Methodology of Pulsed Photoacoustics and Its Application to Probe Photosystems and Receptors

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Principle of Pulsed Photoacoustics
2.1. Molecular Volume Change by Electrostriction and Structural Conformational Changes
2.2. Quantum Yield of Photoreaction
2.3. Enthalpy Change and Entropy Change of Photoreaction
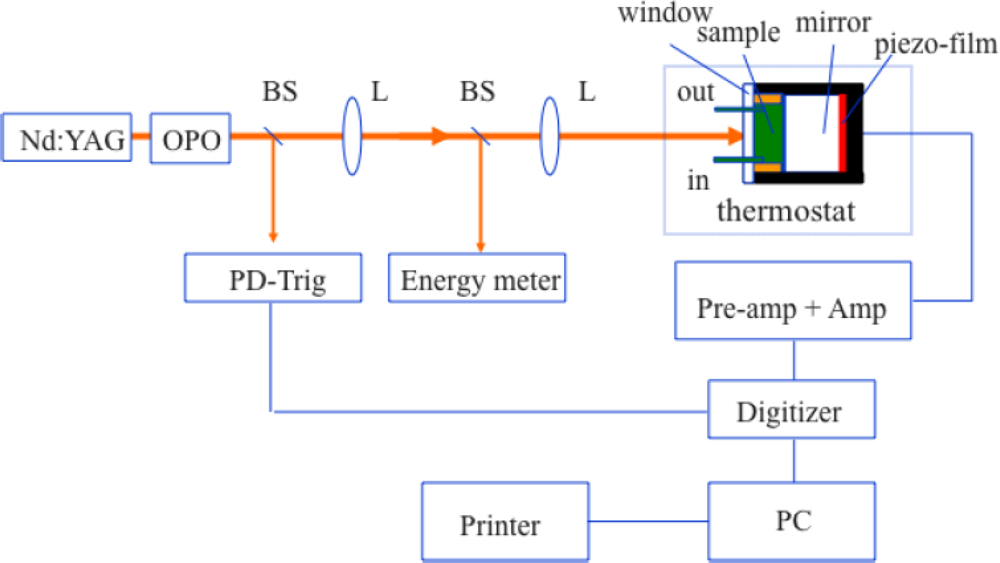
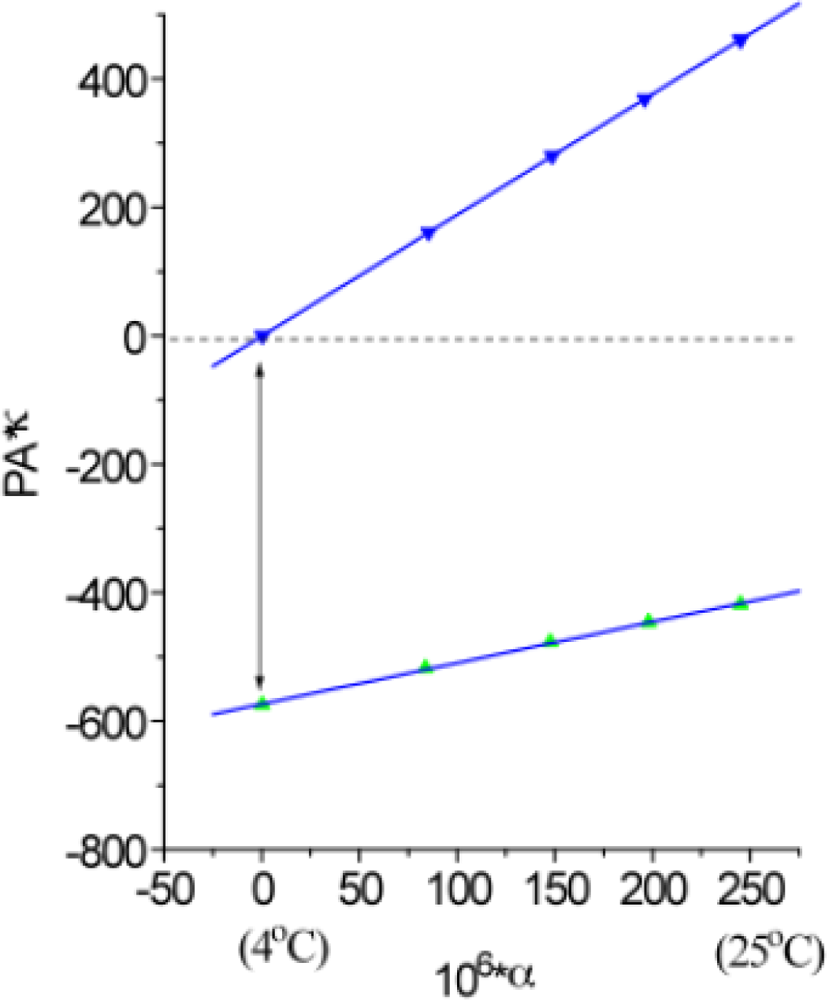
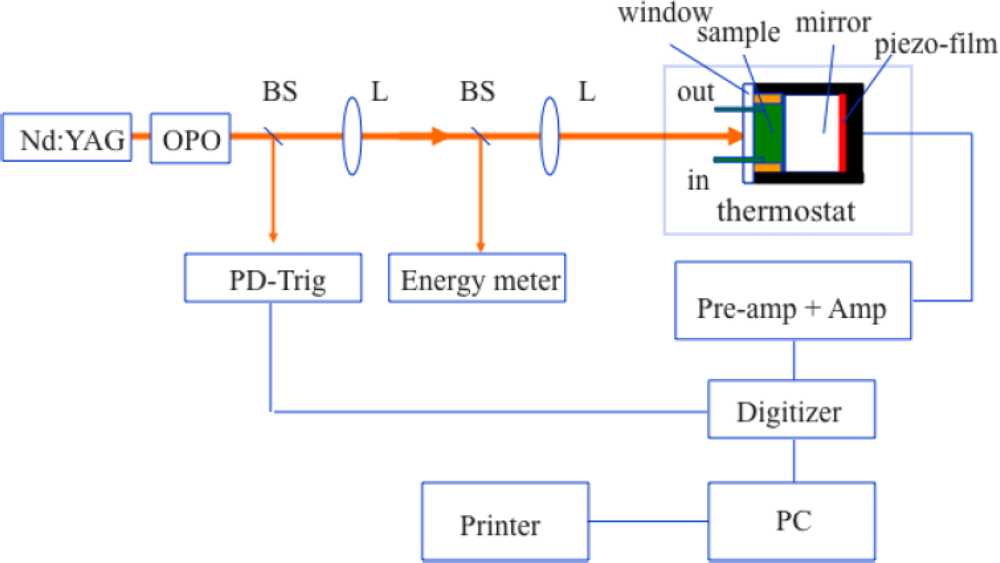
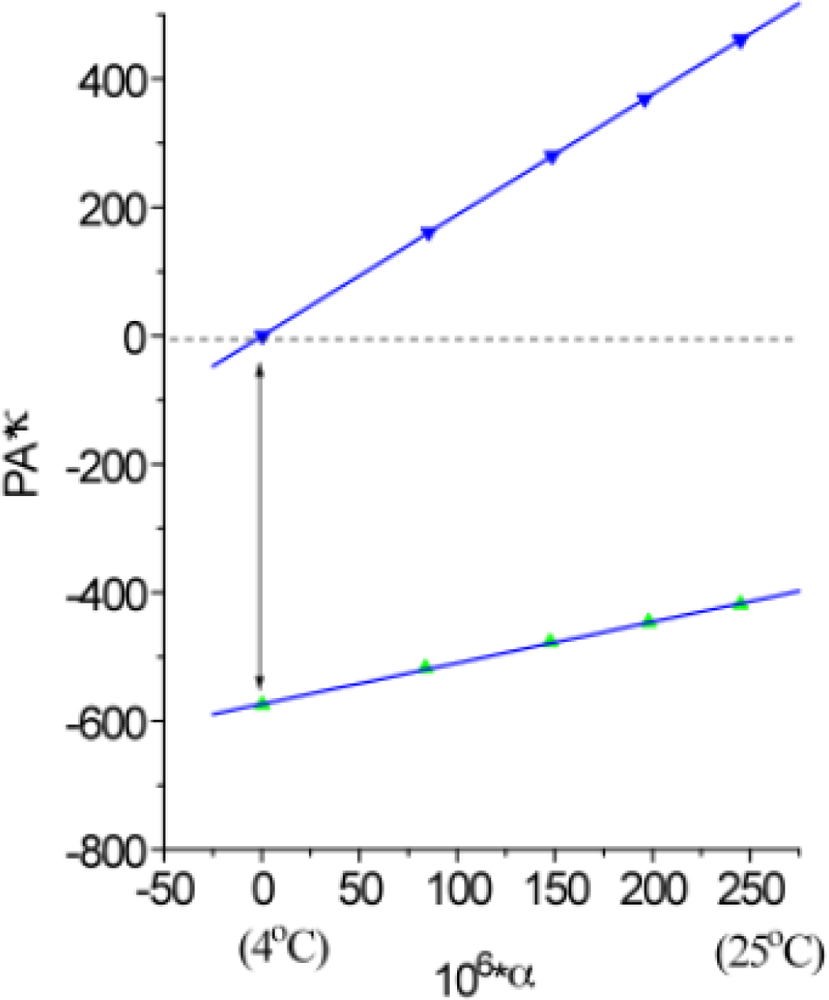
2.4. Measuring the Reaction Enthalpy and Volume Changes
3. Photoacoustic Studies of Photosystems
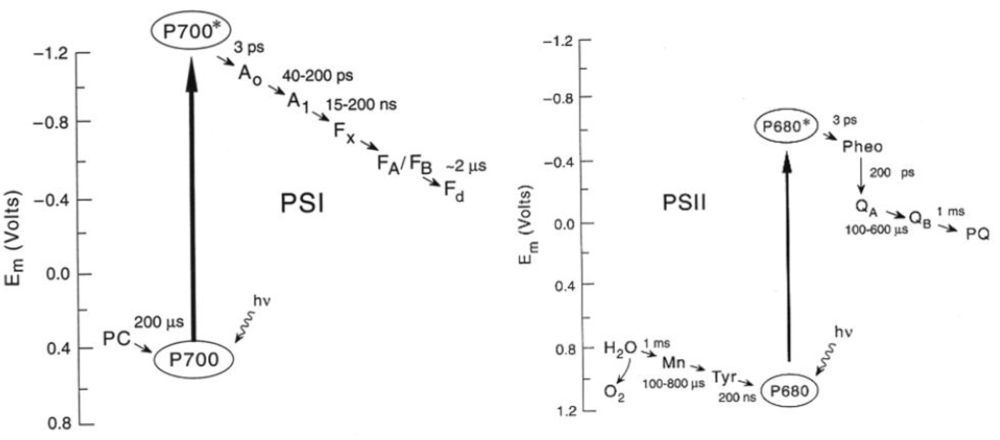
3.1. Structure and Function of Photosystems
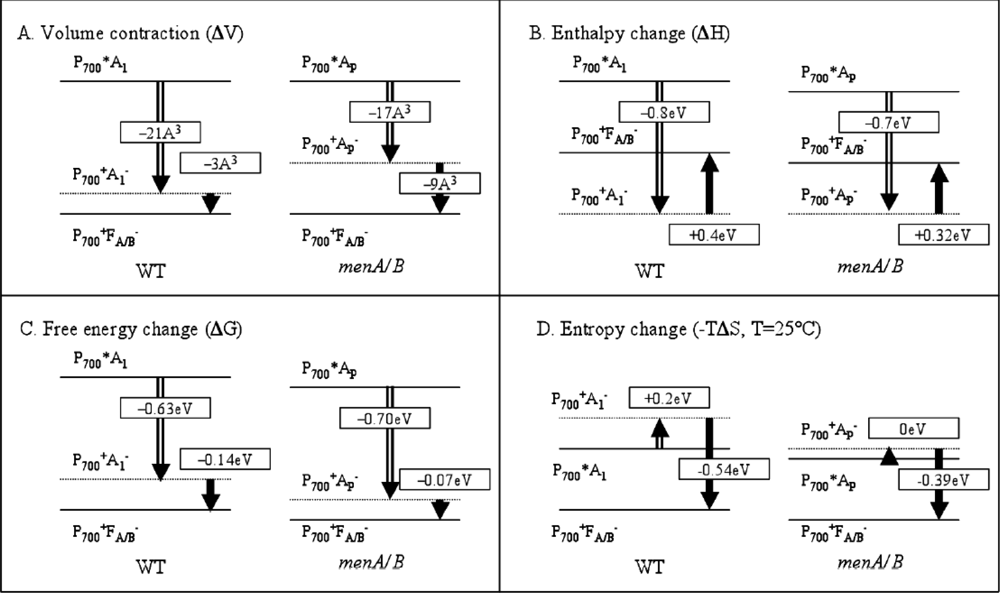
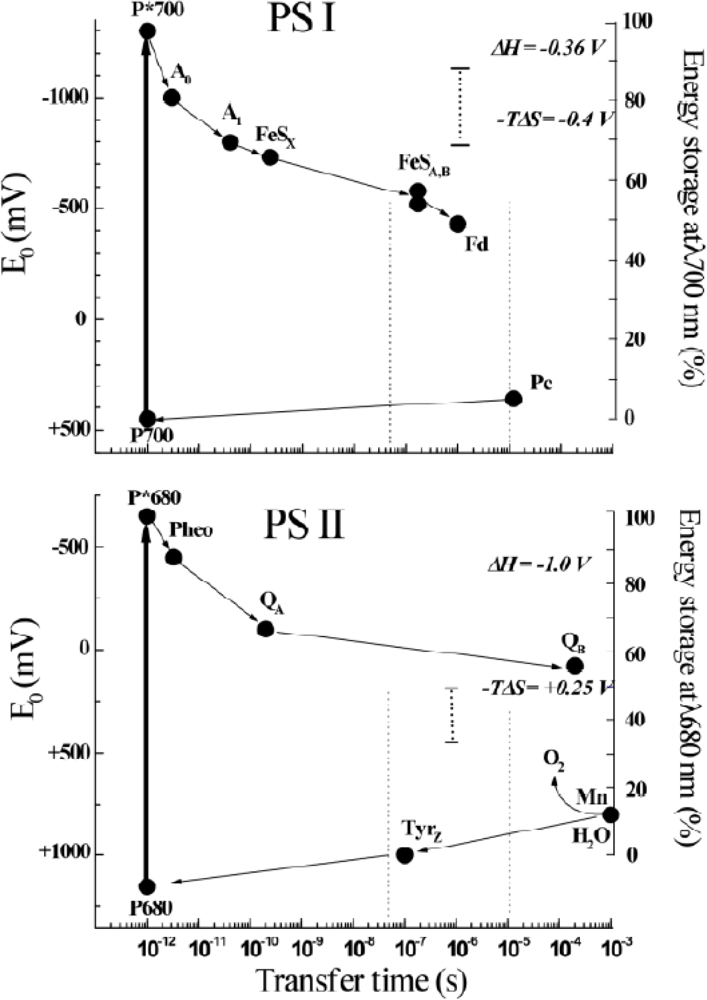
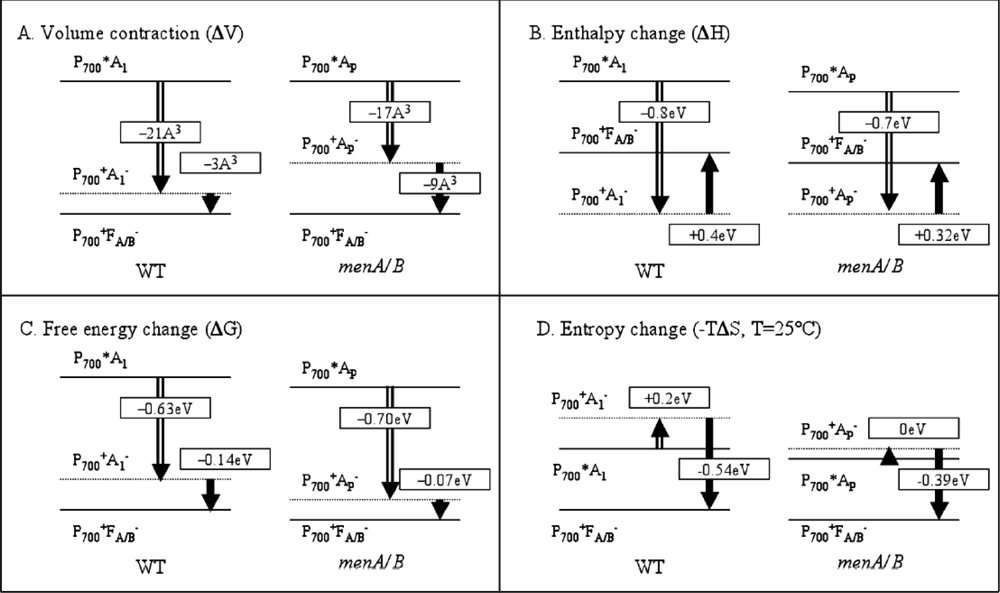
3.2. Photoacoustic Studies of Photosystems
4. Photoacoustic Studies of Receptors
4.1. Structure and Function of G Protein-Coupled Receptors
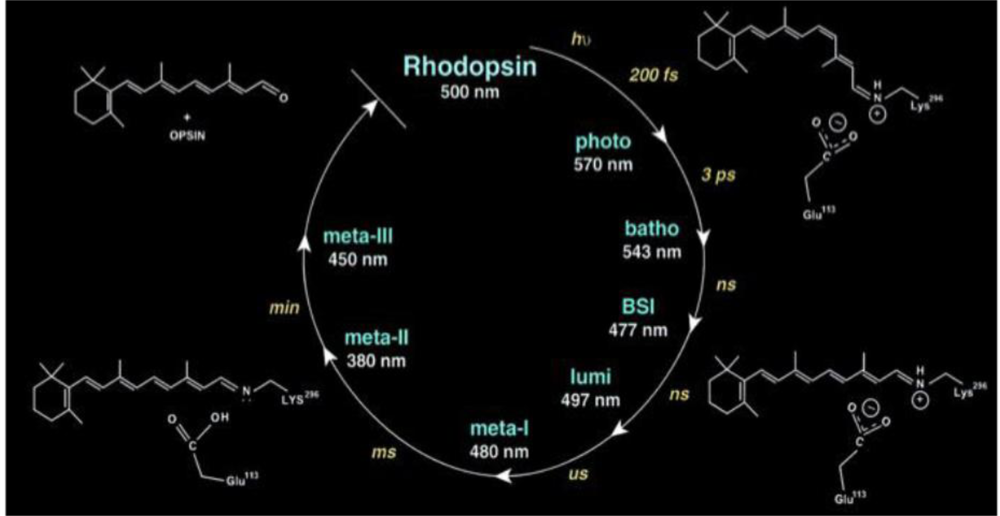
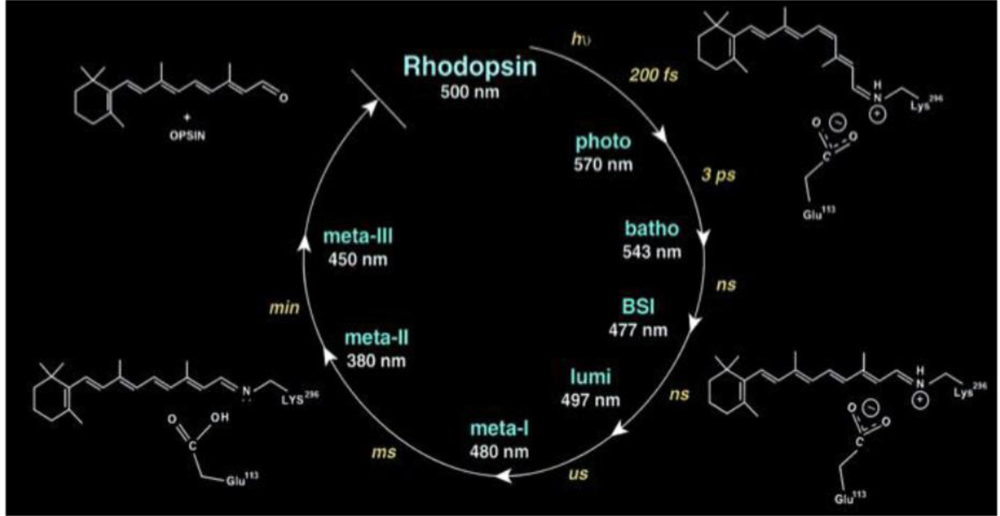
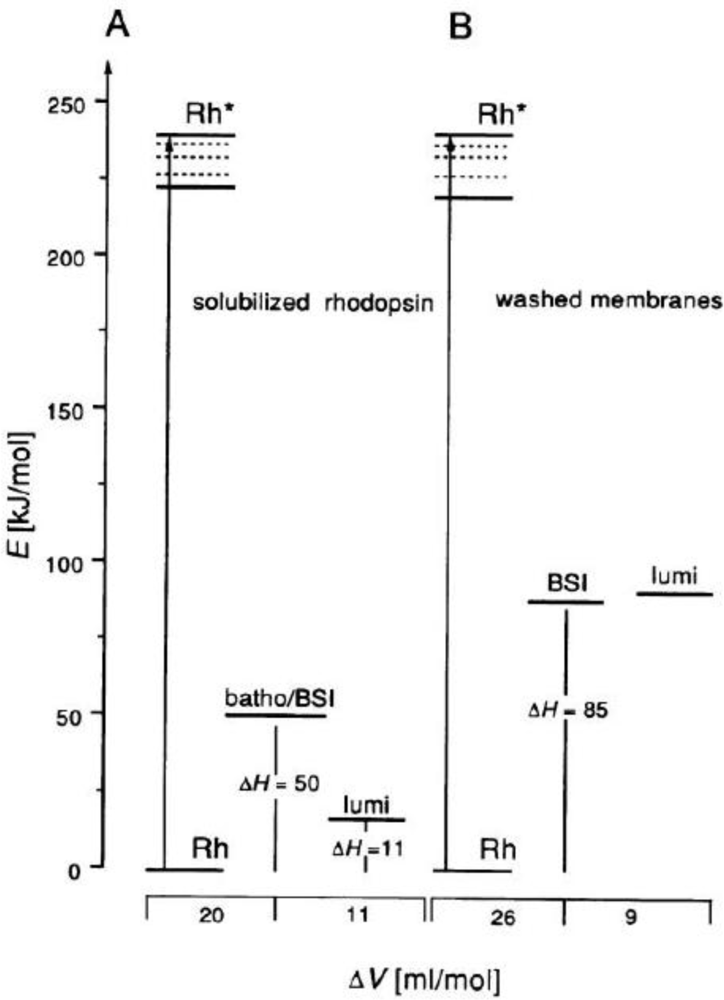
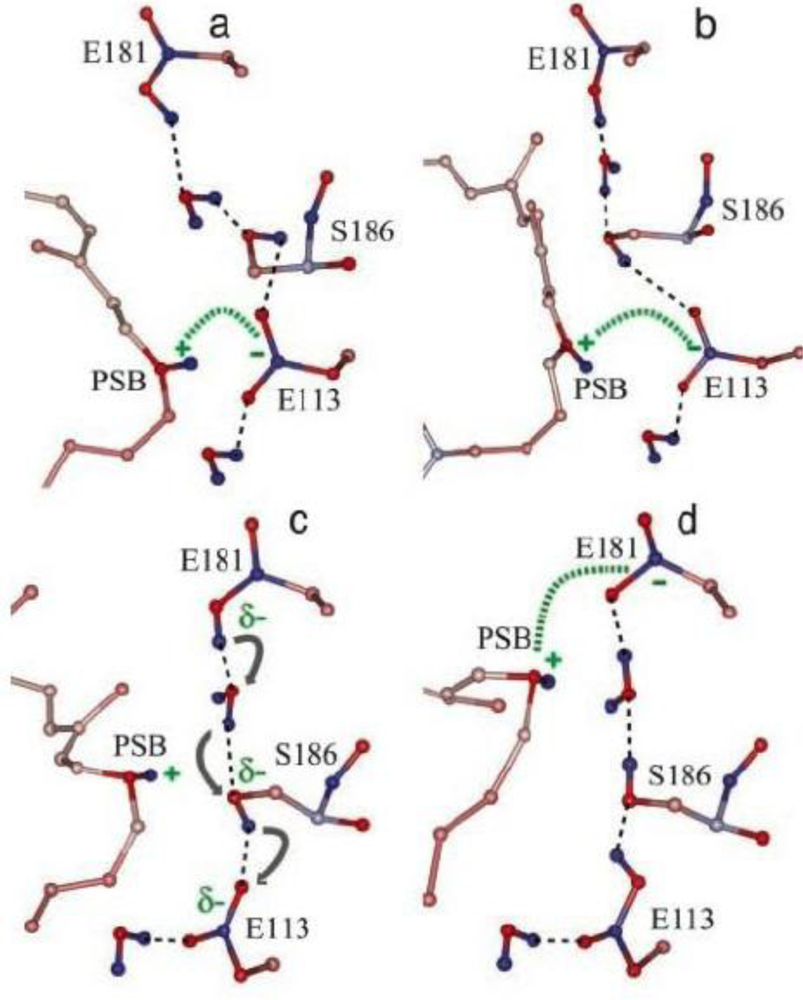
4.2. Photoacoustic Studies of Rhodopsins
5. Conclusions
5.1. Potential Problems and Future Efforts
5.2. Concluding Remarks
Abbreviations
| α’ | thermal expansivity/heat capacity × density |
| ΔG | free energy change |
| ΔH | enthalpy change |
| ΔS | entropy change |
| ΔVRC | volume change of the reaction |
| ΔVY, ΔVS | volume changes per reaction center |
| Φ | quantum yield |
| κ | compressibility |
| A | absorbancy of solution |
| A1 | quinone electron acceptor of photosystem I |
| Amp | amplifier |
| AP | plastoquinone acceptor in menA and menB null photosystem I |
| Batho | bathorhodopsin |
| BS | beam splitter |
| BSI | blue-shift intermediate |
| E181 | counter-ion switch in Meta I state in rhodopsin |
| Ea | photon energy |
| F | piezo film sensitivity |
| FA, FB, FX | iron sulfur clusters of photosystem I |
| GPCR | G protein-coupled receptor |
| I(t) | impulse response of the system |
| L | lens |
| Lumi | lumirhodopsin |
| menA | gene of phytyl transferase in the phylloquinone biosynthetic pathway |
| menB | gene of napthoate synthase in the phylloquinone biosynthetic pathway |
| Meta I | metarhodopsin I state |
| Meta II | metarhodopsin II state |
| N | number of reaction centers |
| OPO | optical parametric oscillator |
| P700 | primary electron donor of photosystem I |
| P680 | primary electron donor of photosystem II |
| PA | photoacoustic |
| Photo | photorhodopsin |
| PS | photosystem |
| QA, QB | quinonoe electron acceptors of photosystem II |
| QRC | heat output |
| TM | transmembrane |
| YZ | tyrosine electron donor of photosystem II |
Acknowledgments
References
- Chen, H.X.; Dibold, G. Production of the photoacoustic effect and transient gratings by molecular volume changes. J. Chem. Phys 1996, 104, 6730–6741. [Google Scholar]
- Feitelson, J.; Mauzerall, D. Enthalpy and electrostriction in the electron-transfer reaction between triplet zinc uroporphyrin and ferricyanide. J. Phys. Chem. B 2002, 106, 9674–9678. [Google Scholar]
