Metaphase FISH on a Chip: Miniaturized Microfluidic Device for Fluorescence in situ Hybridization

Abstract
: Fluorescence in situ Hybridization (FISH) is a major cytogenetic technique for clinical genetic diagnosis of both inherited and acquired chromosomal abnormalities. Although FISH techniques have evolved and are often used together with other cytogenetic methods like CGH, PRINS and PNA-FISH, the process continues to be a manual, labour intensive, expensive and time consuming technique, often taking over 3 5 days, even in dedicated labs. We have developed a novel microFISH device to perform metaphase FISH on a chip which overcomes many shortcomings of the current laboratory protocols. This work also introduces a novel splashing device for preparing metaphase spreads on a microscope glass slide, followed by a rapid adhesive tape-based bonding protocol leading to rapid fabrication of the microFISH device. The microFISH device allows for an optimized metaphase FISH protocol on a chip with over a 20-fold reduction in the reagent volume. This is the first demonstration of metaphase FISH on a microfluidic device and offers a possibility of automation and significant cost reduction of many routine diagnostic tests of genetic anomalies.1. Introduction
During the last decade microfluidic techniques have evolved into a new genre of research areas targeting integration of laboratory protocols into miniaturized devices called lab on chip (LOC) or micro total analysis systems (μtas) [1–5]. The ability to control small sample volumes on micro-sized devices is immensely appealing for techniques involving handling of ultra small volumes of cells or other analytical samples [6]. This has spurred an exponential growth in the number of research articles published in the field of LOC systems targeting complex biological protocols to benefit from the low volume, high throughput and low cost features provided by the microfluidic devices [6–9]. We have created a novel metaphase FISH chip to benefit from the low dead volumes associated with microfluidic devices as FISH protocol reagents and commercial probes are more expensive than gold (over $400 for 100 μL of probe used in this work) [10,11].
FISH is a sensitive diagnostic cytogenetic tool routinely used to visualize numerical and structural chromosomal aberrations [12–20]. The traditional FISH protocol includes steps like immobilization of interphase nuclei or metaphase chromosomes, probe labeling, RNAse treatment, denaturation of chromosomal and probe DNA, hybridization, post hybridization wash and image processing. FISH is routinely used by cytogeneticists in applications ranging from chromosome labeling and mapping, identification of gene expression sites, tissue analysis, mRNA synthesis tracking, tumour genetic alterations monitoring, identifying infections from viruses and other diagnostic pathological applications including cancer e.g., leukemia [21,22].
Interphase and Metaphase FISH are two types of commonly used chromosomal FISH techniques. Each has their specific applications and advantages. Interphase FISH is used to identify numerical abnormalities as well as specific structural abnormalities. Thus, Interphase FISH depends on a specific probe, e.g., to detect known translocations, microdeletions or specific chromosomes and hence can only be used to address questions for which DNA probes are available. Lack of conformity of interphase FISH is a major disadvantage when using this technique for prenatal diagnostics [23]. On the other hand, metaphase FISH can be used to visualize the insertion, deletion or other rearrangement involving a specific region of the genome, with a resolution determined by the probe used [17]. Metaphase FISH can be performed on samples with unknown translocations by targeting all the chromosomes using multi-color FISH probes [24], derived from plasmids, cosmids, bacterial artificial chromosomes (BAC) and yeast artificial chromosomes (YAC) [18]. Recently Interphase FISH was demonstrated on a microfluidic device which highlights the obvious benefits of miniaturizing and automating the FISH protocol in a microfluidic system [10,11,25]. But metaphase FISH protocol has been elusive owing to the difficulty of handling chromosomes on a chip and fixing the chromosomes on a closed microfluidic device [25]. As a result, in spite of FISH being a very powerful cytogenetic tool, it continues to be an expensive and reasonably time consuming method. There is clearly a need for replacing the traditional method with a fast and low-cost method making this technique widely available and easy to handle.
Our efforts have been directed at designing a miniaturized protocol for performing metaphase FISH in a controlled manner on a microfluidic device. It is widely known that the results of classical banding techniques, FISH analysis or Comparative Genomic Hybridization (CGH) are dependent on the quality of the metaphase spreads [26]. Hence the result of any FISH analysis depends on consistency of spreading of chromosomes [27]. During the 90s metaphase spreading techniques were widely investigated and many labs developed their own version of optimized standard methods [28–34]. This led to a number of theories on what determines the quality of metaphase spreads on the glass slide, including the distance and the angle of dropping of the fixed cells onto the slide, the diameter of the pipette, the evaporation of the fixative, the temperature of the slide, the hypotonic treatment of the chromosomes, and the whole air drying process [27,35,36]. Renewed interest has seen a number of attempts towards making devices for preparation of chromosome spreads, which rely on controlled angle and conveyors [37], temperature gradient and humidity [26] and most recently the chromosome dropper tool which relies on dropping angle and height for fixing metaphase spreads on glass slides [38]. But none of these three devices are remotely close to the miniaturized size we are targeting or give any new insights into the mechanism of spreading compared to what was already published in the 90s. Through literature review and our own preliminary experiments we found that the basis of chromosome spreading is rooted in the optimum rate of evaporation of the fixative from the glass slide. Hliscs et al. suggests that the mechanism of chromosome spreading is a slow process, which leads to stretching of chromosomes via flattening [39]. Spurbeck et al. mentioned that in the process of spreading the fixative evaporates, leading to build up of surface tension causing the metaphase cell to flatten, which eventually leads to bursting of the cell membrane and spreading of the chromosomes [27]. Henegariu et al. also concluded that dropping of chromosomes from a height doesn’t improve the spreading [26]. Hence, we concluded that in order to realize a micro-splashing device which produces reliable metaphase spreads on a glass slide sufficient for conducting routine FISH analysis; we need to incorporate a mechanism to allow for optimum evaporation of the fixative leading to stretching of the chromosomes and flattening of the cells. This could be aided by environmental factors like temperature of the slide and humidity but the focus has to be on maintaining an optimum rate of fixative evaporation.
