5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one

Department of Chemistry, Faculty of Science and Technology, Airlangga University, Surabaya 60115, Indonesia
*
Author to whom correspondence should be addressed.
Molbank 2018, 2018(3), M1013; https://doi.org/10.3390/M1013
Submission received: 2 August 2018
/
Revised: 19 August 2018
/
Accepted: 20 August 2018
/
Published: 23 August 2018
(This article belongs to the Collection Molecules from Catalytic Processes)
Abstract
:The title compound was prepared by a two-step reaction. The first step was the formation of a chalcone derivative using Claisen–Schmidt condensation, which was followed by the Michael addition of the formed chalcone with 1,3-dimethylbarbituric acid. The structure of the prepared compound was established by spectral data: FTIR, HRESIMS, 1H- and 13C-NMR.
1. Introduction
Compounds possessing a dihydropyrimidine (DHPM) core attract the interest of researchers, either due to their wide spectrum bioactivities or from a synthesis point of view. The core of dihydropyrimidine can be constructed from a C–C–C and N–C–N scaffold, which may be formed by a Biginelli reaction [1] or cyclocondensation between an enone and urea or its analog. In general, preparation of a dihydropyrimidine derivative through cyclocondensation can be achieved by a reaction between chalcone as the source of the C–C–C unit and urea or its analog as the source of the N–C–N unit.
The molecular structure of dihydropyrimidine from a Biginelli product has a close resemblance to Hantsch 1,4-dihydropyridine, both being aza-analogues of nifedipine, which is well known as a calcium channel modulator [2]. Furthermore, DHPM derivatives are also known to exhibit antihypertensive [3], potassium channel antagonistic [4], antifilarial [5], anti-HIV [6,7], and antitumor [8] activities.
In continuing of our research, we intended to synthesize a 5,7-diphenyl-5,8-dihydropyrido [2,3-d]pyrimidine-2,4(1H,3H)-dione (3) derivative through a Hantzsch cyclocondensation-type reaction constructed from a chalcone derivative (serving as the C–C–C unit) and 1,3-dimethylbarbituric acid (serving as the dioxopyrimidine unit) in the presence of ammonium chloride using triethylamine (TEA) acting as a Lewis base catalyst. Unfortunately, based on the spectroscopic evidence, the reaction stopped at the Michael addition process and did not proceed further to cyclocondensation. In this paper, we describe the synthesis and the characterization of the title compound.
2. Results
The title compound was synthesized in a two-step reaction. The synthesis was started by chalcone preparation employing Claisen–Schmidt condensation according to the procedure reported by Suwito et al., (2014) [9]. The next step was Michael addition between the chalcone with 1,3-dimethylbarbituric acid using TEA as a catalyst. The reaction process is displayed in Figure 1 below. In this article, we discuss only the preparation and structural characterization of compound 2 because compound 1 is already known. The title compound 2 was obtained as a white solid (171 mg; 34%).
5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethyl-pyrimidine-2,4,6-trione: White solid (171.7 mg, 34%); Rf 0.512 (CHCl3:ethyl acetate:n-hexane = 2:1:1); HRESIMS [M − H]− calculated for C23H22N2O6Br 501.0661, found 501.0774; IR (DRS, KBr, cm−1): 3080 (C–H aromatic), 2958 (m, CH aliphatic), 1745 (str, C=O ketone), 1618 (str, C=O amide), 1581.63 (str, C=C aromatic), 1203 (str, C–O–C ether), and 717 (C–Br); 1H-NMR (400 MHz, DMSO-d6) δH (ppm) 7.92 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.9 Hz, 1H), 6.77 (dd, J = 8.9 Hz, J = 2.9 Hz, 1H), 6.61 (d, J = 2.9 Hz, 1H), 4.43 (ddd, J = 8.3 Hz, J = 6.2 Hz, J = 4.4 Hz, 1H), 3.90 (dd, J = 18.3 Hz, J = 8.3 Hz, 1H), 3.79 (d, J = 4.4 Hz, 1H), 3.65 (s, 3H), 3.64 (s, 3H), 3.49 (dd, J = 18.3 Hz, J = 6.2 Hz, 1H), 2.93 (s, 3H), 2.92 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δC (ppm)197.8, 168.7, 168.3, 153.3, 151.9, 151.3, 136.1, 132.3, 130.5, 128.2, 127.9, 115.2, 113.3, 112.1, 56.3, 55.9, 53.2, 40.5, 37.1, 28.3, 28.2.