- Braslavsky, S.E.; Heibel, G.E. Time-resolved photothermal and photoacoustic methods applied to photoinduced processes in solution. Chem. Rev 1992, 92, 1381–1410. [Google Scholar]
- Borsarelli, C.D.; Braslavsky, S.E. Enthalpy, volume, and entropy changes associated with the electron transfer reaction between the 3MLCT state of Ru(bpy)32+ and methyl viologen cation in aqueous solutions. J. Phys. Chem. A 1999, 103, 1719–1727. [Google Scholar]
- Crovetto, L.; Martinez-Junza, V.; Braslavsky, S.E. Entropy changes drive the electron transfer reaction of triplet flavin mononucleotide from aromatic amino acids in cation-organized aqueous media. A laser-induced optoacoustic study. Photochem. Photobiol 2006, 82, 281–290. [Google Scholar]
- Davies, K.W.; Maivald, D.; Grabowski, J.J. A photoacoustic calorimetric characterization of the reaction enthalpy and volume for the preparation of a reactive intermediate from CpMn(CO)3. J. Photochem. Photobiol. A 2008, 197, 335–341. [Google Scholar]
- Andres, G.O.; Martinez-Junza, V.; Crovetto, L.; Braslavsky, S.E. Photoinduced electron transfer from tetrasulfonated porphyrin to benzoquinone revisited. The structural volume-normalized entropy change correlates with Marcus reorganization energy. J. Phys. Chem. A 2006, 110, 10185–10190. [Google Scholar]
- Hou, H.J.M.; Shen, G.; Boichenko, V.A.; Golbeck, J.H.; Mauzerall, D. Thermodynamics of charge separation of photosystem I in the menA and menB null mutants of Synechocystis sp. PCC 6803 determined by pulsed photoacoustics. Biochemistry 2009, 48, 1829–1837. [Google Scholar]
- Yruela, I.; Churio, M.S.; Gensch, T.; Braslavsky, S.E.; Holzwarth, A.R. Optoacoustic and singlet oxygen near-IR emission study of the isolated D1-D2-cyt b-559 reaction center complex of photosystem II. Protein movement associated with charge separation. J. Phys. Chem 1994, 98, 12789–12795. [Google Scholar]
- Losi, A.; Yruela, I.; Reus, M.; Holzwarth, A.R.; Braslavsky, S.E. Structural changes upon excitation of D1-D2-Cyt b559 photosystem II reaction centers depend on the β-carotene content. Photochem. Photobiol. Sci 2003, 2, 722–729. [Google Scholar]
- Rizzi, A.C.; van Gastel, M.; Liddell, P.A.; Palacios, R.E.; Moore, G.F.; Kodis, G.; Moore, A.L.; Moore, T.A.; Gust, D.; Braslavsky, S.E. Entropic changes control the charge separation process in triads mimicking photosynthetic charge separation. J. Phys. Chem. A 2008, 112, 4215–4223. [Google Scholar]
- Malkin, S. The photoacoustic effect in leaves and its applications. In Probing Photosynthesis; Yunus, M., Pathre, U., Mohanty, P., Eds.; Taylor & Francis: Hampshire, UK, 2000; pp. 484–524. [Google Scholar]
- Delosme, R. On some aspects of photosynthesis revealed by photoacoustic studies: A critical evaluation. Photosynth. Res 2003, 76, 289–301. [Google Scholar]
- Mesquita, R.C.; Mansanares, A.M.; da Silva, E.C.; Barja, P.R.; Miranda, L.C.M.; Vargas, H. Open photoacoustic cell: Applications in plant photosynthesis studies. Instrum. Sci. Technol 2006, 34, 33–58. [Google Scholar]