In order to account for that, we devised a novel splashing device with open chamber, which allows for easy evaporation of the fixative. The device provides 11 mm dropping height with two inlets—one for cold water and one for the fixed mitotic cells suspension. Apart from the splashing device, a novel metaphase FISH protocol was developed using a microFISH device. This microFISH device provides the possibility of replacing the traditional Coupling Jar and turning it into a miniaturized microfluidic device. We have looked into possible replacements of glass slides with various common polymers but found them unfavorable for FISH protocol on mainly three counts. Firstly, the spreading of the metaphase chromosomes on the glass slides is highly aided by the surface properties of the glass like contact angle and free surface energy. Secondly, polymers exhibit auto-fluorescent behaviour which hinders the analysis due to interference with the FISH probe signals. Finally, some polymers like PMMA couldn’t withstand the fixative used for fixing the metaphase chromosomes and cracked on contact. Hence, we found it ideal to continue using the traditional microscope glass slides as substrate which also offers the benefits of easy integration into existing workplace protocols in cytogenetic labs. In order to minimize the time of fabrication of the microFISH device, an easy protocol for rapid bonding based assembly has been developed. With this the glass slide can be turned into a microFISH device in a matter of seconds by using an adhesive tape-based stencil and bonding technique (described later in Fabrication section). The ease of operation and handling in this protocol has been targeted to allow non-technical personnel to easily conduct these tests. The rapid fabrication protocol leaves room for conducting multiple tests simultaneously. In addition, the novel protocol allows for an over 20-fold reduction in reagent volume, which is significant considering the costs of commercially available probes. We hope that this first demonstration of metaphase FISH on a chip, will spur renewed efforts in automating metaphase FISH on a chip leading to wider access to low cost, reliable and fast genetic diagnostics in clinical environments. Although the presented device has been developed for metaphase FISH, we feel that the protocol could also be applied for Interphase FISH.
2. Experimental Section
2.1. Materials and Chemicals
Polymethylmethacrylate (PMMA) sheets procured from Nordplast (Denmark) were used to fabricate the Splashing device; Polydimethylsiloxane (commonly known as PDMS, Sylgard 184, Dow Corning, USA) was used to fabricate the microFISH device. Glass slides (SuperFrost), syringes and silicone tubing were obtained from traditional suppliers (Sigma-Aldrich, VWR). SU8-2075 resist was ordered from Microchem (Germany). Double-sided adhesive tape AR100 (50 μm thickness) was procured from Adhesive Research Inc. (Ireland). All the reagents used in the FISH experiment were purchased from Invitrogen (Paisley, UK). 1 μL of RNAse (10 μg/μL) stock solution was mixed with 99 μL of 2 × SSC for removal of RNA. The probe was purchased from Kreatech Diagnostics (Amsterdam, The Netherlands) and it was used as recommended by the manufacturer. 20 × SSC (sodium citrate, sodium chloride) was purchased from G-Biosciences (USA) and further dilutions were made using Milli-Q water. The stock of absolute ethanol was used to make dilutions (50%, 60%, 90%, 99% ethanol). 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen) was used as a counterstain used for colouring the chromosomes. Blood sample of a female patient was received from the Wilhelm Johannsen Centre for Functional Genome Research, Department of Cellular and Molecular Medicine. Cold water used for spreading chromosomes was kept at 4 °C.
