Oxidant Sensing by Protein Kinases A and G Enables Integration of Cell Redox State with Phosphoregulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
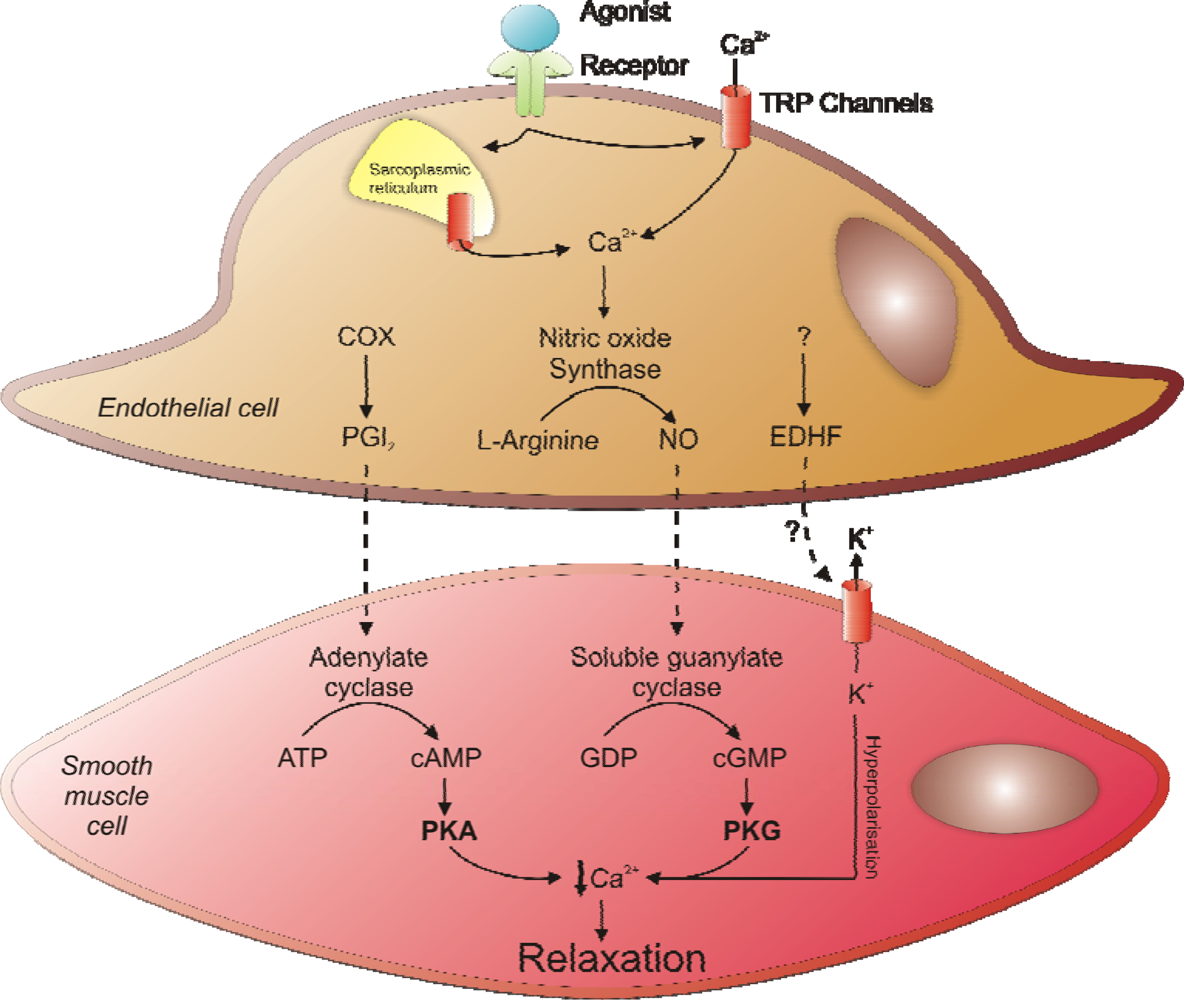
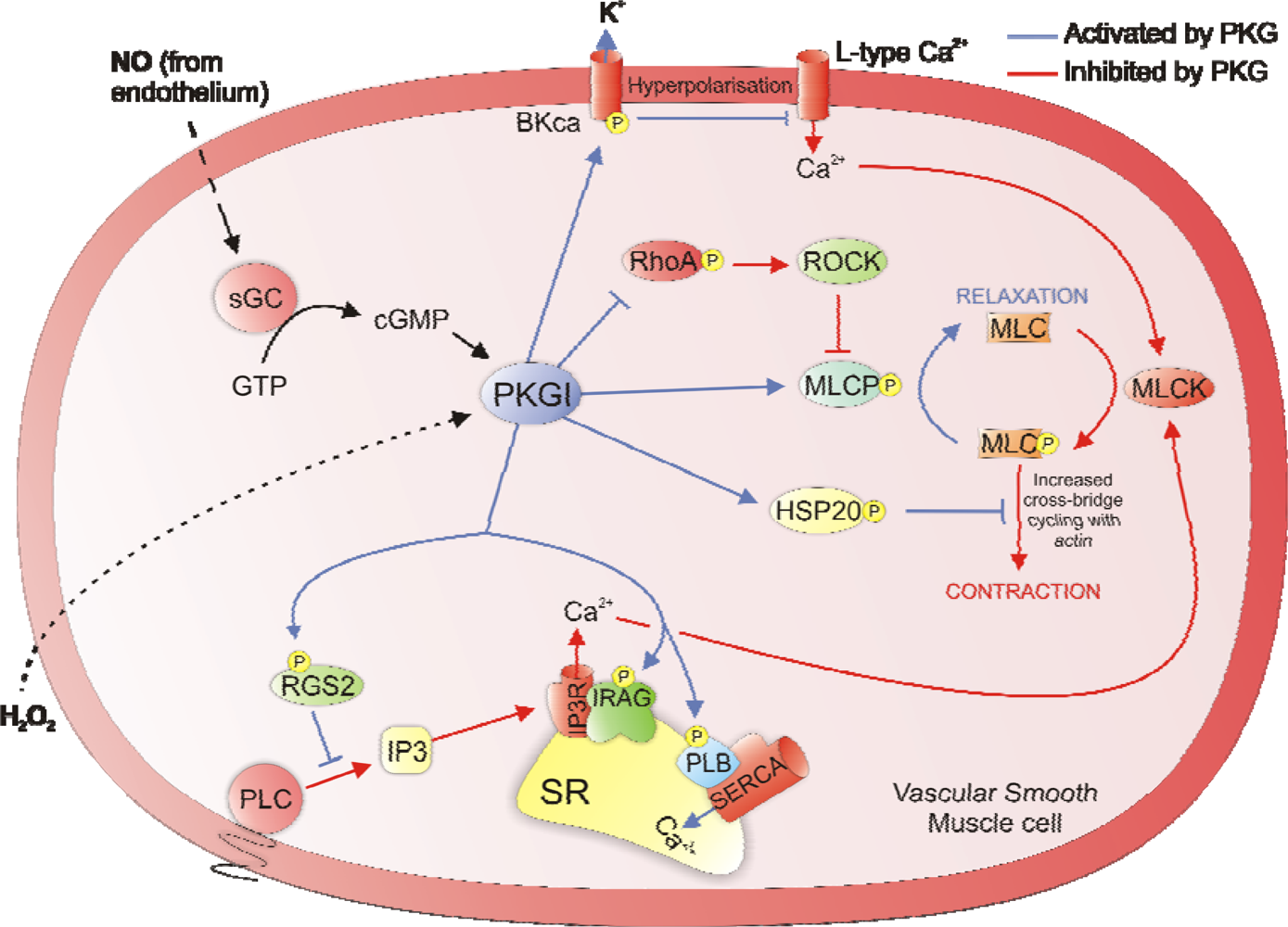
2. Regulation of Vascular Smooth Muscle Tone
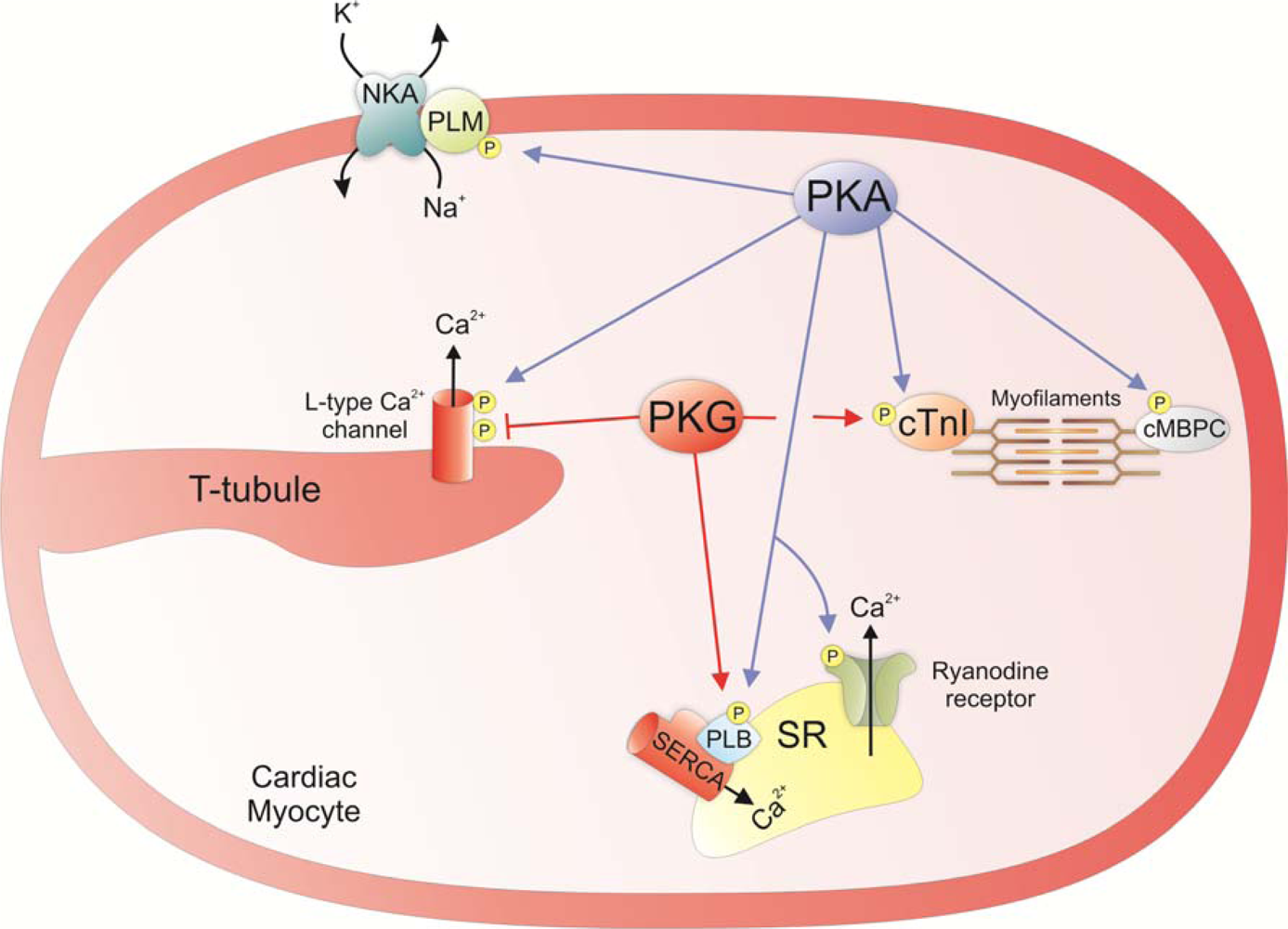
3. Regulation of Cardiac Contractility
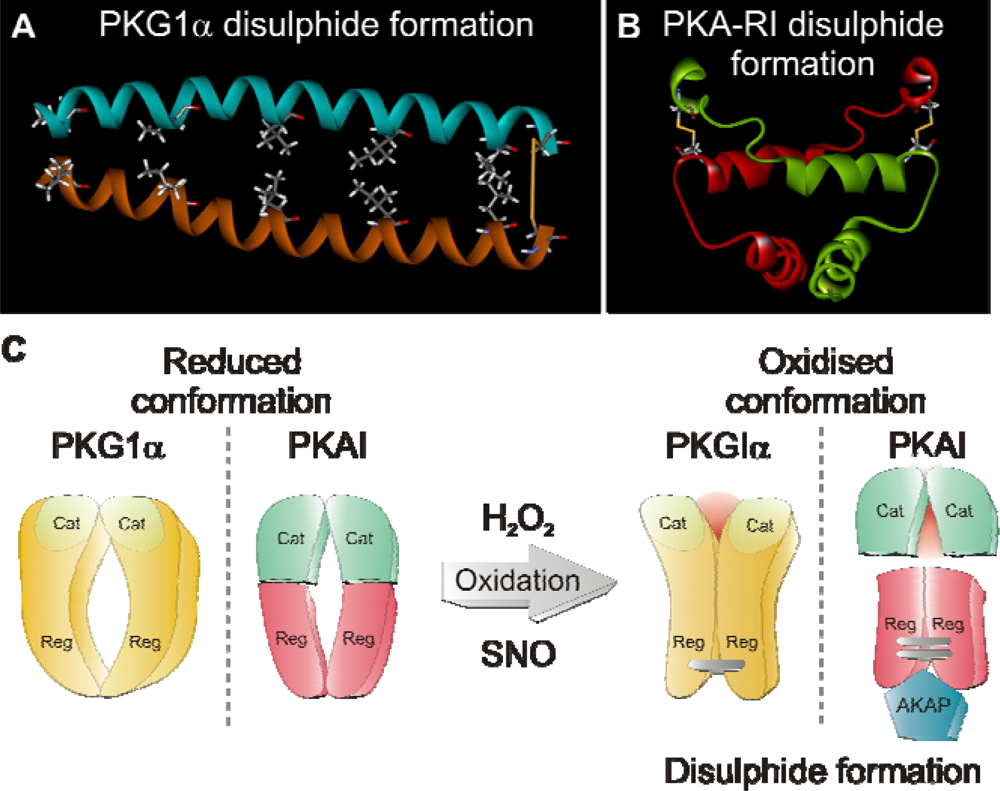
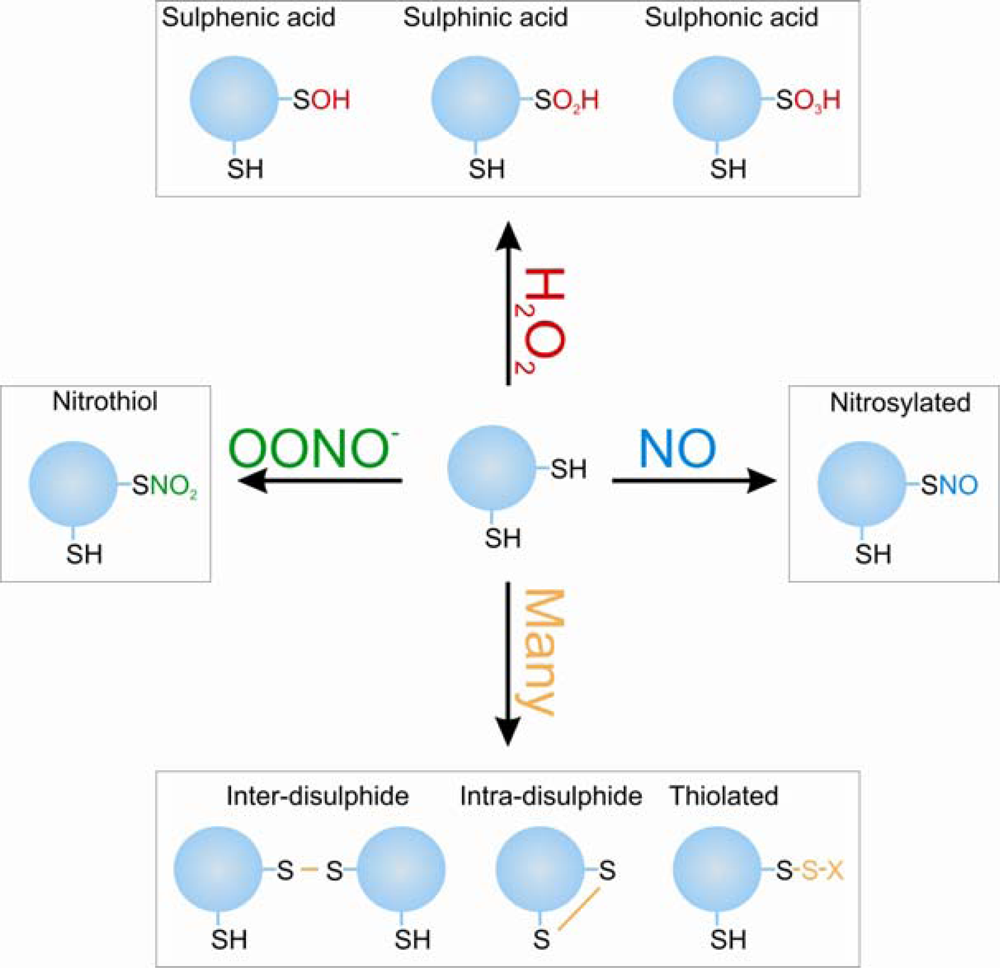
4. Redox Regulation of PKAI and PKG1α Activity
5. Conclusion
Acknowledgments
References
- Germino, F.W. The management and treatment of hypertension. Clin. Cornerstone 2009, 9, S27–S33. [Google Scholar]
- Marchick, M.R.; Kline, J.A.; Jones, A.E. The significance of non-sustained hypotension in emergency department patients with sepsis. Intensive Care Med 2009, 35, 1261–1264. [Google Scholar]
- O'Brien, J.M., Jr.; Ali, N.A.; Aberegg, S.K.; Abraham, E. Sepsis. Am. J. Med 2007, 120, 1012–1022. [Google Scholar]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schroder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schroder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem 2006, 281, 21827–21836. [Google Scholar]
- Vainio, H. Chemoprevention of cancer: lessons to be learned from beta-carotene trials. Toxicol. Lett 2000, 112–113, 513–517. [Google Scholar]
- Miller, E.R., III; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med 2005, 142, 37–46. [Google Scholar]
- MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002, 360, 23–33.
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar]
- Bindoli, A.; Fukuto, J.M.; Forman, H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox. Signal 2008, 10, 1549–1564. [Google Scholar]
- Frisard, M.; Ravussin, E. Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process. Endocrine 2006, 29, 27–32. [Google Scholar]
- Wolin, M.S.; Gupte, S.A.; Oeckler, R.A. Superoxide in the vascular system. J. Vasc. Res 2002, 39, 191–207. [Google Scholar]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox. Signal 2006, 8, 691–728. [Google Scholar]
- Leitch, J.M.; Yick, P.J.; Culotta, V.C. The right to choose: multiple pathways for activating copper,zinc superoxide dismutase. J. Biol. Chem 2009, 284, 24679–24683. [Google Scholar]
- DeYulia, G.J., Jr.; Carcamo, J.M.; Borquez-Ojeda, O.; Shelton, C.C.; Golde, D.W. Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 5044–5049. [Google Scholar]
- Roy, S.; Khanna, S.; Sen, C.K. Redox regulation of the VEGF signaling path and tissue vascularization: Hydrogen peroxide, the common link between physical exercise and cutaneous wound healing. Free Radic. Biol. Med 2008, 44, 180–192. [Google Scholar]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol 2008, 30, 279–289. [Google Scholar]