Two peaks of the molecular negative ion at m/z = 501.0774 and 503.0763 of the HRESIMS spectrum indicate that the prepared compound contains a bromine atom with the molecular formula C23H22N2O6Br and possesses 13 degrees of unsaturation (Supplementary Materials, Figure S1). The analysis of IR spectra showed the existence of C–H aromatic, C–H aliphatic, C=O ketone, C=O amide, C=C aromatic, Calkyl–O–Caryl ether, and C–Br bonds which were indicated consecutively by peaks at νmax (cm−1) 3080, 2958, 1745, 1618, 1581, 1203, and 717, respectively (Supplementary Materials, Figure S2).
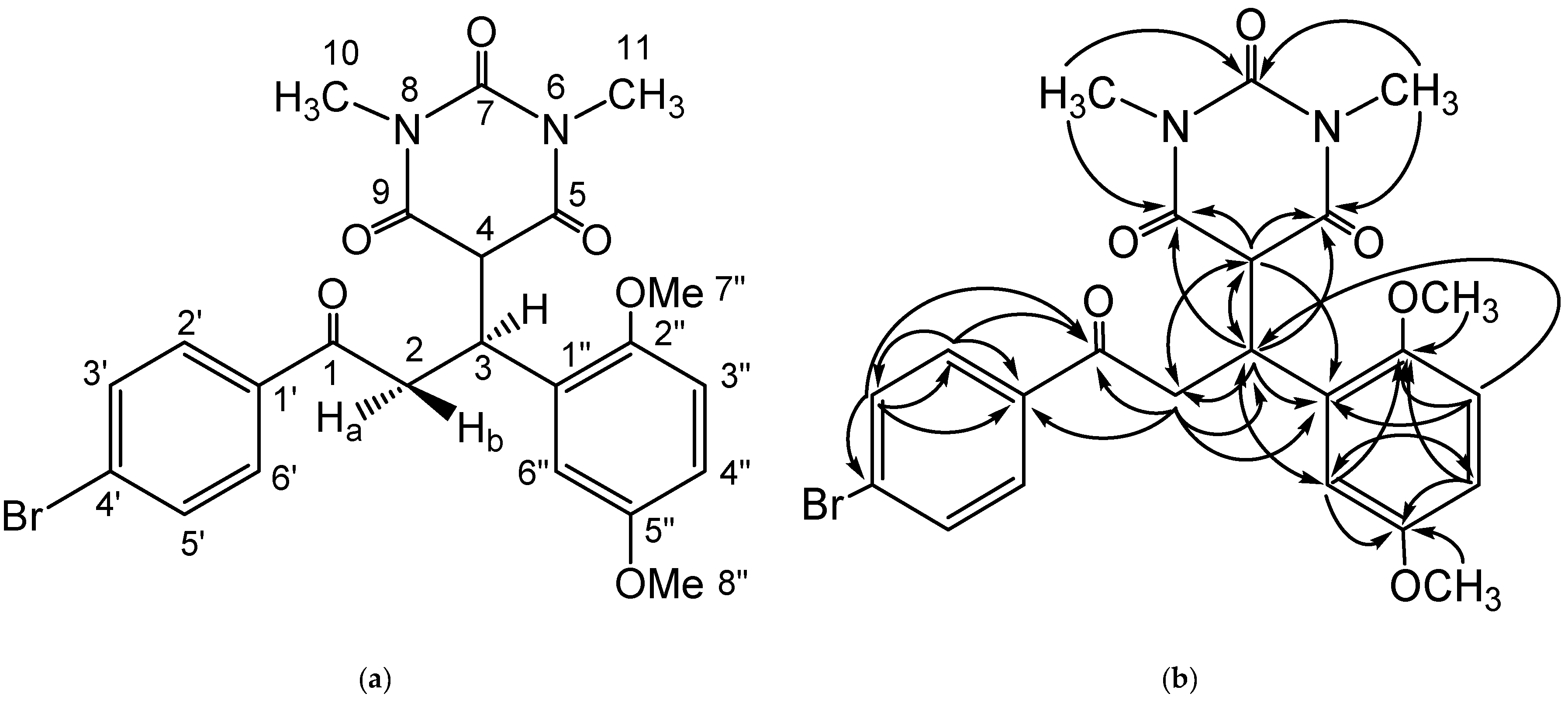
Spectroscopic data of 1H-NMR (Table 1) showed an ABMX spin system which represented a –CH2–CH–CH– fragment of the scaffold of the chalcone adduct with an active methylene. For this system, two signals at δH 3.90 (dd, 1H) and 3.49 ppm (dd, 1H) represented the existence of methylene diastereotopic fragment neighboring with a carbonyle group. Both signals showed a geminal coupling (J = 18.3 Hz). Protons appearing at δH 3.90 ppm (Ha) and 3.49 ppm (Hb) showed consecutively trans coupling (J = 8.3 Hz), and cis coupling (J = 6.2 Hz) with a proton at δH 4.43 ppm. The signal at δH 4.43 ppm (ddd) represented a benzylic proton attached to a diastereotopic methylene, while the barbiturate fragment is exhibited by the existence of a vicinal coupling (J = 4.4 Hz) with a signal at δH 3.79 ppm. This signal represented a proton flanked by the two carbonyl groups of a barbiturate fragment. The existence of an aromatic ring was determined by five aromatic signals: two signals at δH 7.92 and 7.75 ppm appeared as a doublet with J = 8.5 Hz and integration of both. Both signals formed an AA’XX’ spin system, which lead to the conclusion of a benzene fragment possessing