Introduction
The reduction of nitro aromatic compounds (NACs) is of considerable interest because NACs are common groundwater contaminants and reduction reactions can play a central role in their environmental fate or cleanup. Sources of NAC contaminants include activities associated with the production or utilization of explosives, dyes, agrichemicals and pesticides.
Zero-valent iron (ZVI) has been demonstrated to participate directly or indirectly in environmentally beneficial reduction reactions affecting the transformation of a variety of common groundwater contaminants, including chlorinated solvents, heavy metals, uranium, nitrate, and nitroaromatic compounds [
1].
The reduction of nitro-organic compounds also may be affected by non-ferrous metals or by surface catalyzed reactions involving ferrous iron (Fe
+2) as the electron donor. Agrawal and Tratnyek [
2] describe the reduction of nitrobenzene on ZVI, reporting rapid reduction to aniline (aminobenzene) with the transient occurrence of nitrosobenzene.
The detailed molecular mechanism of the NACs reduction by Fe(0) is not yet fully understood. Formally, the overall nitro-to-amino reduction is a six-electron-transfer reduction half-reaction:
where ‘Ar’ represents an aromatic structure, though it could also represent an aliphatic or N-heterocyclic structure. However, the actual reduction of the nitro to amino group by iron comprises a variety of pathways including coordination by iron centers, transformation of nitro group, condensation of NACs, etc. The first 2e
- transfer step in the nitro reduction reaction is proposed to be the formation of nitroso group [
3].
This step merits careful consideration as it describes the initial reaction mechanism as well as the generation of products for subsequent reactions.
To our knowledge, the reduction of NACs by zero-valent iron has never been studied by means of quantum chemical methods. Among the very few such studies devoted to the adsorption of much smaller systems on the iron surface, the following works seem to be representative.
To study the atomic hydrogen adsorption on the Fe(110) surface an extended Hückel tight-binding (EHT) method has been applied recently [
4]. The unit cell geometry was optimized using the atom-superposition-and-electron-delocalization-molecular-orbital (ASED-MO) model core potential. This work was inspired by the wide interest in studying the hydrogen-metal interaction due to its importance for catalysis and material science. The metal surface was approximated by periodic, seven-layer slab using the 87-atom unit cell. The advantage of this approach is its transparency and simplicity which, for example, allows one to avoid the convergence problem associated with the Hartree–Fock or Density Functional Theory (DFT) methods. This EHT study is closely related to the recent
ab initio calculation of the H
2 adsorption on a 32-atom cluster [
5].
Mortenson et al. [
6] applied DFT periodic slab calculations to study nitrogen adsorption in order to gain some insight into microscopic reaction steps for activation of N
2 on the Fe(111), (100), and (110) faces. To model these faces, the six-, four-, and three-layer slabs were used. Unit cells contain 12, 10, and 9 atoms, respectively. Activation of N
2 by Fe was also considered in the cluster DFT calculations using two-atom iron clusters [
7].
Applying first-principle, quantum chemical approaches to study of the NACs adsorption on the iron surface presents numerous computational challenges. The system requires a top layer of cluster (or surface unit cell) that is at least equal in size to the NAC molecule. This means that the corresponding surface unit cell or cluster top layer should be even larger than that used in the EHT study of the H/Fe(110) system [
4]. For the first-principle periodic approaches the requirements are even stricter since the unit cell should be further extended to exclude artificial adsorbate-adsorbate interaction arisen in such methods for small surface unit cells.
The aim of this work is to obtain some insight into the process of the NACs adsorption on the metal iron by means of the Neglect of Diatomic Differential Overlap for Metal Compounds (NDDO/MC) semi-empirical method [
8]. This method has been parameterized to reproduce the experimental geometry and binding energy for molecules like HNO, HNO
2, FeO and FeN2 [
9]. It makes this approach reliable for the study of the nitro group interaction with zero-valent iron despite the approximations used in NDDO/MC. As a prototype NAC we considered nitrobenzene (NB).
Unprotected metallic iron certainly will oxidize or corrode under aquifer conditions to form a variety of iron oxides, hydroxides, or other minerals, depending on water chemistry. Thus, nitro contaminants under field conditions would encounter clean iron for a very short period or time and/or at a very limited surface area. Also under investigation are the interactions of the nitro contaminants with the products of iron corrosion in the presence of ferrous iron which would be supplied by iron corrosion. Characterization of the reactions at metal iron surfaces establishes a benchmark for comparative purposes.
The Fe(110) crystal face is chosen to be the actual surface on which the NB reduction is realized. In case of bcc crystal lattice, this face is the closest packed and the most stable. Because of that it is worth to expect that the real granular iron surface contains the high percent of the (110) ‘patches’.
Method
In this work all calculations have been performed in the cluster approximation using NDDO/MC semiempirical Hamiltonian [
8]. The NDDO/MC method is based on the approximation of zero diatomic differential overlap which results in neglecting of 3-, 4- and part of 2-center two-electron integrals to be appeared in the Fock matrix [
10]. Retained two-electron integrals are calculated explicitly with Slater-type orbitals. Despite that NDDO/MC is a much faster method than
ab initio one due to its quadratic dependence on basis set versus quartic one for the latter.
In order to determine how well the NDDO/MC method predicts the properties of the nitro compounds few simple molecules of this class have been treated. In
Table 1 the bond lengths and valence angles calculated by NDDO/MC are listed along with those from experimental data [
11,
12]. The N-O bond length is predicted with the accuracy of about 0.02 Å. The O-N-O angle is accurate to
Table 1.
Bond length (Å) and valence angles (degree) of the nitro compounds as revealed by the NDDO/MC calculations
Table 1.
Bond length (Å) and valence angles (degree) of the nitro compounds as revealed by the NDDO/MC calculations
| Compound | | NDDO/MC | Experiment1 |
|---|
| NO | N-O | 1.171 | 1.151 |
| NO2 | N-O | 1.224 | 1.193 |
| O-N-O | 146.7 | 134.4 |
| HNO | N-O | 1.200 | 1.239 |
| N-H | 1.080 | 1.020 |
| O-N-H | 112.2 | 114.4 |
| CH3NO | N-O | 1.196 | 1.22 |
| | C-N | 1.479 | 1.49 |
| | C-H | 1.084 | |
| | C-N-O | 124.9 | 112.6 |
| CH3NO2 | N-O | 1.248 | 1.224 |
| | C-N | 1.573 | 1.489 |
| | C-H | 1.088 | |
| | C-N-O | 113.4 | 117.4 |
| C6H6NO2 | N-O | 1.257 | 1.208 |
| | C-N | 1.593 | 1.486 |
| | C-N-O | 114.3 | 118.0 |
| C6H6NO2- | N-O | 1.280 | |
| | C-N | 1.653 | |
| | C-N-O | 117.0 | |
| C6H6NO22- | N-O | 1.342 | |
| | C-N | 1.587 | |
| | C-N-O | 121.0 | |
| C6H6NO | N-O | 1.200 | |
| | C-N | 1.460 | |
| | C-N-O | 134.0 | |
within 8-12 degree. The C-N bond lengths are about 0.08 Å systematically overestimated. NDDO/MC reproduces the electron structure of the nitroaromatics in accordance with that given by
ab initio and other semiempirical methods like MNDO.
