1. Introduction
Doxorubicin is an anthracycline ring antibiotic that is widely used as a cancer therapeutic. Common treatment side effects include nausea and vomiting, loss of appetite, diarrhea, and swelling [
1], but development of a system capable of selectively delivering the drug to the intended target cells may be able to reduce these complications.
There are many approaches to drug delivery via drug/drug carrier combinations, such as encapsulation, hydrogel formation, nanoaggregation, and micellar delivery. For doxorubicin delivery, encapsulation and micellar delivery have received increased attention because this system can protect and carry the drug directed to its intended target. Brannon-Peppas and Blanchette [
2] reported that tumor volumes in mice treated with an encapsulated drug conjugate were lower than tumor volumes in those treated with the conjugate alone. Janes
et al. [
3] modified chitosan nanoparticles to carry the doxorubicin. Wang
et al. [
4] encapsulated the chitosan, which has poor solubility in water, to increase the efficiency of drug delivery. Chan
et al. [
5] synthesized chitosan-g-poly(ethylene glycol) as an alternative drug delivery system with the intention that conjugation to poly(ethylene glycol) would increase the solubility of chitosan. There are several studies on micellar delivery of doxorubicin. Xiangyang
et al. [
6] used
N-succinyl-
N-octylchitosan micelles as doxorubicin carriers with effective anti-tumor activity. Zhang
et al. [
7] attached 10-hydroxycamptothecin, a hydrophobic anticancer drug, to amphiphilic
N-alkyl-
N-trimethylchitosan derivatives, resulting in increased drug solubility and controlled release.
Drug carriers usually have some chemical functional group used to detect their cancer cell targets. Yang
et al. [
8] used isocyanate terminated linear polyethylene glycol to carry drugs to cancer cells. Doxorubicin can be effectively delivered via conjugation to chitosan nanoparticles [
9]. Son
et al. [
10] studied the tumor-targeting properties of glycol-chitosan nanoaggregates
in vivo using fluorescein isothiocyanate-conjugated glycol-chitosan nanoaggregates and doxorubicin-conjugated glycol-chitosan (GC-DOX). Okabe
et al. [
11] studied
P-glycoprotein efflux transporters, which confer drug resistance by decreasing the intracellular accumulation of anticancer drugs. Based on these previous experiments, we concluded that doxorubicin can be conjugated to a biopolymer that selectively targets cancer cells.
Quantum mechanical molecular simulation can be used to study drug delivery. Certain techniques, specifically semi-empirical and
ab initio methods, are useful for simulating molecular interactions for drug delivery applications. Zavodinsky and Mikhailenko [
12] simulated reactions between carbon nanoclusters and molecular oxygen using a semi-empirical PM3 method and found that reactivity depended on the structure of clusters and on the positions of the reactive carbon atoms. Propylene oxide structure optimization can be performed using semi-empiracal and
ab initio methods [
13]. The predicted bond lengths and bond angles agreed with experimental data. Simulations of the effect of hydrophobic molecules on aqueous solutions of amphiphilic block copolymers found that hydrophobic molecules will dissolve in micellar solutions [
14]. This effect has also been investigated in detail by cryogenic transmission electron microscopy (TEM).
Cummins and Gready [
15] used semi-empirical AM1 and PM3 methods to describe molecular interactions. Their method was applied to calculate the free energy of the enzymatic reduction of dihydrofolate (DHF) with nicotinamide adenine dinucleotide phosphate (NADPH) via
Escherichia coli dihydrofolate reductase. The free energy change of this reaction agreed with the experimental results.
Ab initio methods have used density functional theory (DFT), which was suitable for simulating reactions [
16,
17].
In this work, the drug delivery doxorubicin with bis-polymer (chitosan) was studied for potential applications as an anticancer therapeutic. Both semi-empirical and ab intio methods were applied to optimize the structures of the relevant molecules and to find the possible mechanism of micelle formation.
3. Results and Discussion
3.1. Structural optimization of doxorubicin and glucosamine(ethylene glycol)
Structures were optimized using molecular modeling methods. Frimand and Jalkanen [
13] simulated propylene oxide with semi-empirical and
ab initio methods, and their predictions were in good agreement with experimental data. Cummins and Gready [
15] used semi-empirical quantum mechanical methods to describe molecular interactions relevant to the enzymatic cleavage of ribose ring phosphate groups from NADPH. The semi-empirical AM1 and PM3 predictions both agreed with experimental results.
In this study, the semi-empirical PM3 method was used to optimize the molecular geometries of doxorubicin and glucosamine(ethylene glycol). Bond lengths and charge structure were both considered geometric parameters and were optimized in this fashion.