2.2. Apparatus
A micromilling machine (Folken Industries, Glendale, USA) was used for milling the splashing device, 50 W CO2 Laser Machine (Synrad Inc., USA) was used for ablating the adhesive tapes, a photolithographic spinner and aligner from Carl-Zeiss were used for fabricating the master mould for PDMS microFISH device and a Zeiss Axio Observer Z1 Fluorescent microscope was used for analysis of the spreads and FISH signals.
2.3. Fabrication
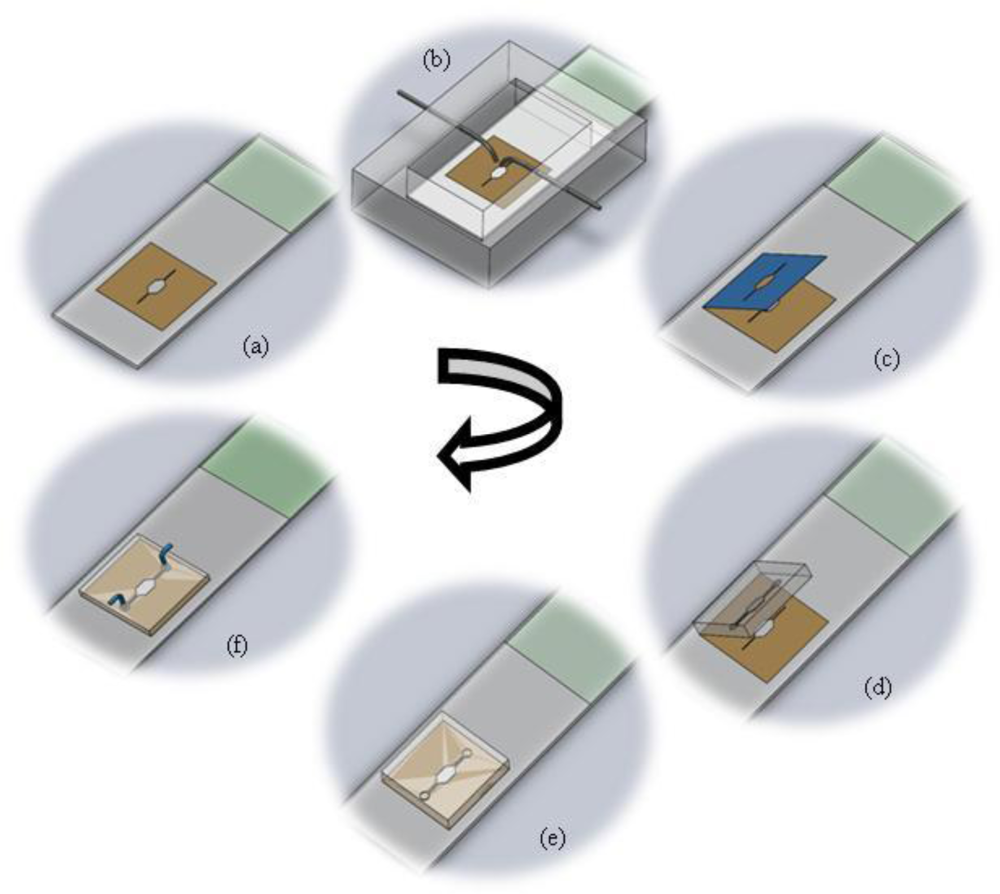
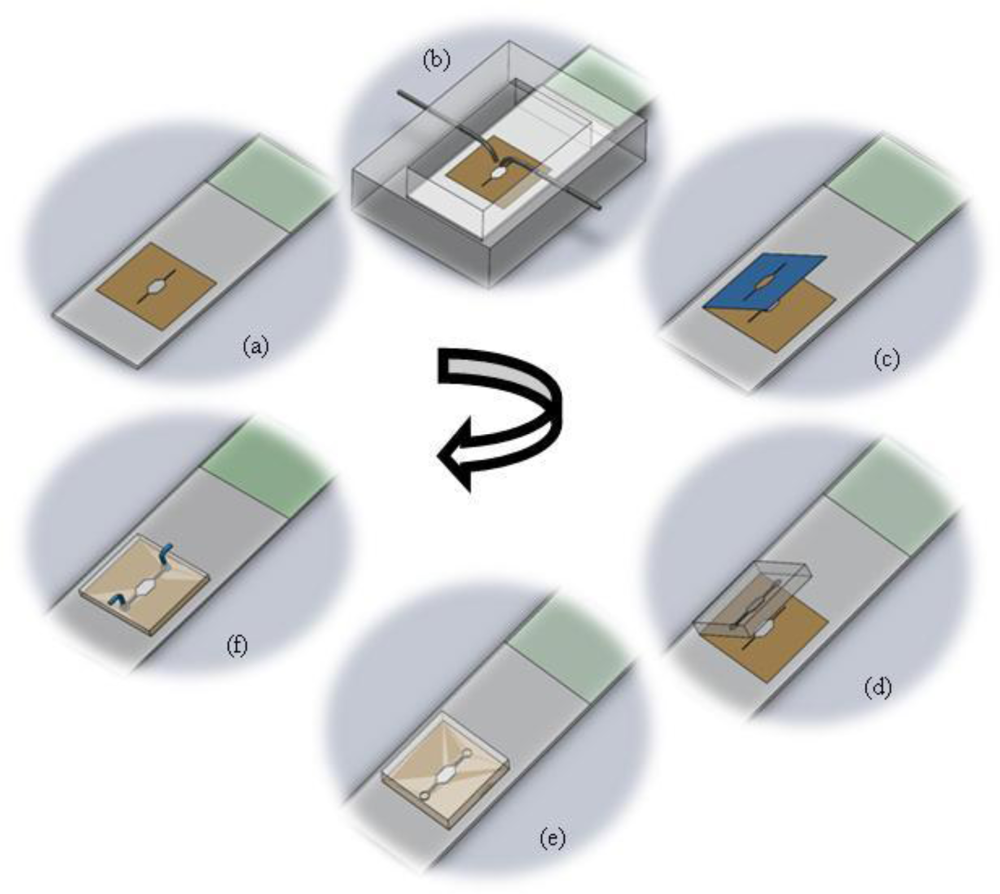
The fabrication protocol has been summarized in Figure 1. The figure details the protocol for splashing metaphase spreads on the glass slide using the splashing device followed by the rapid fabrication of the microFISH device to perform the metaphase FISH analysis protocol. The details of the fabrication protocol will be explained in the following subsections.
2.3.1. Glass Slide with Stencil
The stencil for localization of metaphase spreads is created using a double-sided medical grade tape. The double-sided adhesive tape is fabricated using a laser ablation process with a CO2 laser [40–42]. The laser is operated at 20 W power at 250 mm/s laser velocity using resolution of 800. The operating parameters are set using the Winmark software (Synrad Inc, USA). The design of the tape corresponds to the design of the microFISH device (Figure 2). When the tape is ablated using the CO2 laser, one side of the tape cover is peeled and the tape is bonded to the centre of the glass slide as shown in Figure 2. This tape-glass slide complex acts as a stencil when the glass slide is used in the splashing chip to create metaphase spreads. The localization of the metaphase spreads is achieved by removing the top cover of the tape which allows selective patterning on the glass slide in the centre where the tape is fully ablated using the CO2 laser for creating the microFISH chamber.
2.3.2. Splashing Device
The splashing device is fabricated in two PMMA layers by a micro-milling process. The bottom part contains the sliding chamber for glass slide insertion and the top lid contains the open chamber for evaporation of the fixative. It also contains the two inlet ports for fixed chromosomes and cold water (Figure 3). The syringes are slightly bent towards the end to focus the water and chromosome suspension on to the centre of the stencil which exposes the glass slide. The two PMMA layers are either bonded together using thermal bonding [42] or screwed together as shown in Figure 3.
2.3.3. Master for PDMS Chip and Moulding of the PDMS MicroFISH Chip
The master for the microFISH device lid was prepared in a cleanroom using a traditional photolithography process. The structures were created on a Silicon wafer in SU-8 photoresist using negative patterning. The recipe for creating the master is shown in Table 1. This master can be used several times for moulding the microFISH device lid (Figure 4). The microFISH device lid is moulded in PDMS using traditional soft lithography techniques described in [43] and peeled away from the Silicon/SU-8 master. The final PDMS microFISH device lid is shown in Figure 4. The lid is then bonded on the glass slide with metaphase spreads for assembling the microFISH device.
2.3.4. Assembly of the MicroFISH Device
The double-sided adhesive tape stencil used for spreading chromosomes is now used as a bonding layer for assembling the microFISH device. Peeling the top cover off the tape provides a silicone bonding layer for the microFISH device lid (Figure 5). The PDMS lid is irreversibly attached to the glass slide using the silicone adhesive layer by gently applying pressure across the PDMS lid (Figure 5). Once the device is assembled the holes are made in the PDMS lid for access ports to create interconnections.
