Biosensing for the Environment and Defence: Aqueous Uranyl Detection Using Bacterial Surface Layer Proteins

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Toxicology of Uranium
1.2. Environmental Effects of Uranium
1.3. Current Sensing Technologies
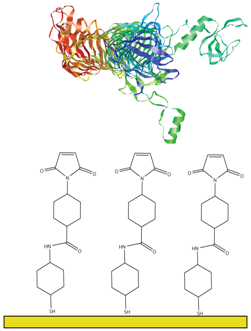
1.4. Bacillus Sphaericus S-layer Proteins
1.5. Electrochemical Biosensors
1.5.1. Electrochemical Impedance Spectroscopy
2. Results and Discussion
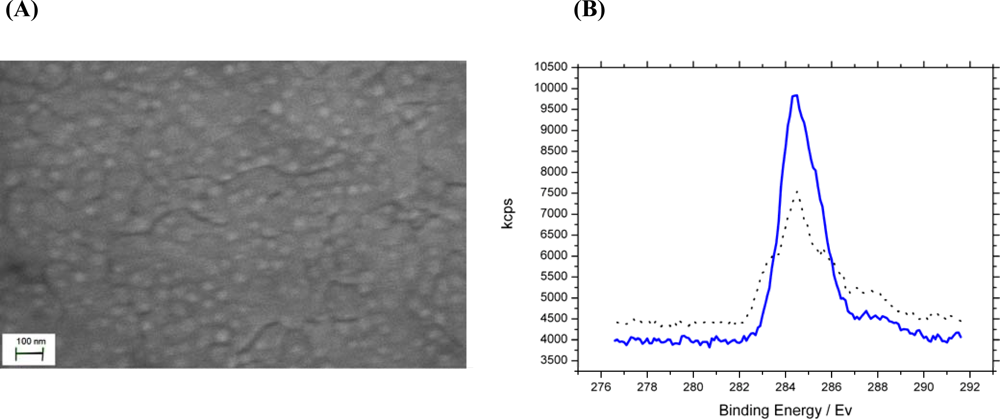
2.1. Surface Preparation
2.2. Analysis of Sensor Fabrication
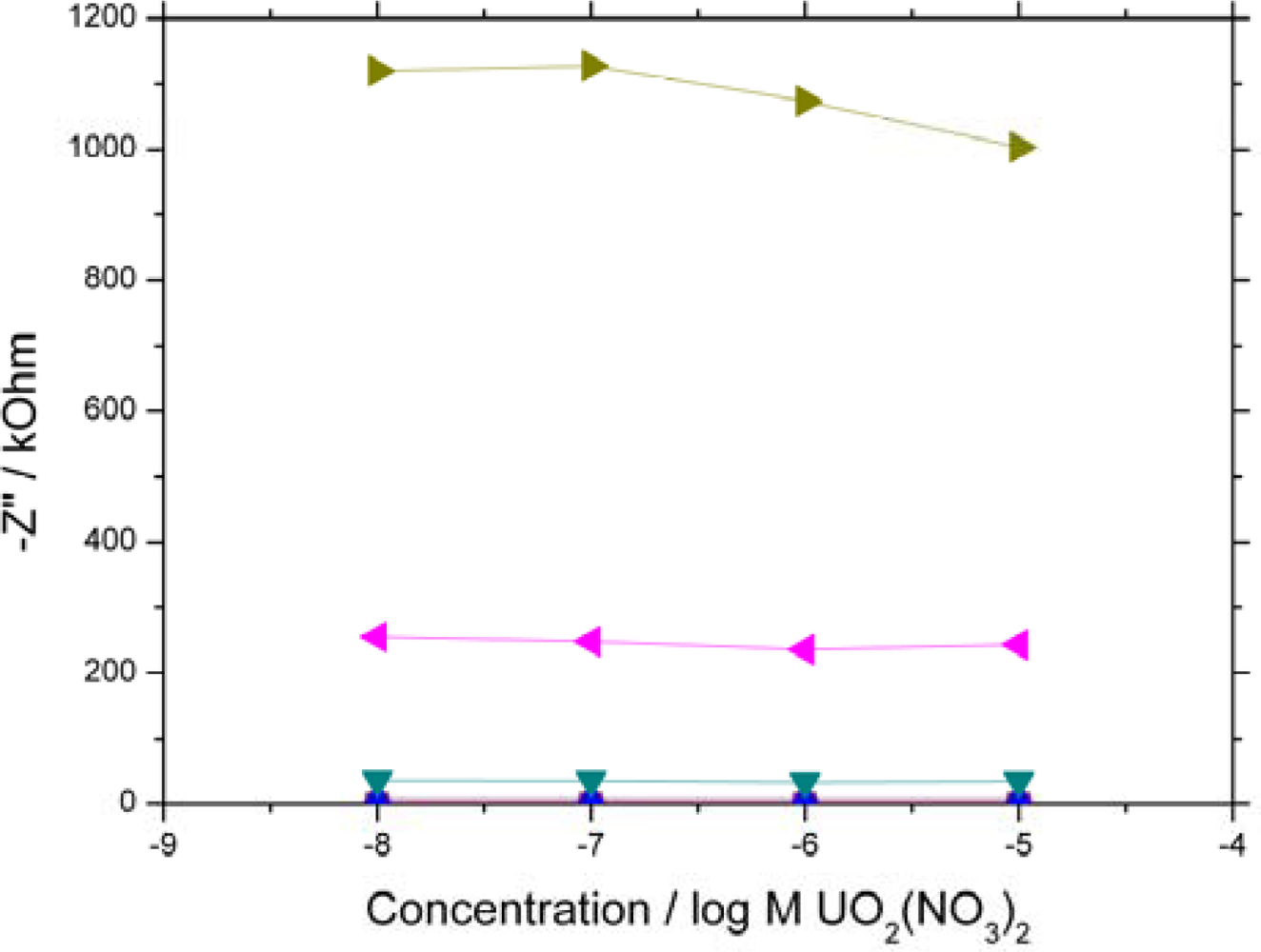
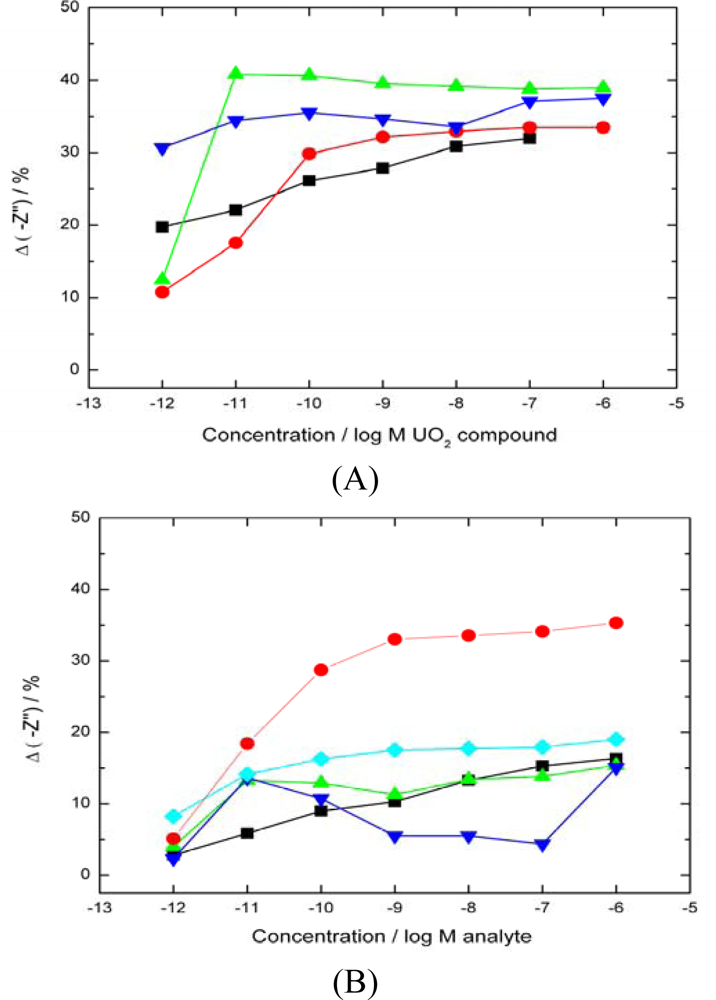
2.3. Binding of UO22+ to the SLP Biosensor
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Electrochemical Setup
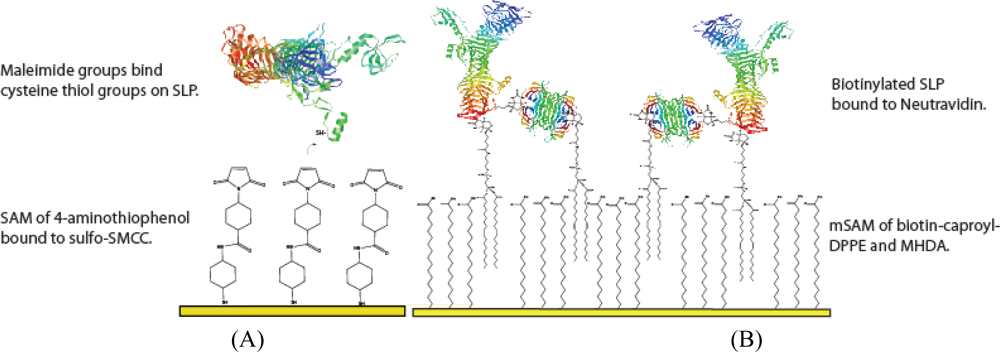
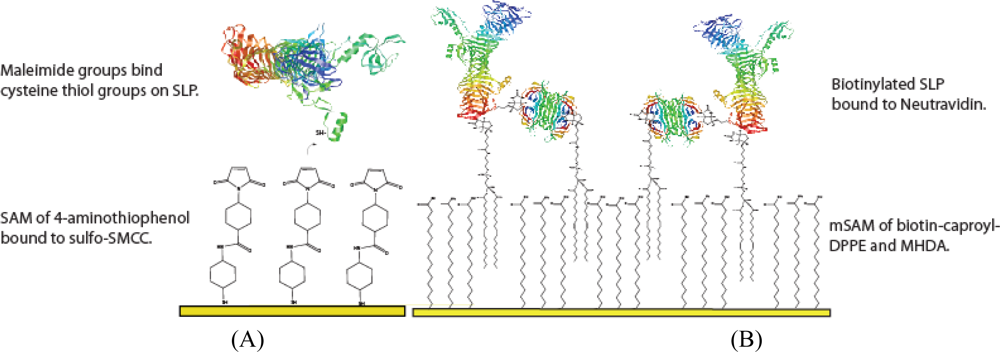
3.3. SLP Tethering Mechanisms
3.3.1. Biotin-Neutravidin mSAM preparation
3.3.1.1. mSAM stability measurements at varying MHDA : biotin-caproyl-DPPE
3.3.2. Bioconjugation layer preparation
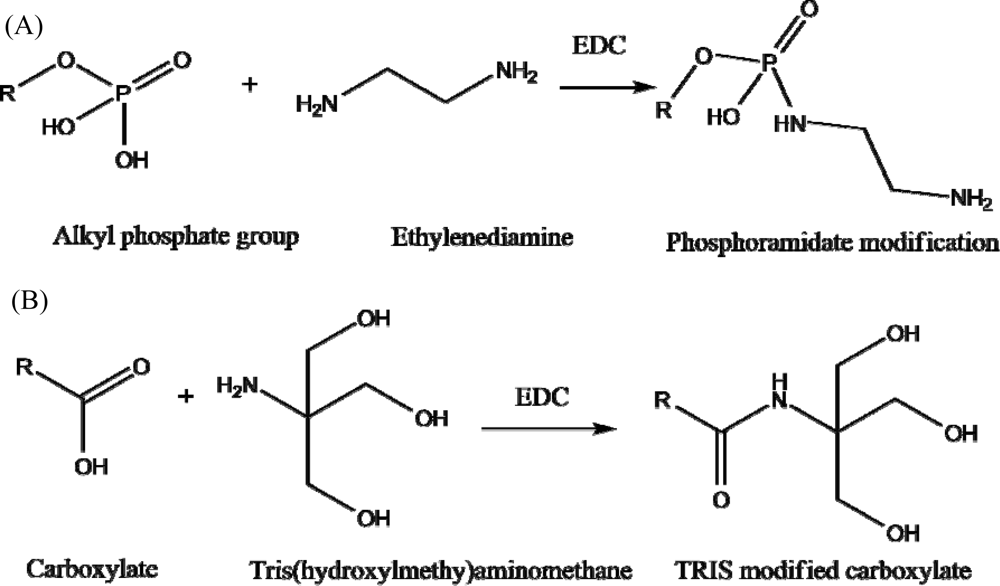
3.4. Blocking S-the SLP Chelating Sites
3.5. Surface and Construction Analysis
4. Conclusions
Acknowledgments
References and Notes
- Klaassen, C.D. Casarett and Doull’s Toxicology, 7th ed; McGraw-Hill: New York, NY, USA, 2008; p. 1275. [Google Scholar]
- Dart, R.C. Medical Toxicology, 3rd ed; Williams&Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Keith-Roach, M.J. The speciation, stability, solubility and biodegradation of organic co-contaminant radionuclide complexes: A review. Sci. Total Environ 2008, 396, 1–11. [Google Scholar]
- Shervedani, R.K.; Farahbakhsh, A.; Bagherzadeh, M. Functionalization of gold cysteamine self-assembled monolayer with ethylenediaminetetraacetic acid as a novel nanosensor. Anal. Chim. Acta 2007, 587, 254–262. [Google Scholar]
- Cherian, S.; Gupta, R.K.; Mullin, B.C.; Thundat, T. Detection of heavy metal ions using protein-functionalized microcantilever sensors. Biosens. Bioelectron 2003, 19, 411–416. [Google Scholar]
- Li, S.; Rosen, B.P.; Borges-Walmsley, M.I.; Walmsley, A.R. Evidence for cooperativity between the four binding sites of dimeric ArsD, an As(III)-responsive transcriptional regulator. J. Biol. Chem 2002, 277, 25992–26002. [Google Scholar]