3.1.1. Doxorubicin
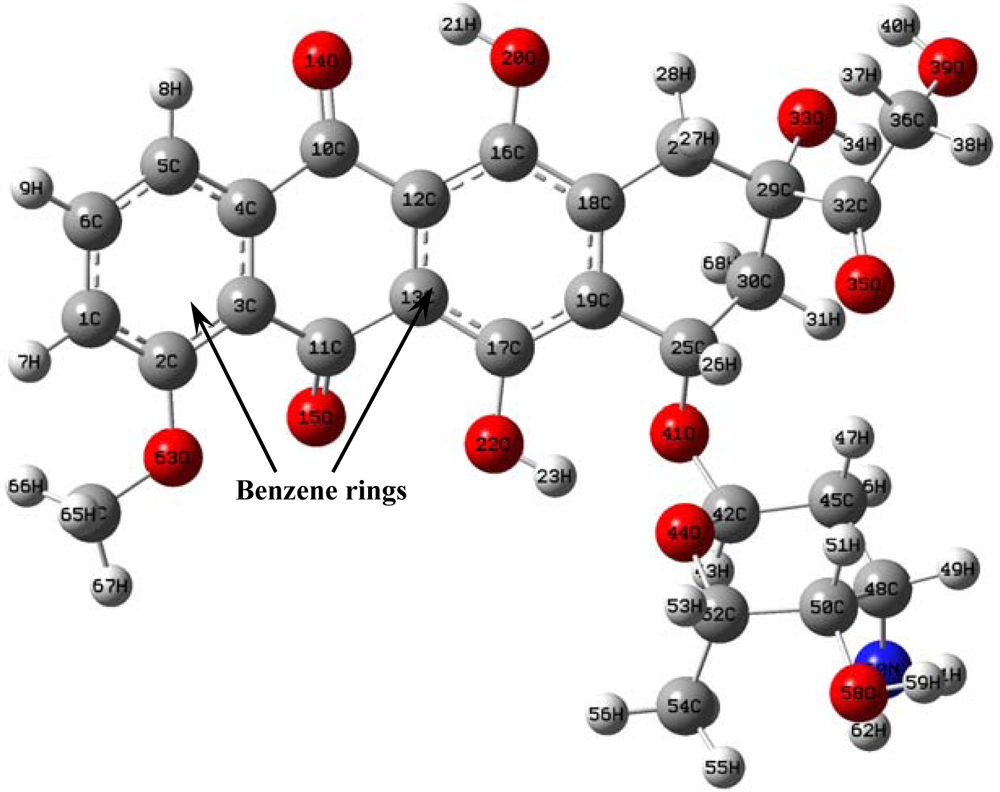
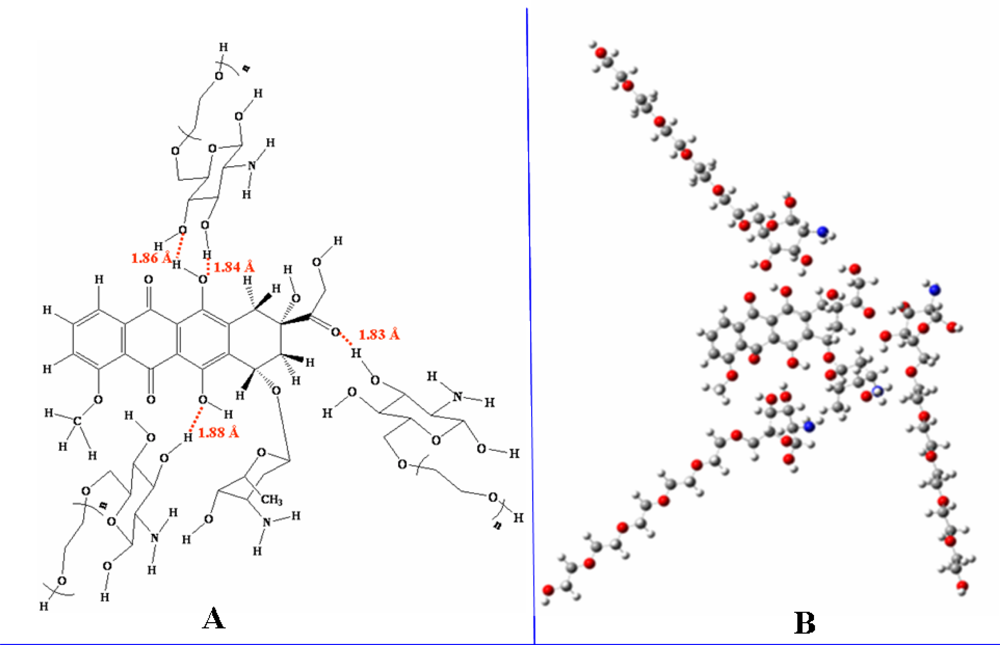
The optimized doxorubicin structures obtained from semi-empirical AM1 and PM3 methods and from the
ab initio HF/6-31G method were identical (
Figure 1).