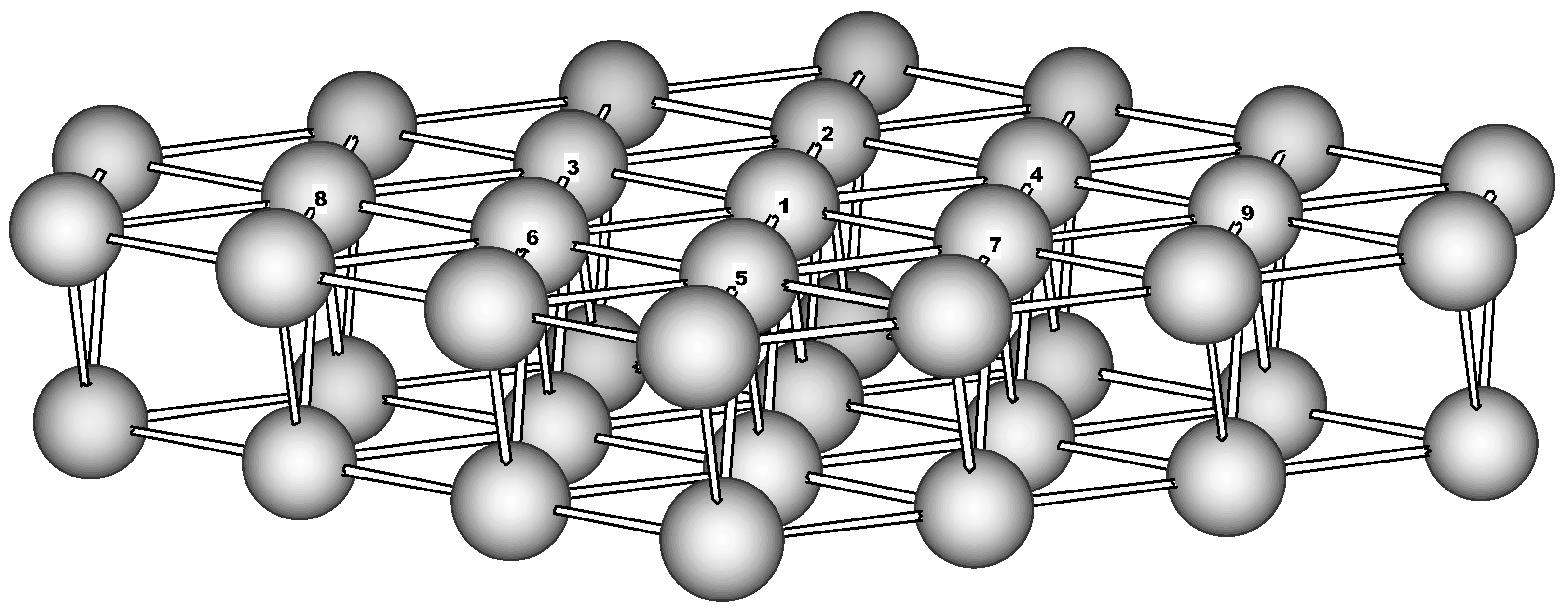
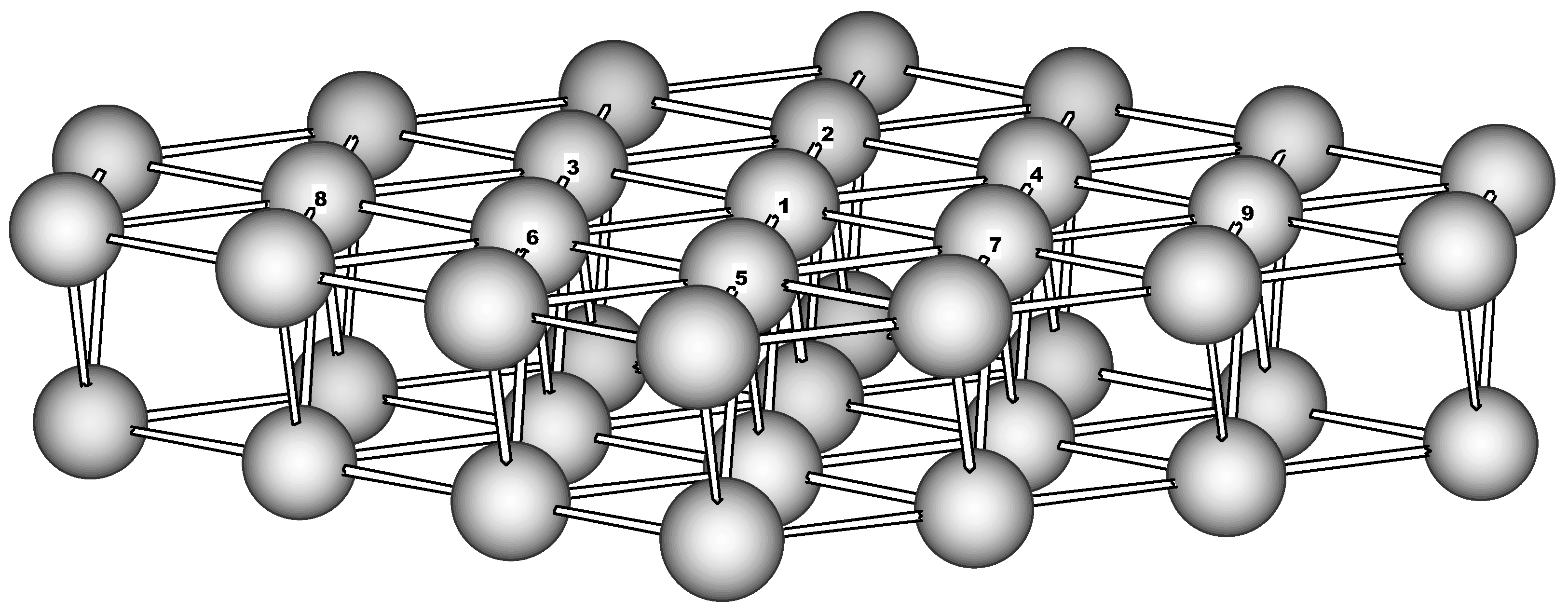
Cluster model. To model the Fe(110) surface a two-layer cluster has been used. It consists of the 23-atom (‘top’) and 16-atom second layer and has the C
2v symmetry (
Figure 1). The boundary for the top layer was chosen in such a way to form an equivalent hexagonal surrounding for every ‘active’ atom (Fe1-Fe9). The second layer brings additional 4 atoms to the coordination sphere of the active atoms. Thus the cluster contains just ‘surface’ atoms having reduced first coordination sphere of 10 atoms. Below this cluster will be referred as Fe
39.