- Bontidean, I.; Berggren, C.; Johansson, G.; Csoregi, E.; Mattiasson, B.; Lloyd, J.R.; Jakeman, K.J.; Brown, N.L. Detection of heavy metal ions at femtomolar levels using protein-based biosensors. Anal. Chem 1998, 70, 4162–4169. [Google Scholar]
- Lucarelli, F.; Marrazza, G.; Mascini, M. Enzyme-based impedimetric detection of PCR products using oligonucleotide-modified screen-printed gold electrodes. Biosens. Bioelectron 2005, 20, 2001–2009. [Google Scholar]
- Yang, Y.; Khoo, S.B. Fabrication of self-assembled monolayer of 8-mercaptoquinoline on polycrystalline gold electrode and its selective catalysis for the reduction of metal ions and the oxidation of biomolecules. Sens. Actuat. B: Chem 2004, 97, 221–230. [Google Scholar]
- Nakajima, A.; Sakaguchi, T. Selective accumulation of heavy metals by microorganisms. Appl. Microbiol. Biotechnol 1986, 24, 59–64. [Google Scholar]
- Bienert, G.P.; Schuessler, M.D.; Jahn, T.P. Metalloids: essential, beneficial or toxic? Major intrinsic proteins sort it out. Trends Biochem. Sci 2008, 33, 20–26. [Google Scholar]
- Pollmann, K.; Raff, J.; Merroun, M.; Fahmy, K.; Selenska-Pobell, S. Metal binding by bacteria from uranium mining waste piles and its technological applications. Biotechnol. Adv 2006, 24, 58–68. [Google Scholar]
- Pollmann, K.; Raff, J.; Schnorpfeil, M.; Radeva, G.; Selenska-Pobell, S. Novel surface layer protein genes in Bacillus sphaericus associated with unusual insertion elements. Microbiology 2005, 151, 2961–2973. [Google Scholar]
- Merroun, M.L.; Raff, J.; Rossberg, A.; Hennig, C.; Reich, T.; Selenska-Pobell, S. Complexation of Uranium by Cells and S-Layer Sheets of Bacillus sphaericus JG-A12. Appl. Environ. Microbiol 2005, 71, 5532–5543. [Google Scholar]
- Sonja, S.-P.; Petra, P.; Vanya, M.; Ivo, B.; Gert, B.; Heino, N. Selective accumulation of heavy metals by three indigenous Bacillus strains, B. cereus, B. megaterium and B. sphaericus, from drain waters of a uranium waste pile. FEMS Microbiol. Ecol 1999, 29, 59–67. [Google Scholar]
