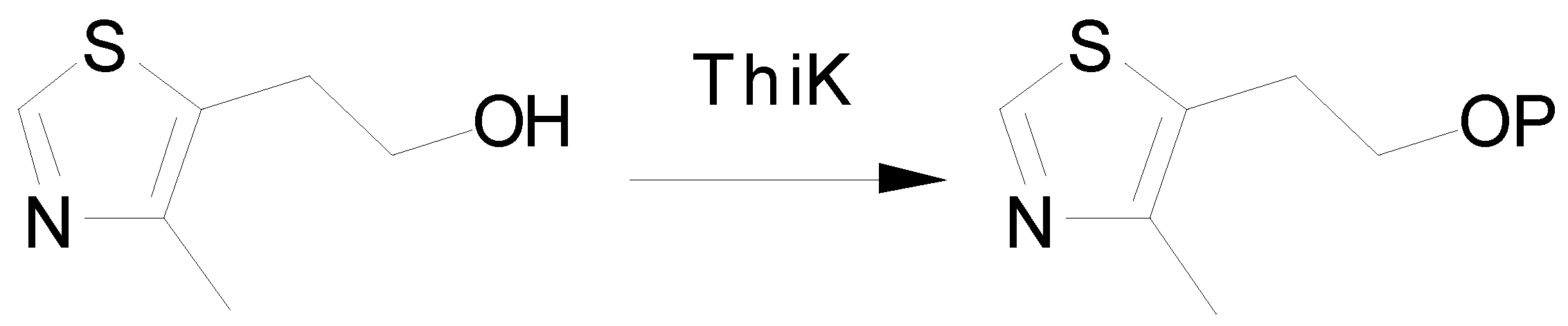
The Mechanism of Phosphoryl Transfer Reaction and the Role of Active Site Residues on the Basis of Ribokinase-Like Kinases
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
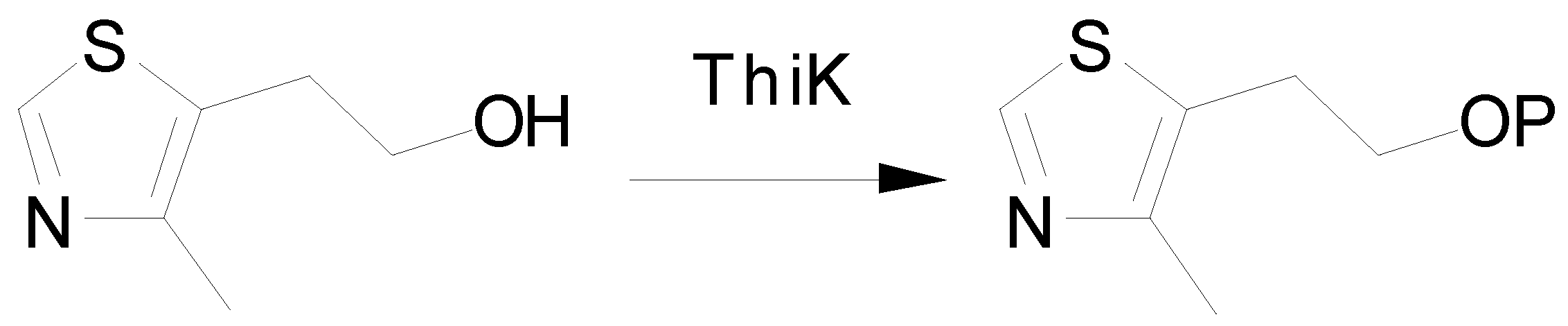
Computational Methods
Preparation of the Starting Geometries
Molecular Mechanics Optimization
Quantum Mechanics Calculations
Static and Dynamic Catalytic Fields
The Reaction Pathway Mapping
Results