Since the metal iron possesses large magnetic moment (2.2 spins per atom for bulk Fe [
13,
14]) a non-trivial question arises concerning spin state to be assumed for the cluster under consideration. For the 39-atom cluster considered the spin multiplicity is chosen in the following way. First, we analyze the wave function associated with the Huckel-type Hamiltonian to find out the one-electron states
Figure 1.
The 39-atom cluster used to model the Fe(110) surface (perspective view).
Figure 1.
The 39-atom cluster used to model the Fe(110) surface (perspective view).
corresponding to the 3d band. Then all the one-electron states were populated starting from the lowest until the 3d band becomes completely occupied. At the same time we keep as many unpaired electrons as possible to form the highest spin multiplicity state. It appears that this state corresponds to the multiplicity of 105 (S=52). This multiplicity corresponds to the magnetization of 2.67 spins per atom on average. It agrees fairly well with the first-principle theoretical predictions of 2.57 [
15] and 2.65 [
16] for the (110) surface of bcc Fe. The unrestricted Hartree-Fock (UHF) calculation for Fe
39 with this multiplicity gives the highest occupied molecular orbital (HOMO) for the alpha-spin electrons having the energy of -4.02 eV. Such energy is close to the electron work function for iron (4.5 eV [
11]). Performing the SCF calculation for Fe
39 we faced the problem of slow convergence (which sometimes takes thousands of iterations to be completed) along with the broken symmetry. The latter is manifested by differences in charge and spin (of the Spin-Density-Wave type) for symmetrical centers of the cluster (
Table 2). The question arises if it can be accepted as physically reasonable solution. As it will be shown below the nitroaromatics-Fe
0 interaction is rather strong and results particularly in substantial charge transfer from metal toward the nitro group. The adsorption species formed reduce the symmetry of substrate. So the charge distribution on the 'naked' metal cluster seems to have a negligible effect on the NACs adsorption on Fe
0 and especially on reduction of nitro aromatics.
Table 2.
Electron (q) and spin (qs) densities on the ‘active’ centers of the ‘naked’ cluster and the cluster with NB adsorbed on it.
Table 2.
Electron (q) and spin (qs) densities on the ‘active’ centers of the ‘naked’ cluster and the cluster with NB adsorbed on it.
| | Fe39 | | NB(A)/Fe39 |
|---|
| | q | qs | | q | qs |
|---|
| Fe1 | 0.39 | 2.97 | | 0.46 | 2.88 |
| Fe2 | 0.24 | 2.99 | | 0.37 | 2.98 |
| Fe3 | 0.28 | 0.97 | | 0.13 | 1.02 |
| Fe4 | -0.43 | 2.03 | | 0.42 | 2.99 |
| Fe5 | 0.24 | 2.99 | | 0.39 | 2.97 |
| Fe6 | 0.30 | 0.97 | | 0.12 | 1.01 |
| Fe7 | -0.43 | 2.03 | | 0.42 | 2.03 |
| Fe8 | 0.14 | 0.93 | | 0.27 | 0.99 |
| Fe9 | -0.48 | 2.01 | | 0.47 | 2.02 |
Surprisingly the spin contamination of UHF function is rather moderate. For example for the naked cluster the <S2> value is about 2756.04 which is very close to the exact value of 2756 for 2S+1=105. All calculations in this work have been performed for this multiplicity.
Results
Considering the interaction of NACs with zero-valent iron some predictions concerning the mechanism of the nitro group reduction can be put forward
a priori on base of electron structure of NACs itself. In
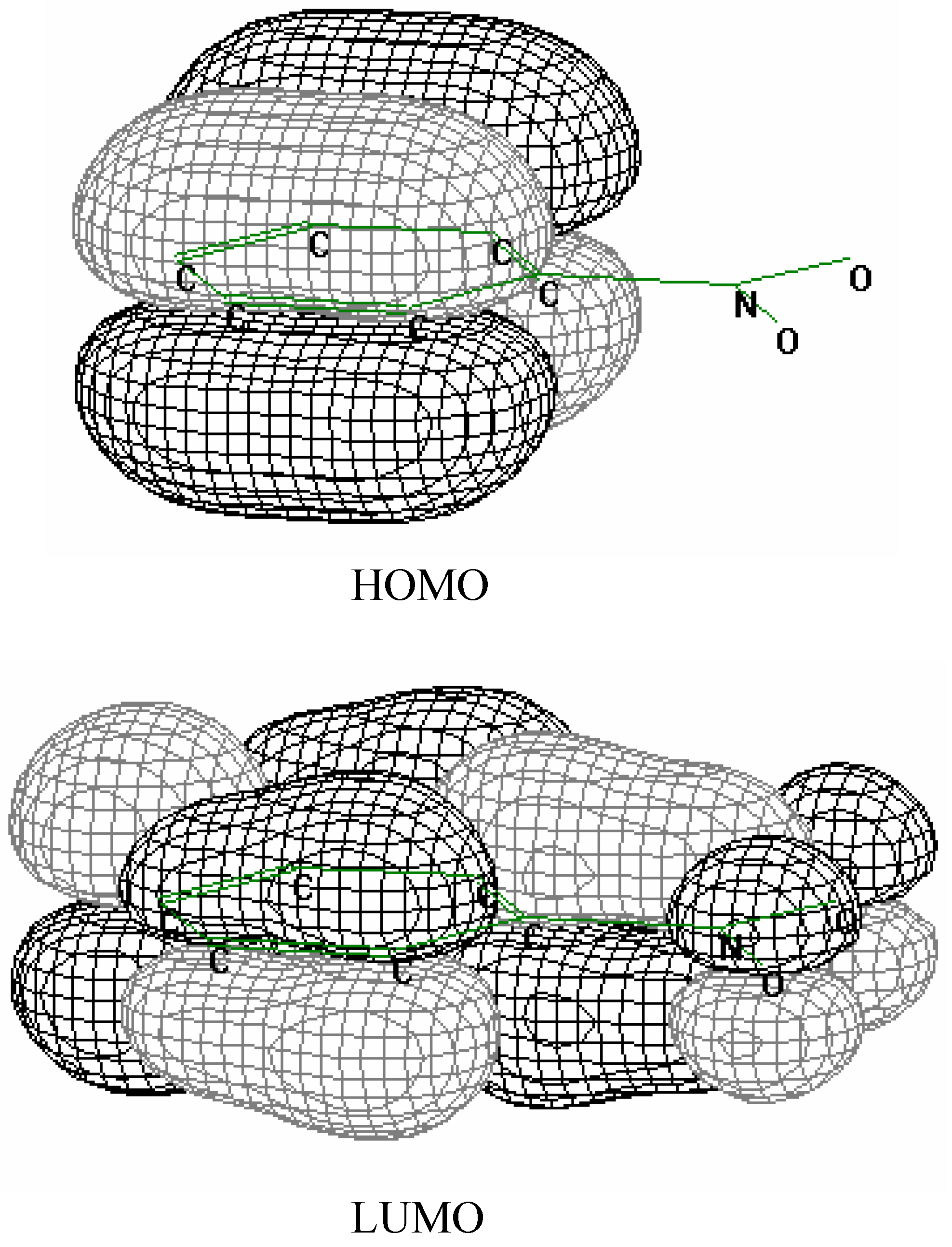
Figure 2 the HOMO and lowest unoccupied molecular orbitals (LUMO) are plotted for nitrobenzene. As seen from
Figure 2 HOMO is localized on the benzene ring unlike LUMO which is the π
* orbital delocalized over the benzene ring and nitro group. Because of that one can expect the N-O bond to be weaken (or even broken) upon the electron transfer into the NACs.