- Hirano, K. Current topics in the regulatory mechanism underlying the Ca2+ sensitization of the contractile apparatus in vascular smooth muscle. J. Pharmacol. Sci 2007, 104, 109–115. [Google Scholar]
- Ivey, M.E.; Osman, N.; Little, P.J. Endothelin-1 signalling in vascular smooth muscle: pathways controlling cellular functions associated with atherosclerosis. Atherosclerosis 2008, 199, 237–247. [Google Scholar]
- Kawanabe, Y.; Nauli, S.M. Involvement of extracellular Ca2+ influx through voltage-independent Ca2+ channels in endothelin-1 function. Cell Signal 2005, 17, 911–916. [Google Scholar]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol 2007, 292, C82–C97. [Google Scholar]
- Heusch, G.; Baumgart, D.; Camici, P.; Chilian, W.; Gregorini, L.; Hess, O.; Indolfi, C.; Rimoldi, O. Alpha-adrenergic coronary vasoconstriction and myocardial ischemia in humans. Circulation 2000, 101, 689–694. [Google Scholar]
- Passmore, J.C.; Joshua, I.G.; Rowell, P.P.; Tyagi, S.C.; Falcone, J.C. Reduced alpha adrenergic mediated contraction of renal preglomerular blood vessels as a function of gender and aging. J. Cell Biochem 2005, 96, 672–681. [Google Scholar]
- Wang, H.; Oestreich, E.A.; Maekawa, N.; Bullard, T.A.; Vikstrom, K.L.; Dirksen, R.T.; Kelley, G.G.; Blaxall, B.C.; Smrcka, A.V.; Phospholipase, C. Epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ. Res 2005, 97, 1305–1313. [Google Scholar]
- Raeymaekers, L.; Nilius, B.; Voets, T.; Missiaen, L.; Van Baelen, K.; Vanoevelen, J.; Wuytack, F. Additional fluxes of activator Ca2+ accompanying Ca2+ release from the sarcoplasmic reticulum triggered by insP3-mobilizing agonists. Novartis Found Symp 2002, 246, 221–227. [Google Scholar]
- Yao, X.; Garland, C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res 2005, 97, 853–863. [Google Scholar]
- Kim, H.R.; Appel, S.; Vetterkind, S.; Gangopadhyay, S.S.; Morgan, K.G. Smooth muscle signalling pathways in health and disease. J. Cell Mol. Med 2008, 12, 2165–2180. [Google Scholar]
- Frolich, J.C. Prostacyclin in hypertension. J. Hypertens. Suppl 1990, 8, S73–S78. [Google Scholar]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol 2006, 147, S193–S201. [Google Scholar]
- Dudzinski, D.M.; Igarashi, J.; Greif, D.; Michel, T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu. Rev. Pharmacol. Toxicol 2006, 46, 235–276. [Google Scholar]
- Cockcroft, J.R. Exploring vascular benefits of endothelium-derived nitric oxide. Am. J. Hypertens 2005, 18, 177S–183S. [Google Scholar]
- Fukao, M.; Mason, H.S.; Britton, F.C.; Kenyon, J.L.; Horowitz, B.; Keef, K.D. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J. Biol. Chem 1999, 274, 10927–10935. [Google Scholar]