- Hu, S.; Lu, Q.; Xu, Y. Biosensors based on direct electron transfer of protein. In Electrochemical Sensors, Biosensors and their Biomedical Applications; Academic Press: San Diego, CA, USA, 2008; p. 531. [Google Scholar]
- Jiang, L.; Yu, Z.; Du, W.; Tang, Z.; Jiang, T.; Zhang, C.; Lu, Z. Development of a fluorescent and colorimetric detection methods-based protein microarray for serodiagnosis of TORCH infections. Biosens. Bioelectron 2008, 24, 376–382. [Google Scholar]
- Chen, H.; Jiang, C.; Yu, C.; Zhang, S.; Liu, B.; Kong, J. Protein chips and nanomaterials for application in tumor marker immunoassays. Biosens. Bioelectron 2009, 24, 3399–3411. [Google Scholar]
- Chan, C.P.Y.; Wan, T.S.M.; Watkins, K.L.; Pelsers, M.M.A.L.; Van der Voort, D.; Tang, F.P.W.; Lam, K.H.K.; Mill, J.; Yuan, Y.; Lehmann, M.; Hempel, A.; Sanderson, J.E.; Glatz, J.F.C.; Renneberg, R. Rapid analysis of fatty acid-binding proteins with immunosensors and immunotests for early monitoring of tissue injury. Biosens. Bioelectron 2005, 20, 2566–2580. [Google Scholar]
- Gupta, R.K.; Dobritsa, S.V.; Stiles, C.A.; Essington, M.E.; Liu, Z.Y.; Chen, C.H.; Serpersu, E.H.; Mullin, B.C. Metallohistins: A new class of plant metal-binding proteins. J. Prot. Chem 2002, 21, 529–536. [Google Scholar]
- Guimarães-Soares, L.; Felícia, H.; João Bebianno, M.; Cássio, F. Metal-binding proteins and peptides in the aquatic fungi Fontanospora fusiramosa and Flagellospora curta exposed to severe metal stress. Sci. Total Envir 2006, 372, 148–156. [Google Scholar]
- Terrance, J.B.; Peter, H.P.; Margit, S.; Anja, K.; Kari, L.; Kirsti, K.; Eero, K.; Markus, H.; Eva, M.E.; Ingrid, S.; Uwe, B.S.; Lorenzo, M.; Maria-Luisa, C.; John, F.N.; Wade, H.B.; John, S.; Emmanuelle, L.; Marc, L.; Isabelle, M.; Sylvie, S.; Pierre, B.; Hélène, O.; Pierre, G.; Markus, M.; Kerstin, S.; Hubert, B.; Rosemary, G.-T.; Joel, D.; Martin, J.B.; Ralph, M.W.; Diane, G.N.; Martin, K.; Susan, F.K.V. Functions of S-layers1. FEMS Microbiol. Rev 1997, 20, 99–149. [Google Scholar]
- Debabov, V.G. Bacterial and archaeal S-layers as a subject of nanobiotechnology. Mol. Biol 2004, 38, 482–493. [Google Scholar]