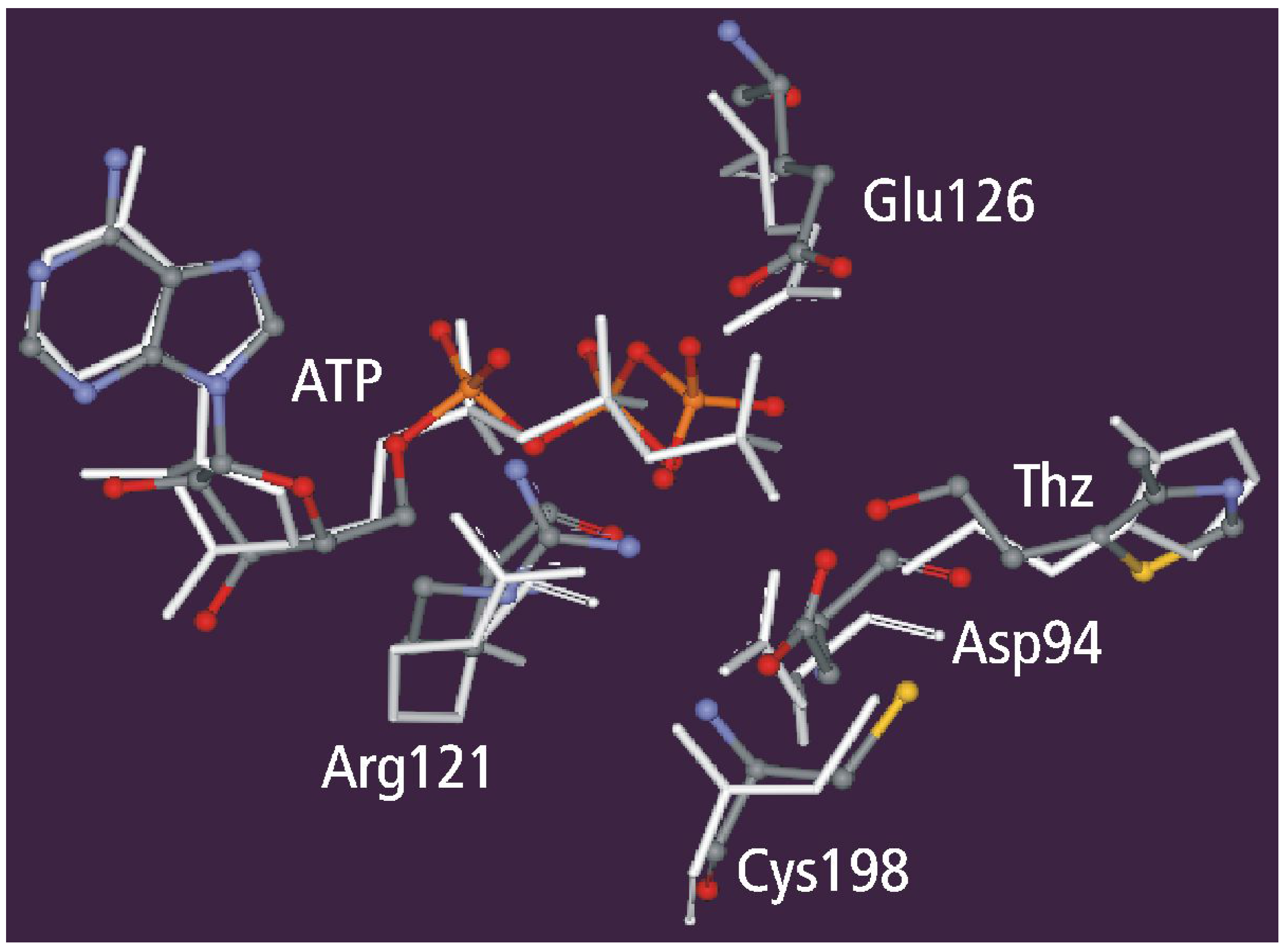
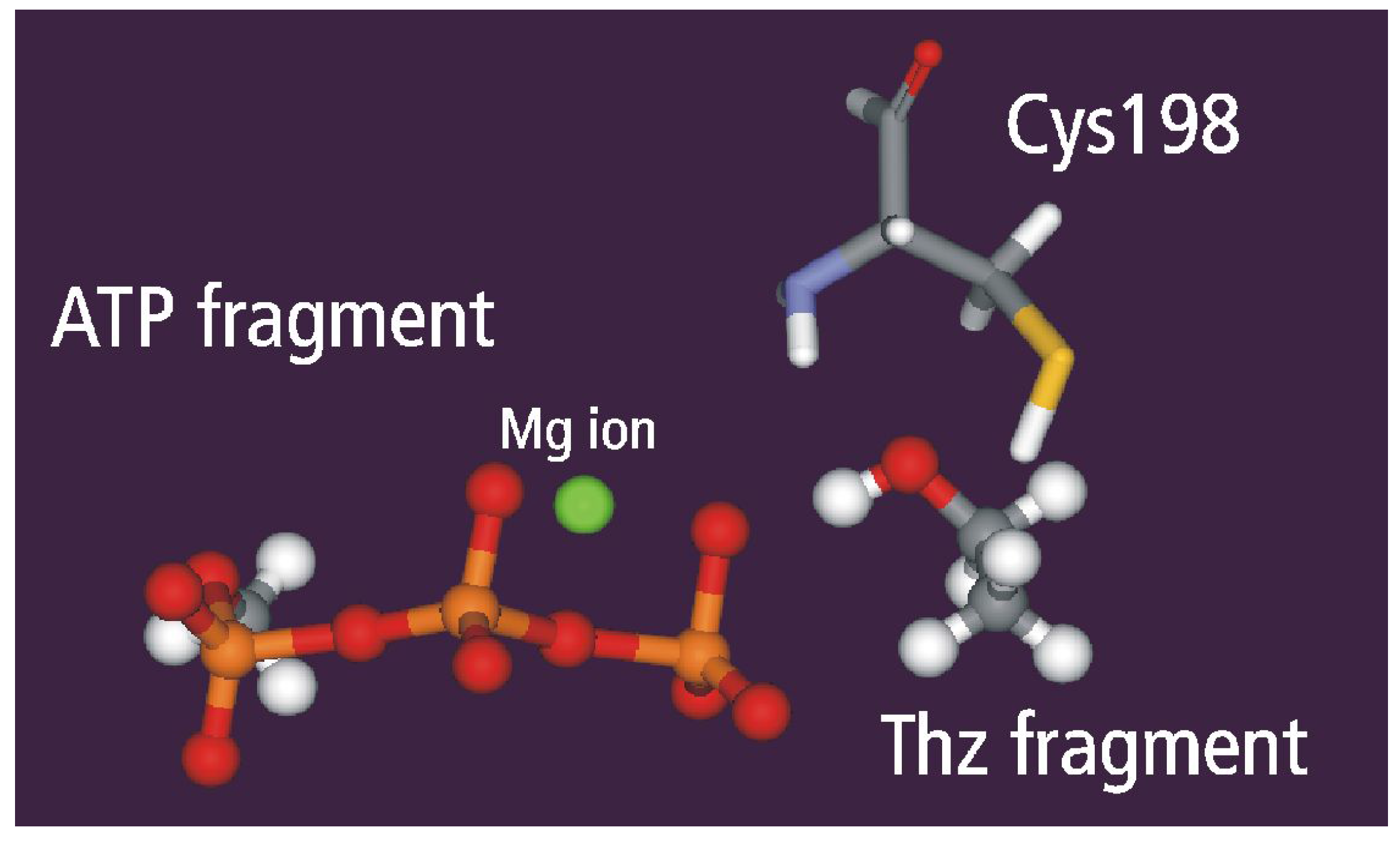
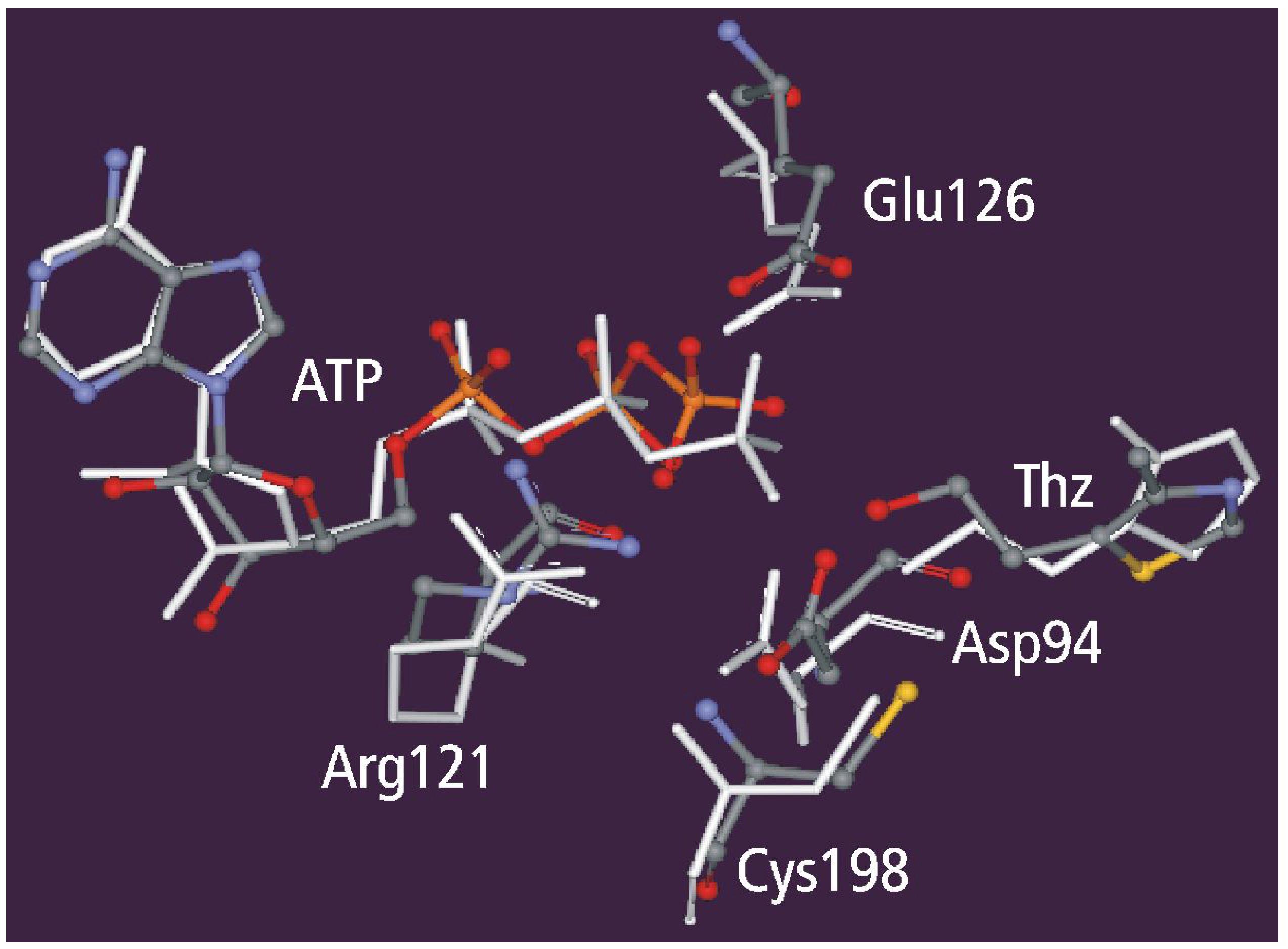
Geometry Optimization
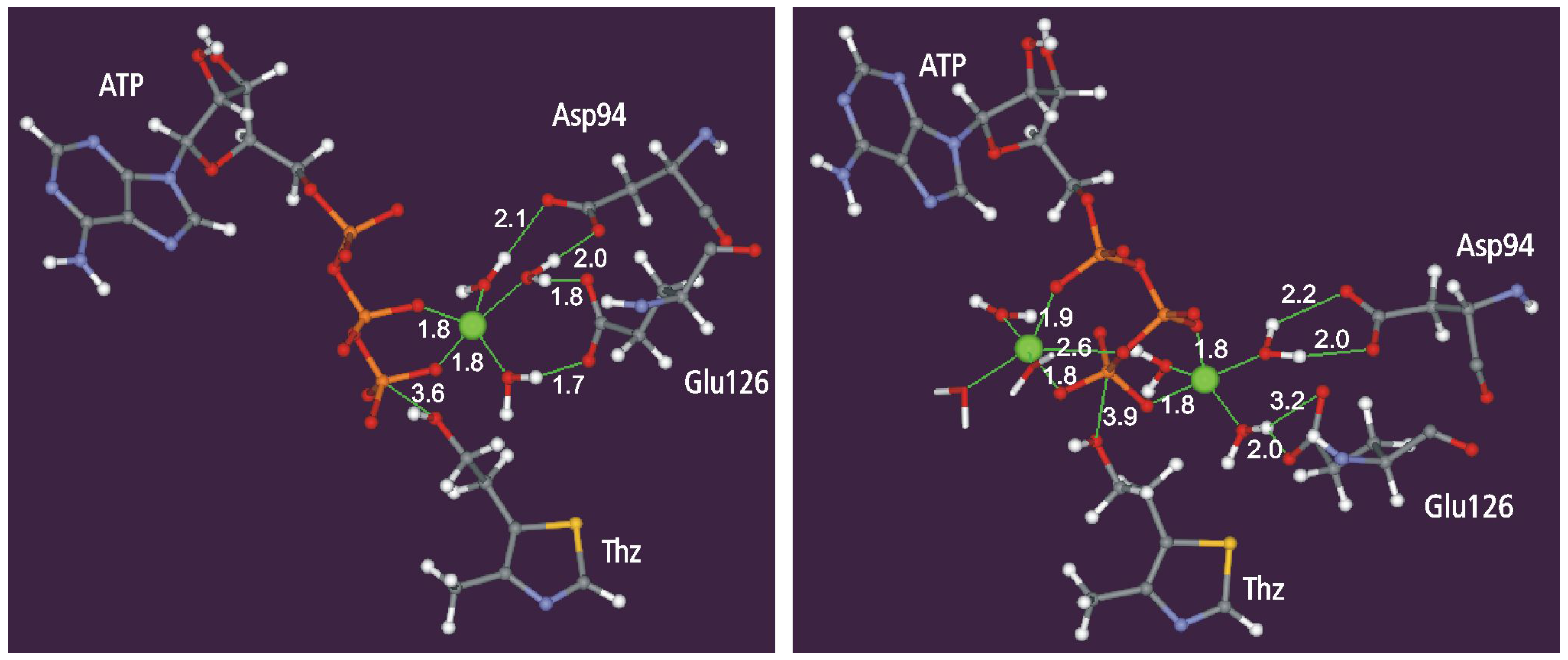
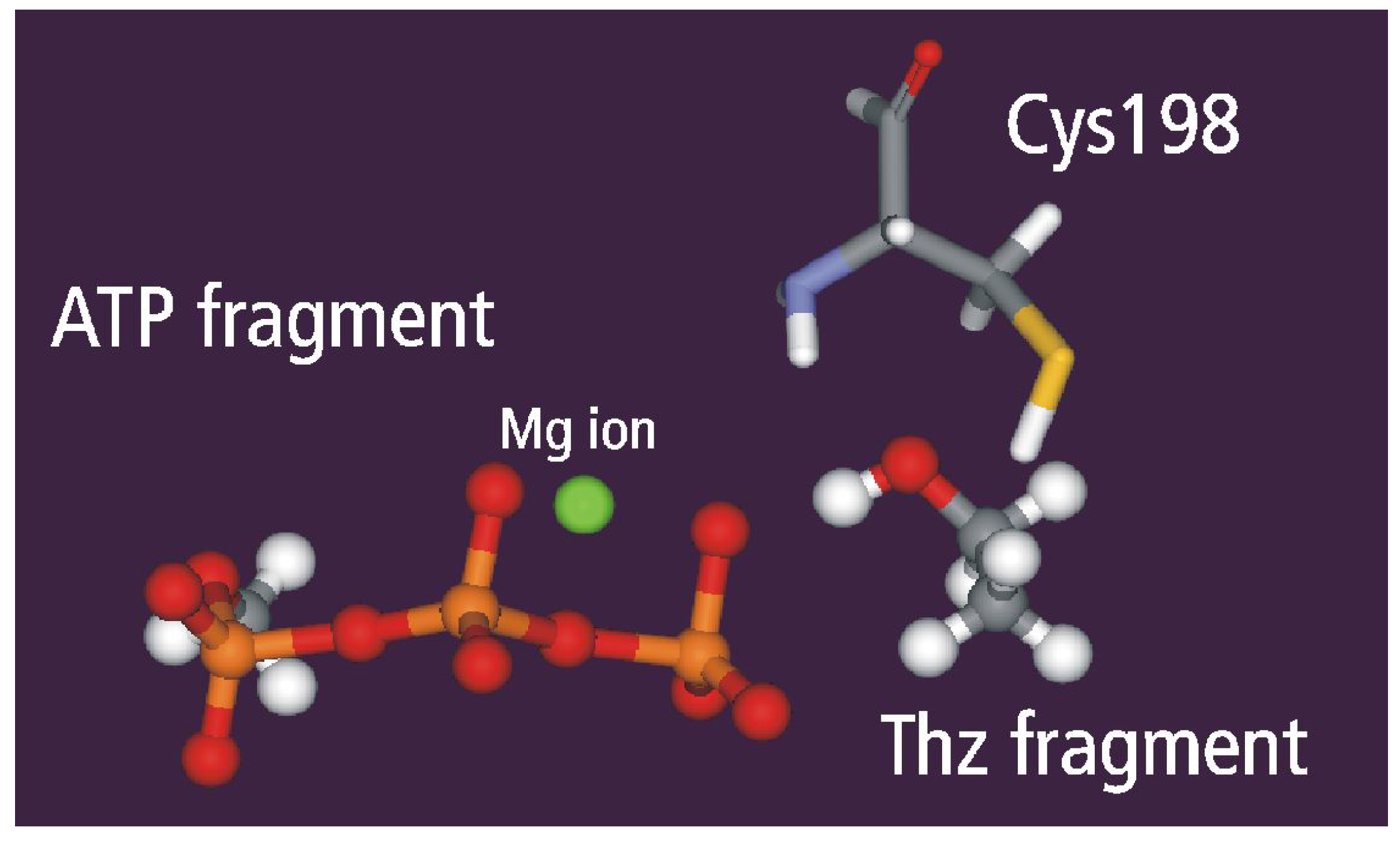
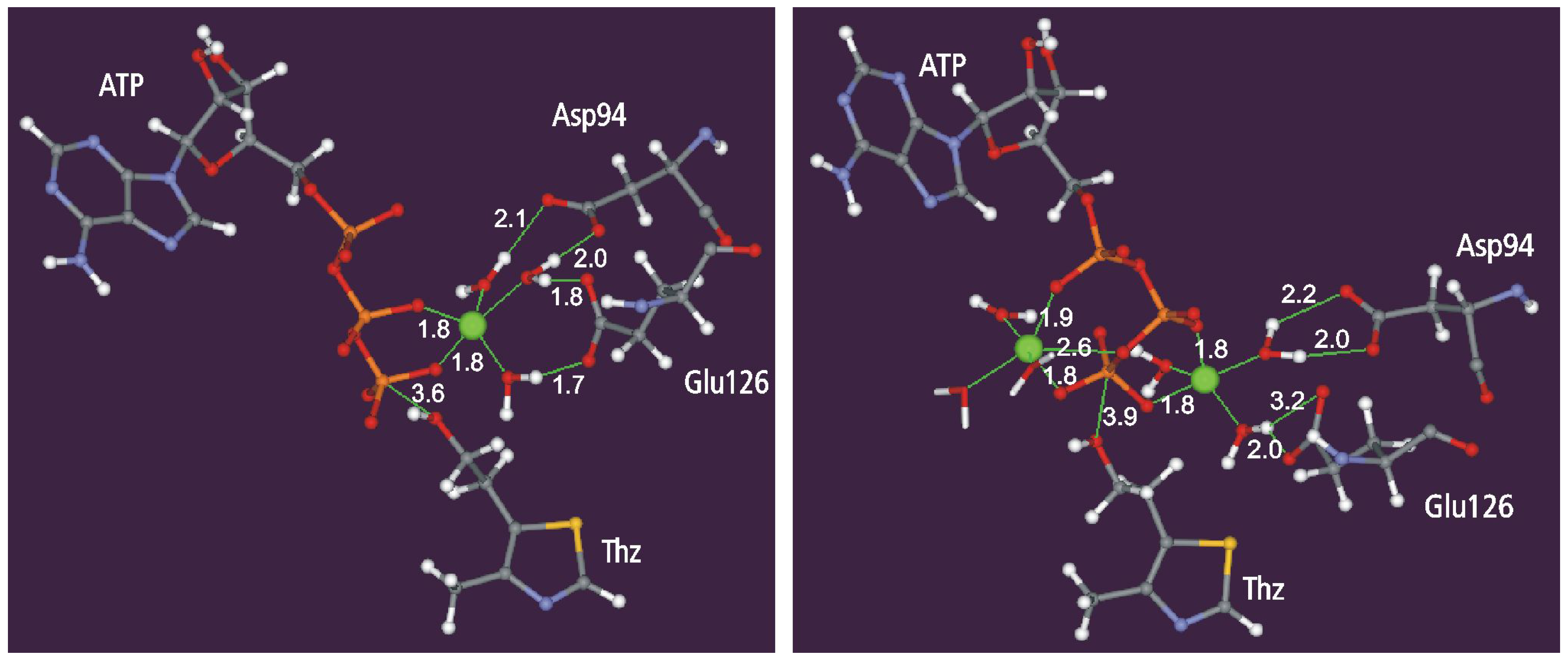
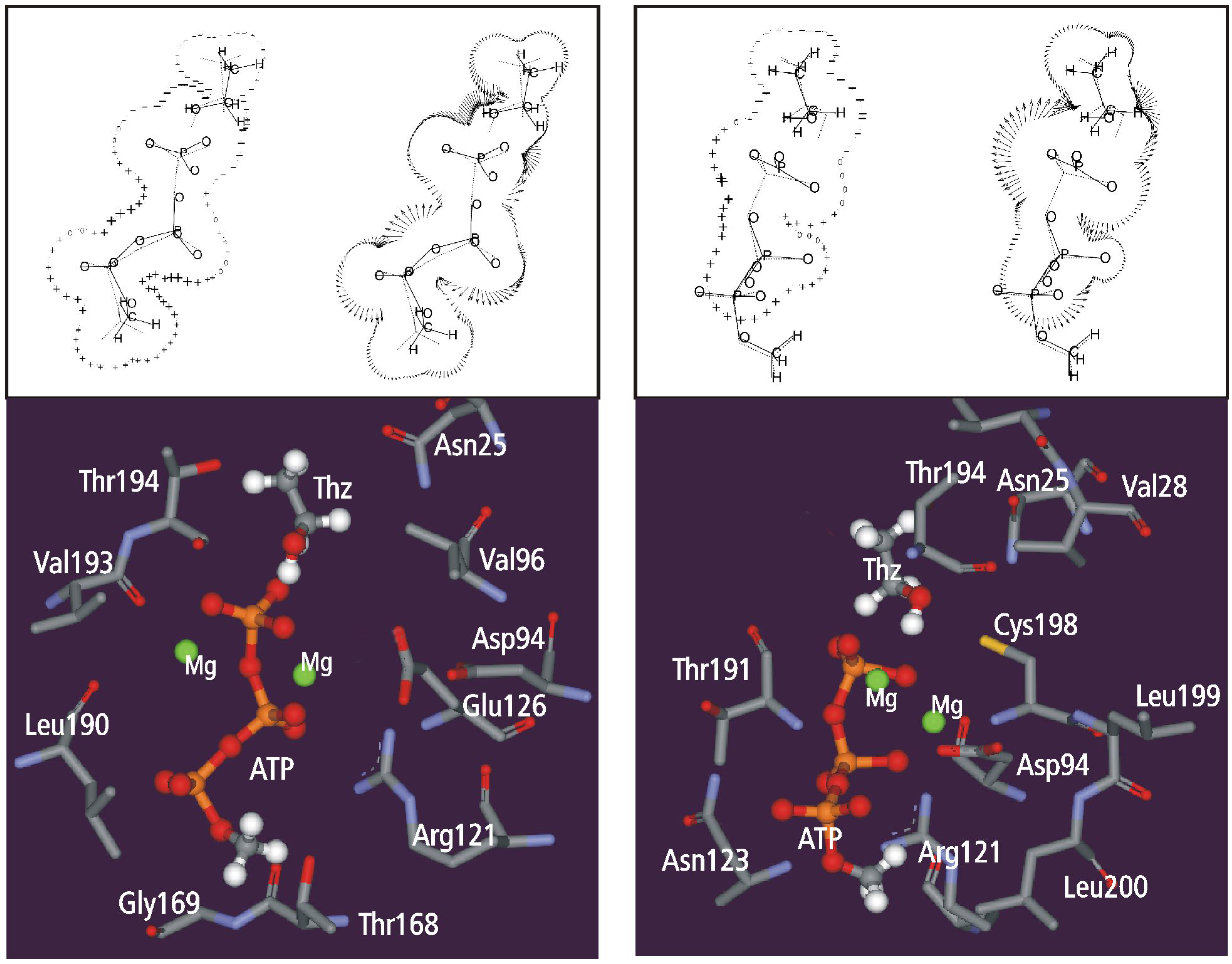
Static and dynamic catalytic fields

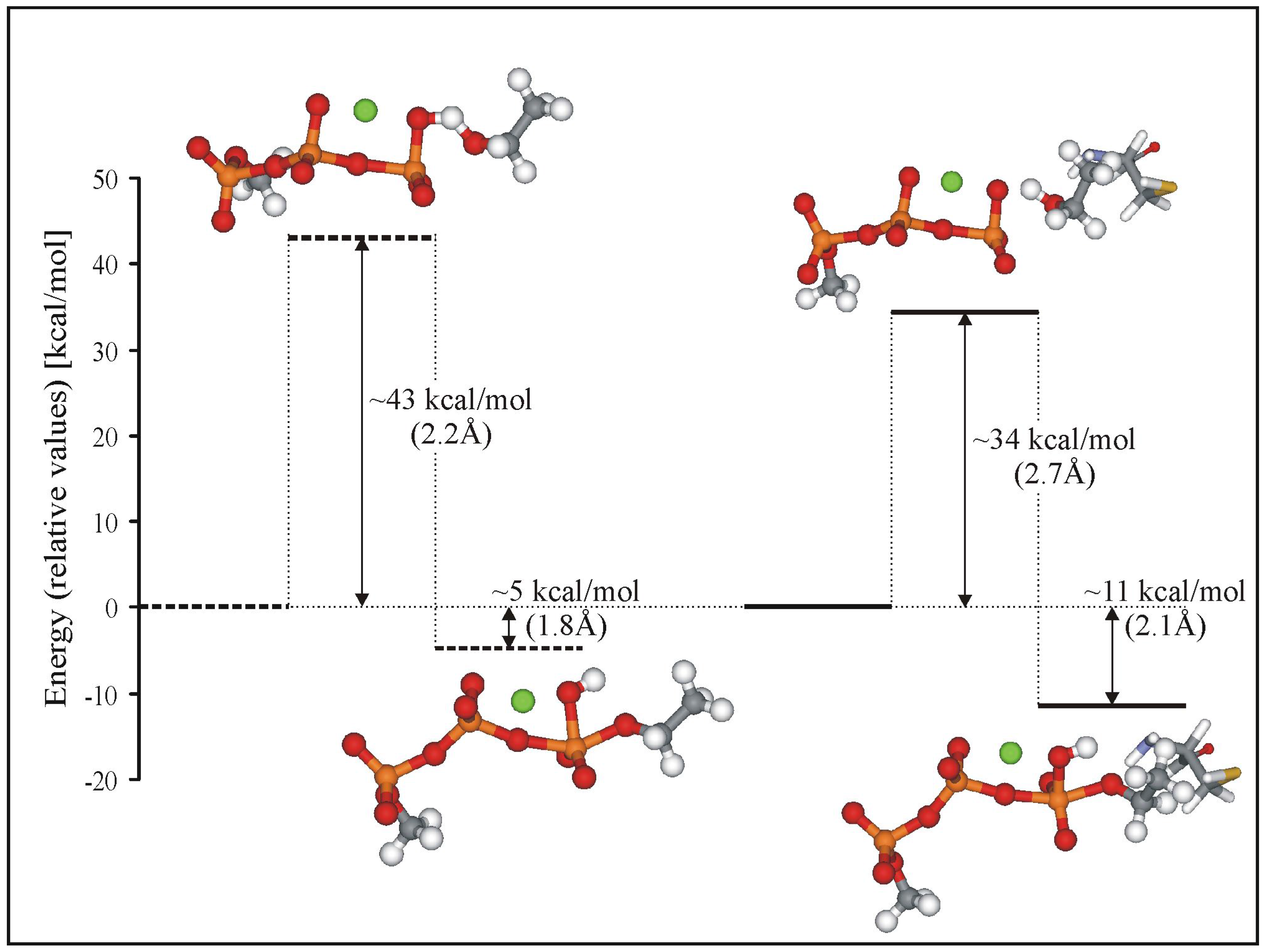
The Reaction Pathway Mapping – Preliminary Results

Discussion and Concluding Remarks
Acknowledgement