- Herbert, S.K.; Han, T.; Vogelmann, T.C. New applications of photoacoustics to the study of photosynthesis. Photosynth. Res 2001, 66, 13–31. [Google Scholar]
- Mauzerall, D. Thermodynamics in photosystem I. In Photosystem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase; Golbeck, J.H., Ed.; Springer: Dordrecht, The Netherlands, 2006; Volume 24, pp. 571–581. [Google Scholar]
- Losi, A.; Wegener, A.A.; Engelhard, M.; Braslavsky, S.E. Enthalpy-entropy compensation in a photocycle: The K-to-L transition in sensory rhodopsin II from Natronobacterium pharaonis. J. Am. Chem. Soc 2001, 123, 1766–1767. [Google Scholar]
- Losi, A.; Wegener, A.A.; Engelhard, M.; Braslavsky, S.E. Thermodynamics of the early steps in the photocycle of Natronobacterium pharaonis halorhodopsin. Influence of medium and of anion substitution. Photochem. Photobiol 2001, 74, 495–503. [Google Scholar]
- Ort, D.R.; Parson, W.W. Enthalpy changes during the photochemical cycle of bacteriorhodopsin. Biophys. J 1979, 25, 355–364. [Google Scholar]
- Ort, D.R.; Parson, W.W. Flash-induced volume changes of bacteriorhodopsin-containing membrane fragments and their relationship to proton movements and absorbance transients. J. Biol. Chem 1978, 253, 6158–6164. [Google Scholar]
- Liu, Y.; Edens, G.J.; Grzymski, J.; Mauzerall, D. Volume and enthalpy changes of proton transfers in the bacteriorhodopsin photocycle studied by millisecond time-resolved photopressure measurements. Biochemistry 2008, 47, 7752–7761. [Google Scholar]
- Schulenber, P.J.; Rohr, M.; Gaertner, W.; Braslavsky, S.E. Photoinduced volume changes associated with the early transformations of bacteriorhodopsin: a laser-induced optoacoustic spectroscopy study. Biophys. J 1994, 66, 838–843. [Google Scholar]
- Schulenberg, P.J.; Gaertner, W.; Braslavsky, S.E. Time-resolved volume changes during the bacteriorhodopsin photocycle: A photothermal beam deflection study. J. Phys. Chem 1995, 99, 9617–9624. [Google Scholar]
- Losi, A.; Michler, I.; Gartner, W.; Braslavsky, S.E. Time-resolved thermodynamic changesphotoinduced in 5,12-trans-locked bacteriorhodopsin. Evidence that retinal isomerization is required for protein activation. Photochem. Photobiol 2000, 72, 590–597. [Google Scholar]
- Strassburger, J.M.; Gartner, W.; Braslavsky, S.E. Volume and enthalpy changes after photoexcitation of bovine rhodopsin: laser-induced optoacoustic studies. Biophys. J 1997, 72, 2294–2303. [Google Scholar]
- Gensch, T.; Strassburger, J.M.; Gartner, W.; Braslavsky, S.E. Volume and enthalpy changes upon photoexcitation of bovine rhodopsin derived from optoacoustic studies by using an equilibrium between bathorhodopsin and blue-shifted intermediate. Israel J. Chem 1998, 38, 231–236. [Google Scholar]
- Kim, J.-W.; Ha, J.-A.; Jung, H.; Ahn, B.-I.; Lee, S.-H.; Choi, J.-G. Kinetic analysis of supported Ni-catalyzed CO2/CH4 reactions using photoacoustic spectroscopy. Phys. Chem. Chem. Phys 2007, 9, 5828–5833. [Google Scholar]