- Dupres, V.E.A. In vivo Imaging of S-Layer Nanoarrays on Corynebacterium glutamicum. Langmuir Lett 2009, 25, 9653–9655. [Google Scholar]
- Nelson, A.; Auffret, N.; Borlakoglu, J. Interaction of hydrophobic organic compounds with mercury adsorbed dioleoylphosphatidylcholine monolayers. BBA-Biomembranes 1990, 1021, 205–216. [Google Scholar]
- Whitehouse, C.; Gidalevitz, D.; Cahuzac, M.; Koeppe, R.E.; Nelson, A. Interaction of Gramicidin Derivatives with Phospholipid Monolayers. Langmuir 2004, 20, 9291–9298. [Google Scholar]
- Hays, H.C.W.; Millner, P.A.; Prodromidis, M.I. Development of capacitance based immunosensors on mixed self-assembled monolayers. Sens. Actuat. B: Chem 2006, 114, 1064–1070. [Google Scholar]
- Billah, M.; Hays, H.C.W.; Millner, P.A. Development of a myoglobin impedimetric immunosensor based on mixed self-assembled monolayer onto gold. Microchim. Acta 2008, 160, 447–454. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundimentals and Applications, 2nd ed; Wiley: Malden, MA, USA, 2001; p. 833. [Google Scholar]
- Wang, J. Analytical Electrochemistry, 3rd ed; Wiley-VCH: Berlin, Germany, 2006; p. 250. [Google Scholar]
- Weygand, M.; Wetzer, B.; Pum, D.; Sleytr, U.B.; Cuvillier, N.; Kjaer, K.; Howes, P.B.; Lösche, M. Bacterial S-Layer Protein Coupling to Lipids: X-Ray Reflectivity and Grazing Incidence Diffraction Studies. Biophys. J 1999, 76, 458–468. [Google Scholar]
- Bergveld, P. A critical evaluation of direct electrical protein detection method. Biosens. Bioelectron 1991, 6, 55–72. [Google Scholar]
- Campuzano, S.; Pedrero, M.; Montemayor, C.; Fatás, E.; Pingarrón, J.M. Characterization of alkanethiol-self-assembled monolayers-modified gold electrodes by electrochemical impedance spectroscopy. J. Electroanal. Chem 2006, 586, 112–121. [Google Scholar]
- Fawcett, W.R.; Kovácová, Z.; Motheo, A.J.; Foss, C.A., Jr. Application of the ac admittance technique to double-layer studies on polycrystalline gold electrodes. J. Electroanal. Chem 1992, 326, 91–103. [Google Scholar]
- Breyer, B.; Bauer, H.H. Electrochemical cells as electrical circuit elements. J. Electroanal. Chem. 12, 411–415.