- Barman, S.A.; Zhu, S.; Han, G.; White, R.E. cAMP activates BKCa channels in pulmonary arterial smooth muscle via cGMP-dependent protein kinase. Am. J. Physiol. Lung Cell Mol. Physiol 2003, 284, L1004–L1011. [Google Scholar]
- Koller, A.; Schlossmann, J.; Ashman, K.; Uttenweiler-Joseph, S.; Ruth, P.; Hofmann, F. Association of phospholamban with a cGMP kinase signaling complex. Biochem. Biophys. Res. Commun 2003, 300, 155–160. [Google Scholar]
- Lalli, M.J.; Shimizu, S.; Sutliff, R.L.; Kranias, E.G.; Paul, R.J. [Ca2+]i homeostasis and cyclic nucleotide relaxation in aorta of phospholamban-deficient mice. Am. J. Physiol 1999, 277, H963–H970. [Google Scholar]
- Fritsch, R.M.; Saur, D.; Kurjak, M.; Oesterle, D.; Schlossmann, J.; Geiselhoringer, A.; Hofmann, F.; Allescher, H.D. InsP3R-associated cGMP kinase substrate (IRAG) is essential for nitric oxide-induced inhibition of calcium signaling in human colonic smooth muscle. J. Biol. Chem 2004, 279, 12551–12559. [Google Scholar]
- Geiselhoringer, A.; Werner, M.; Sigl, K.; Smital, P.; Worner, R.; Acheo, L.; Stieber, J.; Weinmeister, P.; Feil, R.; Feil, S.; Wegener, J.; Hofmann, F.; Schlossmann, J. IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. Embo. J 2004, 23, 4222–4231. [Google Scholar]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.X.; Allescher, H.D.; Korth, M.; Wilm, M.; Hofmann, F.; Ruth, P. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 2000, 404, 197–201. [Google Scholar]
- Komalavilas, P.; Lincoln, T.M. Phosphorylation of the inositol 1,4,5-trisphosphate receptor. Cyclic GMP-dependent protein kinase mediates cAMP and cGMP dependent phosphorylation in the intact rat aorta. J. Biol. Chem 1996, 271, 21933–21938. [Google Scholar]
- Osei-Owusu, P.; Sun, X.; Drenan, R.M.; Steinberg, T.H.; Blumer, K.J. Regulation of RGS2 and second messenger signaling in vascular smooth muscle cells by cGMP-dependent protein kinase. J. Biol. Chem 2007, 282, 31656–31665. [Google Scholar]
- Obst, M.; Tank, J.; Plehm, R.; Blumer, K.J.; Diedrich, A.; Jordan, J.; Luft, F.C.; Gross, V. NO-dependent blood pressure regulation in RGS2-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol 2006, 290, R1012–R1019. [Google Scholar]
- Sawada, N.; Itoh, H.; Yamashita, J.; Doi, K.; Inoue, M.; Masatsugu, K.; Fukunaga, Y.; Sakaguchi, S.; Sone, M.; Yamahara, K.; Yurugi, T.; Nakao, K. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun 2001, 280, 798–805. [Google Scholar]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem 2000, 275, 21722–21729. [Google Scholar]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J. Biol. Chem 2004, 279, 34496–34504. [Google Scholar]
- Somlyo, A.V. Cyclic GMP regulation of myosin phosphatase: a new piece for the puzzle? Circ. Res 2007, 101, 645–647. [Google Scholar]