The adsorption of NB on the Fe
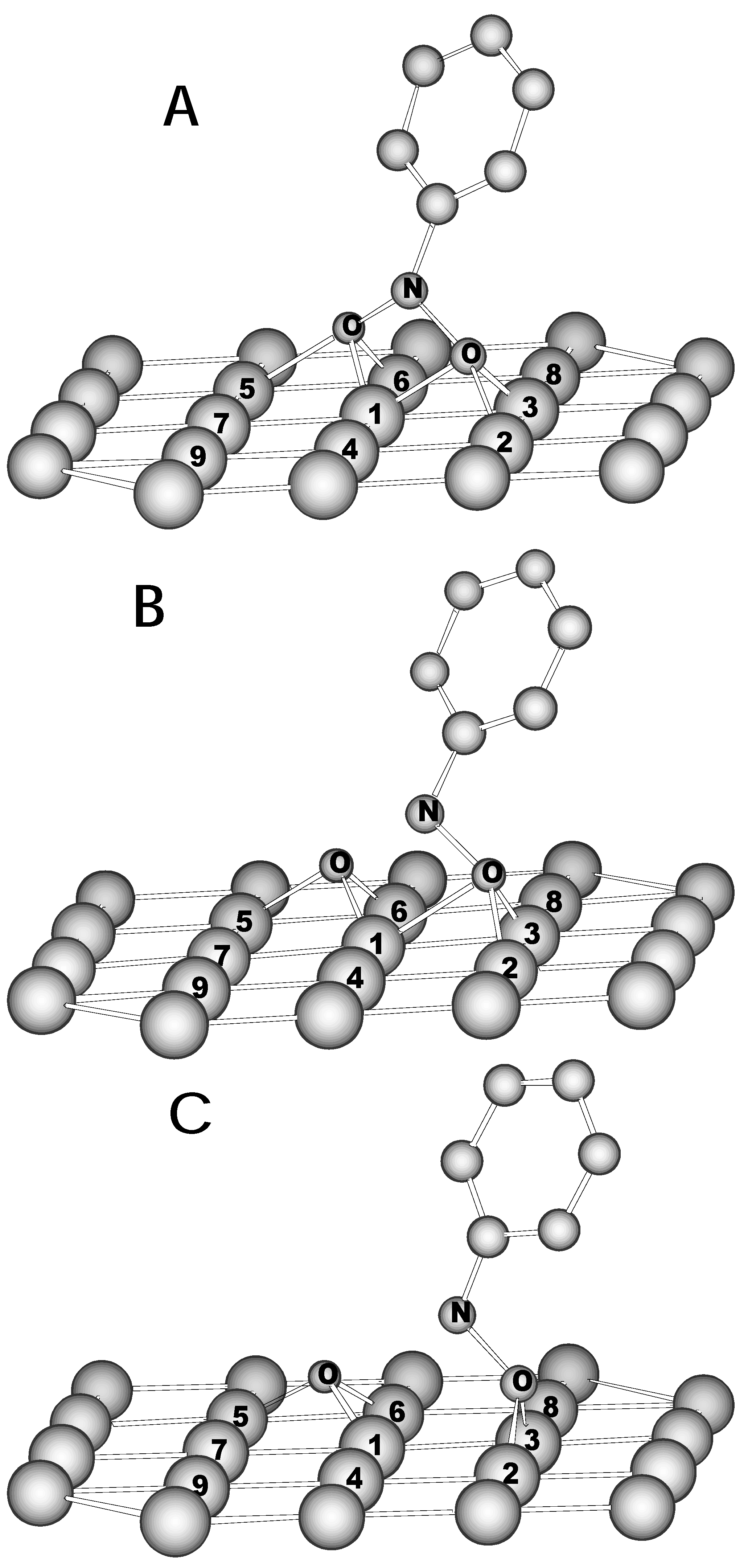
39 cluster has been treated at the UHF level. It is evident that NB can interact with iron surface forming either planar or upright adsorption species. One could anticipate that planar complexes are likely to be formed on surface by analogy with π-bonded structure of ferrocene. We have tested both ways of interactions and found that upright-bonded species are more than 80 kcal/mol preferred. The geometry optimization (geometry of cluster itself and the NB ring were kept frozen) of NB on the cluster yields the molecularly adsorbed NB which is shown in
Figure 3A (this structure is designated as
A). The modification of the NB geometry upon associative
Figure 2.
Frontier orbitals of nitrobenzene.
Figure 2.
Frontier orbitals of nitrobenzene.
