Anisotropy effects in H++H2 collisions

1
Laboratorio Asociado al CIEMAT de Fisica Atomica y Molecular en Plasmas de Fusion. Departamento de Quimica, Universidad Autonoma de Madrid, 28049-Madrid, Spain
2
Instituto de Estructura de la Materia CSIC, Serrano 113 bis, 28006-Madrid, Spain
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2002, 3(3), 142-161; https://doi.org/10.3390/i3030142
Submission received: 1 January 2001
/
Accepted: 2 January 2002
/
Published: 28 March 2002
(This article belongs to the Special Issue Charge Transfer in Ionic and Molecular Systems)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The effect of the anisotropy of the interaction potential on ion-diatom non-adiabatic collisions is analized by considering the in uence of the anisotropy on orientation averaged total cross sections for charge transfer in H++H2(X1) collisions. We discuss the possibility of employing simplified methods such as an isotropic approximation, where only the electronic energies and interactions of a single orientation are necessary. The use of several isotropic calculations to evaluate the orientation averaged cross section is analized.
1 Introduction
Charge transfer (CT) in ion-H2 collisions is usually described with methods based on the sudden approximation [1], which is generaly applied for molecular rotation. At low energies, quantal calculations are carried out employing the infinite order sudden approximation (IOSA) (see e.g. [2]). At higher energies, semiclassical methods are used and the sudden approximation can also be applied for molecular vibration; at sufficiently high energy, the simple Franck-Condon approach is appropriate (see reviews of refs. [3] and [4]). A semiclassical treatment with rectilinear trajectories and the sudden approximation for rotation and vibration (SEIKON) has been used in several publications [5, 6, 7, 8], and the low-v limit of this approach has been studied in a recent work [9]. In practice, these calculations require the evaluation of the potential energy surfaces and non-adiabatic couplings for a large set of relative orientations of the H2 internuclear axis with respect to the ion position vector, R; this is one reason of the relatively few calculations carried out for ion-molecule collisions.
In recent years, several experiments [10, 11] have studied the dissociation of molecules after collision with ions as a function of the molecular orientation. Nevertheless, the main results of ion-molecule beam experiments are orientation-averaged cross sections (OACS); in fact, these are the only results available for single electron capture processes in ion-H2 collisions. The calculation of OACS requires an extensive calculation of molecular data, as well as a cumbersome 2D interpo-lation of potential energy surfaces and dynamical couplings. For H2 collisions with multicharged ions, CT takes place via non-adiabatic transitions at relatively large internuclear distances, where anisotropy effects are unimportant and, a simplified isotropic approximation is appropriate. In this approximation, energies and couplings are taken to be independent of the orientation of the H2 internuclear axis. The situation is different for singly charged ions-H2 collisions; in this case, transitions leading to CT occur at short R. We consider in this paper H++H2 collisions, which are the benchmark of these reactions, and are otherwise important in tokamak divertor plasmas (see [12, 13] and references cited therein). Early semiclassical calculations of ref. [14] reported a sizeable anisotropy effect on H++H2 CT cross sections, but the validity of this conclusion is limited by the Franck-Condon approximation employed to evaluate the vibronic dynamical couplings.
In ref. [7] it was shown that anisotropy effects on dinamical couplings are less important for H++H2 than for Li++H2 at energies E > 0.1 keV; results for Li++H2 collisions [7] pointed out large differences between cross sections calculated using the isotropic approximation for different orientations, and between them and the OACS. However, it was also shown that an average of isotropic results yields cross sections in good agreement with the OACS. This finding, which implies a significant simplification of the computation, has not been checked for other systems. Moreover, the isotropic approximation is the basis of IOSA treatments, where calculations for fixed molecular orientation are carried out, followed by an average of the ensuing cross sections; this is the method employed in IOSA treatments of CT in H++H2 [15,13].
As concluded in ref. [9], for H++H2 at E < 0.1 keV, a calculation beyond the sudden vibrational approximation is required. The first aim of the present work is to study anisotropy effects in this energy range. We perform an eikonal vibronic close-coupling calculation, similar to that employed in [9], using the diatomics in molecules (DIM) potentials [16] because, as in refs. [17, 14, 15, 18, 13], it allows the evaluation of energies and interactions along each trajectory without using 2D interpolation, which is very useful when many orientations are considered. Although the DIM aproximation limits the precission of our results, the aim of the present paper is to study anisotropy effects rather than obtaining very precise cross sections.
The paper is organized as follows: In section 2 we explain the semiclassical vibronic close-coupling method employed, the use of DIM wavefunctions, and the calculation of OACS. In section 3 we present our main results, and discuss the use of simplified methods to evaluate OACS. Our conclusions are summarized in section 4. Atomic units are used unless otherwise stated.
2 Method
2.1 Basic equations.
Our method involves the following basic assumptions: 1) The semiclassical approximation with rectilinear nuclear trajectories. 2) The sudden approximation for rotation of the diatomic. 3) The expansion of the wave function for the remaining degrees of freedom in terms of a vibronic set. 4) The use of DIM electronic wavefunctions.
In the semiclassical approach the ion-diatomic relative motion is described classically with straight-line trajectories R = b + vt, where R is the H+ position vector with respect to the center of the H2 internuclear axis. The remaining degrees of freedom are treated quantally, and the corresponding wave function Ψ(r1, r2, ρ, t) is solution of the semiclassical equation:
![Ijms 03 00142 i001]() where ρ is the target internuclear vector in the laboratory frame and r1,2 the electronic coordinates. Hi is the Hamiltonian of the form:
where ρ is the target internuclear vector in the laboratory frame and r1,2 the electronic coordinates. Hi is the Hamiltonian of the form:
![Ijms 03 00142 i002]() where MH is the mass of the proton and
where MH is the mass of the proton and
![Ijms 03 00142 i003]() being γ the angle between vectors R and ρ, Ti the electronic kinetic energy operators, Vi the attraction potential of electron i by the three H nuclei, and Vnuc the nuclear repulsion potential. An indication of the validity of the semiclassical approximation for this system was given in [7], where practically identical quantal and semiclassical total cross sections were obtained in a calculation for E > 0.05 eV at the Franck-Condon level.
being γ the angle between vectors R and ρ, Ti the electronic kinetic energy operators, Vi the attraction potential of electron i by the three H nuclei, and Vnuc the nuclear repulsion potential. An indication of the validity of the semiclassical approximation for this system was given in [7], where practically identical quantal and semiclassical total cross sections were obtained in a calculation for E > 0.05 eV at the Franck-Condon level.