References
- Zhang, R. G.; Grembecka, J.; Vinokour, E.; Collart, F.; Dementieva, I.; Minor, W.; Joachimiak, A. Structure of Bacillus subtilis YXKO — A member of the UPF0031 family and a putative kinase. J. Struct. Biol. 2002, 139, 161–170. [Google Scholar] [CrossRef]
- Ito, S.; Fushinobu, S.; Yoshioka, I.; Koga, S.; Matsuzawa, H.; Wakagi, T. Structural Basis for the ADP-Specificity of a Novel Glucokinase from a Hyperthermophilic Archaeon. Structure 2001, 9, 205–214. [Google Scholar] [CrossRef]
- Mathews, I. I.; Erion, M. D.; Ealick, S. E. Structure of Human Adenosine Kinase at 1.5Å Resolution. Biochemistry 1998, 37, 15607–15620. [Google Scholar]
- Schumacher, M. A.; Scott, D. M.; Mathews, I. I.; Ealick, S. E.; Roos, D. S.; Ullman, B.; Brennan, R. G. Crystal Structures of Toxoplasma gondii Adenosine Kinase Reveal a Novel Catalytic Mechanism and Prodrug Binding. J. Mol. Biol. 2000, 296, 549–567. [Google Scholar]
- Sigrell, J. A.; Cameron, A. D.; Jones, T. A.; Mowbray, S. L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8Å resolution: insights into a new family of kinases tructures. Structure 1998, 6, 183–193. [Google Scholar]
- Li, M. H.; Kwok, F.; Chang, W. R.; Lau, C. K.; Zhang, J. P.; Lo, S. C. L.; Jiang, T.; Liang, D. C. Crystal structure of brain pyridoxal kinase, a novel member of the ribokinase superfamily. J. Biol. Chem. 2002, 277, 46385–46390. [Google Scholar] [CrossRef] [PubMed]