Some geometric parameters corresponding to this optimal structure are given in
Table 1.The HF method predicted a more optimal structure than either the AM1 or the PM3 methods, but all three predictions demonstrated the same basic trends.
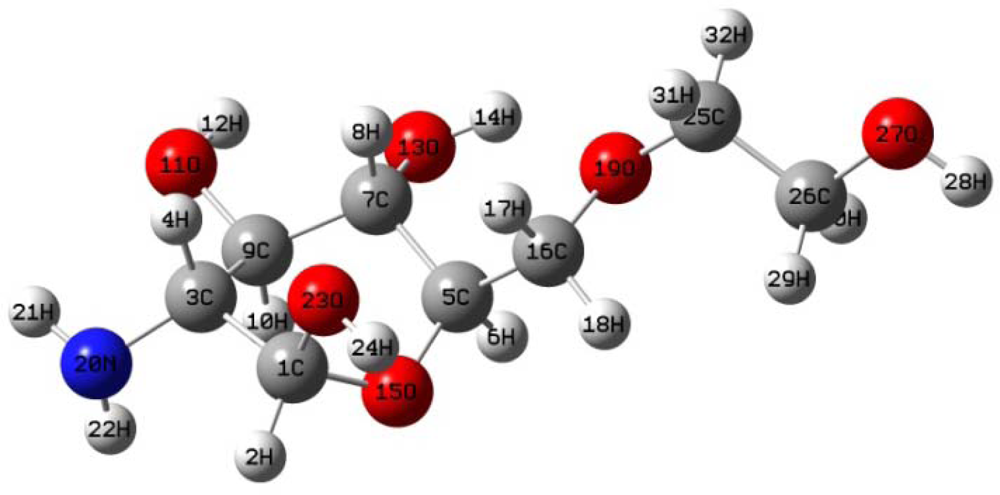
3.1.2. Glucosamine(ethylene glycol)
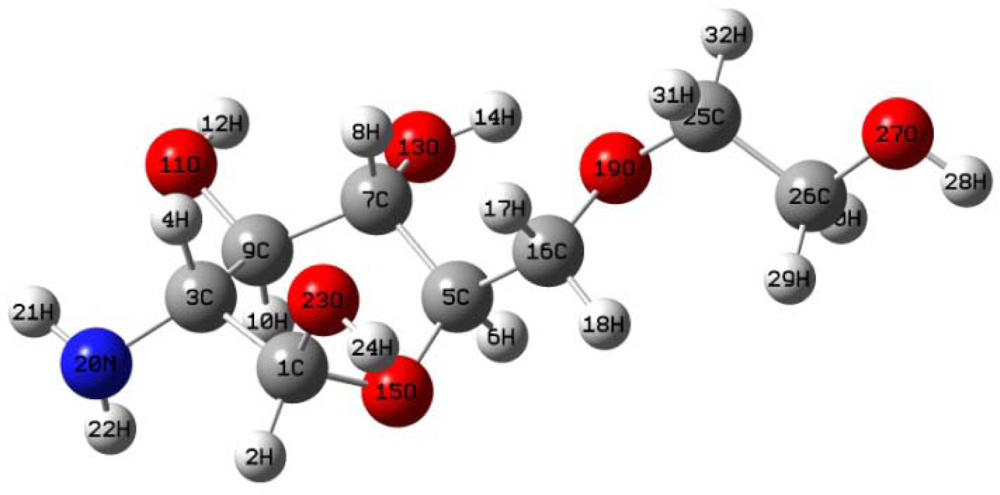
The optimal structure of glucosamine(ethylene glycol) was simulated using the same three aforementioned methods (AM1, PM3 and HF/6-31G). All three methods yielded nearly identical structures (
Figure 2). The relevant geometric structural parameters from each method are given in
Table 2.
All three methods predicted the same optimum structure, so the AM1, PM3, and HF methods can all be used for structure optimization. The optimized molecular structures for glucosamine(ethylene glycol) and doxorubicin were then used to study the two molecules’ interactions.
3.2. Drug release from capsules
The release of doxorubicin from its oligoglucosamine(ethylene glycol) capsule requires breaking the oligmeric bonds of oligoglucosamine(ethylene glycol). The probability of reaction can be estimated from the difference in total molecular energies between the products and the reactants. Simulations revealed that the capsule’s polymer bonds can be broken under acidic, normal, or basic conditions. These simulations involved two steps. First, molecular structures were optimized using the semi-empirical PM3 method. Second, the molecular energies were calculated via ab initio HF/B3LYP/6-31G method.