2.3.5. Interconnects
The access holes in the microFISH device lid are made on the top side in the PDMS lid. The holes are made using a 22 gauge syringe needle (0.7 mm outer diameter). The interconnects are formed by inserting an 18 gauge syringe (1.2 mm outer diameter) with a bigger outer diameter than the interconnection holes, which forms a tight seal into the microFISH device. The syringes are connected to silicone tubing with inner diameter 1 mm and outer diameter 3 mm to provide the world-to-microFISH device contacts (Figure 6). In order to test the device, the silicone tubings are connected with syringe pumps (not shown in the figure) which provides the pressure needed to actuate the reagents and probes through the microFISH device.
2.4. Procedures
2.4.1. Splashing Protocol
The chromosome suspensions used for creating the metaphase spreads were prepared by standard methods [22]. Peripheral blood lymphocyte cultures were prepared and treated with 75 mM KCl hypotonic solution for 5 minutes during the harvesting stage. Claussen et al. signified the importance of proper hypotonic treatment for achieving good quality metaphase spreads [31]. The hypotonic treatment causes the mitotic cells to swell leading to the chromosomes moving to more peripheral locations, which allows for evenly spreading of the chromosomes during fixative evaporation. After the mitotic cells are treated with the hypotonic solution they are fixed using the fixative (methanol/acetic acid, 3:1). The fixative is added drop by drop to the cells all the while mixing thoroughly on a vortex. To compare the spreads achieved using the traditional dropping method and the splashing device, control slides were prepared using the manual dropping method and test slides were prepared using the splashing device. Before dropping the cell suspension on the control slides, the slides are kept in distilled water at 4 °C. On the other hand, the test slides were prepared using the splashing device, where one drop of glacial water was dropped on to the slide followed by a drop of the fixed mitotic cell suspension through the two dedicated inlets (Figure 7). The slides were allowed to air dry in the open chamber of the splashing device. As a final step, both test and control slides were heated at 75 °C degrees for 3 min, which improves the fixation of the chromosomes to the slide.
2.4.2. FISH Protocol
The FISH protocol was conducted on both the control and test slides using the microFISH device. After assembling the microFISH device using the glass slides with chromosome spreads, the optimized FISH protocol was conducted on all the slides. The inlet of the microFISH device was connected with a syringe pump for treating the metaphase spreads with the FISH reagents and probes. The specific temperatures associated with the FISH protocol were provided by a hot plate setup. Firstly, RNAse (10 μg/μL) was delivered through the inlet. Following the RNAse treatment, the microFISH device was kept in the humidity chamber at 37 °C for one hour. Later 2 × SSC was loaded at room temperature for washing the metaphase spreads. The microFISH device was then dehydrated with increasing percentages of ethanol (70%, 80% and 99%), and the device was heated at 75 °C to denature the chromosomal DNA for 5 minutes. Simultaneously, probe DNA was denatured at 75 °C for 5 minutes in the water bath. Then the probe was entered into the device and the inlet and outlet were closed to avoid evaporation of the probe. The microFISH device was incubated in the humidity chamber at 37 °C overnight for hybridization. Subsequently, 50% formamide was delivered as post hybridization wash at 42 °C. Final washing was performed with 0.1 × SSC at 60 °C, 4 × SSC and 1 × PBS at room temperature. The details of the conventional FISH protocol and optimized FISH protocol can be found in Table 2.
2.4.3. Analysis
DAPI was applied to the hybridized targets on the test slides in microFISH chamber and in case of control samples, slides were mounted with cover slips. The Zeiss AxioObserver Z1 fluorescent microscope was used to view and analyze the samples for counting the metaphase spreads and the FISH hybridization signals. The chromosomes spreads on the control and test slides were counted manually using the traditional method.
3. Results and Discussion
3.1. Splashing Device and Metaphase Spreads
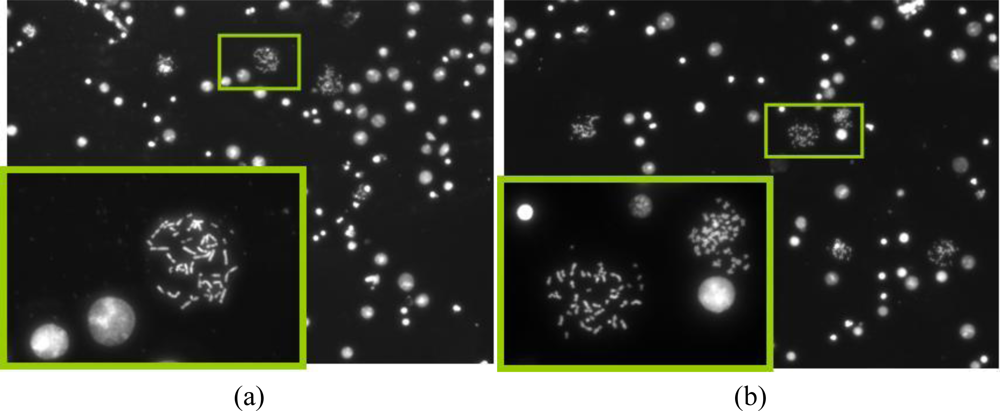
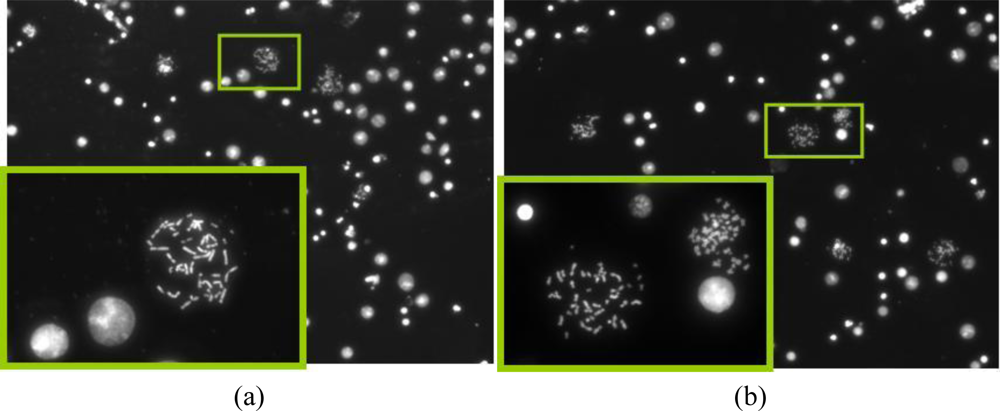
The motivation behind the splashing device was to create a microfluidic device which can provide reliable and sufficient number of metaphase spreads on a glass slide. After spreading the chromosomes on the slide using the traditional method and the splashing device, the slides were stained with DAPI. The metaphase spreads were counted manually in the fluorescence microscope at 20× magnification. Figures 8 and 9 show the images of the metaphase spreads obtained using the conventional method and splashing device respectively. In order to validate the splashing device protocol, we conducted tests using two different cell suspension samples. Table 3 shows the comparison of average spreads obtained using the two techniques.