two substituents at para position. While three other signals—appearing at δH 6.85 (d, J = 8.9 Hz, 1H), δH 6.77 (dd, J = 8.9 Hz, J = 2.9 Hz, 1H), and δH 6.61 (d, J = 2.9 Hz, 1H)—build an ABX spin system and represent a three substituted aromatic ring at position 1,2, and 4. Furthermore, the two signals at δH 3.65 ppm (s, 3H) and δH 3.64 (s, 3H) ppm indicated a methoxy proton attached at an aromatic ring, while the signals at δH 2.93 ppm (s, 3H) and δH 2.92 ppm (s, 3H) were signals of a methyl proton attached at the nitrogen atom of the barbiturate fragment (Supplementary Materials, Figure S3). The 13C-NMR spectra (Table 1) showed 21 signals and represented all carbon atoms of the prepared compound (Supplementary Materials, Figure S4)
The scaffold of the Michael adduct was assigned by the HMBC experiment which showed a correlation of the proton at C-3 with some carbon atoms; the methylene carbon (δC 40.5 (C-2)), methyne carbon (δC 53.2 (C-4)), amide carbonyl of N,N′-dimethylbarbiturate ring (δC 168.3 (C-9)), and (δC 168.7 (C-5)). The existence of a methylene group at C-2 was proved by a correlation of a proton with carbonyl ketone (δC 197.9 (C-1)), C-3 (δC 37.1), and a long-range correlation of the C-2 proton with C-4 (δC 53.2) and C-1″ (δC 128.3). Additionally, the CH position of C-4 was assigned by the correlation of the C-4 proton with the amide carbonyl of the 1,3-dimethylpyrimidine ring (δC 168.3 (C-9)) and (δC 168.7 (C-5)), and the proton correlation with C-3 (δC 37.1) and C-2 (δC 40.5). The proton–carbon correlations of the HMBC experiment were suitable with the molecular structure of the prepared compound and are displayed in Figure 2 and Figure S5 (Supplementary Materials). Based on the structure elucidation, the prepared compound is a new compound.
