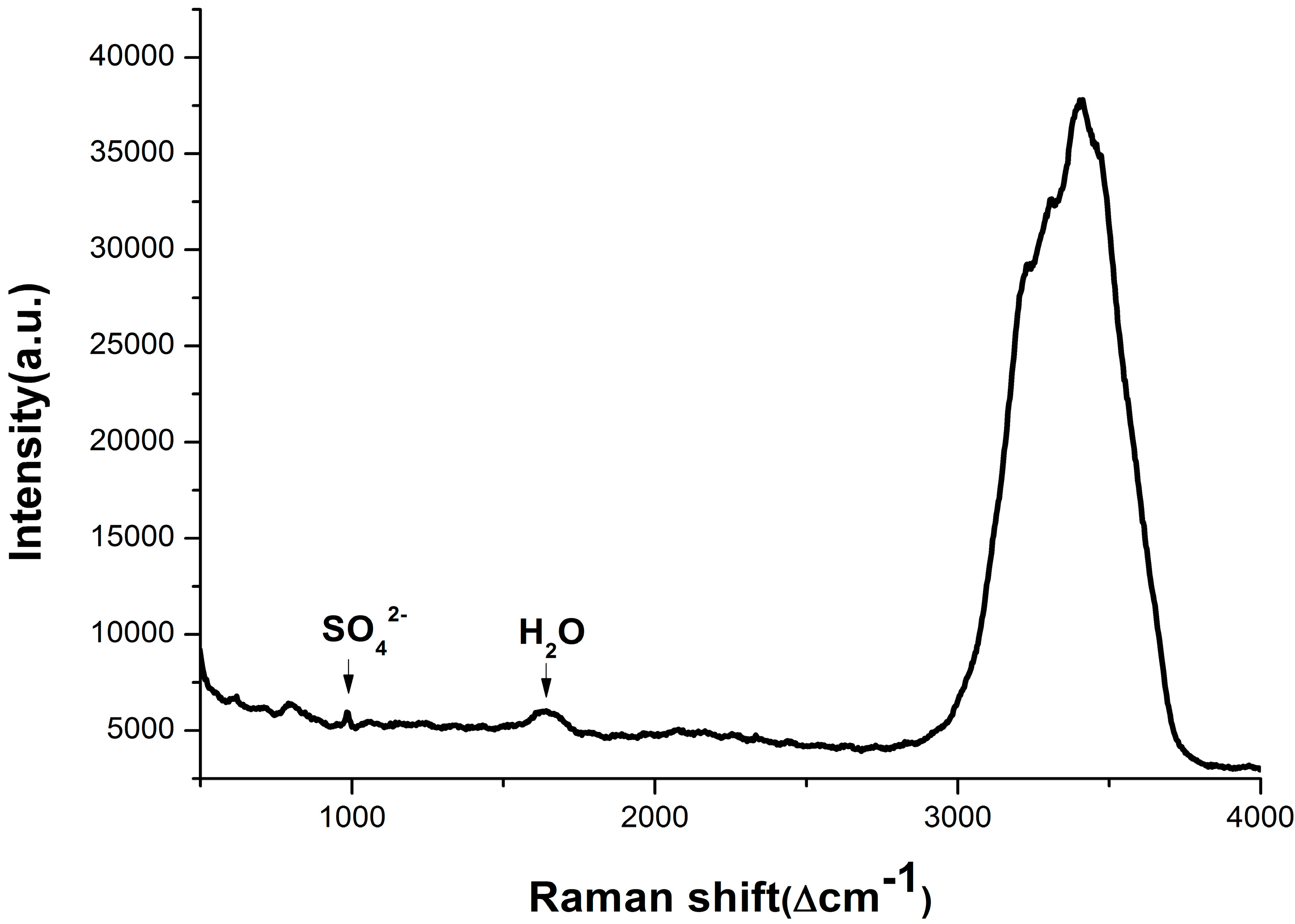

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Sampling Depth (cm) | Concentrations of SO42− (mmol/L) | Relative Deviation │CRaman − CLC│/CLC | |
|---|---|---|---|---|
| CRaman | CLC | |||
| A-12-a | 20–60 | 27.1 | 28.5 | 4.91% |
| B-12-a | 60–100 | 26.1 | 23.4 | 11.54% |
| C-12-a | 100–140 | 25.1 | 23.6 | 6.36% |
| D-12-a | 140–180 | 23.1 | 22.7 | 1.76% |
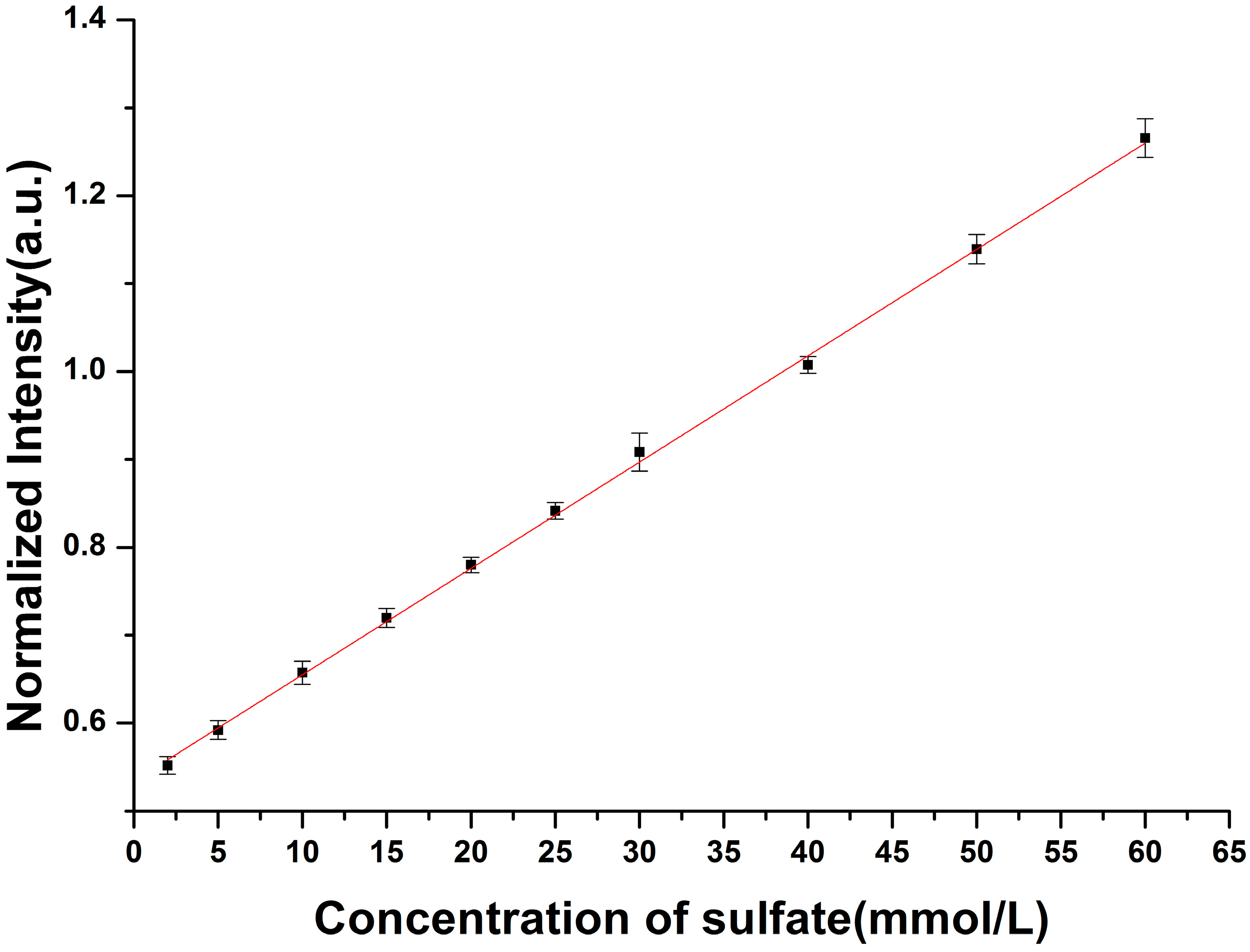
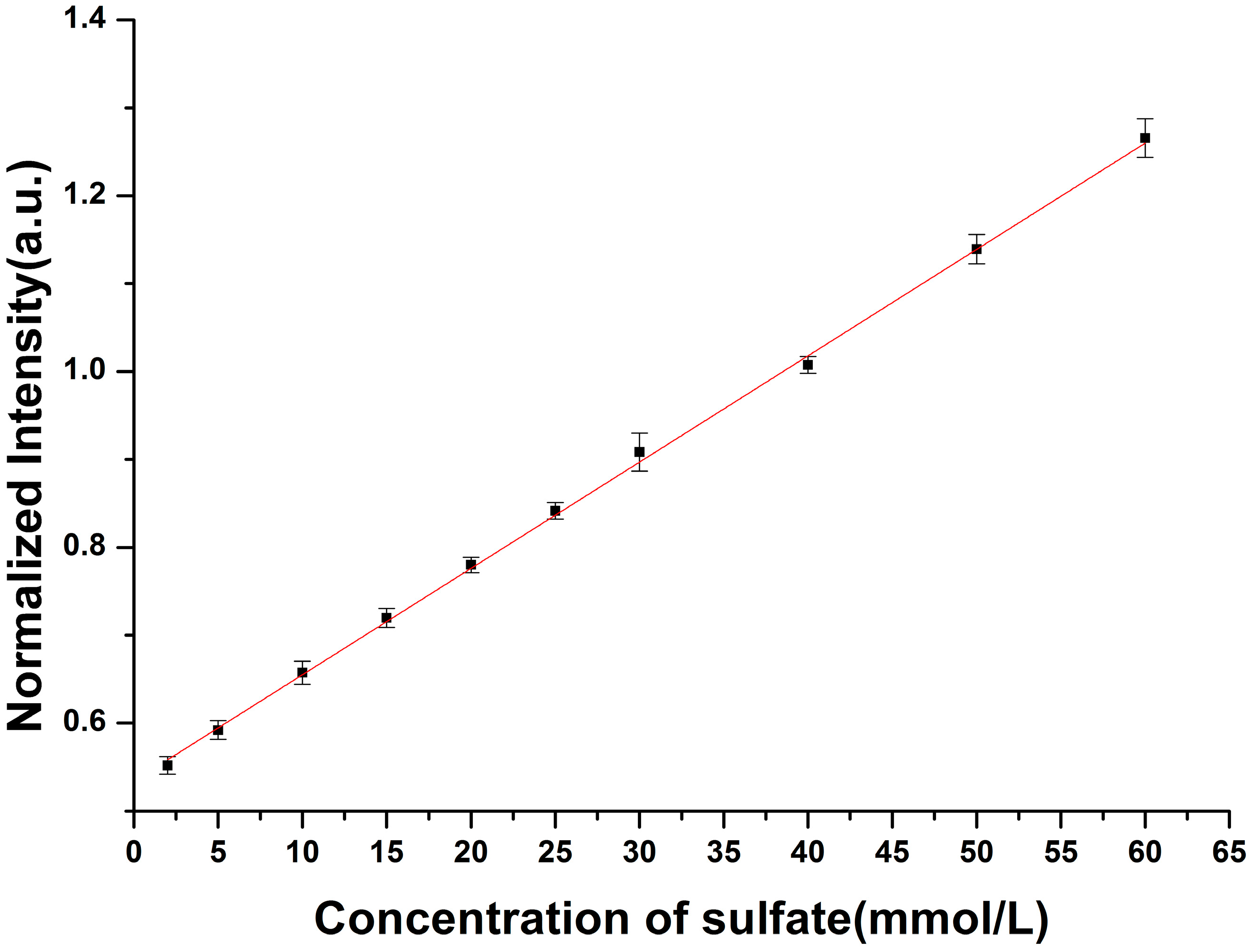
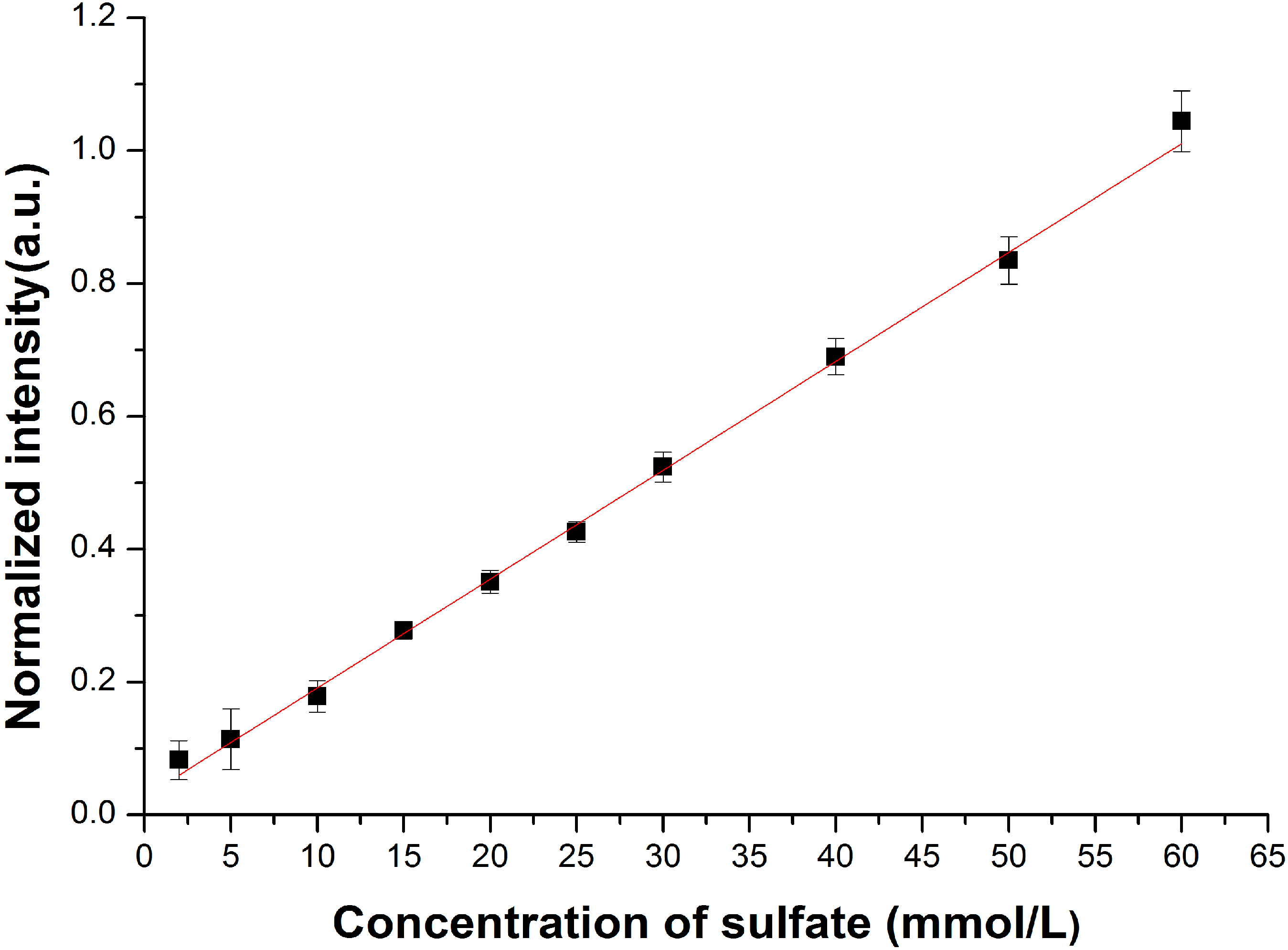
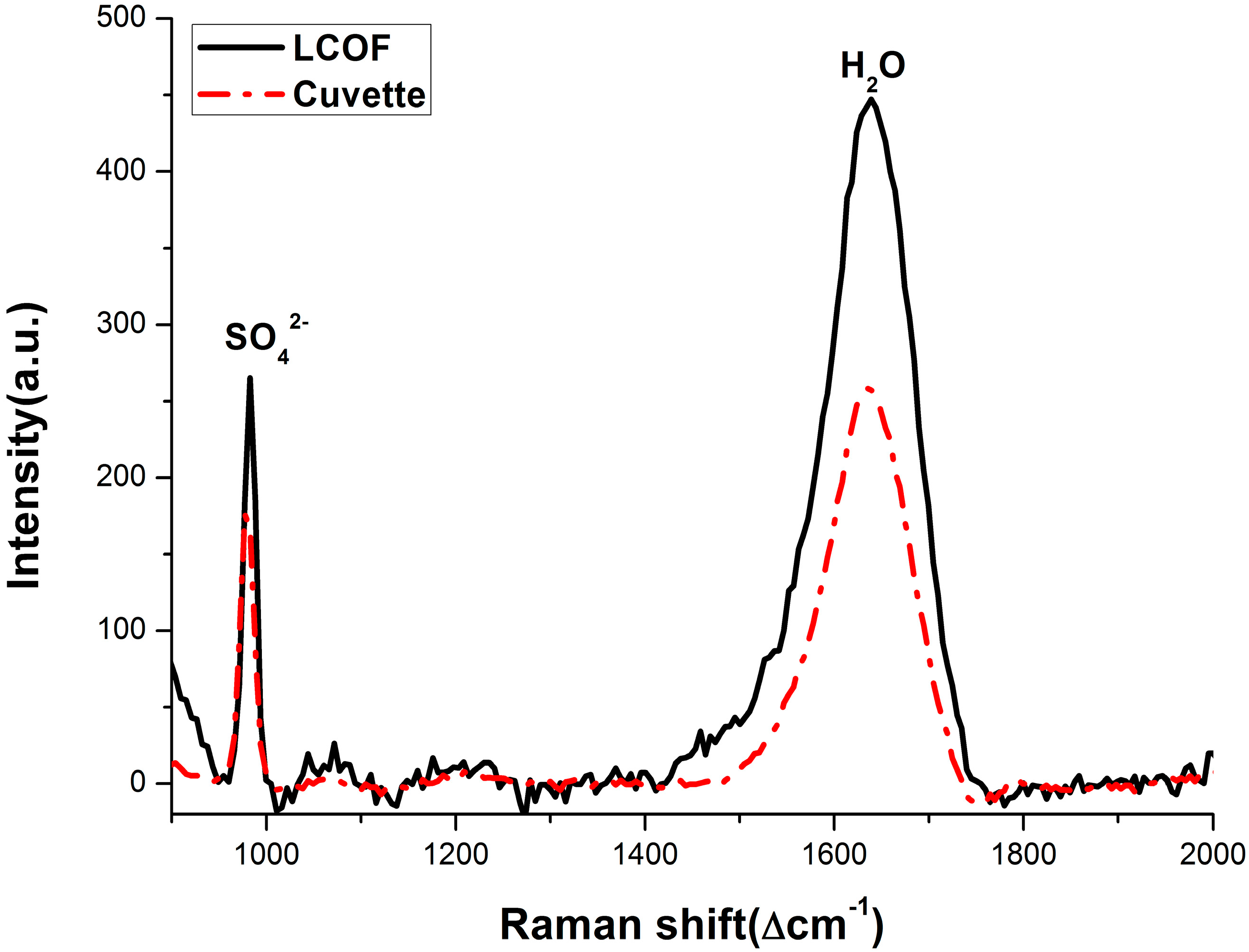
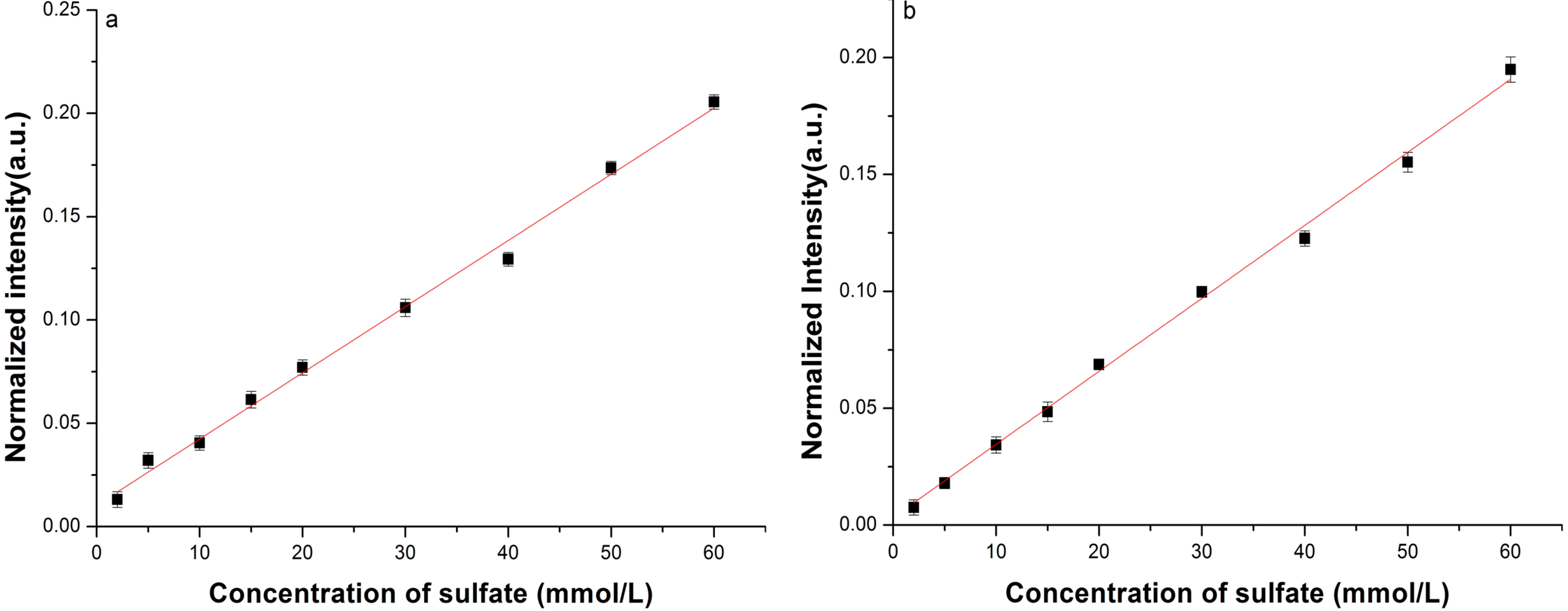
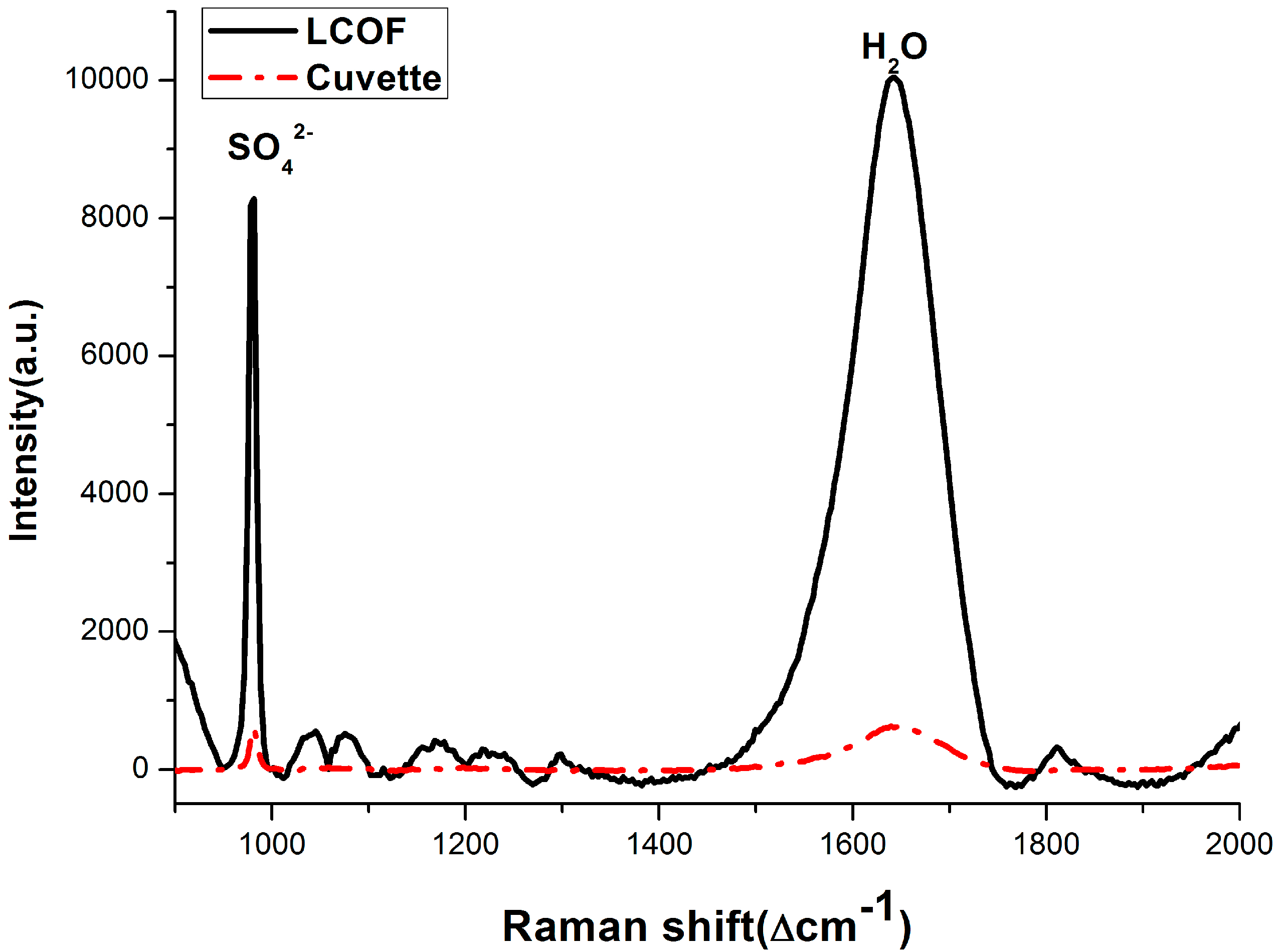
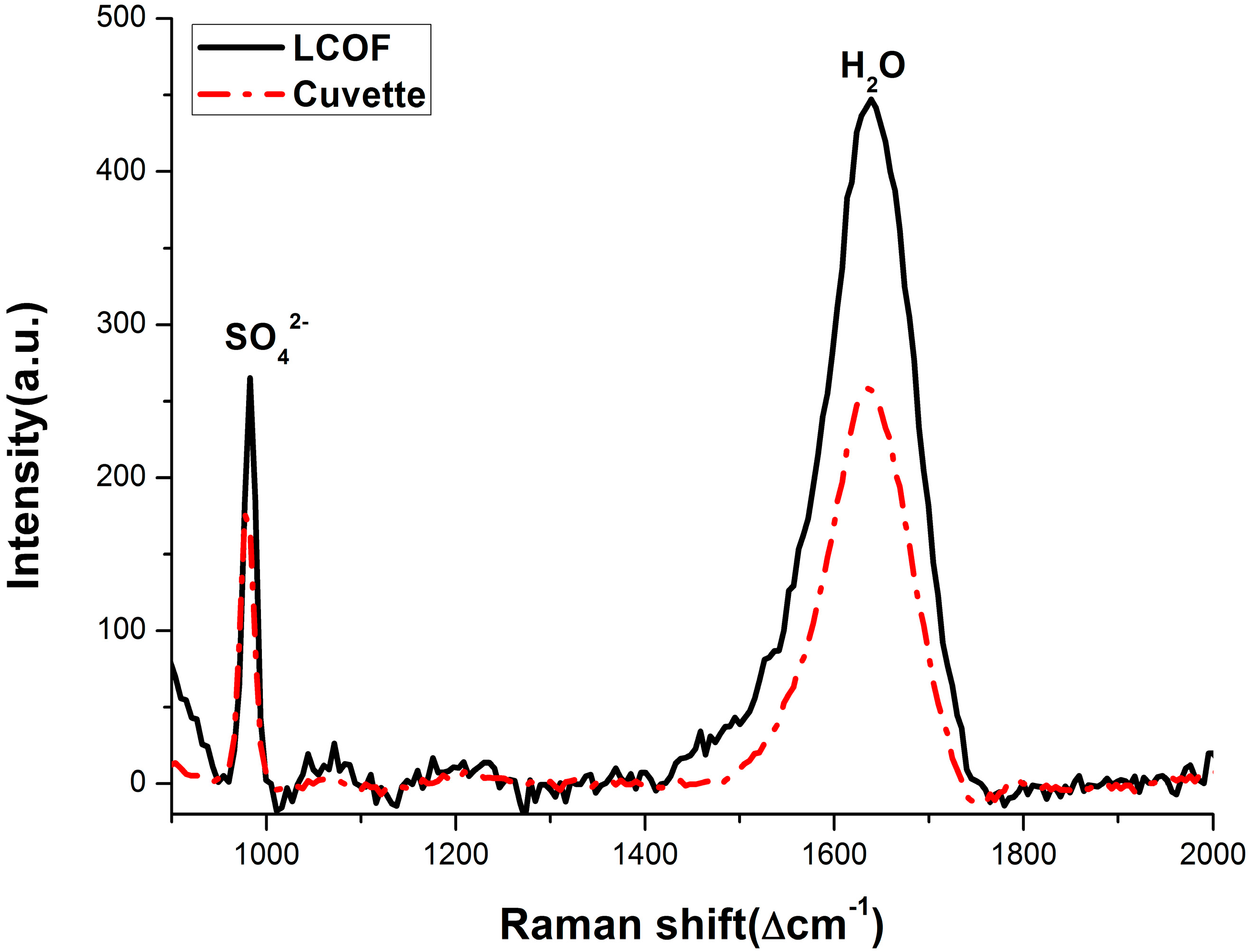
3.2. Enhancement for Raman Signal of SO42− with LCOF
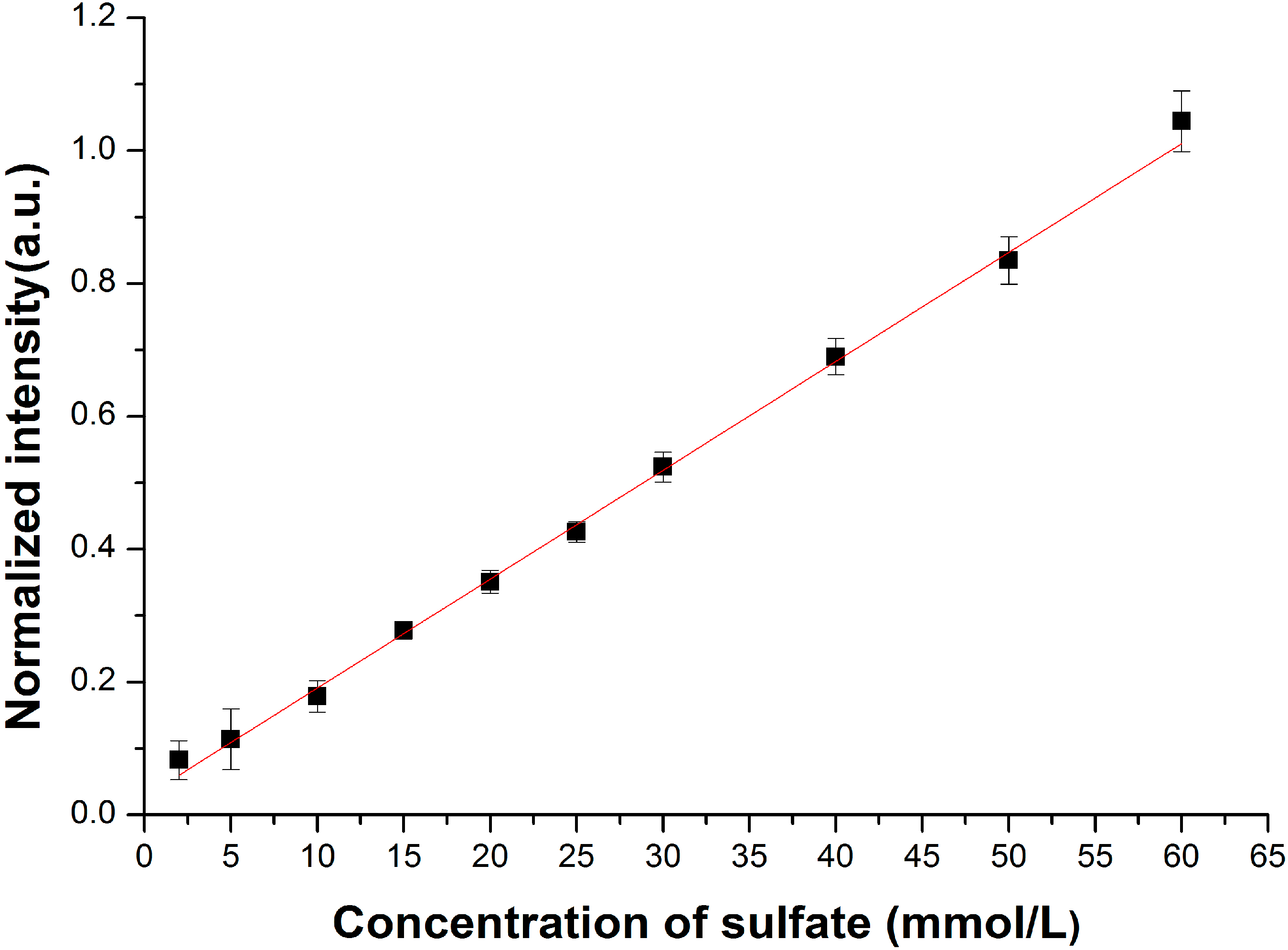
| Fitted Linear Function | LOD (mmol/L) | |
|---|---|---|
| Conventional Raman setup | R* = 0.012C + 0.534 | 2.15 |