Even though the average numbers of spreads obtained by using the splashing device are comparatively lower, we could obtain sufficiently good chromosome spreads in order to perform the FISH analysis. The lower chromosome spread counts may be due to the splashing conditions such as height, temperature and humidity as described in the introduction. While in average the number of chromosome spreads was significantly lower in the splashing device, it must be noted that in certain samples, we found the number of chromosome spreads to be higher compared to the control slides. Considering that the splashing experiments were not studied in detail, we believe that the splashing protocol can be optimized by altering and controlling the various other factors like temperature, humidity, etc.
Moreover, the simple fabrication protocol of the splashing device using micro milling of PMMA sheets allows us to control the height of splashing if needed by addition of more PMMA layers to increase the height of the splashing syringe. The integrated stencil and bonding layer of the adhesive tape allows for localization of the metaphase spreads on the glass slide in the splashing device. Thus, the splashing device not only provides an ability to reliably create metaphase spreads on glass slide but also provides a possibility of automating the image analysis procedure in the future due to ease of finding the metaphase spread using software and an automated stage. A number of companies (Zeiss, BioView and Vysis) and research groups are working on optimizing the data acquisition protocols as this continues to be a major issue after the FISH protocol [44,45]. Automation of the analysis procedure is currently an on-going work where we are aiming to divide the microFISH device chamber into smaller segments to create smaller FISH microchambers corresponding to the microscope objective.
3.2. FISH Protocol Results
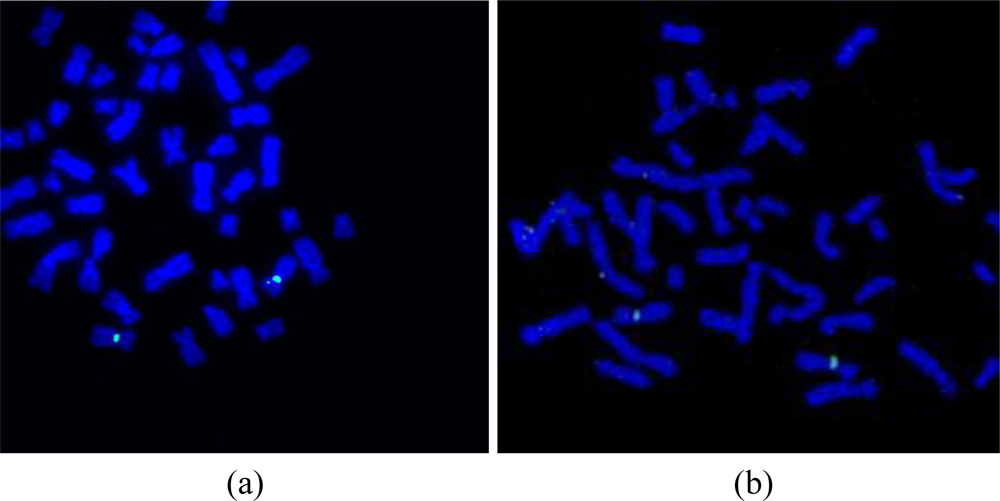
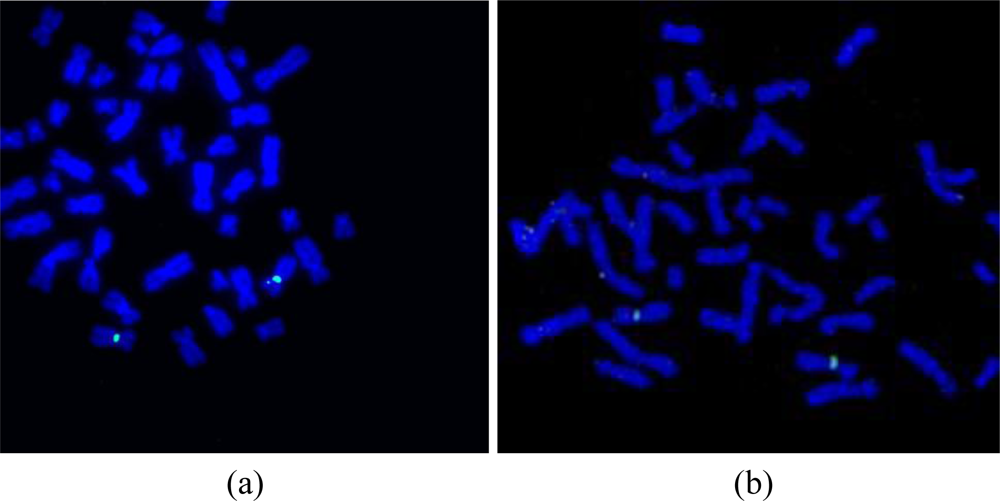
After the preparation of the chromosome spreads, FISH analysis was performed on the peripheral blood lymphocyte chromosomes using an X chromosome centromeric probe. A female patient sample was used in order to show the validation of the microFISH device protocol and the results obtained with XX chromosomes (Figure 9(b)) confirm the successful FISH analysis result. The conventional FISH protocol was also performed on the slides to compare and evaluate the microFISH device efficiency (Figure 9(a)). As can be seen in Table 2, the reagent volume needed in traditional FISH analysis is 327 mL, but now has been reduced to only 14 mL, which is a major step in reducing the costs associated with FISH analysis. As mentioned earlier, the most expensive reagent in traditional FISH analysis is the probe (costing approx. $90 per test). In the microFISH protocol the volume of this has been reduced to half (highlighted in yellow in the Table 2). This alone cuts the cost associated with conducting the routine FISH analysis in half. Considering the cost of fabricating these microFISH devices is less than $2, these successful FISH results will give a major push towards making genetic analysis a routine screening test.