3. Materials and Methods
3.1. General
All reagents and solvents, purchased from E. Merck (Darmstadt, Germany) or Sigma Aldrich (St. Louis, MO, USA), were used without further purification. Reaction progress was monitored by thin-layer chromatography on silica gel GF254 aluminum sheets (0.25 mm) using various developing systems. Spots were detected under UV light (λ 254 nm). IR spectrum was recorded in KBr powder with the diffuse reflectance method on Fourier-transform Infrared spectrometer Shimadzu IRTracer100 (Kyoto, Japan). The mass spectrum was recorded on an High-resolution mass spectrometer, Waters LCT Premier XE (Santa Clara, CA, USA). NMR spectra (1H-, 13C-NMR, HMQC and HMBC) were recorded using the JEOL JNM-ECS400 (Tokyo, Japan) with CDCl3 as a solvent and internal standard.
3.2. Synthesis of Chalcone Derivative
The synthesis of chalcone derivative was conducted following the procedure reported by Suwito et al. [9]. A mixture of 6 mmol 4′-bromoacetophenone, 6 mmol 2,5-dimetoxybenzaldehyde, and 30 mL ethanol was placed in a three neck round bottom flask (equipped with a reflux condenser, thermometer, and dropping funnel), stirred, and cooled under 10 °C. To the reaction mixture, 6 mL NaOH 40% solution was added dropwise and the temperature was kept under 10 °C, stirred for 1 h, and then stirred at room temperature for a further 5 h. The precipitate was filtered off and recrystallized using aqueous ethanol.
3.3. Synthesis of the Target Compound
A mixture of 1.2 mmol of 1,3-dimethylbarbituric acid, 1 mmol of chalcone derivative, 1 mmol of ammonium chloride, 100 μL TEA, and 5 mL methanol was placed in a round bottom flask, and refluxed at 80 °C for 7 h. The reaction mixture was then cooled at room temperature, and 10 mL water was added. The precipitate was then filtered off, washed with a cold aqueous ethanol solution, and recrystallized using aqueous ethanol.
4. Conclusions
In conclusion, we have successfully synthesized a new compound, that is 5-[3-(4-bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one through Michael addition from a chalcone derivative.
Supplementary Materials
The following are available online: the HRESIMS, FTIR, 1H-NMR, 13C-NMR, HMQC, HMBC spectra are reported in the Supplementary Materials as Figures S1–S6, respectively, and the structure refinement in Table S1.
Author Contributions
H.S. brought the idea, managed the research, and wrote the manuscript. R.H.P.S. performed the synthesis, K.U.H. performed the structure elucidation, while A.N.K corrected the manuscript. All the authors have read the draft.
Funding
This research was funded by Penelitian Unggulan Fakultas 2018 Research Grant No. 1898/UN3.1.8/LT/2018.
Acknowledgments
The authors acknowledge Lembaga Penelitian & Inovasi and the Faculty of Science & Technology, Airlangga University for the funding support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kappe, C.O.; Stadler, A. The Biginelli dihydropyrimidine synthesis. Org. React. 2004, 63, 1–116. [Google Scholar]
- Jadhav, V.B.; Holla, H.V.; Tekale, S.U.; Pawar, R.P. Bioactive dihydropyrimidines: An overview. Chem. Sin. 2012, 3, 1213–1228. [Google Scholar]
- Ozair, A.; Khan, S.A.; Siddiqui, N.; Ahsan, W.; Verma, S.P.; Gilani, S.J. Antihypertensive activity of newer 1,4-dihydro-5-pyrimidine carboxamides: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2010, 45, 5113–5119. [Google Scholar]
- Lloyd, J.; Finlay, H.J.; Atwal, K.; Kover, A.; Prol, J.; Yan, L.; Rao, B.; Vaccaro, W.; Huynh, T.; Huang, C.S.; et al. Dihydropyrazolopyrimidines containing benzimidazoles as K(V)1.5 potassium channel antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 5469–5473. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Mishra, M.; Saxena, N.; Yadav, G.P.; Maulk, P.R.; Sahoo, M.K.; Gaur, R.L.; Murthy, P.K.; Tripathi, R.P. Synthesis of 2-sulfanyl-6-methyl-1,4-dihydropyrimidines as a new class of antifilarial agents. Eur. J. Med. Chem. 2008, 43, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, C.; Ok, T.; So, W.; Jo, M.; Seo, M.; Kim, Y.; Sohn, J.H.; Park, Y.; Moon, K.J.; et al. A novel 3,4-dihydropyrimidin-2(1H)-one: HIV-1 replication inhibitors with improved metabolic stability. Bioorg. Med. Chem. Lett. 2012, 22, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, F.E.; De Clerq, E. Synthesis and anti-HIV-1 activity evaluation of 5-alkyl-2-alkylthio-6-(arylcarbonyl or alpha-cyanoarylmethyl)-3,4-dihydropyrimidin-4(3H)-ones as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 2007, 50, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Ulaganathan, V.; Rath, O.; Prokopcov, H.; Dallinger, D.; Kappe, C.O.; Kozielski, F. Structural basis for inhibition of Eg5 by dihydropyrimidines: Stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J. Med. Chem. 2010, 53, 5676–5683. [Google Scholar] [CrossRef] [PubMed]
- Suwito, H.; Jumina; Mustofa; Pudjiastuti, P.; Fanani, M.Z.; Kimata-Ariga, Y.; Katahira, R.; Kawakami, T.; Fujiwara, T.; Hase, T.; et al. Design and synthesis of chalcone derivatives as inhibitors of the ferredoxin—Ferredoxin-NADP+ reductase interaction of Plasmodium falciparum: Pursuing new antimalarial agents. Molecules 2014, 19, 21473–21488. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The reaction process for the synthesis of the title compound.