Equation (1) must be solved with the initial condition:
![Ijms 03 00142 i004]() with
where Di is a plane-wave translation factor [19] that describes the translation of the H2 electrons with respect to the origin of electronic coordinates and Φiχiµ the wave function for the H2 vibronic initial state. Analogously, a reaction exit channel has the form:
with
where Di is a plane-wave translation factor [19] that describes the translation of the H2 electrons with respect to the origin of electronic coordinates and Φiχiµ the wave function for the H2 vibronic initial state. Analogously, a reaction exit channel has the form:
![Ijms 03 00142 i005]() with
Using the sudden approximation for rotation, the collisional wavefunction is written as:
with
Using the sudden approximation for rotation, the collisional wavefunction is written as:
![Ijms 03 00142 i006]() It can be shown (see ref. [5]) that, assuming that all M values are equally probable in the initial condition, the probability for transition to all rotational states of a given vibronic state ψnµ can be expressed as an orientation averaged magnitude:
It can be shown (see ref. [5]) that, assuming that all M values are equally probable in the initial condition, the probability for transition to all rotational states of a given vibronic state ψnµ can be expressed as an orientation averaged magnitude:
![Ijms 03 00142 i007]() With
With
![Ijms 03 00142 i008]()

ψiµ = Φi(r1, r2; ρ)χiµ(ρ)Di(r1, r2, t)exp(−iEit),

ψnν = Φn(r1, r2; ρ)χnν(ρ)Dn(r1, r2, t)exp(−iEft),



2.2 Vibronic close-coupling expansion.
In the DIM method (see [16]), one considers the three valence bond products of the form:
![Ijms 03 00142 i009]() We have employed these states to define the electronic part of the vibronic basis. Since the functions φ1,2,3 are independent of R, the dynamical couplings (φi|∂/∂R|φj) and (φi|∂/∂ρ|φj) vanish, so this is a diabatic basis. In practice, we have checked that transitions from φ1 and φ2 to φ3 are negligible, so we have only kept the states φ1 and φ2 in our calculation.
We have employed these states to define the electronic part of the vibronic basis. Since the functions φ1,2,3 are independent of R, the dynamical couplings (φi|∂/∂R|φj) and (φi|∂/∂ρ|φj) vanish, so this is a diabatic basis. In practice, we have checked that transitions from φ1 and φ2 to φ3 are negligible, so we have only kept the states φ1 and φ2 in our calculation.

In the present calculation, we have expanded the function ψ in the vibronic basis {φ1(r; R, ρ)χν (ρ),φ2(r; R, ρ)χν (ρ)}, where χν are vibrational functions of the H2 electronic ground state; they were calculated by diagonalizing the vibrational Hamiltonian in a Gaussian basis set, as explained in [20]. Explicitly:
![Ijms 03 00142 i010]() where we have included 14 vibrational states for each electronic state. The coefficients ajν are obtained for each nuclear trajectory and molecular orientation, by solving the system of differential equations:
where we have included 14 vibrational states for each electronic state. The coefficients ajν are obtained for each nuclear trajectory and molecular orientation, by solving the system of differential equations:
![Ijms 03 00142 i011]() with
with
![Ijms 03 00142 i012]() In the present calculation, we do not include electron translation factors because they introduce small corrections at the low velocities we are interested in. Besides, although in general these factors are required to remove spurious asymptotic couplings, the matrix elements (14) vanish when R → ∞.
In the present calculation, we do not include electron translation factors because they introduce small corrections at the low velocities we are interested in. Besides, although in general these factors are required to remove spurious asymptotic couplings, the matrix elements (14) vanish when R → ∞.



An alternative set of adiabatic vibronic states, Φjν, is obtained by diagonalizing the Hamiltonian matrix of eq. (14). Calling U (t) the adiabatic-diabatic transformation matrix which fulfills:
![Ijms 03 00142 i013]() the transition amplitudes in the adiabatic basis, cjν , are related to the diabatic ones, ajν, solutions of the system of differential equations (13), by:
the transition amplitudes in the adiabatic basis, cjν , are related to the diabatic ones, ajν, solutions of the system of differential equations (13), by:
![Ijms 03 00142 i014]()


2.3 Orientation averaged cross sections.
The OACS for charge transfer and H2 vibrational excitation are, respectively:
![Ijms 03 00142 i015]() where, from eqs. (10) and (12):
where, from eqs. (10) and (12):
![Ijms 03 00142 i016]() Probabilities for charge transfer or excitation to individual vibrational states can be obtained from the coefficients cjν in the adiabatic basis (eq. (16)):
Probabilities for charge transfer or excitation to individual vibrational states can be obtained from the coefficients cjν in the adiabatic basis (eq. (16)):
![Ijms 03 00142 i017]()