4. Conclusions
We have successfully demonstrated the first metaphase FISH on a microfluidic device. This work also presents an alternative method for slide preparation and hybridization of FISH probes. The achievements gained in the present study will be used in further improvement of the methodology and aim to develop a completely automated system for performing miniaturized FISH on chip. The micro splashing device designed for spreading metaphase chromosomes on a glass slide provides more reliable and easy alternative for creation of metaphase spreads. The rapid and easy assembly protocol for the microFISH device allows for quick transformation of a simple glass slide into a microFISH device, which makes it an ideal solution for integration into existing work routines at cytogenetic labs. Our current efforts are focused on further miniaturization of this device, which will offer significant benefits with respect to sample preparation and reduction in reagent costs. We are also working towards improving the chromosome spreading protocol by further miniaturization of the splashing chamber and localized micro-spotting of fixed cells, optimizing the hybridization efficiency using temperature and electro-kinetic effects, automating the protocol by incorporation of reagent reservoirs and improving the analysis by developing automated software for data acquisition and metaphase spread recognition. In the longer run, we envisage to create a chromosome total analysis system providing a more efficient and cheaper solution to the traditional protocol and enable faster diagnosis.
Acknowledgments
The authors would like to thank FTP and Lundbeck for their financial support. Authors would also like to acknowledge the collaborators at Panum and Kennedy Institute and especially thank Elisabeth Larsen for technical assistance and Asli Silahtaroglu for providing samples for conventional FISH analysis and useful technical discussions, Jaya Kandi for schematic 3D diagrams and Peder Skafte-Pedersen for assistance with microscope for image acquisition. Wilhelm Johannsen Centre for Functional Genome Research is established by The Danish National Research Foundation.
References
- Reyes, DR; Iossifidis, D; Auroux, PA; Manz, A. Micro Total Analysis Systems. 1. Introduction, Theory, and Technology. Anal. Chem 2002, 74, 2623–2636. [Google Scholar]
- Auroux, PA; Iossifidis, D; Reyes, DR; Manz, A. Micro Total Analysis Systems. 2. Analytical Standard Operations and Applications. Anal. Chem 2002, 74, 2637–2652. [Google Scholar]
- Vilkner, T; Janasek, D; Manz, A. Micro Total Analysis Systems. Recent Developments. Anal. Chem 2004, 76, 3373–3385. [Google Scholar]
- Dittrich, PS; Tachikawa, K; Manz, A. Micro Total Analysis Systems. Latest Advancements and Trends. Anal. Chem 2006, 78, 3887–3907. [Google Scholar]
- West, J; Becker, M; Tombrink, S; Manz, A. Micro Total Analysis Systems: Latest Achievements. Anal. Chem 2008, 80, 4403–4419. [Google Scholar]
- Castillo, J; Tanzi, S; Dimaki, M; Svendsen, W. Manipulation of Self-Assembly Amyloid Peptide Nanotubes by Dielectrophoresis. Electrophoresis 2008, 29, 5026–5032. [Google Scholar]
- Shah, P; Dimaki, M; Svendsen, WE. A Novel Passive Microfluidic Device for Preprocessing Whole Blood for Point of Care Diagnostics. Proceedings of the 15th International Conference on Solid-State Sensors, Actuators and Microsystem Transducers, Denver, CO, USA, 21–25 June, 2009; pp. 417–420.
- Dimaki, M; Clausen, CH; Lange, J; Shah, P; Jensen, LB; Svendsen, W. A Microfabricated Platform for Chromosome Separation and Analysis. Proceedings of the 17th International Vacuum Congress/13th International Conference on Surface Science/International Conference on Nanoscience and Technology, Stockholm, Sweden, 2–6 July, 2007; Johansson, LSO, Andersen, JN, Gothelid, M, Helmersson, U, Montelius, L, Rubel, M, Setina, J, Wernersson, LE, Eds.;
- Crane, MM; Chung, K; Stirman, J; Lu, H. Microfluidics-Enabled Phenotyping, Imaging, and Screening of Multicellular Organisms. Lab Chip 2010, 10, 1509–1517. [Google Scholar]
- Sieben, VJ; Marun, CSD; Pilarski, PM; Kaigala, GV; Pilarski, LM; Backhouse, CJ. FISH and Chips: Chromosomal Analysis on Microfluidic Platforms. IET Nanobiotechnol 2007, 1, 27–35. [Google Scholar]
- Sieben, VJ; Debes-Marun, CS; Pilarski, LM; Backhouse, CJ. An Integrated Microfluidic Chip for Chromosome Enumeration Using Fluorescence in situ Hybridization. Lab Chip 2008, 8, 2151–2156. [Google Scholar]