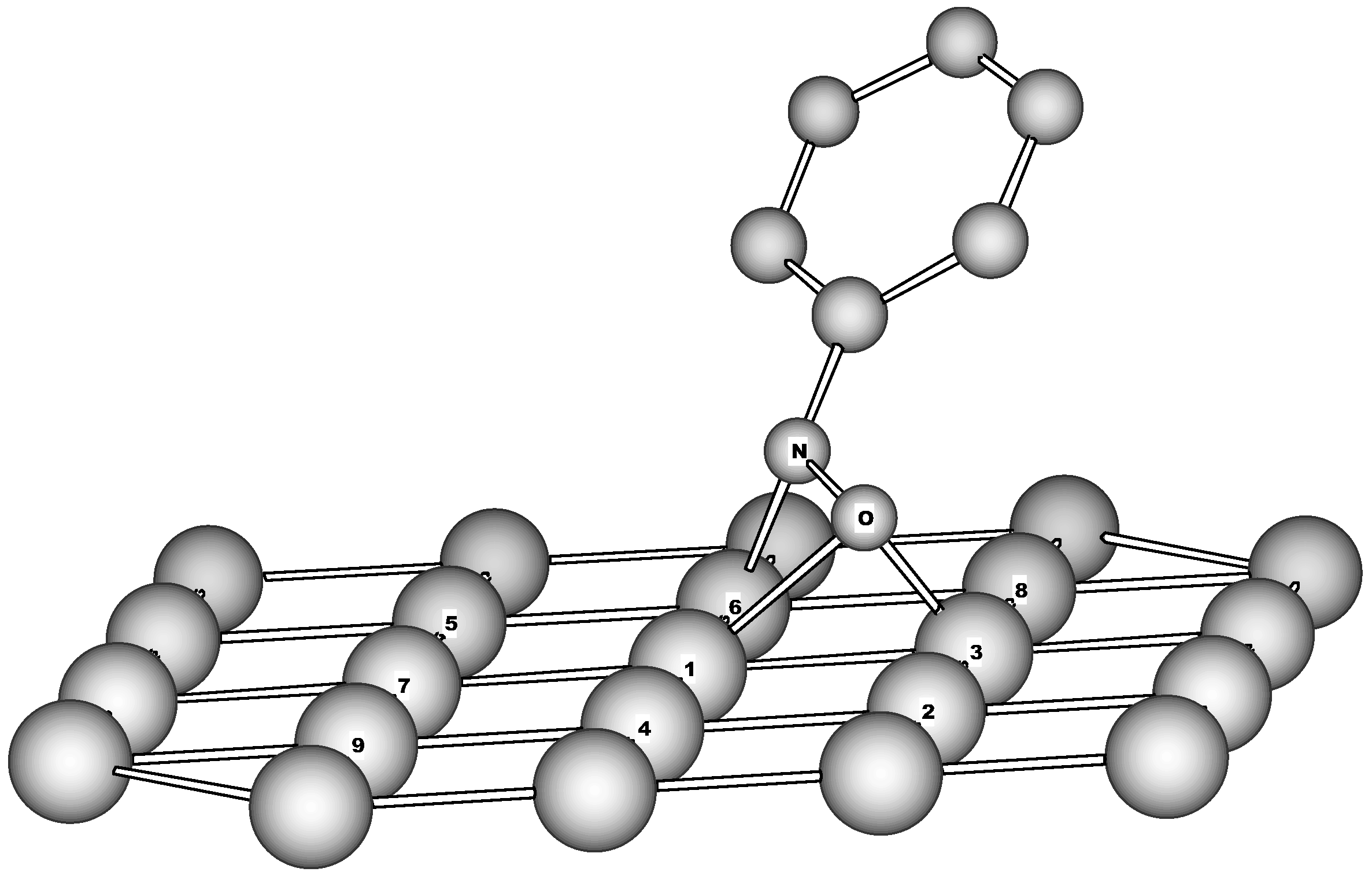
Figure 3.
Adsorption of nitrobenzene on Fe(110) as revealed by NDDO/MC cluster calculations. The only top layer of the cluster is shown for simplicity. (A) Molecular adsorption of NB. Calculated distances (Å) and valence angles (degrees): N-C 1.452, N-O 1.586, N-O’ 1.560, N-Fe1 2.099, O-Fe1 1.898, O-Fe2 2.024, O-Fe3 1.632, O’-Fe1 1.923, O’-Fe5 2.069, O’-Fe6 1.630, O-O’ 2.699, O-N-C 116.1, O’-N-C 118.6 (B) Transition state for dissociation of nitro group. Calculated distances (Å) and valence angles (degrees): N-C 1.403, N-O 1.531, N-O’ 2.080, N-Fe1 2.265, O-Fe1 2.222, O-Fe2 1.979, O-Fe3 1.634, O’-Fe1 1.925, O’-Fe5 1.888, O’-Fe6 1.602, O-O’ 3.082, O-N-C 111.3, O’-N-C 124.5 (C) Dissociative adsorption of NB on Fe(110). Calculated distances (Å) and valence angles (degrees): N-C 1.357, N-O 1.535, N-O’ 2.780, N-Fe1 2.478, O-Fe1 2.646, O-Fe2 1.826, O-Fe3 1.627, O’-Fe1 1.891, O’-Fe5 1.636, O’-Fe6 1.619, O-O’ 3.717, O-N-C 106.9, O’-N-C 135.3
Figure 3.
Adsorption of nitrobenzene on Fe(110) as revealed by NDDO/MC cluster calculations. The only top layer of the cluster is shown for simplicity. (A) Molecular adsorption of NB. Calculated distances (Å) and valence angles (degrees): N-C 1.452, N-O 1.586, N-O’ 1.560, N-Fe1 2.099, O-Fe1 1.898, O-Fe2 2.024, O-Fe3 1.632, O’-Fe1 1.923, O’-Fe5 2.069, O’-Fe6 1.630, O-O’ 2.699, O-N-C 116.1, O’-N-C 118.6 (B) Transition state for dissociation of nitro group. Calculated distances (Å) and valence angles (degrees): N-C 1.403, N-O 1.531, N-O’ 2.080, N-Fe1 2.265, O-Fe1 2.222, O-Fe2 1.979, O-Fe3 1.634, O’-Fe1 1.925, O’-Fe5 1.888, O’-Fe6 1.602, O-O’ 3.082, O-N-C 111.3, O’-N-C 124.5 (C) Dissociative adsorption of NB on Fe(110). Calculated distances (Å) and valence angles (degrees): N-C 1.357, N-O 1.535, N-O’ 2.780, N-Fe1 2.478, O-Fe1 2.646, O-Fe2 1.826, O-Fe3 1.627, O’-Fe1 1.891, O’-Fe5 1.636, O’-Fe6 1.619, O-O’ 3.717, O-N-C 106.9, O’-N-C 135.3
adsorption on Fe
39 confirmed the idea about leading role of nitro group in the NACs reduction by iron. It appeared that the NB ring forms an angle of 36 degree with respect to its upright position (
Figure 3A). Another prediction arisen from the LUMO structure of NB was also confirmed: the N-O bonds become substantially elongated (1.56 for adsorbed NB vs. 1.26 Å for isolated NB). Otherwise, the N-C bond for
A is more than 0.1 Å shorter than that of isolated NB. These changes seem to be determined by transfer of almost 2 electrons from cluster toward nitro group (
Table 3). And as it might be anticipated the charge density transferred is localized mostly on oxygen atoms. The spin density is also transferred into NB but in contrast to the charge density is delocalized over the whole molecule. When adsorbed on Fe(110) the NB molecule proceeds in fact as a triplet-state charge-transfer intermediate.