- Rivera-Gandía, J.; Cabrera, C.R. Self-assembled monolayers of 6-mercapto-1-hexanol and mercapto-n-hexyl-poly(dT)18-fluorescein on polycrystalline gold surfaces: An electrochemical impedance spectroscopy study. J. Electroanal. Chem 2007, 605, 145–150. [Google Scholar]
 ) and after (
) and after (
 ) chemical modification of phosphate and carboxylate groups. The data show a 30.2% carbon C 1s to gold 4f peak ratio increase confirming successful modification of analyte binding sites.
) and after (
) chemical modification of phosphate and carboxylate groups. The data show a 30.2% carbon C 1s to gold 4f peak ratio increase confirming successful modification of analyte binding sites.
) chemical modification of phosphate and carboxylate groups. The data show a 30.2% carbon C 1s to gold 4f peak ratio increase confirming successful modification of analyte binding sites.
) and after (
) chemical modification of phosphate and carboxylate groups. The data show a 30.2% carbon C 1s to gold 4f peak ratio increase confirming successful modification of analyte binding sites.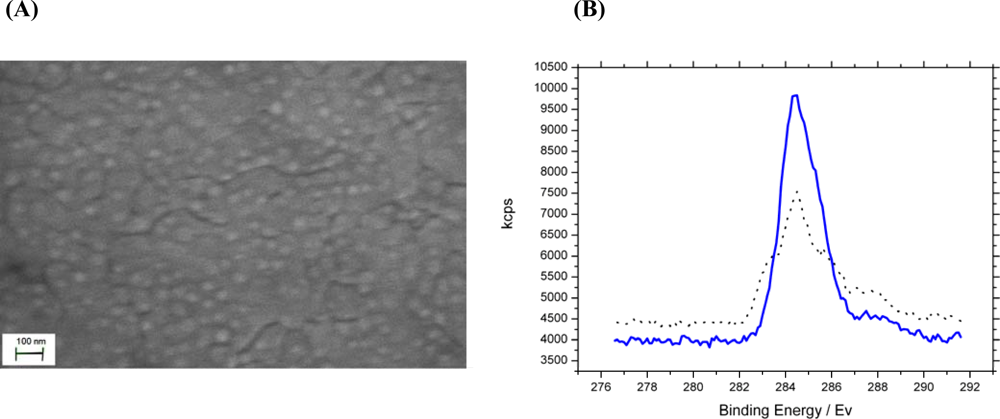
 1 kHz,
1 kHz,
 100 Hz,
100 Hz,
 10 Hz,
10 Hz,
 1 Hz,
1 Hz,
 0.1 Hz) are shown. 10 kHz – 100 Hz all overlay each other close to zero due to the system exhibiting high resistive and low capacitive behaviour at high frequency.
1 kHz,
100 Hz,
10 Hz,
1 Hz,
0.1 Hz) are shown. 10 kHz – 100 Hz all overlay each other close to zero due to the system exhibiting high resistive and low capacitive behaviour at high frequency.
0.1 Hz) are shown. 10 kHz – 100 Hz all overlay each other close to zero due to the system exhibiting high resistive and low capacitive behaviour at high frequency.
1 kHz,
100 Hz,
10 Hz,
1 Hz,
0.1 Hz) are shown. 10 kHz – 100 Hz all overlay each other close to zero due to the system exhibiting high resistive and low capacitive behaviour at high frequency.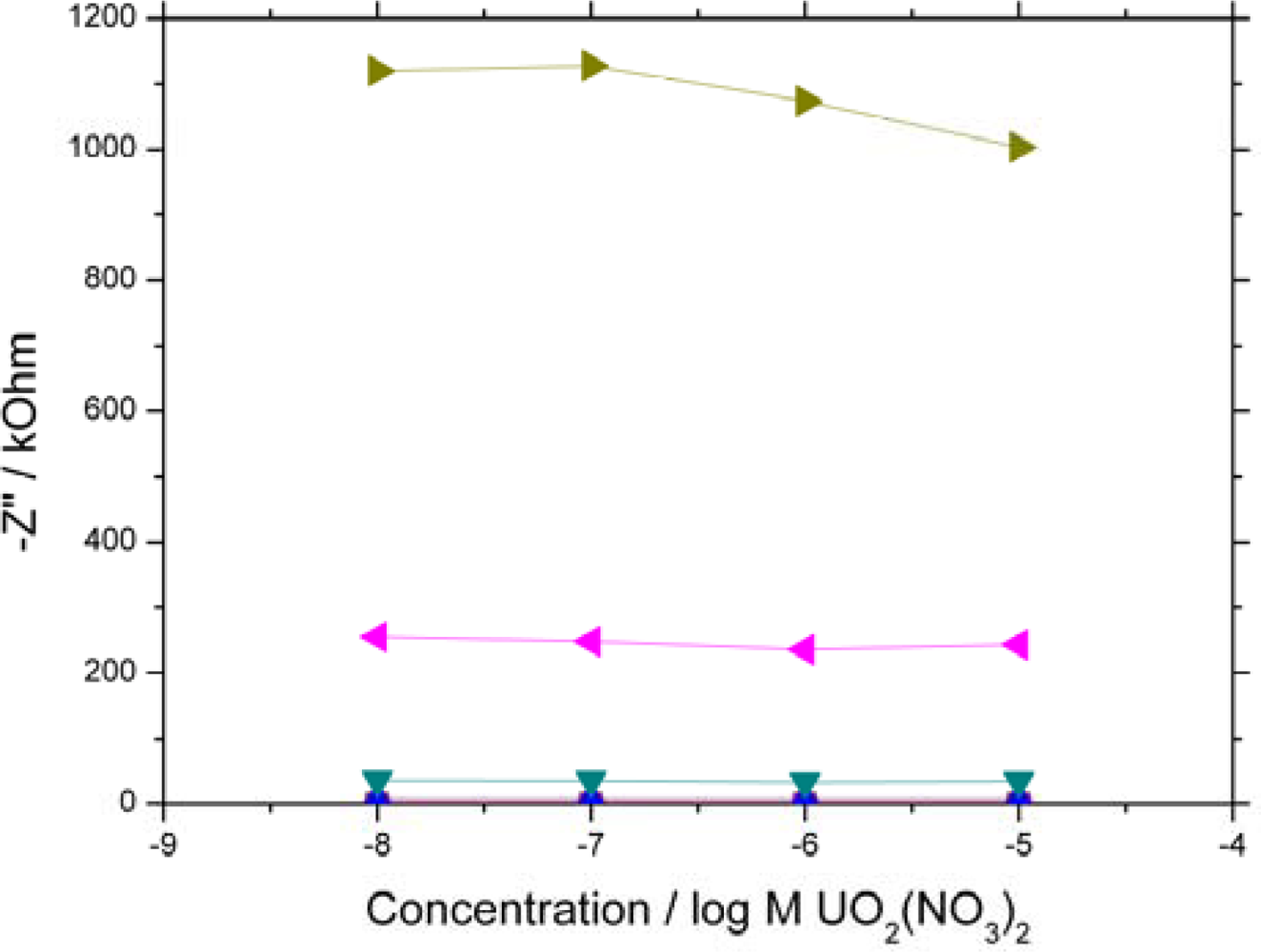