- Tepperberg, J; Pettenati, MJ; Rao, PN; Lese, CM; Rita, D; Wyandt, H; Gersen, S; White, B; Schoonmaker, MM. Prenatal Diagnosis Using Interphase Fluorescence in situ Hybridization (FISH): 2-Year Multi-Center Retrospective Study and Review of the Literature. Prenatal Diag 2001, 21, 293–301. [Google Scholar]
- Leung, WC; Waters, JJ; Chitty, L. Prenatal Diagnosis by Rapid Aneuploidy Detection and Karyotyping: A Prospective Study of the Role of Ultrasound in 1589 Second-Trimester Amniocenteses. Prenatal Diag 2004, 24, 790–795. [Google Scholar]
- Evans, MI; Henry, GP; Miller, WA; Bui, TH; Snidjers, RJ; Wapner, RJ; Miny, P; Johnson, MP; Peakman, D; Johnson, A; Nicolaides, K; Holzgreve, W; Ebrahim, SAD; Babu, R; Jackson, L. International, Collaborative Assessment of 146,000 Prenatal Karyotypes: Expected Limitations If Only Chromosome-Specific Probes and Fluorescent in situ Hybridization Are Used. Hum. Reprod 1999, 14, 1213–1216. [Google Scholar]
- Ward, BE; Gersen, SL; Carelli, MP; McGuire, NM; Dackowski, WR; Weinstein, M; Sandlin, C; Warren, R; Klinger, KW. Rapid Prenatal-Diagnosis of Chromosomal Aneuploidies by Fluorescence in situ Hybridization—Clinical Experience with 4,500 Specimens. Amer. J. Hum. Genet 1993, 52, 854–865. [Google Scholar]
- Jiang, F; Katz, RL. Use of Interphase Fluorescence in situ Hybridization as a Powerful Diagnostic Tool in Cytology. Diagn. Mol. Pathol 2002, 11, 47–57. [Google Scholar]
- Laan, M; Kallioniemi, OP; Hellsten, E; Alitalo, K; Peltonen, L; Palotie, A. Mechanically Stretched Chromosomes as Targets for High-Resolution Fish Mapping. Genome Res 1995, 5, 13–20. [Google Scholar]
- Korenberg, JR; Yangfeng, T; Schreck, R; Chen, XN. Using Fluorescence in situ Hybridization (FISH) in Genome Mapping. Trends Biotech 1992, 10, 27–32. [Google Scholar]
- Andreeff, M; Pinkel, D. Introduction to Flourescence in situ Hybridization: Principles and Clinical Applications; Wiley-Liss: New Jersey, NJ, USA, 1999; pp. 223–238. [Google Scholar]
- Weise, A; Liehr, T. Fluorescence in situ Hybridization for Prenatal Screening of Chromosomal Aneuploidies. Expert Rev. Mol. Diagn 2008, 8, 355–357. [Google Scholar]
- Jain, KK. Current Status of Fluorescent in situ Hybridization. Med. Device Technol 2004, 15, 14–17. [Google Scholar]
- Tanas, MR; Goldblum, JR. Fluorescence in situ Hybridization in the Diagnosis of Soft Tissue Neoplasms: A Review. Adv. Anat. Pathol 2009, 16, 383–391. [Google Scholar]
- Lee, C; Lemyre, E; Miron, PM; Morton, CC. Multicolor Fluorescence in situ Hybridization in Clinical Cytogenetic Diagnostics. Curr. Opin. Pediatr 2001, 13, 550–555. [Google Scholar]
- Liehr, T; Starke, H; Heller, A; Kosyakova, N; Mrasek, K; Gross, M; Karst, C; Steinhaeuser, U; Hunstig, F; Fickelscher, I; Kuechler, A; Trifonov, V; Romanenko, SA; Weise, A. Multicolor Fluorescence in situ Hybridization (FISH) Applied to FISH-Banding. Cytogenet. Genome Res 2006, 114, 240–244. [Google Scholar]
- Zanardi, A; Bandiera, D; Bertolini, F; Corsini, CA; Gregato, G; Milani, P; Barborini, E; Carbone, R. Miniaturized FISH for screening if Onco-Hematological Malignancies. BioTechniques 2010, 49, 497–504. [Google Scholar]
- Henegariu, O; Heerema, NA; Wright, LL; Bray-Ward, P; Ward, DC; Vance, GH. Improvements in Cytogenetic Slide Preparation: Controlled Chromosome Spreading, Chemical Aging and Gradual Denaturing. Cytometry 2001, 43, 101–109. [Google Scholar]
- Spurbeck, JL; Zinsmeister, AR; Meyer, KJ; Jalal, SM. Dynamics of Chromosome Spreading. Am. J. Med. Genet 1996, 61, 387–393. [Google Scholar]
- Barch, MJ; Lawce, HJ; Arsham, MS. Peripheral Blood Culture, 2nd ed; Raven Press: New York, NY, USA, 1991; pp. 17–30. [Google Scholar]
- Chattopadhyay, S; Marinduque, B; Gilbert, F. Ovarian-Cancer—Protocol for the Preparation of Banded Metaphases from Tumor-Tissue. Cancer Genet. Cytogenet 1992, 59, 210–212. [Google Scholar]
- Gibas, LM; Grujic, S; Barr, MA; Jackson, LG. A Simple Technique for Obtaining High-Quality Chromosome Preparations from Chorionic Villus Samples Using FDU Synchronization. Prenatal Diag 1987, 7, 323–327. [Google Scholar]
- Lawce, HJ; Brown, MG. Harvesting, Slide-Making and Chromosome Elongation Techniques, 2nd ed; Raven Press: New York, NY, USA, 1991; pp. 31–106. [Google Scholar]
- Miron, PM. Preparation, Culture and Analysis of Amniotic Fluid Samples; John Wiley & Sons: New York, NY, USA, 1995; pp. 831–836. [Google Scholar]
- Sasai, K; Tanaka, R; Kawamura, M; Honjo, K; Matsunaga, N; Nakada, T; Homma, K; Fujimura, H. Chromosome Spreading Techniques for Primary Gastrointestinal Tumors. J. Gastroenterol 1996, 31, 505–511. [Google Scholar]