| LCOF-Raman setup | R* = 0.016C + 0.027 | 1.50 |
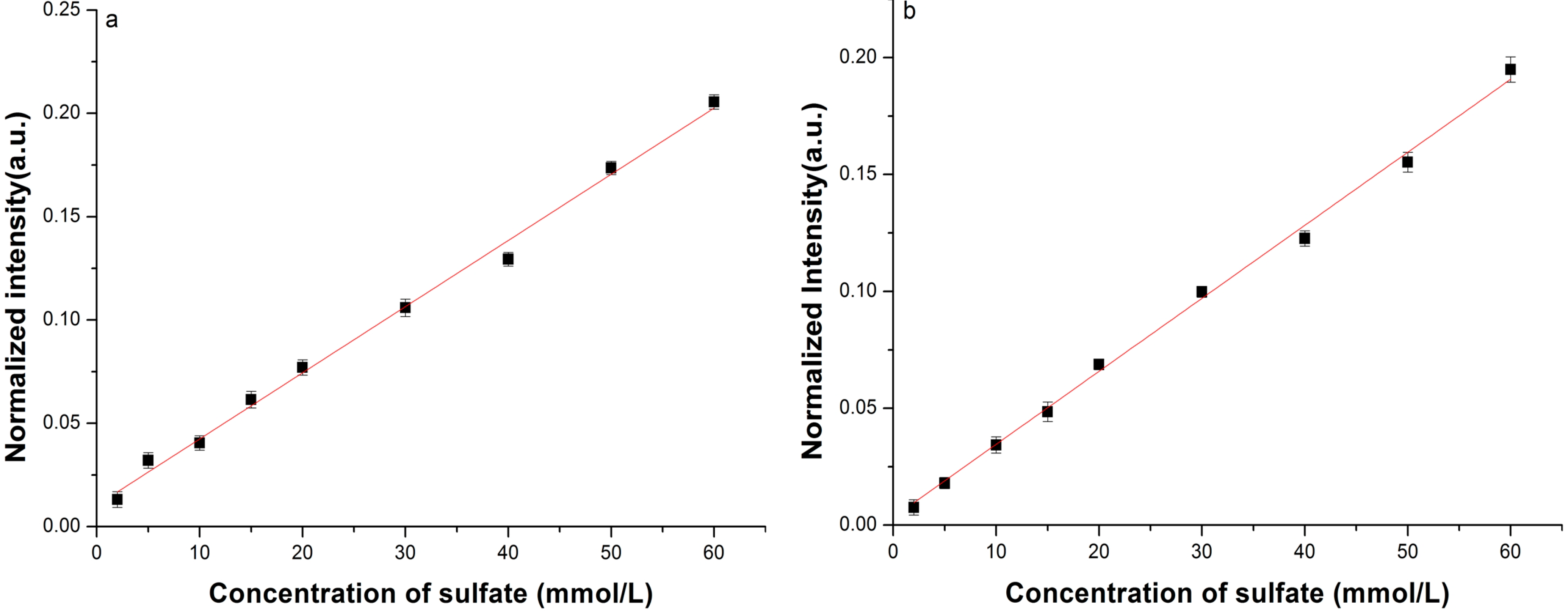
| Fitted Linear Function | LOD (mmol/L) | |
|---|---|---|
| Conventional Raman setup | R* = 0.0032C + 0.010 | 1.80 |
| LCOF-Raman setup | R *= 0.0031C + 0.0034 | 0.35 |
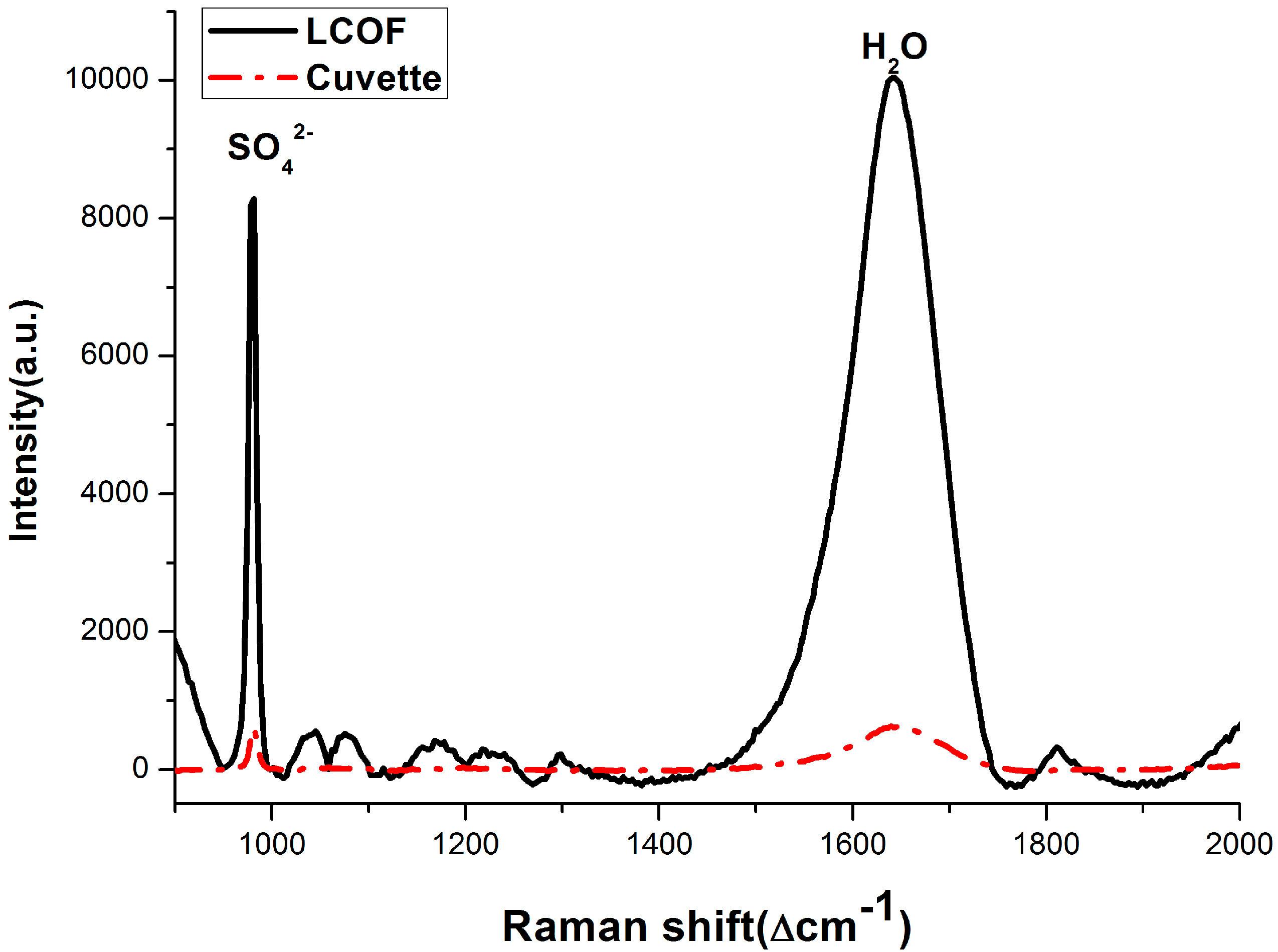
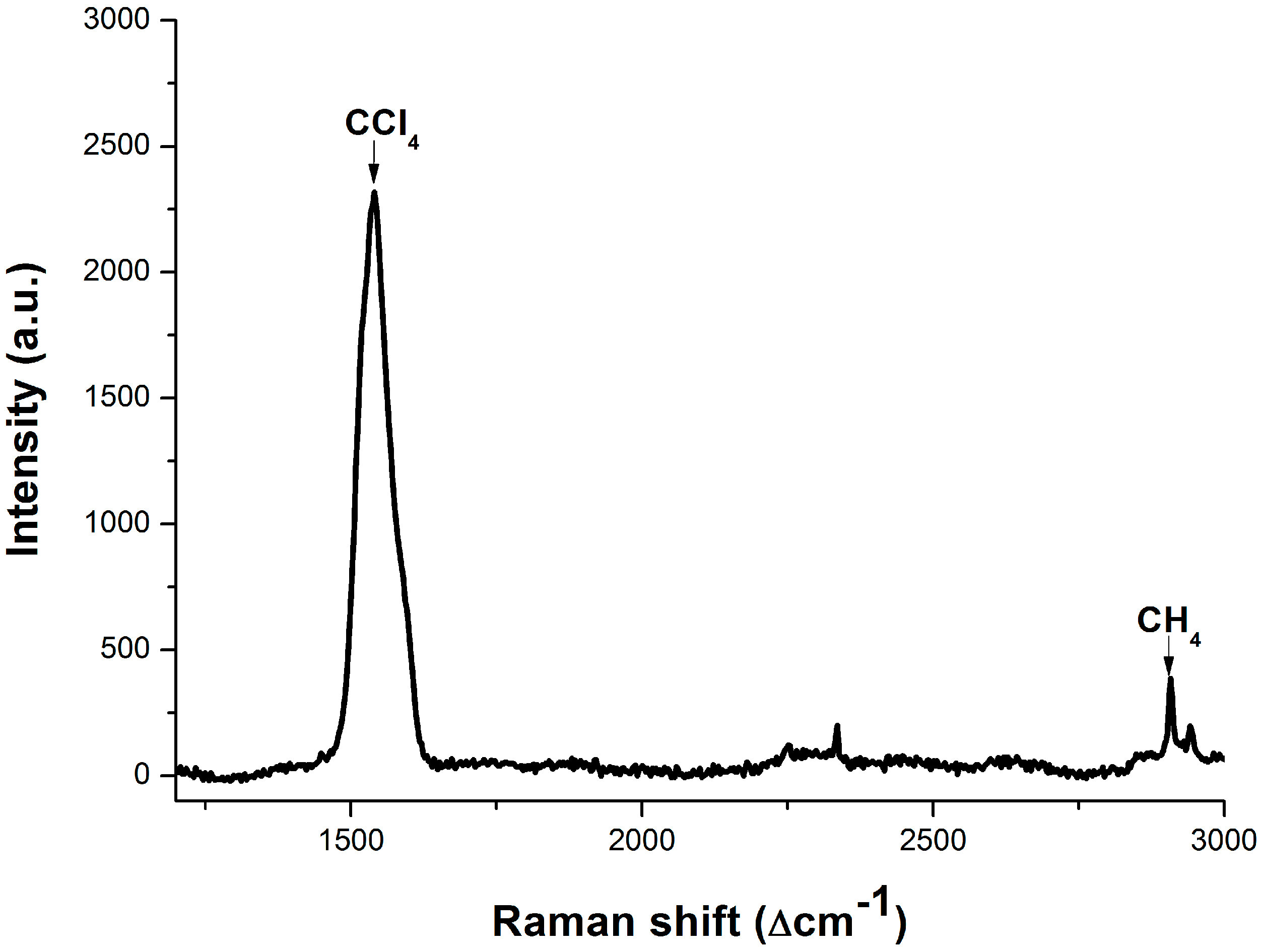
3.3. Dissolved Methane Detection Aided with an Enrichment Process
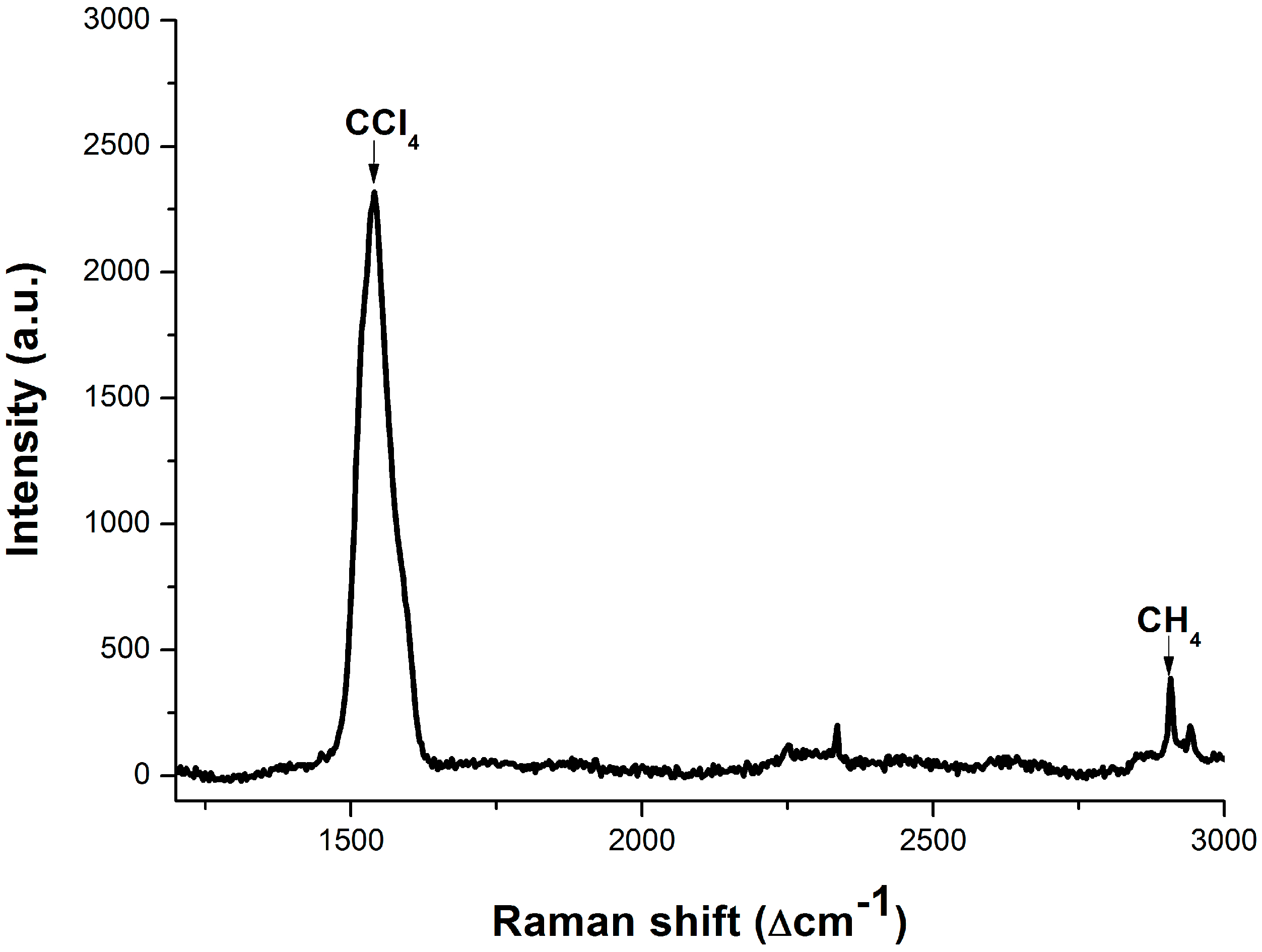
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prieur, D.; Erauso, G.; Jeanthon, C. Hyperthermophilic life at deep-sea hydrothermal vents. Planet. Space Sci. 1995, 43, 115–122. [Google Scholar] [CrossRef]
- Simoneit, B.R.T.; Schoell, M.; Dias, R.F.; de Auuino Neto, F.R. Unusual carbon isotope compositions of biomarker hydrocarbons in a Permian tasmanite. Geochim. Cosmochim. Acta 1993, 57, 4205–4211. [Google Scholar] [CrossRef]