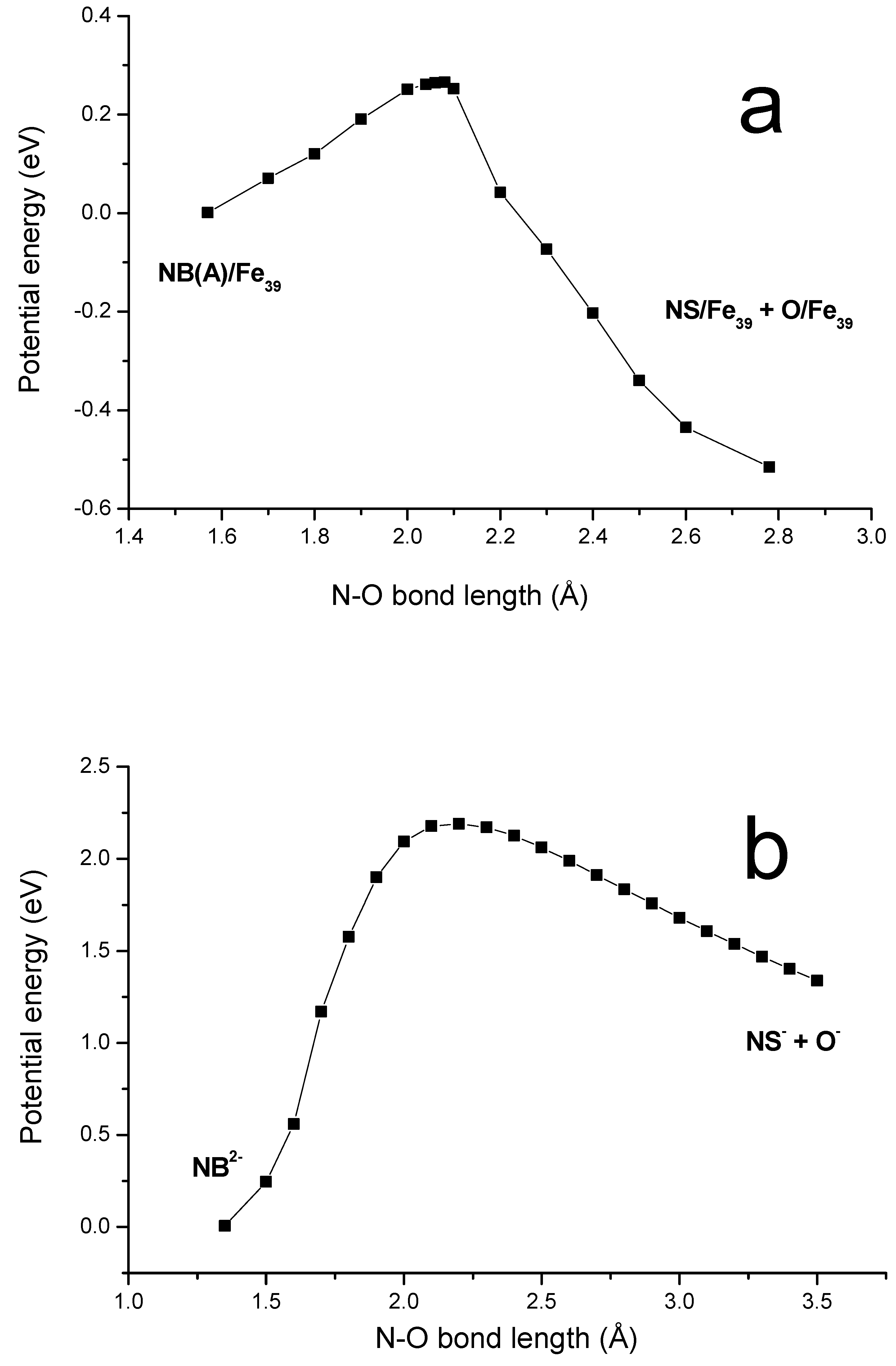
Taking into account substantial weakening of N-O bonds for adsorbed NB here comes the suggestion about subsequent modification of NB on iron surface. One may predict that one or both N-O bonds are to be broken to form NB-reduced species. In order to model the dissociation of nitro group on iron surface the potential energy profile has been calculated for a number of N-O’ distances (O’ here and below designates runaway oxygen atom). Along this reaction coordinate the geometry of NB was optimized keeping above-mentioned geometry restrictions. The saddle point (
Figure 3B) obtained relates to nitrobenzene on Fe
39 with almost broken N-O bond (2.08 Å). This species will be referred to as
B. The energy minimum beyond the barrier (it is designated as
C) corresponds to the nitrosobenzene (NS) molecule and atomic oxygen adsorbed on the surface (
Figure 3C). The potential energy profile is plotted in
Figure 4a. The starting point corresponds to nitrobenzene which is associatively adsorbed on surface. The height of barrier for the N-O bond cleavage on Fe(110) is estimated to be just 6.0 kcal/mol when going from
A to
C species. The dissociative-adsorption species
Table 3.
Adsorption nergy and population analysis for nitro group of NB and NS adsorbed on Fe(110) as predicted by the NDDO/MC calculations for the 39-atom iron cluster
Table 3.
Adsorption nergy and population analysis for nitro group of NB and NS adsorbed on Fe(110) as predicted by the NDDO/MC calculations for the 39-atom iron cluster
| Species | Electron density (a.u) | Spin density (a.u.) | Eads(eV) 1 |
|---|
| | N | O | O’ | NB | | N | O | O’ | NB | | |
|---|
| | | | | | | | | | | | |
| NB isolated | 0.67 | -0.27 | -0.27 | 0.0 | | 0.0 | 0.0 | 0.0 | 0.0 | | - |
| NB(A)/Fe39 | -0.08 | -0.69 | -0.73 | -1.88 | | 0.34 | 0.27 | 0.23 | 1.12 | | 6.16 |
| NB(B)/Fe39 | -0.45 | -0.66 | -0.61 | -1.99 | | 0.09 | 0.03 | 0.82 | 0.97 | | 5.90 |
| NB(C)/Fe39 | -0.51 | -0.65 | -1.00 | -2.45 | | 0.01 | 0.04 | 0.44 | 0.072 | | 6.68 |
| NS/Fe39 | -0.67 | -0.49 | - | -1.35 | | -0.01 | 0.02 | - | 0.03 | | 5.72 |
| O/Fe39 | - | - | -0.98 | - | | - | 0.01 | - | - | | 6.80 |
Figure 4.