3.2.1. Acidic conditions
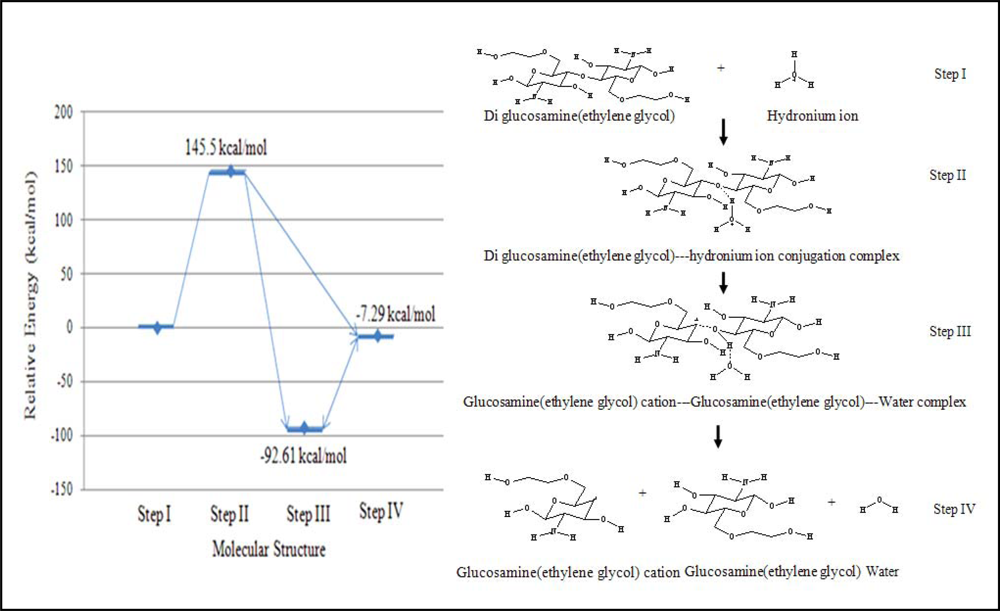
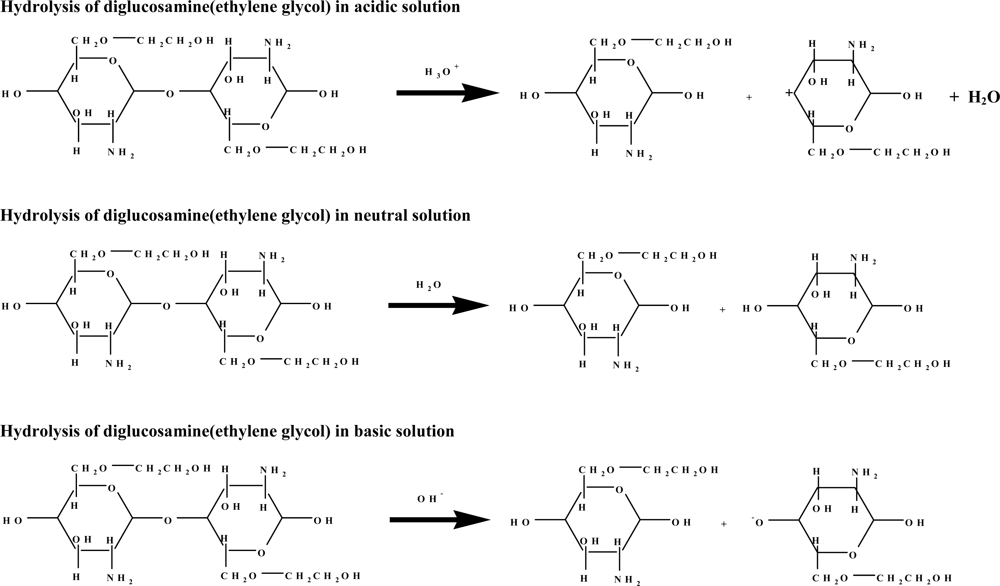
The following assumptions were made to simulate polymeric bond breaking under acidic conditions: (1) hydrolysis of diglucosamine(ethylene glycol) was used to represent cleavage of glucosamine(ethylene glycol) oligomers, and (2) H3O+ (hydronium ion) was used to represent acidic conditions.