- Vairavamurthy, M.A.; Orr, W.L.; Manowitz, B. Geochemical transformations of sedimentary sulfur: An introduction. In ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1995; pp. 1–15. [Google Scholar]
- Henrichs, S.M.; Reeburgh, W.S. Anaerobic mineralization of marine sediment organic matter: Rates and the role of anaerobic processes in the oceanic carbon economy. Geomicrobiol. J. 1987, 5, 191–237. [Google Scholar] [CrossRef]
- Jorgensen, B.B.; Kasten, S. Sulfur Cycling and Methane Oxidation. In Marine Geochemistry; Horst, D.S., Matthias, Z., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 271–309. [Google Scholar]
- Whitman, W.B.; Bowen, T.L.; Boone, D.R. The methanogenic bacteria. In The Prokaryotes; Dworkin, M., Balows, A., Eds.; Springer US: New York, NY, USA, 2006; pp. 165–207. [Google Scholar]
- Borowski, W.S.; Paull, C.K.; Ussler, W. Carbon cycling within the upper methanogenic zone of continental rise sediments; an example from the methane-rich sediments overlying the Blake Ridge gas hydrate deposits. Mar. Chem. 1997, 57, 299–311. [Google Scholar] [CrossRef]
- Claypool, G.E.; Kaplan, I.R. The origin and distribution of methane in marine sediments. In Natural Gases in Marine Sediments; Isaac, R.K., Ed.; Springer US: New York, NY, USA, 1974; pp. 99–139. [Google Scholar]
- Borowski, W.S.; Paull, C.K.; Ussler, W. Marine pore-water sulfate profiles indicate in situ methane flux from underlying gas hydrate. Geology 1996, 24, 655–658. [Google Scholar] [CrossRef]
- Borowski, W.S.; Paull, C.K.; Ussler, W. Global and local variations of interstitial sulfate gradients in deep-water, continental margin sediments: Sensitivity to underlying methane and gas hydrates. Mar. Geol. 1999, 159, 131–154. [Google Scholar] [CrossRef]
- Ussler, W.; Paull, C.K. Rates of anaerobic oxidation of methane and authigenic carbonate mineralization in methane-rich deep-sea sediments inferred from models and geochemical profiles. Earth Planet. Sci. Lett. 2008, 266, 271–287. [Google Scholar] [CrossRef]