- Beall, A.; Bagwell, D.; Woodrum, D.; Stoming, T.A.; Kato, K.; Suzuki, A.; Rasmussen, H.; Brophy, C.M. The small heat shock-related protein, HSP20, is phosphorylated on serine 16 during cyclic nucleotide-dependent relaxation. J. Biol. Chem 1999, 274, 11344–11351. [Google Scholar]
- Beall, A.C.; Kato, K.; Goldenring, J.R.; Rasmussen, H.; Brophy, C.M. Cyclic nucleotide-dependent vasorelaxation is associated with the phosphorylation of a small heat shock-related protein. J. Biol. Chem 1997, 272, 11283–11287. [Google Scholar]
- Wong, W.; Scott, J.D. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol 2004, 5, 959–970. [Google Scholar]
- Hoshi, N.; Langeberg, L.K.; Scott, J.D. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat. Cell Biol 2005, 7, 1066–1073. [Google Scholar]
- Vo, N.K.; Gettemy, J.M.; Coghlan, V.M. Identification of cGMP–dependent protein kinase anchoring proteins (GKAPs). Biochem. Biophys. Res. Commun 1998, 246, 831–835. [Google Scholar]
- Cha, B.; Kim, J.H.; Hut, H.; Hogema, B.M.; Nadarja, J.; Zizak, M.; Cavet, M.; Lee-Kwon, W.; Lohmann, S.M.; Smolenski, A.; Tse, C.M.; Yun, C.; de Jonge, H.R.; Donowitz, M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem 2005, 280, 16642–16650. [Google Scholar]
- Eckly-Michel, A.; Martin, V.; Lugnier, C. Involvement of cyclic nucleotide-dependent protein kinases in cyclic AMP-mediated vasorelaxation. Br. J. Pharmacol 1997, 122, 158–164. [Google Scholar]
- Casteels, R.; Wuytack, F.; Raeymaekers, L.; Himpens, B. Ca(2+)-transport ATPases and Ca(2+)-compartments in smooth muscle cells. Z. Kardiol 1991, 80, 65–68. [Google Scholar]
- Mundina-Weilenmann, C.; Vittone, L.; Rinaldi, G.; Said, M.; de Cingolani, G.C.; Mattiazzi, A. Endoplasmic reticulum contribution to the relaxant effect of cGMP- and cAMP-elevating agents in feline aorta. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H1856–H1865. [Google Scholar]
- Komalavilas, P.; Penn, R.B.; Flynn, C.R.; Thresher, J.; Lopes, L.B.; Furnish, E.J.; Guo, M.; Pallero, M.A.; Murphy-Ullrich, J.E.; Brophy, C.M. The small heat shock-related protein, HSP20, is a cAMP-dependent protein kinase substrate that is involved in airway smooth muscle relaxation. Am. J. Physiol. Lung Cell Mol. Physiol 2008, 294, L69–L78. [Google Scholar]
- Murthy, K.S.; Zhou, H.; Grider, J.R.; Makhlouf, G.M. Inhibition of sustained smooth muscle contraction by PKA and PKG preferentially mediated by phosphorylation of RhoA. Am. J. Physiol. Gastrointest Liver Physiol 2003, 284, G1006–G1016. [Google Scholar]
- Azam, M.A.; Yoshioka, K.; Ohkura, S.; Takuwa, N.; Sugimoto, N.; Sato, K.; Takuwa, Y. Ca2+-independent, inhibitory effects of cyclic adenosine 5′-monophosphate on Ca2+ regulation of phosphoinositide 3-kinase C2alpha, Rho, and myosin phosphatase in vascular smooth muscle. J. Pharmacol. Exp. Ther 2007, 320, 907–916. [Google Scholar]