- Campobasso, N.; Mathews, I. I.; Begley, T. P.; Ealick, S. E. Crystal Structure of 4-Methyl-5-β-Hydroxyethylthiazole Kinase from Bacillus subtilis at 1.5Å Resolution. Biochemistry 2000, 39, 7868–7877. [Google Scholar] [CrossRef]
- Cheng, G.; Bennett, E. M.; Begley, T. P.; Ealick, S. E. Crystal Structure of 4-Amino-5-Hydroxymethyl-2-Methylpyrimidinephosphate Kinase from Salmonella typhimurium at 2.3Å resolution. Structure 2002, 10, 225–235. [Google Scholar] [CrossRef]
- Herberg, F. W.; Doyle, M. L.; Cox, S.; Taylor, S. S. Dissection of the nucleotide and metal-phosphate binding sites in cAMP-dependent protein kinase. Biochemistry 1999, 38, 6352–6360. [Google Scholar]
- Pichierri, F.; Matsuo, Y. Mechanism of tyrosine phosphorylation catalyzed by the insulin receptor tyrosine kinase: a semiempirical PM3 study. J. Mol. Struc.-Theochem 2003, 622, 257–267. [Google Scholar]
- Biosym Technologies Inc. (2000) Insight II 2000 and Discover version 2.9. San Diego.