The calculation of OACS requires the evaluation of the transition probabilities Pα(ρˆ, b, v) for several molecular orientations in the laboratory reference frame or, equivalently, for several orientations of the vector v with respect to ρˆ. A simple approach to average the cross sections of eq. (17) was proposed in [5], where six relative orientations of v and ρˆ were considered, leading, for ion-H2 collisions, to the expression:
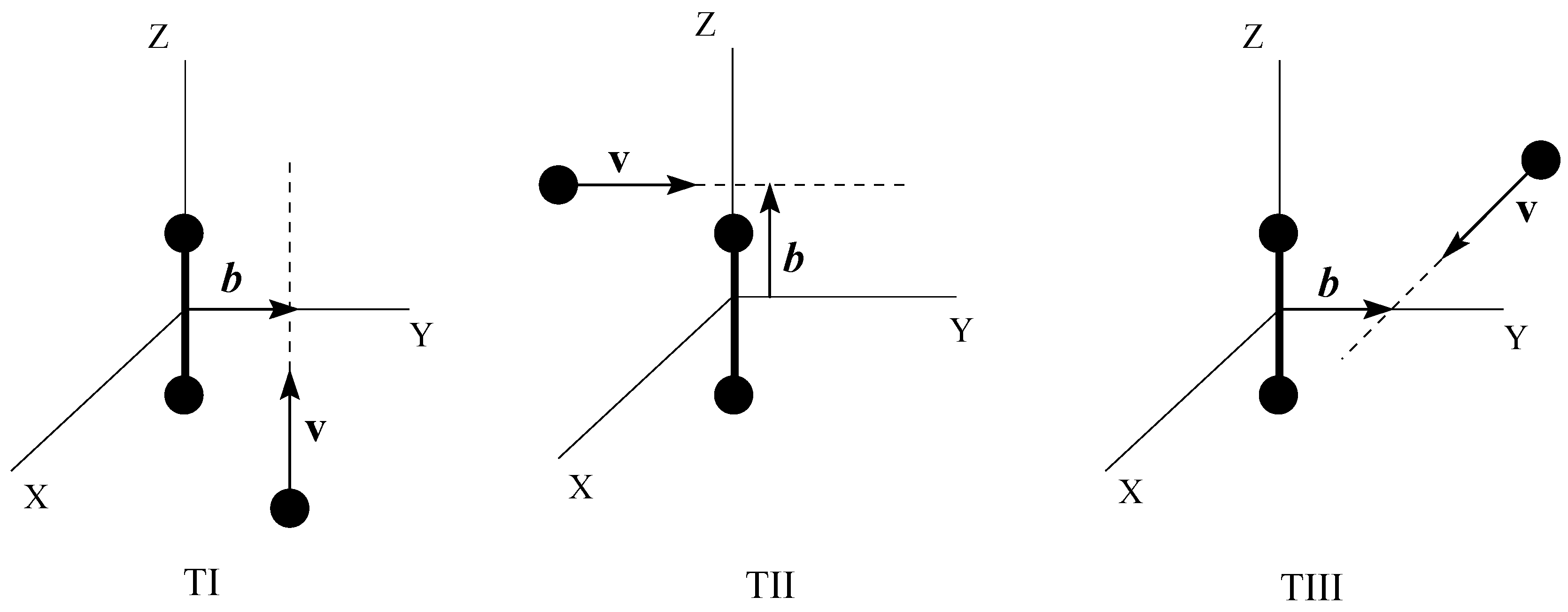
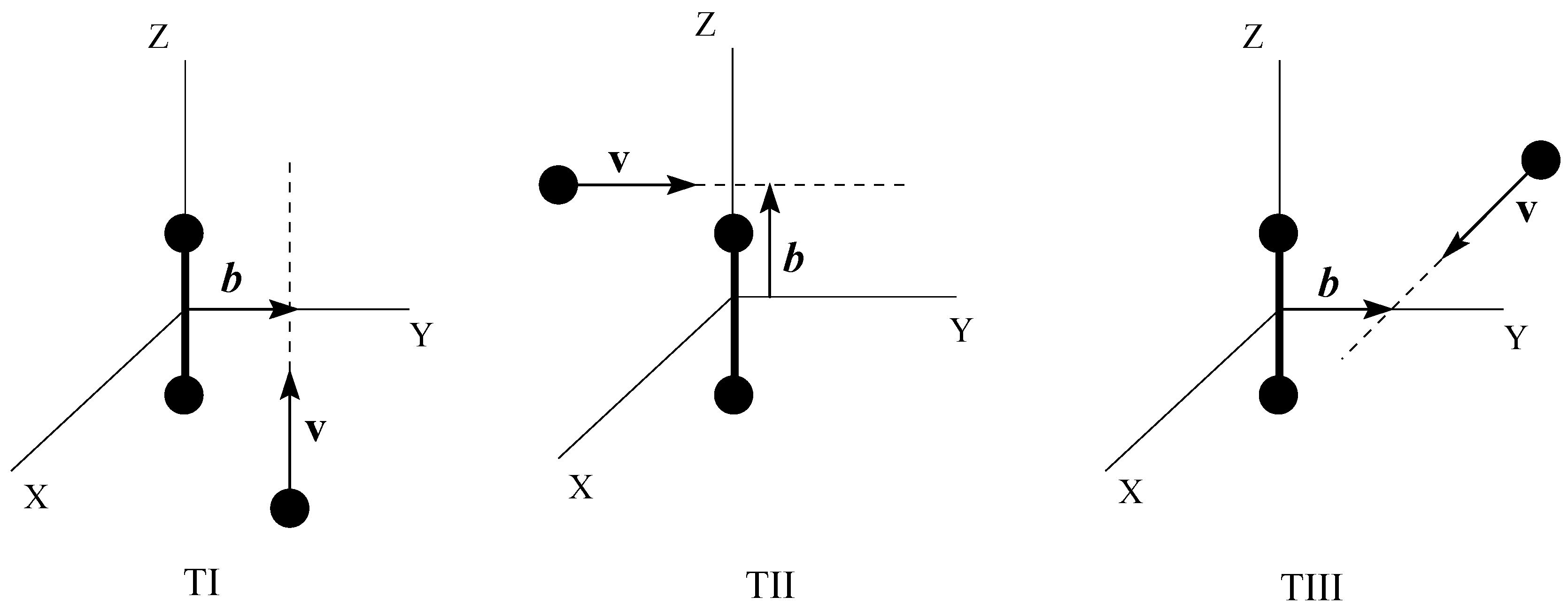
![Ijms 03 00142 i018]() Where are the transition probabilities for the process α in the collisions where the trajectory orientations are defined as (see fig. 1):
Where are the transition probabilities for the process α in the collisions where the trajectory orientations are defined as (see fig. 1):

- Trajectory I: b ⊥ ρ, v || ρ.
- Trajectory II: b || ρ, v ⊥ ρ.
Figure 1.
Geometry of the trajectories TI, TII and TIII used to evaluate the OACS in eq. (21).
- 3.
- Trajectory III: b ⊥ ρ, v ⊥ ρ.