- Murakami, N.; Abe, R.; Ohtani, B. In situ observation of photocatalytic reaction by photoacoustic spectroscopy: Detection of heat of exothermic photocatalytic reaction. Chem. Phys. Lett 2008, 451, 316–320. [Google Scholar]
- Serpa, C.; Schabauer, J.; Piedade, A.P.; Monteiro, C.J.P.; Pereira, M.M.; Douglas, P.; Burrows, H.D.; Arnaut, L.G. Photoacoustic measurement of electron injection efficiencies and energies from excited sensitizer dyes into nanocrystalline TiO2 films. J. Am. Chem. Soc 2008, 130, 8876–8877. [Google Scholar]
- Zhang, H.F.; Maslov, K.; Stoica, G.; Wang, L.V. Functional photoacoustic microscopy for high-resolution and noninvasive in vivo imaging. Nature Biotechnology 2006, 24, 848–851. [Google Scholar]
- Xu, M.; Wang, L.V. Photoacoustic imaging in biomedicine. Rev. Sci. Instrum 2006, 77, 041101–041122. [Google Scholar]
- Xu, L.; Lowney, D.; McNally, P.J.; Gomez, F.E.; Stopford, J. Non-destructive measurement of deep embedded defects in silicon using photoacoustic microscope (PAM). Mater. Res. Soc. Sym. Proc 2006, 914, 451–455. [Google Scholar]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar]
- Renger, G.; Christen, G.; Karge, M.; Eckert, H.J.; Irrgang, K.D. Application of the Marcus theory for analysis of the temperature dependence of the reactions leading to photosynthetic water oxidation: results and implications. J. Biol. Inorg. Chem 1998, 3, 360–366. [Google Scholar]
- Turro, C.; Zaleski, J.M.; Karabatsos, Y.M.; Nocera, D.G. Bimolecular electron transfer in the Marcus inverted region. J. Am. Chem. Soc 1996, 118, 6060–6067. [Google Scholar]
- Mayer, J.M.; Rhile, I.J.; Larsen, F.B.; Mader, E.A.; Markle, T.F.; Dipasquale, A.G. Models for proton-coupled electron transfer in photosystem II. Photosynth, Res 2006, 87, 3–20. [Google Scholar]
- LeBard, D.N.; Kapko, V.; Matyushov, D.V. Energetics and kinetics of primary charge separation in bacterial photosynthesis. J. Phys. Chem. B 2008, 112, 10322–10342. [Google Scholar]
- Edens, G.J.; Gunner, M.R.; Xu, Q.; Mauzerall, D. The enthalpy and entropy of reaction for formation of P+QA− from excited reaction centers of Rhodobacter sphaeroides. J. Am. Chem. Soc 2000, 122, 1479–1485. [Google Scholar]
- Feitelson, J.; Mauzerall, D. Photoacoustic evaluation of volume and entropy changes in energy and electron transfer. Triplet state porphyrin with oxygen and naphthoquinone-2-sulfonate. J. Phys. Chem 1996, 100, 7698–7703. [Google Scholar]
- Hou, H.J.M.; Mauzerall, D. The A−FX to FA/B step in Synechocystis 6803 photosystem I is entropy driven. J. Am. Chem. Soc 2006, 128, 1580–1586. [Google Scholar]
- Xu, Q.; Gunner, M.R. Temperature dependence of the free energy, enthalpy, and entropy of P+QA− charge recombination in Rhodobacter sphaeroides R-26 reaction centers. J. Phys. Chem. B 2000, 104, 8035–8043. [Google Scholar]
- Levskaya, A; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signaling using a light-switchable protein interaction. Nature 2009, 461, 997–1001. [Google Scholar]