 uranyl nitrate response from a 7 days old electrode,
uranyl nitrate response from a 7 days old electrode,
 natural uranyl nitrate response
natural uranyl nitrate response
 uranyl acetate response). The data shows no differentiation between uranyl compounds as all are able to bind with the UO22+ in the +6 oxidation state. (B)–Response of biosensor to a range of interfering divalent cations (▪ nickel nitrate,
uranyl acetate response). The data shows no differentiation between uranyl compounds as all are able to bind with the UO22+ in the +6 oxidation state. (B)–Response of biosensor to a range of interfering divalent cations (▪ nickel nitrate,
 caesium sulphate,
caesium sulphate,
 cadmium nitrate,
cadmium nitrate,
 cobalt chloride,
cobalt chloride,
 average uranyl response).
uranyl nitrate response from a 7 days old electrode,
natural uranyl nitrate response
uranyl acetate response). The data shows no differentiation between uranyl compounds as all are able to bind with the UO22+ in the +6 oxidation state. (B)–Response of biosensor to a range of interfering divalent cations (▪ nickel nitrate,
caesium sulphate,
cadmium nitrate,
cobalt chloride,
average uranyl response).
average uranyl response).
uranyl nitrate response from a 7 days old electrode,
natural uranyl nitrate response
uranyl acetate response). The data shows no differentiation between uranyl compounds as all are able to bind with the UO22+ in the +6 oxidation state. (B)–Response of biosensor to a range of interfering divalent cations (▪ nickel nitrate,
caesium sulphate,
cadmium nitrate,
cobalt chloride,
average uranyl response).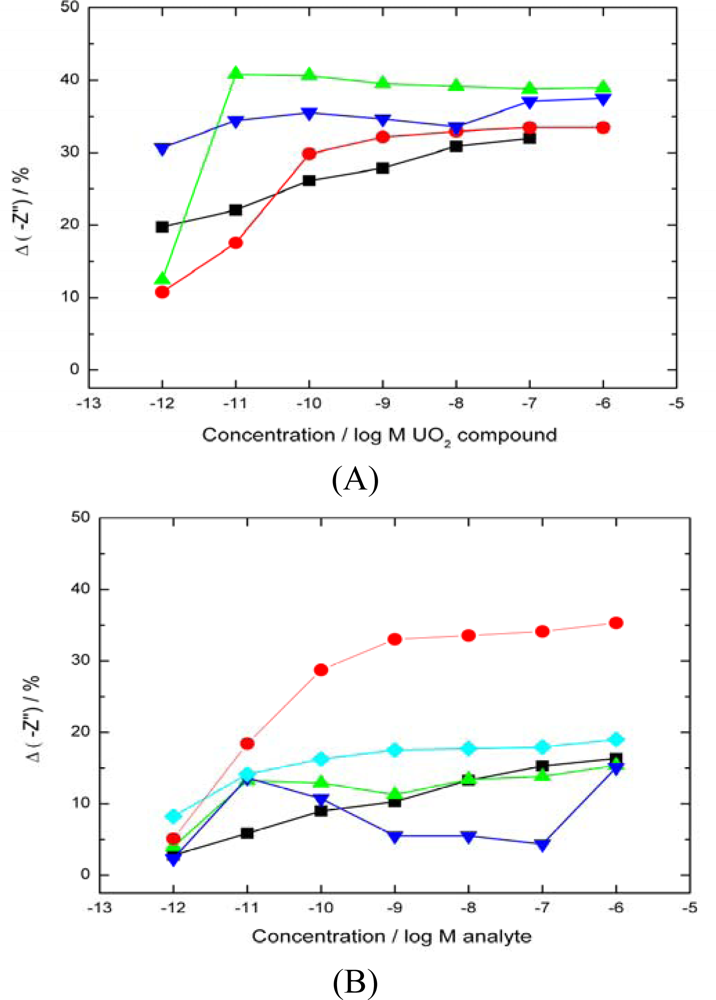
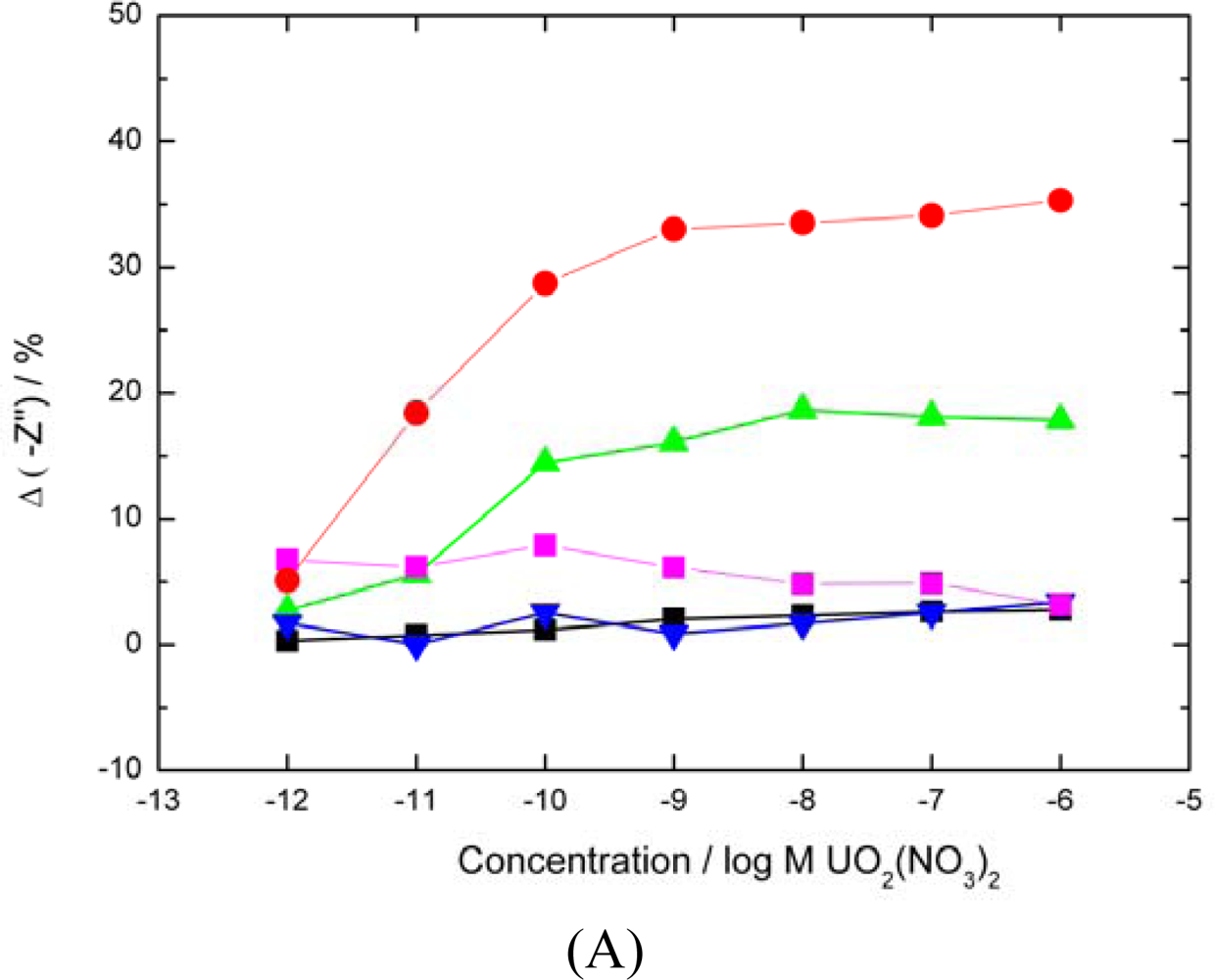
 Casein sensor response,
Casein sensor response,