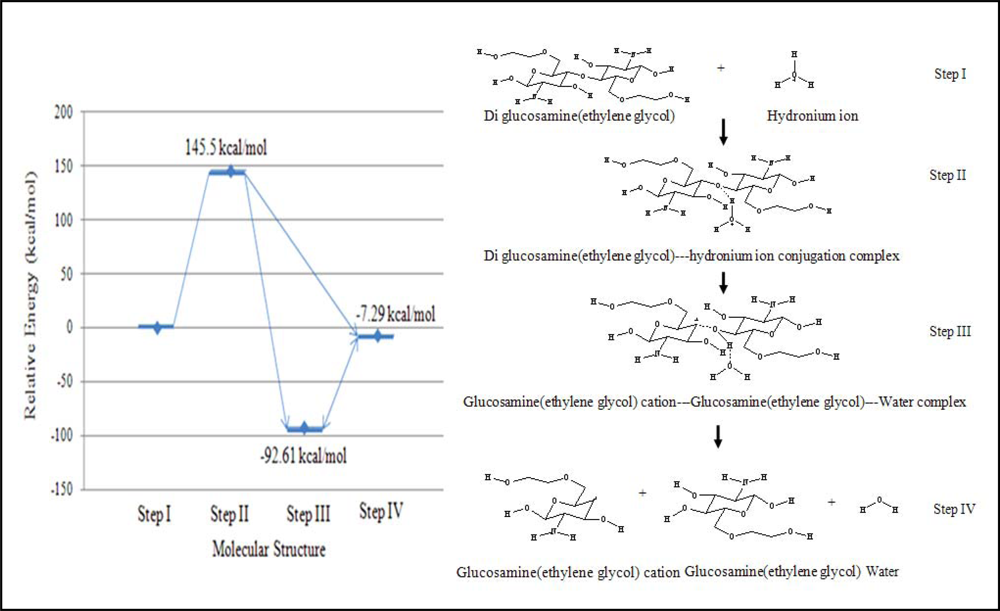
The reaction was also assumed to have four continuous steps. First, the hydronium ion complexes with diglucosamine(ethylene glycol) at the polymeric bond. Second, the hydrogen atom of the hydronium ion is attracted to the high electron density of the polymeric bond’s oxygen atom, forming the transition state complex. Third, the reaction generates a ternary complex among glucosamine(ethylene glycol), the glucosamine(ethylene glycol) cation, and water. Finally, these three product molecules separate. The relative energy of each step in the acid hydrolysis of diglucosamine(ethylene glycol) was calculated (
Figure 3).
The acid hydrolysis reaction was simulated using B3LYP/6-31G//PM3 level calculations. The probability of reaction is related to the relative energy of the reaction. If the net change in energy for the reaction is negative, then product formation is favored. If the relative energy change is more negative, then the reaction products are more highly favored. The energies of each step were calculated relative to the molecular energy of step I, which represents the reactants. The energy of the transition state (step II) was 145.51 kcal/mol greater than that of the first step because the hydronium ion requires energy to react with glucosamine(ethylene glycol). Two distinct structures can be considered as the reaction’s final products. The first structure (step III) was the ternary product complex of glucosamine(ethylene glycol), a glucosamine(ethylene glycol) cation, and water, and this complex was predicted to have energy of –92.61 kcal/mol relative to the reactants in step I. The second structure (step IV), in which none of the product molecules interact, had a relative energy of –7.29 kcal/mol. Thus, the simulation predicted that the product complex from step III would be formed because it was of lower energy and was more stable than the non-interacting of product structure from step IV.
3.2.2. Neutral conditions
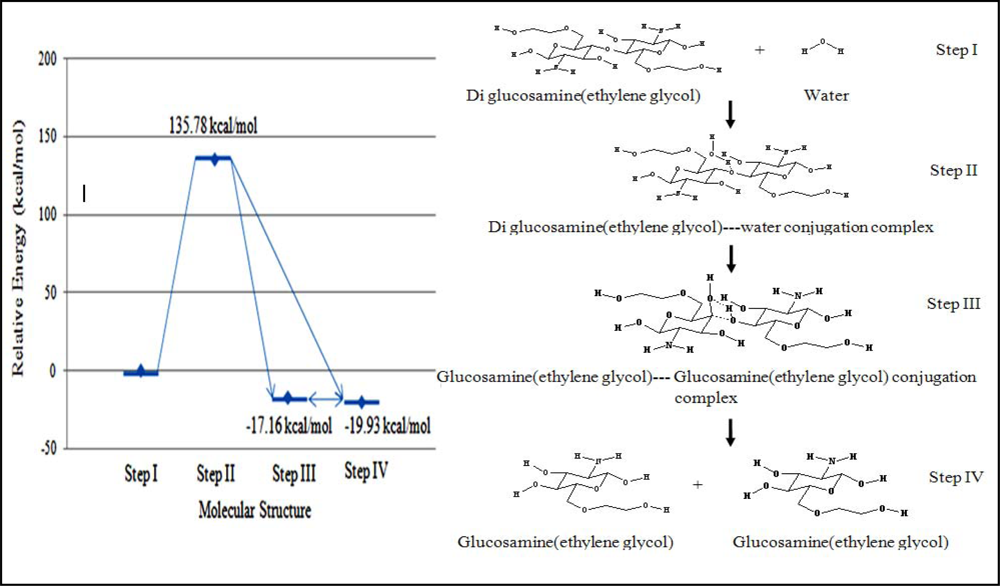
It was again assumed that diglucosamine(ethylene glycol) was an acceptable reactant surrogate for a glucosamine(ethylene glycol) oligomer. Since the simulated solution was of neutral pH, H2O was used as the nucleophile.