- Arnaut, L.G.; Caldwell, R.A.; Elbert, J.E.; Melton, L.A. Recent advances in photoacoustic calorimetry: theoretical basis and improvements in experimental design. Rev. Sci. Instrum 1992, 63, 5381–5389. [Google Scholar]
- Rudzki, J.E.; Goodman, J.L.; Peters, K.S. Simultaneous determination of photoreaction dynamics and energetics using pulsed, time-resolved photoacoustic calorimetry. J. Am. Chem. Soc 1985, 107, 7849–7854. [Google Scholar]
- Amunts, A.; Drory, O.; Nelson, N. The structure of a plant photosystem I supercomplex at 3.4 A. resolution. Nature 2007, 447, 58–63. [Google Scholar]
- Jordan, P.; Fromme, P.; Witt, H.T.; Klukas, O.; Saenger, W.; Krauss, N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 A resolution. Nature 2001, 411, 909–917. [Google Scholar]
- Loll, B.; Kern, J.; Saenger, W.; Zouni, A.; Biesiadka, J. Towards complete cofactor arrangement in the 3.0 A resolution structure of photosystem II. Nature 2005, 438, 1040–1044. [Google Scholar]
- Ferreira, K.N.; Iverson, T.M.; Maghlaoui, K.; Barber, J.; Iwata, S. Architecture of the photosynthetic oxygen-evolving center. Science 2004, 303, 1831–1838. [Google Scholar]
- Brettel, K. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta 1997, 1318, 322–373. [Google Scholar]
- Dekker, J.P.; Van Grondelle, R. Primary charge separation in photosystem II. Photosynth. Res 2000, 63, 195–208. [Google Scholar]
- Brettel, K.; Leibl, W. Electron transfer in photosystem I. Biochim. Biophys. Acta 2001, 1507, 100–114. [Google Scholar]
- Gobets, B.; van Grondelle, R. Energy transfer and trapping in photosystem I. Biochim. Biophys. Acta 2001, 1507, 80–99. [Google Scholar]
- Canaani, O.; Barber, J.; Malkin, S. Evidence that phosphorylation and dephosphorylation regulate the distribution of excitation energy between the two photosystems of photosynthesis in vivo: Photoacoustic and fluorimetric study of an intact leaf. Proc. Natl. Acad. Sci. USA 1984, 81, 1614–1618. [Google Scholar]
- Mauzerall, D.C. Determination of oxygen emission and uptake in leaves by pulsed, time resolved photoacoustics. Plant Physiol 1990, 94, 278–283. [Google Scholar]
- Nitsch, C.; Braslavsky, S.E.; Schatz, G.H. Laser-induced optoacoustic calorimetry of primary processes in isolated photosystem I and photosystem II particles. Biochim. Biophys. Acta 1988, 934, 201–212. [Google Scholar]
- Mauzerall, D.; Feitelson, J.; Prince, R. Wide band time-resolved photoacoustic study of electron transfer reactions: Difference between measured enthalpies and redox free energies. J. Phys. Chem 1995, 99, 1090–1093. [Google Scholar]
- Arata, H.; Parson, W.W. Enthalpy and volume changes accompanying electron transfer from P-870 to quinones in Rhodopseudomonas sphaeroides reaction centers. Biochim. Biophys. Acta 1981, 636, 70–81. [Google Scholar]
- Delosme, R.; Beal, D.; Joliot, P. Photoacoustic detection of flash-induced charge separation in photosynthetic systems. Spectral dependence of the quantum yield. Biochim. Biophys. Acta 1994, 1185, 56–64. [Google Scholar]