- Zhang, X.; Walz, P.M.; Kirkwood, W.J.; Hester, K.C.; Ussler, W.; Peltzer, E.T.; Brewer, P.G. Development and deployment of a deep-sea Raman probe for measurement of pore water geochemistry. Deep Sea Res. Part I Oceanogr. Res. Pap. 2010, 57, 297–306. [Google Scholar] [CrossRef]
- Brewer, P.G.; Malby, G.; Pasteris, J.D.; White, S.N.; Peltzer, E.T.; Wopenka, B.; Freeman, J.; Brown, M.O. Development of a laser Raman spectrometer for deep-ocean science. Deep Sea Res. Part I Oceanogr. Res. Pap. 2004, 51, 739–753. [Google Scholar] [CrossRef]
- Reeburgh, W.S. Oceanic Methane Biogeochemistry. Chem. Rev. 2007, 107, 486–513. [Google Scholar] [CrossRef] [PubMed]
- Paull, C.K.; Ussler, W. History and Significance of Gas Sampling During DSDP and ODP Drilling Associated with Gas Hydrates. In Natural Gas Hydrates: Occurrence, Distribution, and Detection; Paull, C.K., Dillon, W.P., Eds.; American Geophysical Union: Washington, DC, USA, 2001; pp. 53–65. [Google Scholar]
- Newman, K.R.; Cormier, M.H.; Weissel, J.K.; Driscoll, N.W.; Kastner, M.; Solomon, E.A.; Robertson, G.; Hill, J.C.; Singh, H.; Camilli, R.; et al. Active methane venting observed at giant pockmarks along the US mid-atlantic shelf break. Earth Planet. Sci. Lett. 2008, 267, 341–352. [Google Scholar] [CrossRef]
- Marinaro, G.; Etiope, G.; Bue, N.L.; Favali, P.; Papatheodorou, G.; Christodoulou, D.; Furlan, F.; Gasparoni, F.; Ferentinos, G.; Masson, M.; et al. Monitoring of a methane-seeping pockmark by cabled benthic observatory (Patras Gulf, Greece). Geo-Mar. Lett. 2006, 26, 297–302. [Google Scholar] [CrossRef]
- Fietzek, P.; Kramer, S.; Esser, D. Deployments of the HydroC™(CO2/CH4) on stationary and mobile platforms-Merging trends in the field of platform and sensor development. In Proceedings of the IEEE OCEANS, Waikoloa, HI, USA, 19–22 September 2011; pp. 1–9.