The molecular energy of each step in the reaction mechanism was calculated relative to that of step I. The relative energy of the transition state was 135.78 kcal/mol. Both steps III and IV were considered for the final product. The first structure (stage III), which had a relative energy of –17.16 kcal/mol, was the complex product containing two molecules of glucosamine(ethylene glycol). The second structure (stage IV), which had a relative energy of –19.93 kcal/mol, was the dissociated glucosamine(ethylene glycol) molecules (
Figure 4).
3.2.3. Basic conditions
Again, diglucosamine(ethylene glycol) was assumed to be an acceptable reactant surrogate for a glucosamine(ethylene glycol) oligomer. As hydrolysis was simulated under basic conditions, OH− was used as the nucleophile.
Two different hydrolysis products can form under basic conditions. Either the hydroxide ion reacts with the polymeric bond forming the desired products or it deprotonates the molecule. In the case of the former reaction, the high electron density on the oxygen of the polymer bond repels the hydroxide ion, hindering nucleophilic attack. In the case of the latter reaction, the hydroxide ion removes a proton from the ethylene glycol moiety of diglucosamine(ethylene glycol) forming a dimeric anion.
3.2.3.1. Hydroxide ion hydrolysis of the polymeric bond
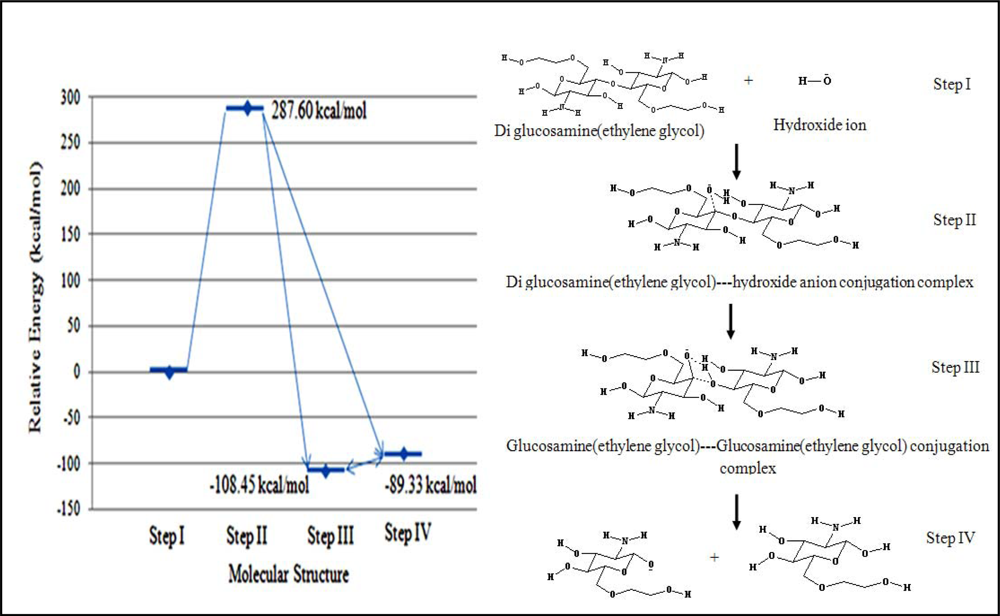
Under basic conditions, a hydroxide ion can act as a nucleophile to hydrolyze the diglucosamine(ethylene glycol) polymeric bond. The relative energy of the transition state was 287.60 kcal/mol. Both steps III and IV were considered for the final product. The first structure, which had a relative energy of –108.43 kcal/mol, was the binary product complex containing two molecules of glucosamine(ethylene glycol). The second structure, which had a relative energy of –89.33 kcal/mol, was dissociated complex consisting of a molecule of glucosamine(ethylene glycol) and glucosamine(ethylene glycol) anion (
Figure 5).
The high activation energy of this reaction suggested that this reaction cannot proceed at an appreciable rate. The high relative energy of the transition state was attributed to the repulsive effect of the high electron density on the polymeric linkage.