- Gunner, M.R.; Dutton, P.L. Temperature and −ΔG° dependence of the electron transfer from BPh.− to QA in reaction center protein from Rhodobacter sphaeroides with different quinones as QA. J. Am. Chem. Soc 1989, 111, 3400–3412. [Google Scholar]
- Hou, J.M.; Boichenko, V.A.; Wang, Y.C.; Chitnis, P.R.; Mauzerall, D. Thermodynamics of electron transfer in oxygenic photosynthetic reaction centers: A pulsed photoacoustic study of electron transfer in photosystem I reveals a similarity to bacterial reaction centers in both volume change and entropy. Biochemistry 2001, 40, 7109–7116. [Google Scholar]
- Hou, J.-M.; Boichenko, V.A.; Diner, B.A.; Mauzerall, D. Thermodynamics of electron transfer in oxygenic photosynthetic reaction centers: Volume change, enthalpy, and entropy of electron transfer reactions in manganese-depleted photosystem II core complexes. Biochemistry 2001, 40, 7117–7125. [Google Scholar]
- Boichenko, V.A.; Hou, J.-M.; Mauzerall, D. Thermodynamics of electron transfer in oxygenic photosynthetic reaction centers: Volume change, enthalpy, and entropy of electron-transfer reactions in the intact cells of the cyanobacterium Synechocystis PCC 6803. Biochemistry 2001, 40, 7126–7132. [Google Scholar]
- Xu, Q.; Gunner, M.R. Trapping conformational intermediate states in the reaction center protein from photosynthetic bacteria. Biochemistry 2001, 40, 3232–3241. [Google Scholar]
- Johnson, T.W.; Shen, G.; Zybailov, B.; Kolling, D.; Reategui, R.; Beauparlant, S.; Vassiliev, I.R.; Bryant, D.A.; Jones, A.D.; Golbeck, J.H. Recruitment of a foreign quinone into the A1 site of photosystem I. I. Genetic and physiological characterization of phylloquinone biosynthetic pathway mutants in Synechocystis sp. pcc 6803. J. Biol. Chem 2000, 275, 8523–8530. [Google Scholar]
- Zybailov, B.; van der Est, A.; Zech, S.G.; Teutloff, C.; Johnson, T.W.; Shen, G.; Bittl, R.; Stehlik, D.; Chitnis, P.R.; Golbeck, J.H. Recruitment of a foreign quinone into the A1 site of photosystem I. II. Structural and functional characterization of phylloquinone biosynthetic pathway mutants by electron paramagnetic resonance and electron-nuclear double resonance spectroscopy. J. Biol. Chem 2000, 275, 8531–8539. [Google Scholar]
- Semenov, A.Y.; Vassiliev, I.R.; van Der Est, A.; Mamedov, M.D.; Zybailov, B.; Shen, G.; Stehlik, D.; Diner, B.A.; Chitnis, P.R.; Golbeck, J.H. Recruitment of a foreign quinone into the A1 site of photosystem I. Altered kinetics of electron transfer in phylloquinone biosynthetic pathway mutants studied by time-resolved optical, EPR, and electrometric techniques. J. Biol. Chem 2001, 275, 23429–23438. [Google Scholar]
- Santabarbara, S.; Heathcote, P.; Evans, M.C. Modelling of the electron transfer reactions in Photosystem I by electron tunnelling theory: the phylloquinones bound to the PsaA and the PsaB reaction centre subunits of PS I are almost isoenergetic to the iron-sulfur cluster FX. Biochim. Biophys. Acta 2005, 1708, 283–310. [Google Scholar]