- Warburton, D. Preparation and Culture of Products of Conception and Other Solid Tissues for Chromosome Analysis; John Wiley & Sons: New York, NY, USA, 1995; pp. 851–856. [Google Scholar]
- Claussen, U; Michel, S; Muhlig, P; Westermann, M; Grummt, UW; Kromeyer-Hauschild, K; Liehr, T. Demystifying Chromosome Preparation and the Implications for the Concept of Chromosome Condensation during Mitosis. Cytogenet. Genome Res 2002, 98, 136–146. [Google Scholar]
- Deng, W; Tsao, SW; Lucas, JN; Leung, CS; Cheung, ALM. A New Method for Improving Metaphase Chromosome Spreading. Cytometry A 2003, 51A, 46–51. [Google Scholar]
- Yamada, K; Kakinuma, K; Tateya, H; Miyasaka, C. Development of an Instrument for Chromosome Slide Preparation. J. Radiat. Res 1992, 33, 242–249. [Google Scholar]
- Qu, YY; Xing, LY; Hughes, ED; Saunders, TL. Chromosome Dropper Tool: Effect of Slide Angles on Chromosome Spread Quality for Murine Embryonic Stem Cells. J. Histotechnol 2008, 31, 75–79. [Google Scholar]
- Hliscs, R; Muhlig, P; Claussen, U. The Spreading of Metaphases Is a Slow Process which Leads to a Stretching of Chromosomes. Cytogenet. Cell Genet 1997, 76, 167–171. [Google Scholar]
- Jensen, MF; Noerholm, M; Christensen, LH; Geschke, O. Microstructure Fabrication with a CO2 Laser System: Characterization and Fabrication of Cavities Produced by Raster Scanning of the Laser Beam. Lab Chip 2003, 3, 302–307. [Google Scholar]
- Bowden, M; Geschke, O; Kutter, JP; Diamond, D. CO2 Laser Microfabrication of an Integrated Polymer Microfluidic Manifold for the Determination of Phosphorus. Lab Chip 2003, 3, 221–223. [Google Scholar]
- Klank, H; Kutter, JP; Geschke, O. CO2-Laser Micromachining and Back-End Processing for Rapid Production of PMMA-Based Microfluidic Systems. Lab Chip 2002, 2, 242–246. [Google Scholar]
- McDonald, JC; Duffy, DC; Anderson, JR; Chiu, DT; Wu, HK; Schueller, OJA; Whitesides, GM. Fabrication of Microfluidic Systems in Poly(Dimethylsiloxane). Electrophoresis 2000, 21, 27–40. [Google Scholar]
- Netten, H; Young, IT; van Vliet, LJ; Tanke, HJ; Vroljik, H; Sloos, WCR. FISH and Chips: Automation of Fluorescent Dot Counting in Interphase Cell Nuclei. Cytometry 1997, 28, 1–10. [Google Scholar]
- Theodosiou, Z; Kasampalidis, LN; Livanos, G; Zervakis, M; Pitas, L; Lyroudia, K. Automated Analysis of FISH and Immunohistochemistry Images: A Review. Cytometry A 2007, 71A, 439–450. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Type | Parameters |
|---|---|---|
| 1 | Spin-coating of SU-8 | Acceleration: 200 rpm/s, Speed: 1,000 rpm, Time 40 seconds |
| 2 | Soft Bake | Temperature: 50 °C, Time: 5 hrs, Ramp up Time: 15 mins |
| 3 | Exposure | Time: 30 seconds Type: Soft |
| 4 | Post Exposure Bake | Temperature: 50 °C, Time: 5 hrs, Ramp up Time: 15 mins |
| 5 | Development | First: 4 minute, Final: 1 minute |
| Conventional protocol | microFISH protocol | |||
|---|---|---|---|---|
| Step No.: | Name of the step | Vol | Vol | Flow rate |
| 1
| RNAse
| 100 μL | 25 μL | 5 μL/min |
| 2
| Wait 60 mins
| No flow | ||
| 3
| Wash 2 × SSC
| 75 mL | 3 mL | 200 μL/min |
| 4
| 70% Alcohol
| 25 mL | 1 mL | 200 μL/min |
| 5
| 90% Alcohol
| 1 mL | 1 mL | 200 μL/min |
| 6
| 100% Alcohol
| 1 mL | 1 mL | 200 μL/min |
| 7
| Drying-Air
| NA | ||
| 8
| 70% formamide
| 25 mL | NA | |
| 9
| 90% Alcohol
| 25 mL | NA | |
| 10
| 100% Alcohol
| 25 mL | NA | |
| 11
| Probe
| 10 μL | 5 μL | 2 μL/min |
| 12
| Hybridisation time
| Overnight | Overnight | No flow |
| 13
| 50% Formamide
| 75 mL | 3 mL | 200 μL/min |
| 14
| 0.1 × SSC
| 25 mL | 3 mL | 200 μL/min |
| 15
| 4 × SSC
| 25 mL | 1 mL | 200 μL/min |
| 16
| 1 × PBS
| 25 mL | 1 mL | 100 μL/min |
| 17 | DAPI | 15 μL | 5 μL | 2 μL/min |
| Total Reagent volume | 327.2 mL | 14.1 mL | ||
| Sample 1 | Sample 2 | |
|---|---|---|
| Control Slide | 41 | 52 |
| Splashing device | 29 | 34 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Vedarethinam, I.; Shah, P.; Dimaki, M.; Tumer, Z.; Tommerup, N.; Svendsen, W.E. Metaphase FISH on a Chip: Miniaturized Microfluidic Device for Fluorescence in situ Hybridization. Sensors 2010, 10, 9831-9846. https://doi.org/10.3390/s101109831
Vedarethinam I, Shah P, Dimaki M, Tumer Z, Tommerup N, Svendsen WE. Metaphase FISH on a Chip: Miniaturized Microfluidic Device for Fluorescence in situ Hybridization. Sensors. 2010; 10(11):9831-9846. https://doi.org/10.3390/s101109831
Chicago/Turabian StyleVedarethinam, Indumathi, Pranjul Shah, Maria Dimaki, Zeynep Tumer, Niels Tommerup, and Winnie E. Svendsen. 2010. "Metaphase FISH on a Chip: Miniaturized Microfluidic Device for Fluorescence in situ Hybridization" Sensors 10, no. 11: 9831-9846. https://doi.org/10.3390/s101109831