- Pelletier, M.J. Analytical Applications of Raman Spectroscopy; Blackwell Science Ltd.: Osney Mead, Oxford, UK, 1999; p. 478. [Google Scholar]
- Altkorn, R.; Koev, I.; van Duyne, R.P.; Litorja, M. Low-loss liquid-core optical fiber for low-refractive-index liquids fabrication, characterization, and application in Raman spectroscopy. Appl. Opt. 1997, 36, 8892–8898. [Google Scholar] [CrossRef]
- Reeburgh, W.S. An improved interstitial water sampler. Limnol. Oceanogr. 1967, 12, 163–165. [Google Scholar] [CrossRef]
- Qi, D.; Berger, A.J. Quantitative analysis of Raman signal enhancement from aqueous samples in liquid core optical fibers. Appl. Spectrosc. 2004, 58, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Altkron, R.; Malinsky, M.D.; van Duyne, R.P.; Koev, I. Intensity considerations in liquid core optical fiber Raman spectroscopy. Appl. Spectrosc. 2001, 55, 373–381. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Z.; Chen, J.; Ye, W.; Guo, J.; Zhang, X.; Zheng, R. Investigation of Two Novel Approaches for Detection of Sulfate Ion and Methane Dissolved in Sediment Pore Water Using Raman Spectroscopy. Sensors 2015, 15, 12377-12388. https://doi.org/10.3390/s150612377
Du Z, Chen J, Ye W, Guo J, Zhang X, Zheng R. Investigation of Two Novel Approaches for Detection of Sulfate Ion and Methane Dissolved in Sediment Pore Water Using Raman Spectroscopy. Sensors. 2015; 15(6):12377-12388. https://doi.org/10.3390/s150612377
Chicago/Turabian StyleDu, Zengfeng, Jing Chen, Wangquan Ye, Jinjia Guo, Xin Zhang, and Ronger Zheng. 2015. "Investigation of Two Novel Approaches for Detection of Sulfate Ion and Methane Dissolved in Sediment Pore Water Using Raman Spectroscopy" Sensors 15, no. 6: 12377-12388. https://doi.org/10.3390/s150612377