- Zhang, D.; Mauzerall, D. Volume and enthalpy changes in the early steps of bacteriorhodopsin photocycle studied by time-resolved photoacoustics. Biophys. J 1996, 71, 381–388. [Google Scholar]
- Sakmar, T.P. Structure and function of G-protein-coupled receptors: lessons from the crystal structure of rhodopsin. In Handbook of Cell Signaling; Bradshaw, R., Dennis, E., Eds.; Academic Press: Oxford, UK, 2004; Volume 1, pp. 139–143. [Google Scholar]
- Seibert, C.; Sakmar, T.P. Small-molecule antagonists of CCR5 and CXCR4: A promising new class of anti-HIV-1 drugs. Curr. Pharm. Des 2004, 10, 2041–2062. [Google Scholar]
- Menon, S.T.; Han, M.; Sakmar, T.P. Rhodopsin: structural basis of molecular physiology. Physiol. Rev 2001, 81, 1659–1688. [Google Scholar]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le, T.I.; Teller, D.C.; Okada, T.; Stenkamp, R.E. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar]
- Shimamura, T.; Hiraki, K.; Takahashi, N.; Hori, T.; Ago, H.; Masuda, K.; Takio, K.; Ishiguro, M.; Miyano, M. Crystal structure of squid rhodopsin with intracellularly extended cytoplasmic region. J. Biol. Chem 2008, 283, 17753–17756. [Google Scholar]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R.; Leslie, A.G.W.; Tate, C.G.; Schertler, G.F.X. Structure of a beta 1-adrenergic G-protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar]
- Rasmussen, S.G.F.; Choi, H.-J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F. Crystal structure of the human beta 2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar]
- Nathans, J. Determinants of visual pigment absorbance: identification of the retinylidene Schiff’s base counterion in bovine rhodopsin. Biochemistry 1990, 29, 9746–9752. [Google Scholar]
- Sakmar, T.P.; Franke, R.R.; Khorana, H.G. Glutamic acid-113 serves as the retinylidene Schiff base counterion in bovine rhodopsin. Proc. Natl. Acad. Sci. USA 1989, 86, 8309–8313. [Google Scholar]
- Zhukovsky, E.A.; Oprian, D.D. Effect of carboxylic acid side chains on the absorption maximum of visual pigments. Science 1989, 246, 928–930. [Google Scholar]
- Boucher, F.; Leblanc, R.M. Photoacoustic spectroscopy of cattle visual pigment at low temperature. Biochem. Biophys. Re.s Commun 1981, 100, 385–390. [Google Scholar]
- Peters, K.S.; Watson, T.; Marr, K. Time-resolved photoacoustic calorimetry: A study of myoglobin and rhodopsin. Annu. Rev. Biophys. Biophys. Chem 1991, 20, 343–362. [Google Scholar]
- Logunov, S.L.; El-Sayed, M.A. Redetermination of the quantum yield of photoisomerization and energy content in the K-intermediate of bacteriorhodopsin photocycle and its mutants by the photoacoustic technique. J. Phys. Chem. B 1997, 101, 6629–6633. [Google Scholar]
- Losi, A.; Braslavsky, S.E.; Gartner, W.; Spudich, J.L. Time-resolved absorption and photothermal measurements with sensory rhodopsin I from Halobacterium salinarum. Biophys. J 1999, 76, 2183–2191. [Google Scholar]
- Darnall, H.J.A. Photochemistry of Vision Photochemistry; Springer-Verlag: New York, NY, USA, 1972. [Google Scholar]