Dissociation of the N-O bond of nitrobenzene. (a) Plot for NB on Fe39. The starting point (taken with zero energy) corresponds to the N-O bond of molecularly adsorbed nitrobenzene. The last one corresponds to the N-O bond dissociated to form nitrosobenzene and oxygen atom adsorbed on iron surface. (b) Potential energy profile calculated for different N-O bond of isolated nitrobenzene having total charge of –2 and spin multiplicity of 3.
Figure 4.
Dissociation of the N-O bond of nitrobenzene. (a) Plot for NB on Fe39. The starting point (taken with zero energy) corresponds to the N-O bond of molecularly adsorbed nitrobenzene. The last one corresponds to the N-O bond dissociated to form nitrosobenzene and oxygen atom adsorbed on iron surface. (b) Potential energy profile calculated for different N-O bond of isolated nitrobenzene having total charge of –2 and spin multiplicity of 3.
of NB is calculated to be about 12 kcal/mol preferred over the associative one. This allows us to predict that on the metal iron surface equilibrium
is shifted to the reductive form of nitrobenzene. For comparison in
Figure 4b there is plotted similar energy profile calculated for dissociation of gaseous NB anion in triplet state:
Such charge and spin was chosen to model surface NB species which are the most close to this anion. For this anion a barrier for N-O bond cleavage is estimated to be about 50 kcal/mol and products lie substantially higher than the NB
2-. This finding shows a crucial role which zero-valent iron surface plays in the reduction of nitrobenzene.
The products of the NB dissociation on Fe(110) deserves to be considered in more detail. The oxygen coming out from broken nitro group occupies the 3-fold position (
Figure 3C) with Fe-O distances ranging from about 1.6 to 1.9 Å. Such Fe-O bond lengths are quite close to that of the FeO molecule (experimental Fe-O distance is 1.57 Å [
17]). As can be seen from
Figure 3C the nitrosobenzene moiety is bound to surface mostly through oxygen center. In order to find out how isolated oxygen atom and nitroso benzene adsorb on iron surface we performed investigation of their interactions with the same cluster. The nitrosobenzene (NS) adsorption species is shown in
Figure 5. It was optimized using restrictions similar to those for NB, i.e. keeping frozen the benzene ring and metal cluster geometry. Unlike the nitroso moiety of NB on Fe
39 the isolated NS molecule binds to surface through oxygen and nitrogen as well (as shown in
Figure 5). The other changes reflect the same trend: the N-O bond becomes substantially elongated while the C-N bond is shortened upon adsorption. The slope of benzene ring is about 54 degree from the upright position. There is also a substantial electron density transfer (1.353 units) toward adsorbate.