An additional approximation is obtained by taking a fixed molecular orientation during the trajectory, assuming:
which leads to a system of differential equations (13), similar, for all nuclear trajectories, to that obtained for H+ collisions with a spherically symmetric target. The solutions of this system are the isotropic coefficients ajν(t, γ0), and we can define isotropic cross sections (v,γ0) as:
![Ijms 03 00142 i020]() with (b, v, γ0) and (b,v,γ0) defined analogously to eq. (18). An approximation to the OACS can be then obtained by averaging the isotropic cross sections (see ref. [15]):
with (b, v, γ0) and (b,v,γ0) defined analogously to eq. (18). An approximation to the OACS can be then obtained by averaging the isotropic cross sections (see ref. [15]):
![Ijms 03 00142 i021]() with
with
![Ijms 03 00142 i022]()
Hjν kμ(R, γ) ≃ Hjν kμ(R, γ0)



3 Results
The Hamiltonian matrix elements in the vibronic basis can be expressed as:
![Ijms 03 00142 i023]() where
where

Hjk(R, γ) = 〈φj|Hel|φk〉r1,r2.
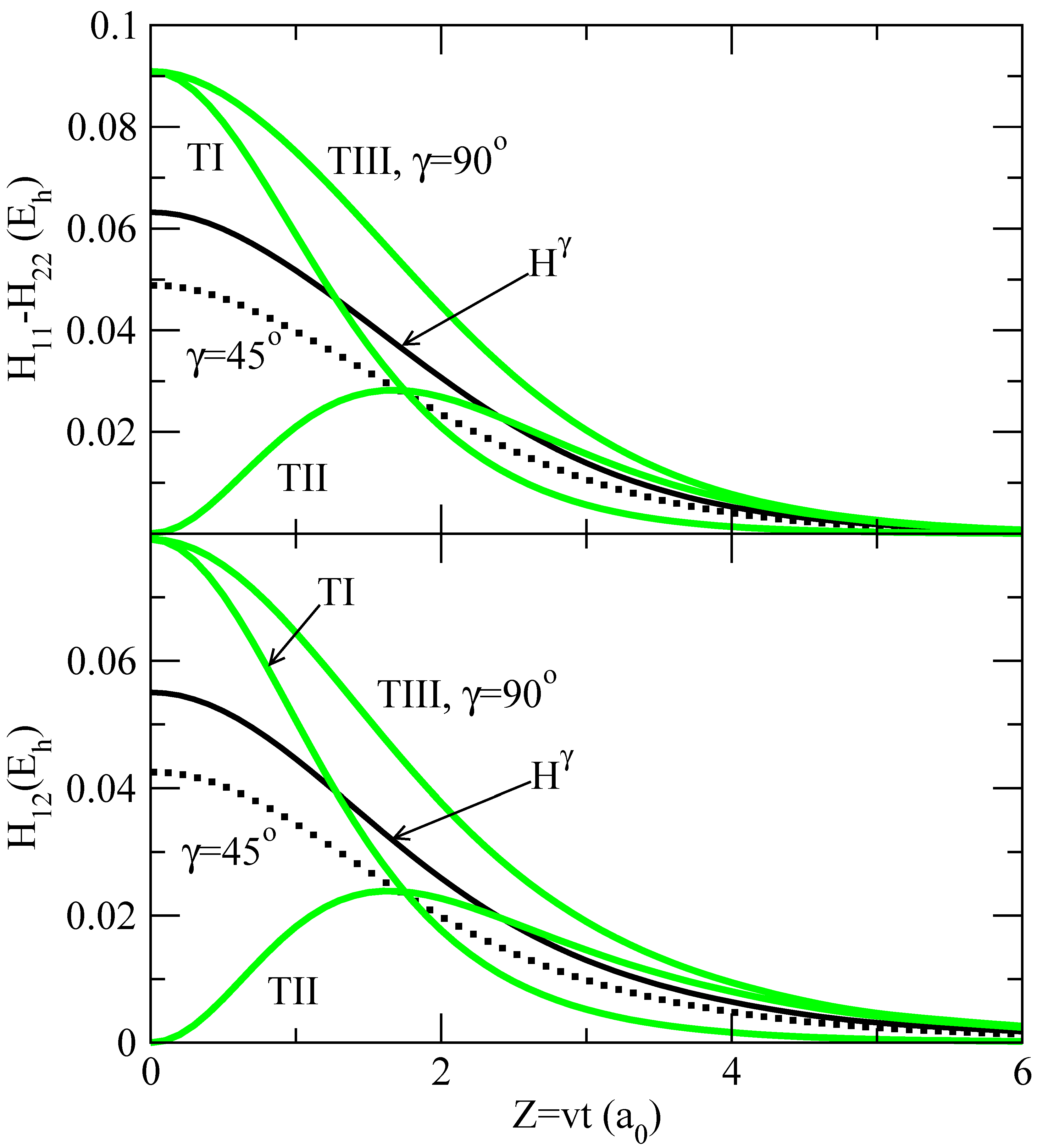
The orientation dependence of the couplings of eq. (13) is therefore a consequence of the dependence of the electronic matrix elements Hjk of eq. (27), which is illustrated in figure 2. There, we have plotted the values of H22 − H11 and H12 along a representative trajectory with b = 1.75 a0, and for t > 0. In the top panel of figure 2, we show the values of the differences: [H22 − H11](R,γ = 90°) and [H22 − H11](R,γ = 45°), while the bottom panel contains H12(R,γ = 90°) and H12(R,γ = 45°). In both cases, to make the illustration more clear, we have subtracted from these couplings the corresponding values for γ = 0. It can be noted that the dependence of the matrix elements Hjk on γ is almost linear. We have also included in figure 2 the orientation averaged couplings
![Ijms 03 00142 i024]() As a consequence of the quasi-linear dependence of Hij on γ, these average values are close to the matrix elements evaluated at γ = 57.3° ≃ 60°, obtained for a linear function Hij (γ).
As a consequence of the quasi-linear dependence of Hij on γ, these average values are close to the matrix elements evaluated at γ = 57.3° ≃ 60°, obtained for a linear function Hij (γ).