- Birge, R.R. Nature of the primary photochemical events in rhodopsin and bacterirhodopsin. Biochim. Biophys. Acta 1990, 1016, 293–327. [Google Scholar]
- Marr, K.; Peters, K.S. Photoacoustic calorimetry study of the conversion of rhodopsin and isorhodopsin to lumirhodopsin. Biochemistry 1991, 30, 1254–1258. [Google Scholar]
- Luedeke, S.; Beck, M.; Yan, E.C.Y.; Sakmar, T.P.; Siebert, F.; Vogel, R. The role of Glu181 in the photoactivation of Rhodopsin. J. Mol. Biol 2005, 353, 345–356. [Google Scholar]
- Yan, E.C.Y.; Ganim, Z.; Kazmi, M.A.; Chang, B.S.W.; Sakmar, T.P.; Mathies, R.A. Resonance raman analysis of the mechanism of energy storage and chromophore sistortion in the primary visual photoproduct. Biochemistry 2004, 43, 10867–10876. [Google Scholar]
- Yan, E.C.Y.; Kazmi, M.A.; Ganim, Z.; Hou, J.-M.; Pan, D.; Chang, B.S.W.; Sakmar, T.P.; Mathies, R.A. Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. USA 2003, 100, 9262–9267. [Google Scholar]
- Birge, R.R.; Knox, B.E. Perspectives on the counterion switch-induced photoactivation of the G protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. USA 2003, 100, 9105–9107. [Google Scholar]
- Yan, E.C.Y.; Epps, J.; Lewis, J.W.; Szundi, I.; Bhagat, A.; Sakmar, T.P.; Kliger, D.S. Photointermediates of the rhodopsin S186A mutant as a probe of the hydrogen-bond network in the chromophore pocket and the mechanism of counterion switch. J. Phys. Chem. C 2007, 111, 8843–8848. [Google Scholar]
- Lewis, J.W.; Szundi, I.; Kazmi, M.A.; Sakmar, T.P.; Kliger, D.S. Proton movement and photointermediate kinetics in rhodopsin mutants. Biochemistry 2006, 45, 5430–5439. [Google Scholar]
- De Vitry, C.; Diner, B.A.; Popot, J.L. Photosystem II particles from Chlamydomonas reinhardtii. Purification, molecular weight, small subunit composition, and protein phosphorylation. J. Biol. Chem 1991, 266, 16614–16621. [Google Scholar]
- Hou., J.-M.; Dejonghe, D.; Shan, J.-X.; Li, L.-B.; Kuang, T.-Y. Orientation of pigments in the isolated photosystem II sub-core reaction center CP47/D1/D2/Cyt b-559 complexes: A linear dichroism study. J. Integr. Plant Biol 2000, 42, 1211–1214. [Google Scholar]
- Krivanek, R.; Dau, H.; Haumann, M. Enthalpy changes during photosynthetic water oxidation tracked by time-resolved calorimetry using a photothermal beam deflection technique. Biophys. J 2008, 94, 1890–1903. [Google Scholar]


© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hou, H.J.M.; Sakmar, T.P. Methodology of Pulsed Photoacoustics and Its Application to Probe Photosystems and Receptors. Sensors 2010, 10, 5642-5667. https://doi.org/10.3390/s100605642
Hou HJM, Sakmar TP. Methodology of Pulsed Photoacoustics and Its Application to Probe Photosystems and Receptors. Sensors. 2010; 10(6):5642-5667. https://doi.org/10.3390/s100605642
Chicago/Turabian StyleHou, Harvey J.M., and Thomas P. Sakmar. 2010. "Methodology of Pulsed Photoacoustics and Its Application to Probe Photosystems and Receptors" Sensors 10, no. 6: 5642-5667. https://doi.org/10.3390/s100605642