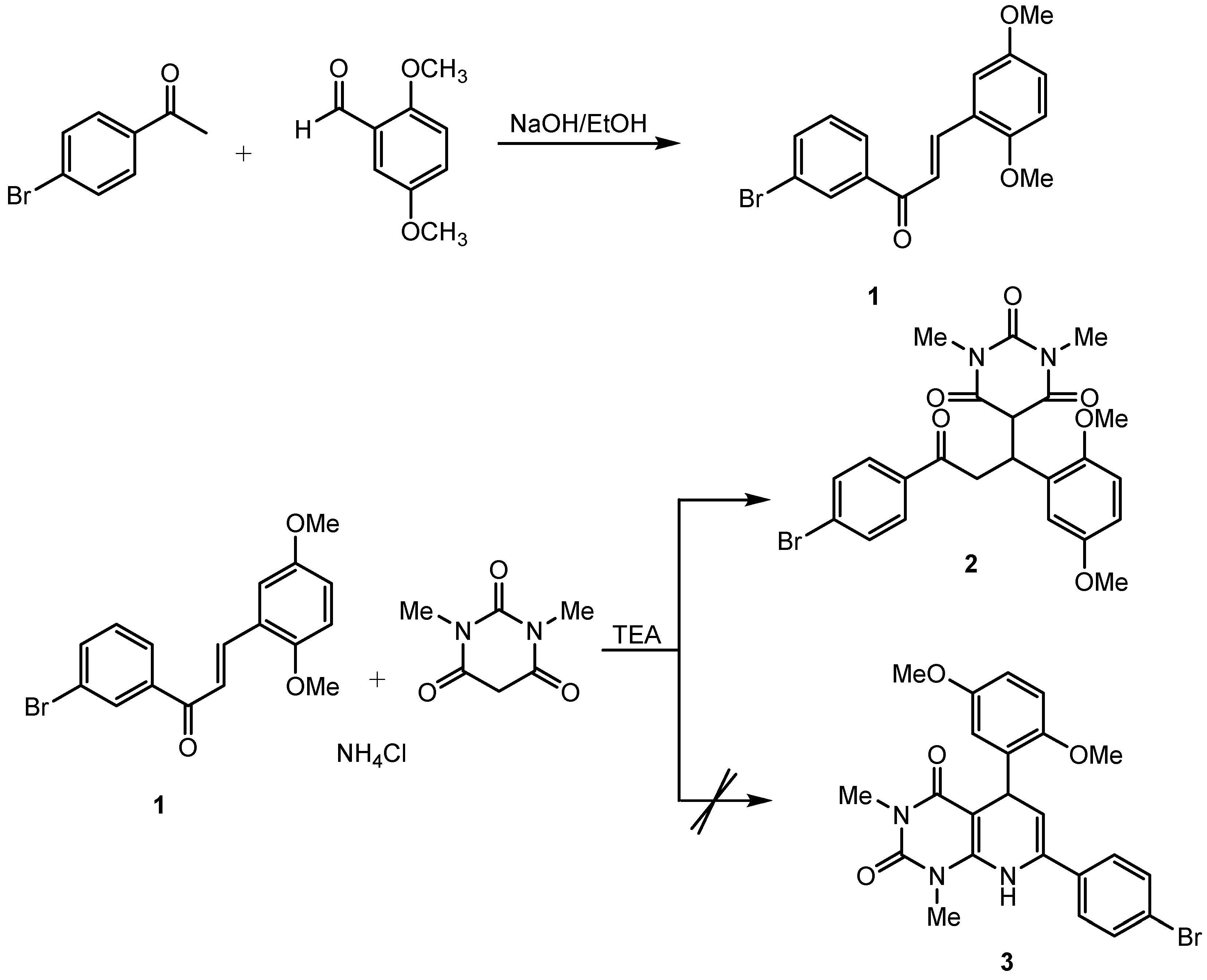
Figure 2.