- Kohler, R.; Hoyer, J. The endothelium-derived hyperpolarizing factor: insights from genetic animal models. Kidney Int 2007, 72, 145–150. [Google Scholar]
- Bellien, J.; Thuillez, C.; Joannides, R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam. Clin. Pharmacol 2008, 22, 363–377. [Google Scholar]
- Chen, G.; Suzuki, H.; Weston, A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol 1988, 95, 1165–1174. [Google Scholar]
- Mombouli, J.V.; Nakashima, M.; Hamra, M.; Vanhoutte, P.M. Endothelium-dependent relaxation and hyperpolarization evoked by bradykinin in canine coronary arteries: enhancement by exercise-training. Br. J. Pharmacol 1996, 117, 413–418. [Google Scholar]
- Takamura, Y.; Shimokawa, H.; Zhao, H.; Igarashi, H.; Egashira, K.; Takeshita, A. Important role of endothelium-derived hyperpolarizing factor in shear stress--induced endothelium-dependent relaxations in the rat mesenteric artery. J. Cardiovasc. Pharmacol 1999, 34, 381–387. [Google Scholar]
- Michaelis, U.R.; Fleming, I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: Epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther 2006, 111, 584–595. [Google Scholar]
- Shimokawa, H.; Matoba, T. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pharmacol. Res 2004, 49, 543–549. [Google Scholar]
- de Wit, C.; Wolfle, S.E. EDHF and gap junctions: important regulators of vascular tone within the microcirculation. Curr. Pharm. Biotechnol 2007, 8, 11–25. [Google Scholar]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszodi, A.; Andersson, K.E.; Krombach, F.; Mayerhofer, A.; Ruth, P.; Fassler, R.; Hofmann, F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. Embo. J 1998, 17, 3045–3051. [Google Scholar]
- Kobayashi, T.; Solaro, R.J. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol 2005, 67, 39–67. [Google Scholar]
- Rockman, H.A.; Koch, W.J.; Milano, C.A.; Lefkowitz, R.J. Myocardial beta-adrenergic receptor signaling in vivo: insights from transgenic mice. J. Mol. Med 1996, 74, 489–495. [Google Scholar]
- Xiao, R.P.; Zhu, W.; Zheng, M.; Cao, C.; Zhang, Y.; Lakatta, E.G.; Han, Q. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol. Sci 2006, 27, 330–337. [Google Scholar]
- Irisawa, H.; Brown, H.F.; Giles, W. Cardiac pacemaking in the sinoatrial node. Physiol. Rev 1993, 73, 197–227. [Google Scholar]
- Clusin, W.T. Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1–H10. [Google Scholar]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar]
- Hovnanian, A. SERCA pumps and human diseases. Subcell Biochem 2007, 45, 337–363. [Google Scholar]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: its physiological implications. Physiol. Rev 1999, 79, 763–854. [Google Scholar]