 BSA sensor response,
BSA sensor response,
 BSA sensor response with carboxylates blocked,
BSA sensor response with carboxylates blocked,
 average uranyl response of SLP biosensor for comparison). The percentage decrease in –Z” was calculated as previously. Sequential analyte injections were performed over a 6 hours period. (▪ A control sensor with no analyte added showed only a 2% drift in base signal over the same period). (B) Modified SLP protein response to UO22+ (
average uranyl response of SLP biosensor for comparison). The percentage decrease in –Z” was calculated as previously. Sequential analyte injections were performed over a 6 hours period. (▪ A control sensor with no analyte added showed only a 2% drift in base signal over the same period). (B) Modified SLP protein response to UO22+ (
 Both carboxylates and phosphates moieties blocked
Both carboxylates and phosphates moieties blocked
 carboxylates only blocked,
carboxylates only blocked,
 phosphates only blocked, ▪ base signal drift over a 6 h period,
phosphates only blocked, ▪ base signal drift over a 6 h period,
 average uranyl response of SLP biosensor for comparison).
Casein sensor response,
BSA sensor response,
BSA sensor response with carboxylates blocked,
average uranyl response of SLP biosensor for comparison). The percentage decrease in –Z” was calculated as previously. Sequential analyte injections were performed over a 6 hours period. (▪ A control sensor with no analyte added showed only a 2% drift in base signal over the same period). (B) Modified SLP protein response to UO22+ (
Both carboxylates and phosphates moieties blocked
carboxylates only blocked,
phosphates only blocked, ▪ base signal drift over a 6 h period,
average uranyl response of SLP biosensor for comparison).
average uranyl response of SLP biosensor for comparison).