- Akola, J.; Jones, R. O. ATP Hydrolysis in Water – A Density Functional Study. J. Phys. Chem. B 2003, 107, 11774–11783. [Google Scholar] [CrossRef]
- Diaz, N.; Field, M. J. Insights into the phosphoryl-transfer mechanism of cAMP-dependent protein kinase from quantum chemical calculations and molecular dynamics simulations. J. Am. Chem. Soc. 2004, 126, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Hata, M.; Hoshino, T.; Tsuda, M. Quantum chemical study on the catalytic mechanism of the C-subunit of cAMP-dependent protein kinase. J. Phys. Chem. B 2002, 106, 5788–5792. [Google Scholar] [CrossRef]
- Kitao, A.; Hirata, F.; Go, N. The effects of solvent on the conformation and the collective motions of protein - normal mode analysis and molecular-dynamics simulations of melittin in water and in vacuum. Chem. Phys. 1991, 158, 447–472. [Google Scholar] [CrossRef]
- Zhou, RH. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins 2003, 53, 148–161. [Google Scholar]
- Gaussian 03, Revision B.04: Frisch, M. J.; et al. Gaussian, Inc.: Pittsburgh PA, 2003.
- Stewart, J. J. P. Optimization of parameters for semiempirical methods. 1. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Peng, C. Y.; Schlegel, H. B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Israel J. Chem. 1993, 33, 449–454. [Google Scholar]
- Rakowski, F.; Grochowski, P.; Lesyng, B. SCC DFT-TB Energy calculations of the phosphoryl transfer reaction in the PKA active site. Cell. Mol. Biol. Lett. 2003, 8, 603–604. [Google Scholar]
- Sokalski, W. A. The physical nature of catalytic activity due to the molecular environment in terms of intermolecular interaction theory: derivation of simplified models. J. Mol. Catalysis 1985, 30, 395–410. [Google Scholar] [CrossRef]
- Sokalski, W. A.; Kedzierski, P.; Grembecka, J.; Dziekonski, P.; Strasburger, K. Computational Molecular Biology; Leszczynski, J., Ed.; Elsevier Science, 1999; Chapter 10: Theoretical tools for analysis and modelling electrostatic effects in biomolecules.; pp. 369–391. [Google Scholar]
- Sokalski, W. A. Nonempirical modeling of the static and dynamic properties of the optimum environment for chemical reactions. J. Mol. Struct. 1986, 138, 77–87. [Google Scholar] [CrossRef]
- Stewart, J. J. P. Special issue - MOPAC - a semiempirical molecular-orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–45. [Google Scholar] [CrossRef]
- Cernohorsky, M.; Kuty, M.; Koca, J. A multidimensional driver for quantum chemistry program MOPAC. Comput. Chem. 1997, 21, 35–44. [Google Scholar] [CrossRef]
- Prokop, M.; Damborsky, J.; Koca, J. TRITON: in silico construction of protein mutants and prediction of their activities. Bioinformatics 2000, 16, 845–846. [Google Scholar] [CrossRef] [PubMed]
- Damborsky, J.; Prokop, M.; Koca, J. TRITON: graphic software for rational engineering of enzymes. Trends Biochem. Sci. 2001, 26, 71–73. [Google Scholar] [CrossRef]
- Shaltiel, S.; Cox, S.; Taylor, S. S. Conserved water molecules contribute to the extensive network of interactions at the active site of protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 484–491. [Google Scholar]
© 2004 by MDPI (http://www.mdpi.org)
Share and Cite
Dyguda, E.; Szefczyk, B.; Sokalski, W.A. The Mechanism of Phosphoryl Transfer Reaction and the Role of Active Site Residues on the Basis of Ribokinase-Like Kinases. Int. J. Mol. Sci. 2004, 5, 141-153. https://doi.org/10.3390/i5040141
Dyguda E, Szefczyk B, Sokalski WA. The Mechanism of Phosphoryl Transfer Reaction and the Role of Active Site Residues on the Basis of Ribokinase-Like Kinases. International Journal of Molecular Sciences. 2004; 5(4):141-153. https://doi.org/10.3390/i5040141
Chicago/Turabian StyleDyguda, Edyta, Borys Szefczyk, and W. Andrzej Sokalski. 2004. "The Mechanism of Phosphoryl Transfer Reaction and the Role of Active Site Residues on the Basis of Ribokinase-Like Kinases" International Journal of Molecular Sciences 5, no. 4: 141-153. https://doi.org/10.3390/i5040141