- Hirota, S.; Pertens, E.; Janssen, L.J. The reverse mode of the Na(+)/Ca(2+) exchanger provides a source of Ca(2+) for store refilling following agonist-induced Ca(2+) mobilization. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 292, L438–L447. [Google Scholar]
- Dong, H.; Jiang, Y.; Triggle, C.R.; Li, X.; Lytton, J. Novel role for K+-dependent Na+/Ca2+ exchangers in regulation of cytoplasmic free Ca2+ and contractility in arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol 2006, 291, H1226–H1235. [Google Scholar]
- Grantham, C.J.; Cannell, M.B. Ca2+ influx during the cardiac action potential in guinea pig ventricular myocytes. Circ. Res 1996, 79, 194–200. [Google Scholar]
- Oceandy, D.; Stanley, P.J.; Cartwright, E.J.; Neyses, L. The regulatory function of plasma-membrane Ca(2+)-ATPase (PMCA) in the heart. Biochem. Soc. Trans 2007, 35, 927–930. [Google Scholar]
- Bezprozvanny, I.; Watras, J.; Ehrlich, B.E. Bell-shaped calcium-response curves of Ins(1,4,5)P3-and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 1991, 351, 751–754. [Google Scholar]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol 2003, 4, 566–577. [Google Scholar]
- Bunemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J. Biol. Chem 1999, 274, 33851–33854. [Google Scholar]
- Gao, T.; Yatani, A.; Dell'Acqua, M.L.; Sako, H.; Green, S.A.; Dascal, N.; Scott, J.D.; Hosey, M.M. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 1997, 19, 185–196. [Google Scholar]
- Shattock, M.J. Phospholemman: its role in normal cardiac physiology and potential as a druggable target in disease. Curr. Opin. Pharmacol 2009, 9, 160–166. [Google Scholar]
- Despa, S.; Tucker, A.L.; Bers, D.M. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation 2008, 117, 1849–1855. [Google Scholar]
- Moir, A.J.; Solaro, R.J.; Perry, S.V. The site of phosphorylation of troponin I in the perfused rabbit heart. The effect of adrenaline. Biochem. J 1980, 185, 505–513. [Google Scholar]
- Robertson, S.P.; Johnson, J.D.; Holroyde, M.J.; Kranias, E.G.; Potter, J.D.; Solaro, R.J. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J. Biol. Chem 1982, 257, 260–263. [Google Scholar]
- Solaro, R.J.; Moir, A.J.; Perry, S.V. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature 1976, 262, 615–617. [Google Scholar]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Martin, L.A.; Hahn, H.S.; Dorn, G.W., 2nd; Klevitsky, R.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res 2005, 97, 1156–1163. [Google Scholar]
- Tong, C.W.; Stelzer, J.E.; Greaser, M.L.; Powers, P.A.; Moss, R.L. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ. Res 2008, 103, 974–982. [Google Scholar]
- Gonzalez, D.R.; Fernandez, I.C.; Ordenes, P.P.; Treuer, A.V.; Eller, G.; Boric, M.P. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Nitric. Oxide 2008, 18, 157–167. [Google Scholar]