Casein sensor response,
BSA sensor response,
BSA sensor response with carboxylates blocked,
average uranyl response of SLP biosensor for comparison). The percentage decrease in –Z” was calculated as previously. Sequential analyte injections were performed over a 6 hours period. (▪ A control sensor with no analyte added showed only a 2% drift in base signal over the same period). (B) Modified SLP protein response to UO22+ (
Both carboxylates and phosphates moieties blocked
carboxylates only blocked,
phosphates only blocked, ▪ base signal drift over a 6 h period,
average uranyl response of SLP biosensor for comparison).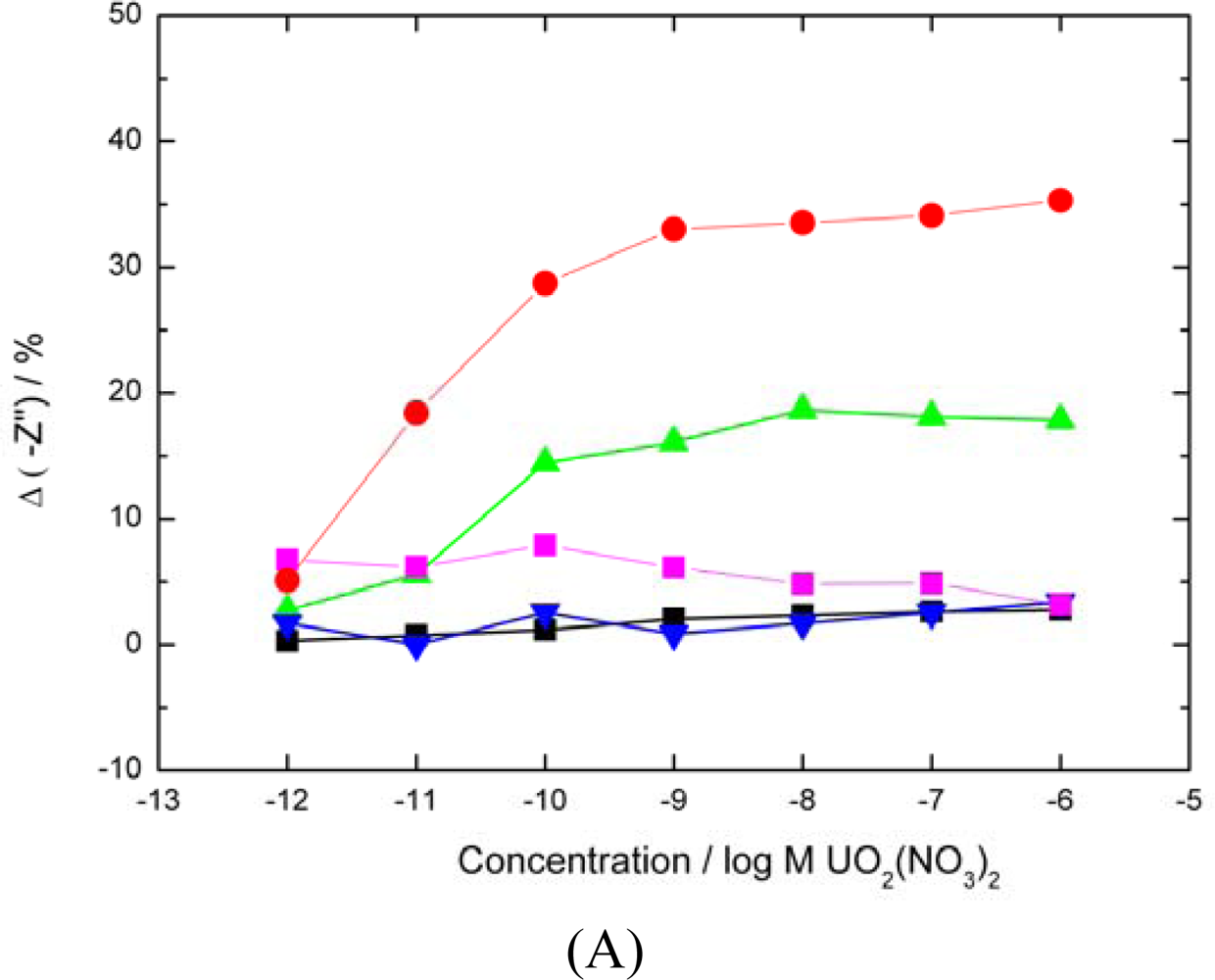

© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Conroy, D.J.R.; Millner, P.A.; Stewart, D.I.; Pollmann, K. Biosensing for the Environment and Defence: Aqueous Uranyl Detection Using Bacterial Surface Layer Proteins. Sensors 2010, 10, 4739-4755. https://doi.org/10.3390/s100504739
Conroy DJR, Millner PA, Stewart DI, Pollmann K. Biosensing for the Environment and Defence: Aqueous Uranyl Detection Using Bacterial Surface Layer Proteins. Sensors. 2010; 10(5):4739-4755. https://doi.org/10.3390/s100504739
Chicago/Turabian StyleConroy, David J.R., Paul A. Millner, Douglas I. Stewart, and Katrin Pollmann. 2010. "Biosensing for the Environment and Defence: Aqueous Uranyl Detection Using Bacterial Surface Layer Proteins" Sensors 10, no. 5: 4739-4755. https://doi.org/10.3390/s100504739