(a) Structure numbering, and (b) HMBC correlations of the prepared compound.

{kind=link}
{kind=link}
Table 1.
NMR data of the target compound in DMSO-d6.
| No. Atom | δH (Mult, J Hz) | δC (ppm) | HMBC |
|---|---|---|---|
| 1 | - | 197.9 | |
| 2 | Ha = 3.90 (dd, 1H, J = 18.3 Hz, J = 8.3 Hz) | 40.5 | C-1, C-3, C-4, C-1′, C-1″ |
| Hb = 3.49 (dd, 1H, J = 18.3 Hz, J = 6.2 Hz) | |||
| 3 | 4.43 (ddd, 1H, J = 8.3 Hz, J = 6.2 Hz, J = 4.4 Hz) | 37.1 | C-2, C-4, C-5, C-9, C-1″, C-6″ |
| 4 | 3.79 (d, 1H, J = 4.4 Hz) | 53.2 | C-2, C-3, C-5, C-9, C-1″ |
| 5 | - | 168.7 | |
| 6 | - | - | |
| 7 | - | 151.3 | |
| 8 | - | - | |
| 9 | - | 168.3 | |
| 10 | 2.93 (s, 3H) | 28.21 | C-9, C-7 |
| 11 | 2.92 (s, 3H) | 28.31 | C-5, C-7 |
| 1′ | - | 127.9 | |
| 2′, 6′ | 7.92 (d, 2H, J = 8.5 Hz) | 132.3 | C-1, C-1′, C-3′ |
| 3′, 5′ | 7.75 (d, 2H, J = 8.4 Hz) | 130.5 | C-1, C-1′, C-2′, C-4′ |
| 4′ | - | 136.1 | |
| 1″ | - | 128.3 | |
| 2″ | - | 153.4 | |
| 3″ | 6.85 (d, 1H, J = 8.9 Hz) | 112.1 | C-3, C-1″, C-2″ |
| 4″ | 6.77 (dd, 1H, J = 8.9 Hz, J = 2.9 Hz) | 113.3 | C-2″, C-5″, C-6″ |
| 5″ | - | 151.9 | |
| 6″ | 6.61 (d, 1H, J = 2.9 Hz) | 115.2 | C-3, C-2″, C-4″, C-5″ |
| 7″ | 3.64 (s, 3H) | 56.3 | C-2″ |
| 8″ | 3.65 (s, 3H) | 55.9 | C-5″ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suwito, H.; Sari, R.H.P.; Ul Haq, K.; Kristanti, A.N. 5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one. Molbank 2018, 2018, M1013. https://doi.org/10.3390/M1013
AMA Style
Suwito H, Sari RHP, Ul Haq K, Kristanti AN. 5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one. Molbank. 2018; 2018(3):M1013. https://doi.org/10.3390/M1013
Chicago/Turabian StyleSuwito, Hery, Ria Hesty Purnama Sari, Kautsar Ul Haq, and Alfinda Novi Kristanti. 2018. "5-[3-(4-Bromophenyl)-1-(2,5-dimethoxyphenyl)-3-oxopropyl]-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-tri-one" Molbank 2018, no. 3: M1013. https://doi.org/10.3390/M1013
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.