- Wegener, J.W.; Nawrath, H.; Wolfsgruber, W.; Kuhbandner, S.; Werner, C.; Hofmann, F.; Feil, R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res 2002, 90, 18–20. [Google Scholar]
- Sabine, B.; Willenbrock, R.; Haase, H.; Karczewski, P.; Wallukat, G.; Dietz, R.; Krause, E.G. Cyclic GMP-mediated phospholamban phosphorylation in intact cardiomyocytes. Biochem. Biophys. Res. Commun 1995, 214, 75–80. [Google Scholar]
- Zhang, Q.; Scholz, P.M.; He, Y.; Tse, J.; Weiss, H.R. Cyclic GMP signaling and regulation of SERCA activity during cardiac myocyte contraction. Cell Calcium 2005, 37, 259–266. [Google Scholar]
- Blumenthal, D.K.; Stull, J.T.; Gill, G.N. Phosphorylation of cardiac troponin by guanosine 3′:5′-monophosphate-dependent protein kinase. J. Biol. Chem 1978, 253, 324–326. [Google Scholar]
- Yuasa, K.; Michibata, H.; Omori, K.; Yanaka, N. A novel interaction of cGMP-dependent protein kinase I with troponin T. J. Biol. Chem 1999, 274, 37429–37434. [Google Scholar]
- Yang, L.; Liu, G.; Zakharov, S.I.; Bellinger, A.M.; Mongillo, M.; Marx, S.O. Protein kinase G phosphorylates Cav1.2 alpha1c and beta2 subunits. Circ. Res 2007, 101, 465–474. [Google Scholar]
- Schroder, F.; Klein, G.; Fiedler, B.; Bastein, M.; Schnasse, N.; Hillmer, A.; Ames, S.; Gambaryan, S.; Drexler, H.; Walter, U.; Lohmann, S.M.; Wollert, K.C. Single L-type Ca(2+) channel regulation by cGMP-dependent protein kinase type I in adult cardiomyocytes from PKG I transgenic mice. Cardiovasc. Res 2003, 60, 268–277. [Google Scholar]
- Brennan, J.P.; Wait, R.; Begum, S.; Bell, J.R.; Dunn, M.J.; Eaton, P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J. Biol. Chem 2004, 279, 41352–41360. [Google Scholar]
- Zick, S.K.; Taylor, S.S. Interchain disulfide bonding in the regulatory subunit of cAMP-dependent protein kinase I. J. Biol. Chem 1982, 257, 2287–2293. [Google Scholar]
- Sarma, G.N.; Kinderman, F.S.; Kim, C.; von Daake, S.; Chen, L.; Wang, B.C.; Taylor, S.S. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure 2010, 18, 155–166. [Google Scholar]
- Monken, C.E.; Gill, G.N. Structural analysis of cGMP-dependent protein kinase using limited proteolysis. J. Biol. Chem 1980, 255, 7067–7070. [Google Scholar]
- Schnell, J.R.; Zhou, G.P.; Zweckstetter, M.; Rigby, A.C.; Chou, J.J. Rapid and accurate structure determination of coiled-coil domains using NMR dipolar couplings: application to cGMP-dependent protein kinase Ialpha. Protein Sci 2005, 14, 2421–2428. [Google Scholar]