3.2.3.2. Hydroxide ion deprotonates ethylene glycol group
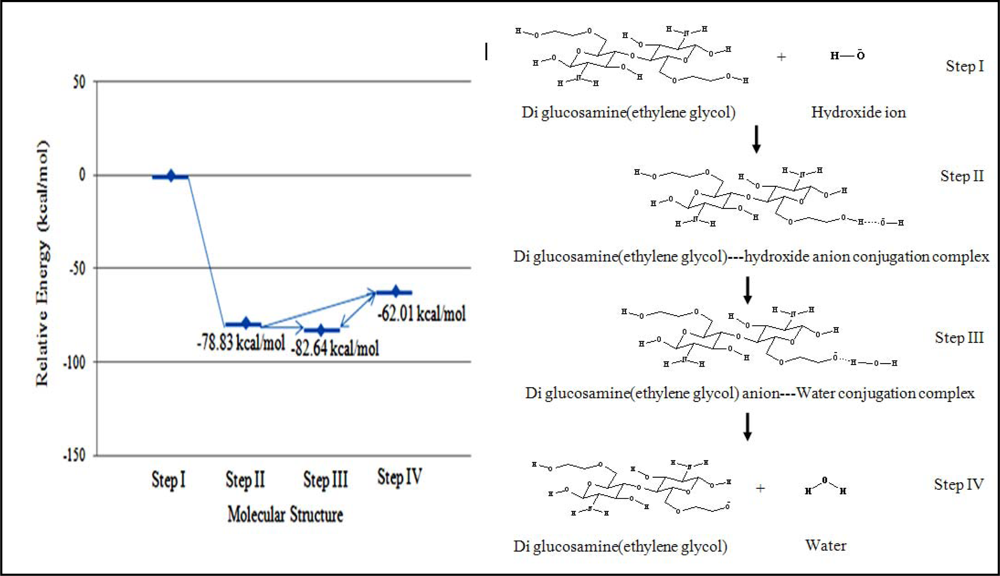
The hydroxide can also deprotonate the ethylene glycol moiety of diglucosamine(ethylene glycol) under base conditions and the relative energy of each mechanistic step was calculated (
Figure 6).
The relative energy of the transition state was –78.83 kcal/mol. This implied that this reaction would dominate under basic conditions because it was much faster than hydrolysis. Two possible products were considered to follow the transition state. The first structure, which had an energy of –82.64 kcal/mol, was the product complex containing a diglucosamine(ethylene glycol) anion and a molecule of water. The second structure, which had an energy of –62.01 kcal/mol, was the dissociated diglucosamine(ethylene glycol) molecule.
The results for all three conditions (acid, neutral, and basic) are summarized in
Table 3. The activation energy was highest for the basic hydrolysis reaction (Basic I). With the exception of neutral hydrolysis conditions, the product complex states (all mechanisms, step III) had relative energies lower than those of the dissociated products (all mechanisms, step IV). This implied that the molecules most likely stay closely associated following reaction. From these stages (step III), the complex molecules can move apart from each other and stay in stable form with optimum structure (stage IV). In the case of a competing side reaction under basic conditions (basic II), the high electron density at the ethylene glycol oxygen is insufficient to repel the hydroxide anion and prevent deprotonation but at hydrogen is mild electron density, which occurs very rapidly. The hydroxide anion is likely to form at hydrogen position of ethylene glycol than of polymer bond.
These predictions agreed with the experimental results of Oungbho
et al. [
18], who found that drug release from chitosan was much more favorable under acidic conditions than under neutral conditions. Under basic conditions, the hydroxide ion is difficult to attack at polymer bond because of very high electron density but deprotonation of the ethylene glycol is much faster than hydrolysis, and so drugs cannot be released from a glucosamine(ethylene glycol) capsule in this manner.
4. Conclusions
The hydrolytic release of doxorubicin from a glucosamine(ethylene glycol) oligomer capsule was simulated under acidic, neutral, and basic conditions. Using diglycosamine(ethylene glycol) as a surrogate substrate, hydrolysis was predicted under acidic and neutral conditions but not under basic conditions. Thus, an acidic environment, such as the stomach, may break the encapsulating polymer bonds. Deprotonation is favored over hydrolysis under basic conditions, so the capsule will not be broken in this case.