In fig. 2, we also include the values of the matrix elements as functions of Z = vt for trajectories I, II and III. From the definition of trajectory III (see also fig. 1), the angle γ ≡ 90° along this trajectory, which leads to couplings identical to the isotropic ones for this value of γ. Couplings along trajectory I join smoothly the isotropic values for γ = 90° at t = 0 with those for γ = 0° at t → ∞, while the inverse situation holds for trajectory II: the values for γ = 0° are reached at t = 0 and those for γ = 90° for t → ∞. From fig. 2, we can also note that the orientation dependence of the couplings is very small at large internuclear distances; as a rule, we can neglect the anisotropy for Z > 4 a0 (R > 4.4 a0)
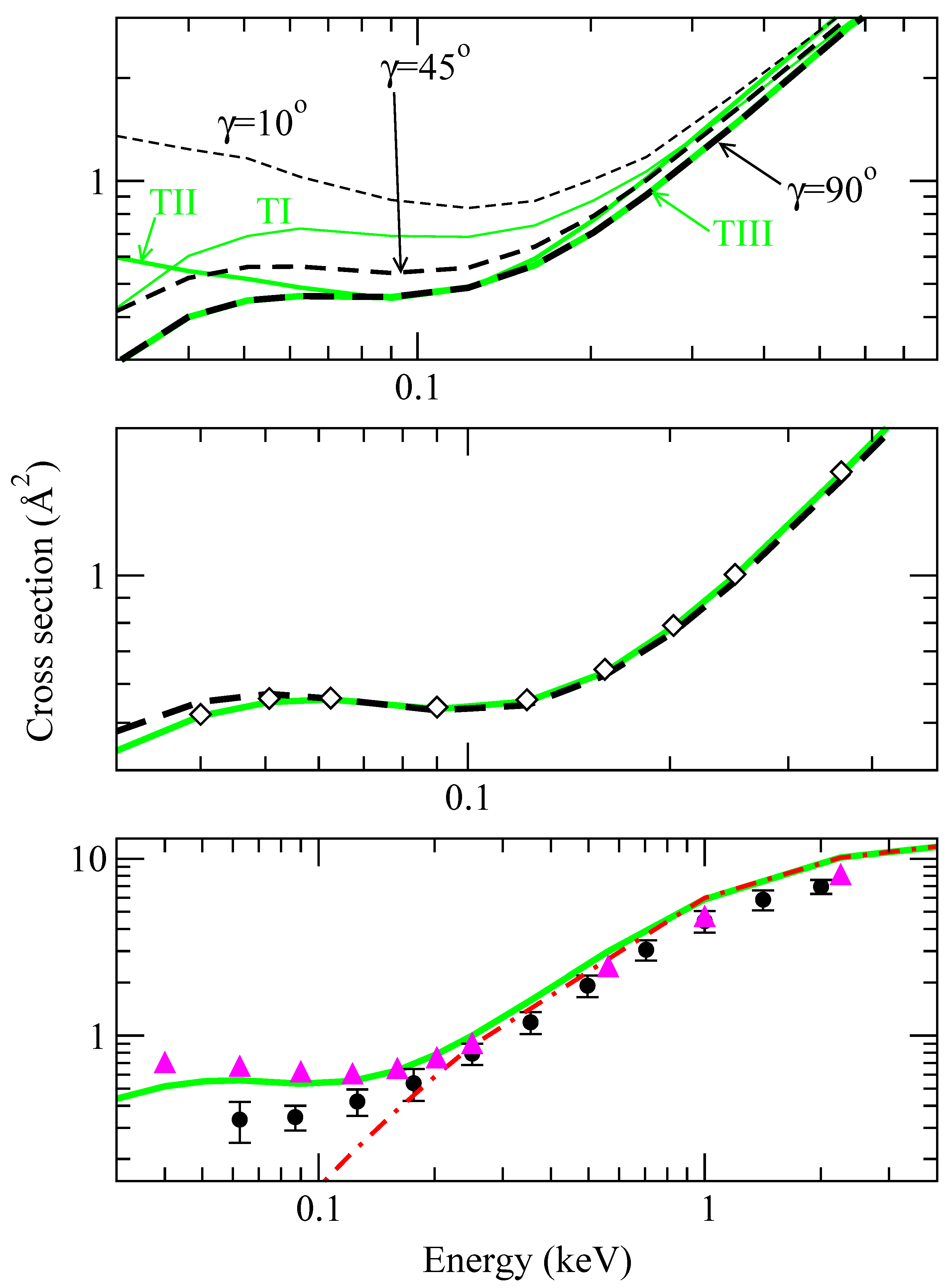
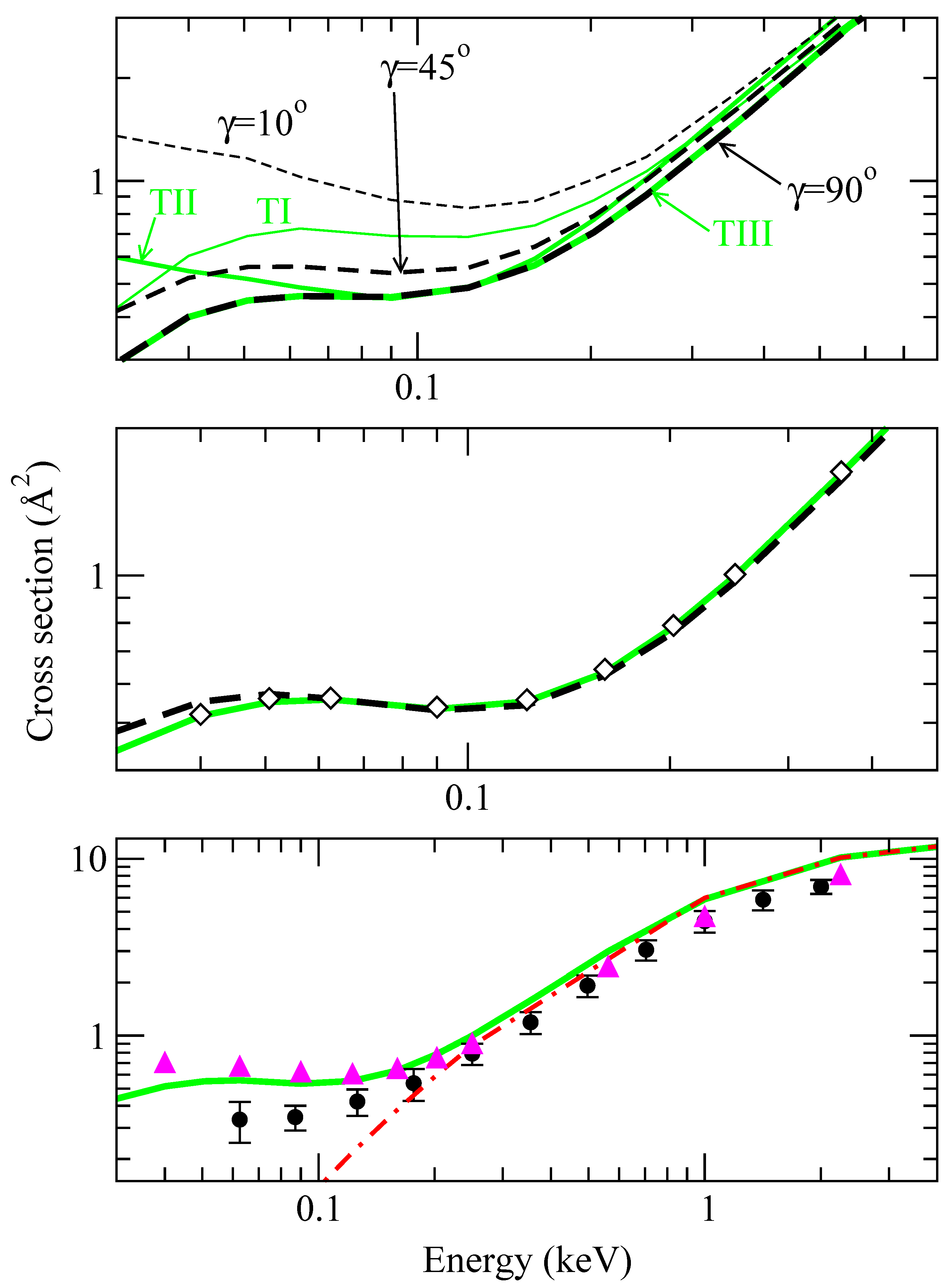
Integration of the system of differential tial equations (13) in the isotropic approximation yields the CT cross sections (v,γ0) (see the top panel in fig. 3); these cross sections do not appreciably depend on γ0 for E > 0.2 keV (differences are of about 10%), in agreement with the conclusions of ref. [7], but the dependence increases as E decreases: (v, γ0 = 10°) is a factor of 5 larger than (v, γ0 = 90°) at E ≃ 40 eV. We have also included in fig. 3 the values of , which exhibit smaller differences than the isotropic ones (at most a factor of 2 between σII and σIII at E = 0.05 keV). As a first result of the present calculation, the anisotropy of the couplings leads to a significant orientation dependence of total CT cross sections.
Figure 2.
Hamiltonian matrix elements Hij(R, γ) as functions of Z(= vt = ) for several values of γ and with γ(t) for trajectories I, II and III. The full lines labelled as Hγ are the average couplings from eq. (28). Top panel: values of [H22 − H11](R, γ) − [H22 − H11](R,γ = 0). Bottom panel: values of H12(R, γ) − H12(R, γ = 0).
Figure 2.
Hamiltonian matrix elements Hij(R, γ) as functions of Z(= vt = ) for several values of γ and with γ(t) for trajectories I, II and III. The full lines labelled as Hγ are the average couplings from eq. (28). Top panel: values of [H22 − H11](R, γ) − [H22 − H11](R,γ = 0). Bottom panel: values of H12(R, γ) − H12(R, γ = 0).