- Banky, P.; Roy, M.; Newlon, M.G.; Morikis, D.; Haste, N.M.; Taylor, S.S.; Jennings, P.A. Related protein-protein interaction modules present drastically different surface topographies despite a conserved helical platform. J. Mol. Biol 2003, 330, 1117–1129. [Google Scholar]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid. Redox. Signal 2005, 7, 1021–1031. [Google Scholar]
- Seo, J.H.; Ahn, Y.; Lee, S.R.; Yeol Yeo, C.; Chung Hur, K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol. Biol. Cell 2005, 16, 348–357. [Google Scholar]
- Burgoyne, J.R.; Eaton, P. Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling. J. Biol. Chem 2009, 284, 29260–29268. [Google Scholar]
- Iesaki, T.; Okada, T.; Shimada, I.; Yamaguchi, H.; Ochi, R. Decrease in Ca2+ sensitivity as a mechanism of hydrogen peroxide-induced relaxation of rabbit aorta. Cardiovasc. Res 1996, 31, 820–825. [Google Scholar]
- Hayabuchi, Y.; Nakaya, Y.; Matsuoka, S.; Kuroda, Y. Hydrogen peroxide-induced vascular relaxation in porcine coronary arteries is mediated by Ca2+-activated K+ channels. Heart Vessels 1998, 13, 9–17. [Google Scholar]
- Yang, Z.W.; Zhang, A.; Altura, B.T.; Altura, B.M. Endothelium-dependent relaxation to hydrogen peroxide in canine basilar artery: a potential new cerebral dilator mechanism. Brain Res. Bull 1998, 47, 257–263. [Google Scholar]
- Morikawa, K.; Fujiki, T.; Matoba, T.; Kubota, H.; Hatanaka, M.; Takahashi, S.; Shimokawa, H. Important role of superoxide dismutase in EDHF-mediated responses of human mesenteric arteries. J. Cardiovasc. Pharmacol 2004, 44, 552–556. [Google Scholar]
- Shimokawa, H.; Morikawa, K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J. Mol. Cell Cardiol 2005, 39, 725–732. [Google Scholar]
- Matoba, T.; Shimokawa, H.; Nakashima, M.; Hirakawa, Y.; Mukai, Y.; Hirano, K.; Kanaide, H.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Invest 2000, 106, 1521–1530. [Google Scholar]
- Matoba, T.; Shimokawa, H.; Morikawa, K.; Kubota, H.; Kunihiro, I.; Urakami-Harasawa, L.; Mukai, Y.; Hirakawa, Y.; Akaike, T.; Takeshita, A. Electron spin resonance detection of hydrogen peroxide as an endothelium-derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler. Thromb. Vasc. Biol 2003, 23, 1224–1230. [Google Scholar]

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Burgoyne, J.R.; Eaton, P. Oxidant Sensing by Protein Kinases A and G Enables Integration of Cell Redox State with Phosphoregulation. Sensors 2010, 10, 2731-2751. https://doi.org/10.3390/s100402731
Burgoyne JR, Eaton P. Oxidant Sensing by Protein Kinases A and G Enables Integration of Cell Redox State with Phosphoregulation. Sensors. 2010; 10(4):2731-2751. https://doi.org/10.3390/s100402731
Chicago/Turabian StyleBurgoyne, Joseph R., and Philip Eaton. 2010. "Oxidant Sensing by Protein Kinases A and G Enables Integration of Cell Redox State with Phosphoregulation" Sensors 10, no. 4: 2731-2751. https://doi.org/10.3390/s100402731