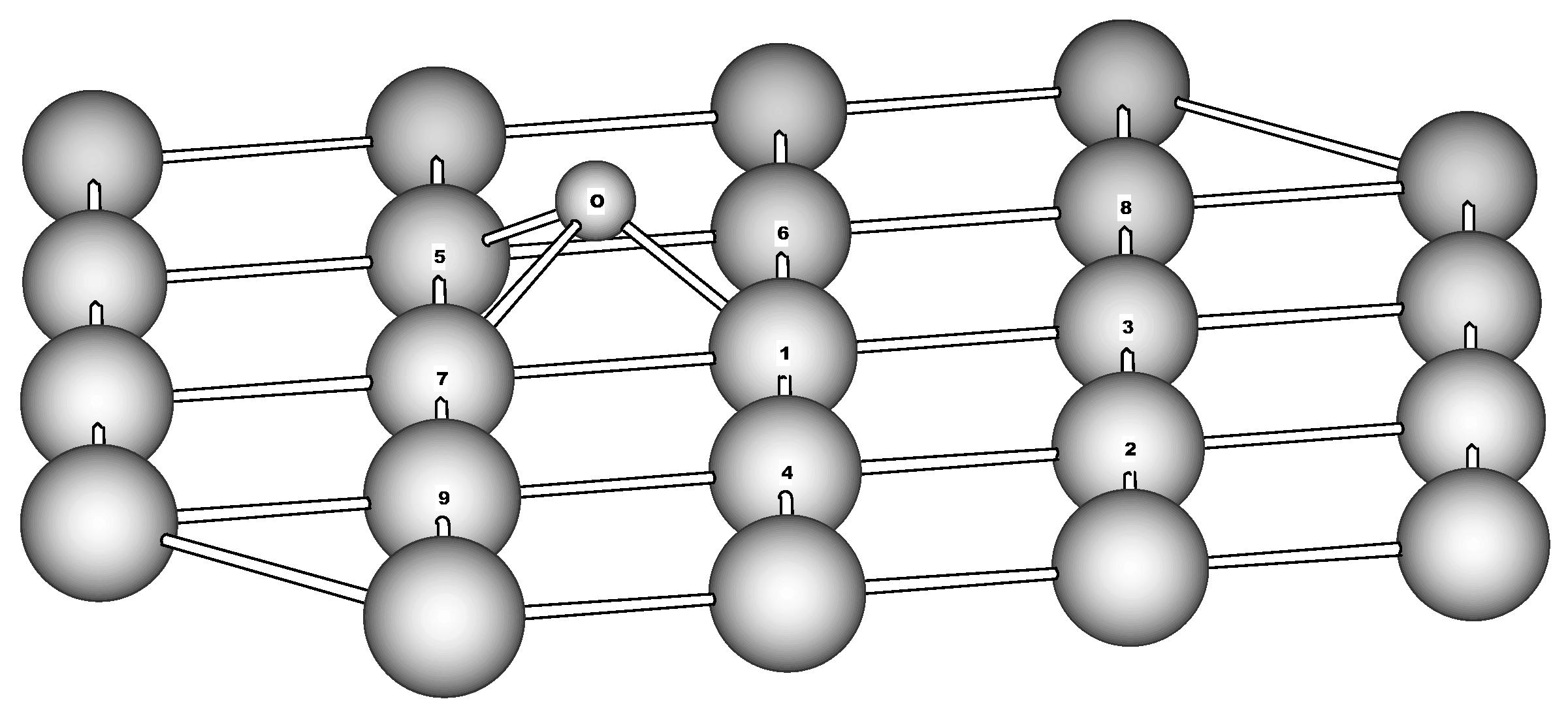
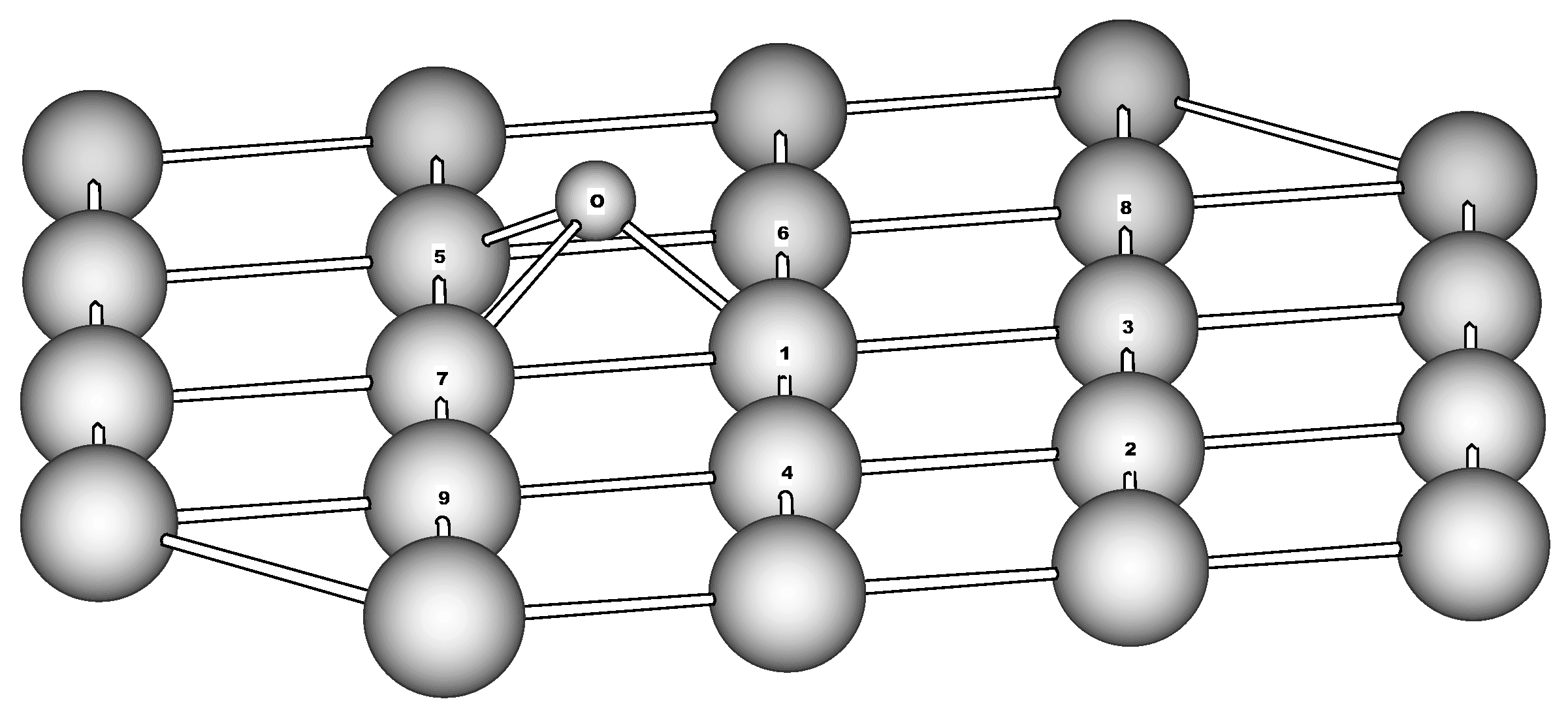
Figure 5.
Adsorption of nitrosobenzene on Fe(110). The only top layer of the cluster is shown for simplicity. Calculated distances (Å) and valence angles (degrees): N-C 1.350, N-O 1.506, N-Fe1 2.314, N-Fe3 2.356, N-Fe6 2.004, O-Fe1 1.994, O-Fe2 2.441, O-Fe3 1.625, O-N-C 123.6
Figure 5.
Adsorption of nitrosobenzene on Fe(110). The only top layer of the cluster is shown for simplicity. Calculated distances (Å) and valence angles (degrees): N-C 1.350, N-O 1.506, N-Fe1 2.314, N-Fe3 2.356, N-Fe6 2.004, O-Fe1 1.994, O-Fe2 2.441, O-Fe3 1.625, O-N-C 123.6
The oxygen adsorption on Fe(110) surface is modeled by structure shown in
Figure 6. The oxygen occupies the 3-fold position just like the runaway oxygen moiety of NB on surface. To check how the oxygen adsorption depends on cluster site chosen we calculated the oxygen adsorption at the center of second (smaller) layer of Fe
39. The oxygen is stabilized in the 3-fold position having Fe-O distances of 1.618, 1.752 and 1.818 Å. The adsorption energy insignificantly deviates from that obtained for oxygen shown in
Figure 6 by small value of 0.05 eV. It is interesting that E
ads is of the same magnitude order as the energy of dissociation for FeO molecule which is 4.2 eV [
17]. This fact seems to be an additional confirmation that the electron transfer into oxygen is the major factor responsible for the adsorption energy with oxygen-containing adsorbate.
Comparing the sum of adsorption energies for separated NS and oxygen atom from one side and Eads for NB(C)/Fe39 from the other one can see that the former is twice as much. We suggest that this difference is determined by the Coulomb repulsion between nitroso and oxygen moieties of NB(C)/Fe39. This repulsion energy is 5.2 eV if one assumes that its largest contribution comes from the oxygen and nitroso group of NB(C)/Fe39. If one subtracts this value from sum of the O and NS adsorption energies the latter becomes equal to 7.3 eV that agrees fairly well with Eads of 6.7 eV for NB(C)/Fe39. This Coulomb repulsion between the oxygen and nitrosobenzene could be a driving force which might cause their migration on surface after dissociation of NB.
Figure 6.
Atomic oxygen adsorption on Fe(110) surface. The only top layer of the cluster is shown for simplicity. Calculated distances (Å) and valence angles (degrees): O-Fe1 1.684, O-Fe5 1.588, O-Fe6 2.320, O-Fe7 2.021
Figure 6.
Atomic oxygen adsorption on Fe(110) surface. The only top layer of the cluster is shown for simplicity. Calculated distances (Å) and valence angles (degrees): O-Fe1 1.684, O-Fe5 1.588, O-Fe6 2.320, O-Fe7 2.021


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}