The OACS obtained using eq. (21) and the simplified methods of eqs. (24) are plotted in the middle panel of fig. 3. It can be noted that the results of averaging over trajectories (eq. (21)) or averaging the isotropic cross sections (eq. (24)) are practically indistinguishable, and the small differences at low v are probably due to the simple six-trajectory expression used to perform the trajectory average. There is a very good agreement between the OACS and the isotropic result with γ0 = 45°, which is repeated in the middle panel (with diamonds) for clarity. This indicates that the calculation of (v,γ0) provides a good approximation to when an intermediate γ0 is employed, such that the coupling matrix H fulfills H(R, γ0) ≃ (R) (see eq. (28)). This supports our ab initio results of ref. [9], obtained with γ0 = 45°.
In the bottom panel of fig. 3, we compare the present OACS with the experimental values of ref. [21]. We have also included in this figure the SEIKON values obtained with DIM potentials and fixed γ0 = 45° and the vibronic close coupling result calculated with ab initio energies and couplings and fixed orientation γ0 = 45°. By comparing the present averaged results, which are practically identical to the isotropic ones obtaned at γ0 = 45°, with the ab initio ones, we obtain that the errors introduced in the total cross section by the DIM approximation are smaller than 25%. The comparison with the SEIKON calculation confirms the result of ref. [7] of a small orientation dependence in the energy range where the SEIKON method is valid.
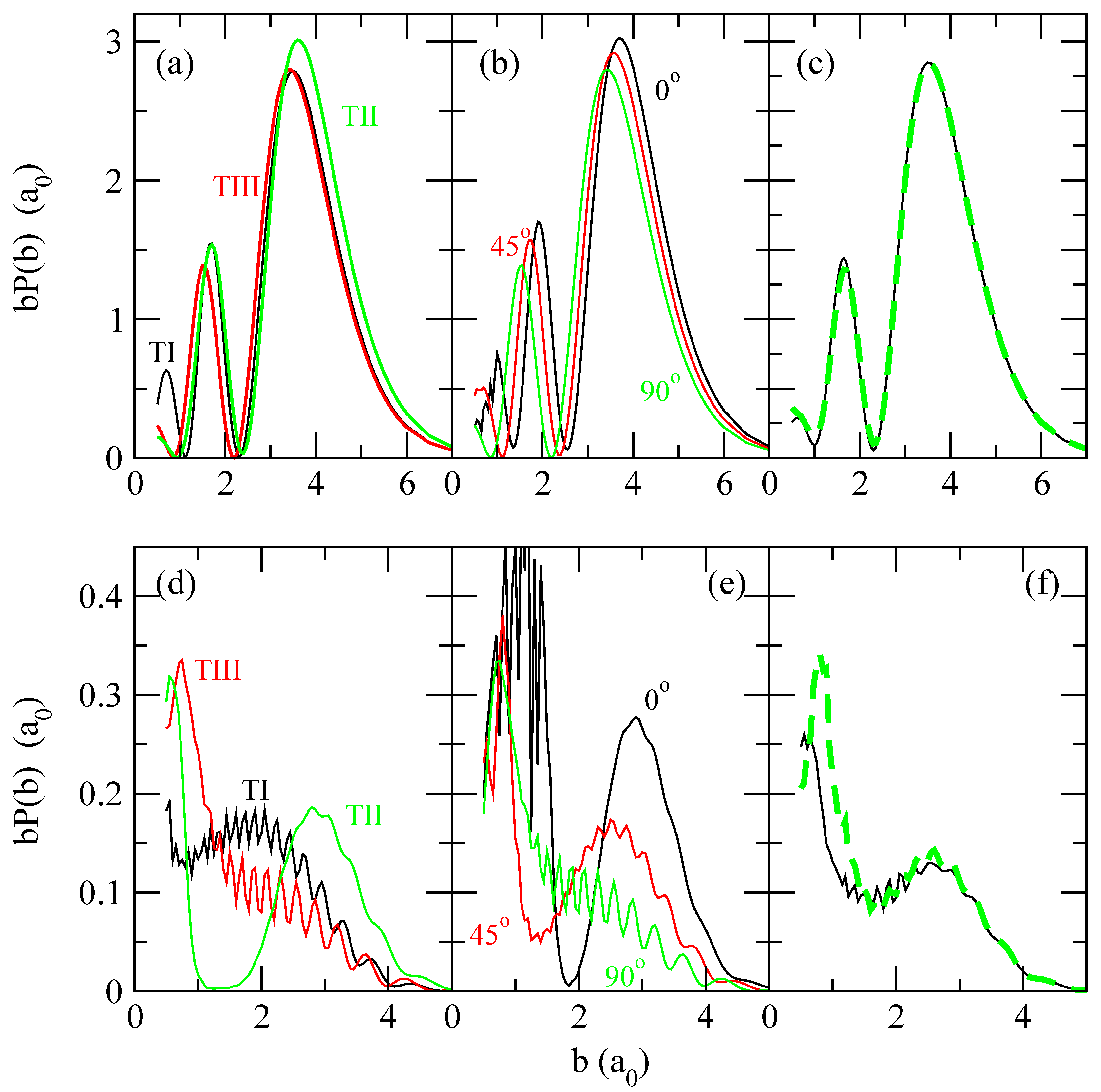
To analyze the results of fig. 3, we have plotted in fig. 4 the values of transition probabilities bPct(b) and b(b) for two impact energies (4 keV and 62.5 eV). At E = 4 keV, the shapes of all curves in fig. 3 are very similar in all calculations, showing a broad maximum for b > 3 a0, where the couplings are practically independent of γ (see fig. 2). When v decreases, the importance of trajectories with small b increases leading to a larger anisotropy of the transition probabilities. The comparison of bPct(b) of trajectory II with those of trajectories I and III at b > 2.0 a0 and E = 62.5 eV shows a shift of the former to larger b. To explain this shift (see also ref. [22]), we assume that, at relatively large b, the interaction matrix elements are dominated by the interaction of the incident H+ ion with the nearest H atom of H2, and a given distance from H+ to the nearest H nucleus is reached at larger b in trajectory II than in I and III (see fig. 1). At low b, the interaction between the incident ion and both H atoms are equally important, yielding different