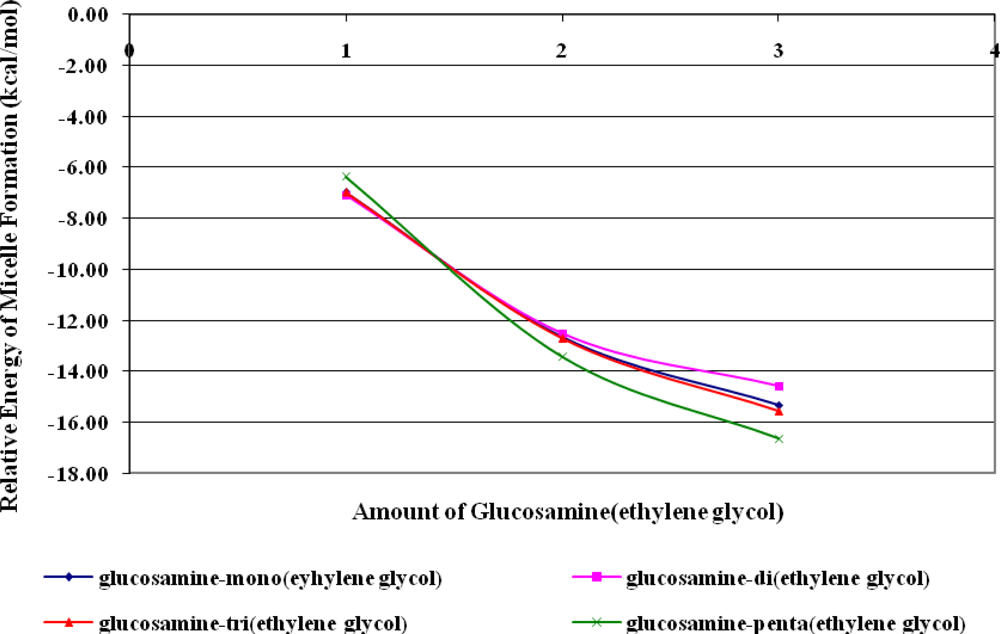
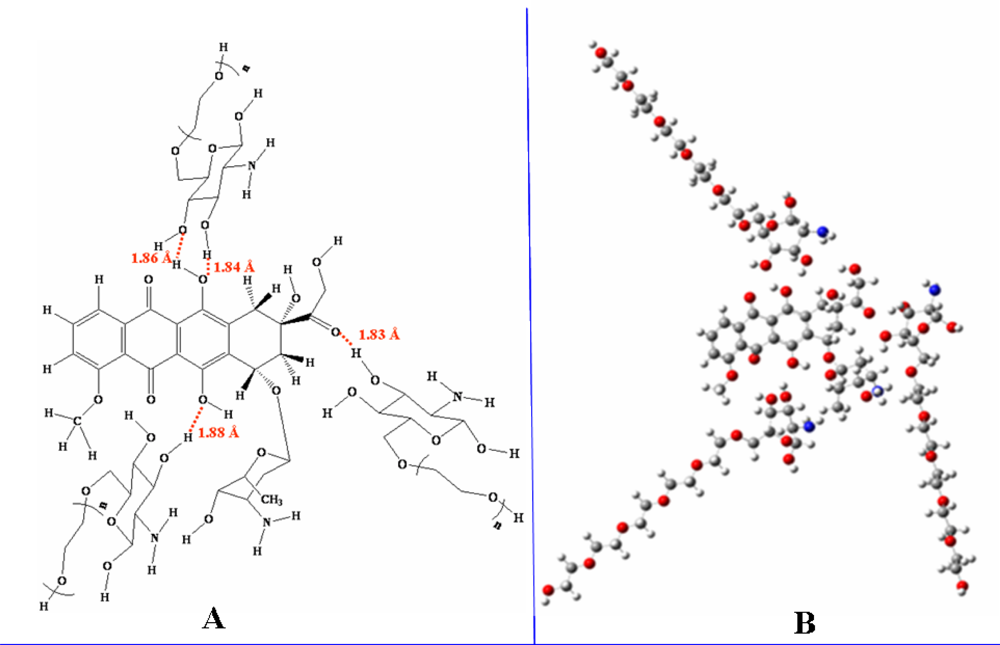
Micelles formation from doxorubicin and glucosamine(n-ethylene glycol) depended on the length of the ethylene glycol side chain. According to B3LYP/6-31G//PM3 predictions, lengthening the ethylene glycol chain stabilized the resulting micelles, thereby facilitating their formation. The number of glucosamine(ethylene glycol) molecules included in the simulation was varied from one to three, and increasing the number of molecules also facilitated micelle formation. Thus, glucosamine(ethylene glycol) and doxorubicin will form complex micellar structures capable of acting as drug delivery systems.
The capsule polymer used to deliver doxorubicin should possess a sufficiently long ethylene glycol chain to promote micelle formation. The length of the ethylene glycol chain also had an effect on polymer hydrolysis. In an acidic solution, increasing of the length of the ethylene glycol side chain facilitated hydrolysis because the side chain pulled H3O+ from solution, thereby reducing the relative energy of the transition state. If, however, the ethylene glycol chain was too long, it reacted with the polymeric bond, forming (diglucosamine-tri(ethylene glycol)). In neutral and basic solutions, as the length of ethylene glycol chain increased, the polymeric bonds became more resistant to hydrolysis because the ethylene glycol side chain sterically hindered nucleophilic attack.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}