shapes of the three curves in fig. 3(d). By comparing the transition probabilities for trajectories I and II in fig. 3(d) with those calculated using the isotropic approximation (fig. 3(e)), and other intermediate values of γ0 not shown in the figure, it can be seen that the former calculation cannot be simplified by taking a constant value of γ along these trajectories, while, as mentioned before, for trajectory III γ ≡ π/2. However, a good agreement is found between the averages shown in the third panel of fig. 4, which explains the good agreement between the OACS of fig. 3 as due to a compensation of differences between the isotropic calculations. The possibility of using a single isotropic calculation to evaluate orientation averaged transition probabilities is checked
Figure 3.
Charge transfer cross sections calculated using different approximations. Top panel: comparison between cross sections obtained for three characteristic trajectories (grey lines, , and with those obtained with the isotropic approximation at three angles (dashed lines, (γ0 = 10°), (γ0 = 45°) and (γ0 = 90°)), as labeled in the figure. Middle panel: comparison between cross sections averaged over trajectories, σct (eq. (21), grey solid line), with those averaged over isotropic results, (eq. (24), dashed line), and with the isotropic result at γ0 = 45° (◇). Bottom panel: comparison between the trajectory average of the middle panel (grey solid line) with an isotropic calculation at γ0 = 45° using ab initio couplings and potentials (solid triangles), a SEIKON calculation with DIM couplings and potentials at γ0 = 45° (dashed-dotted line) and the experimental results of [21]. Note the different scales in the panels.
Figure 3.
Charge transfer cross sections calculated using different approximations. Top panel: comparison between cross sections obtained for three characteristic trajectories (grey lines, , and with those obtained with the isotropic approximation at three angles (dashed lines, (γ0 = 10°), (γ0 = 45°) and (γ0 = 90°)), as labeled in the figure. Middle panel: comparison between cross sections averaged over trajectories, σct (eq. (21), grey solid line), with those averaged over isotropic results, (eq. (24), dashed line), and with the isotropic result at γ0 = 45° (◇). Bottom panel: comparison between the trajectory average of the middle panel (grey solid line) with an isotropic calculation at γ0 = 45° using ab initio couplings and potentials (solid triangles), a SEIKON calculation with DIM couplings and potentials at γ0 = 45° (dashed-dotted line) and the experimental results of [21]. Note the different scales in the panels.
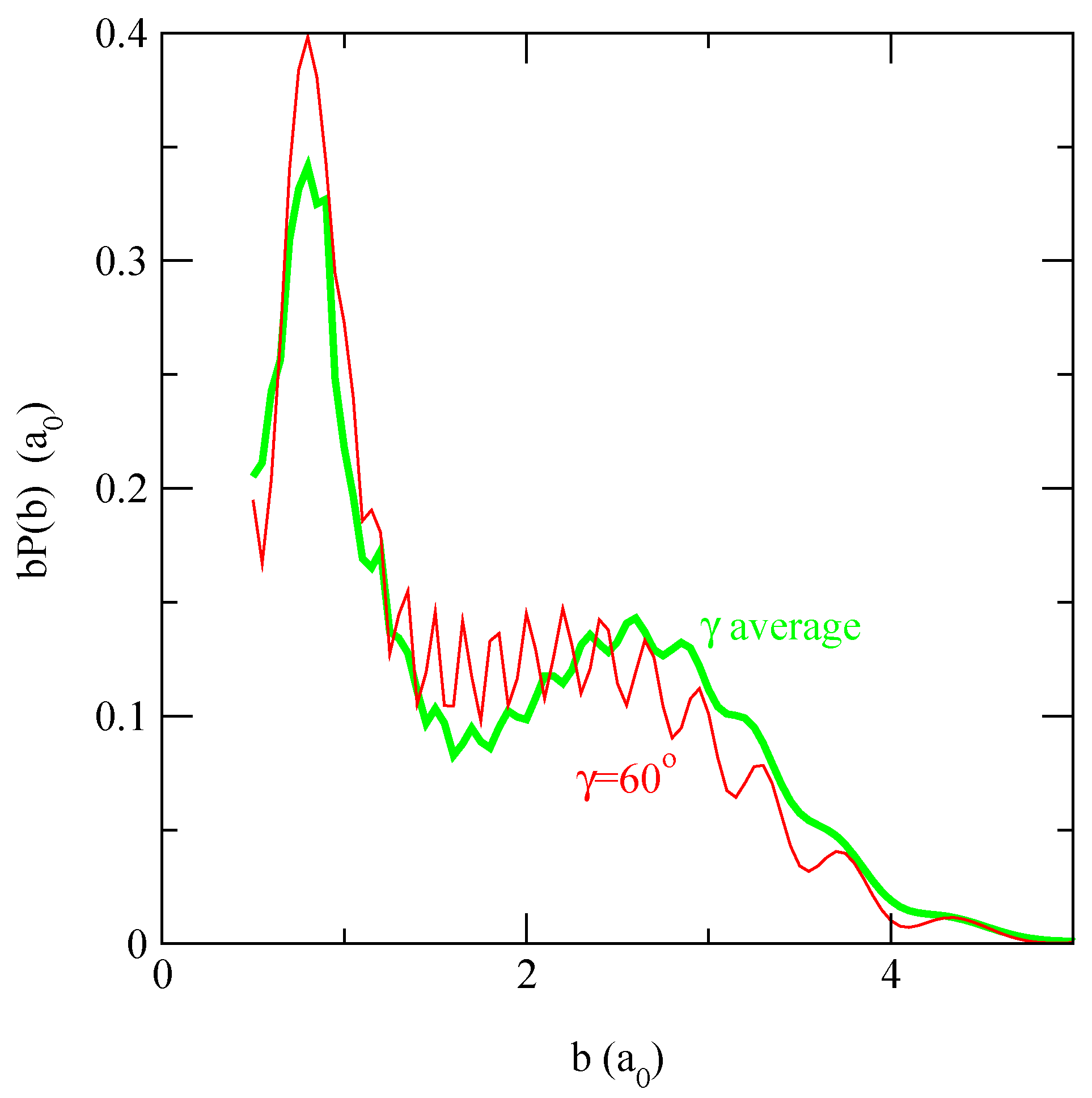
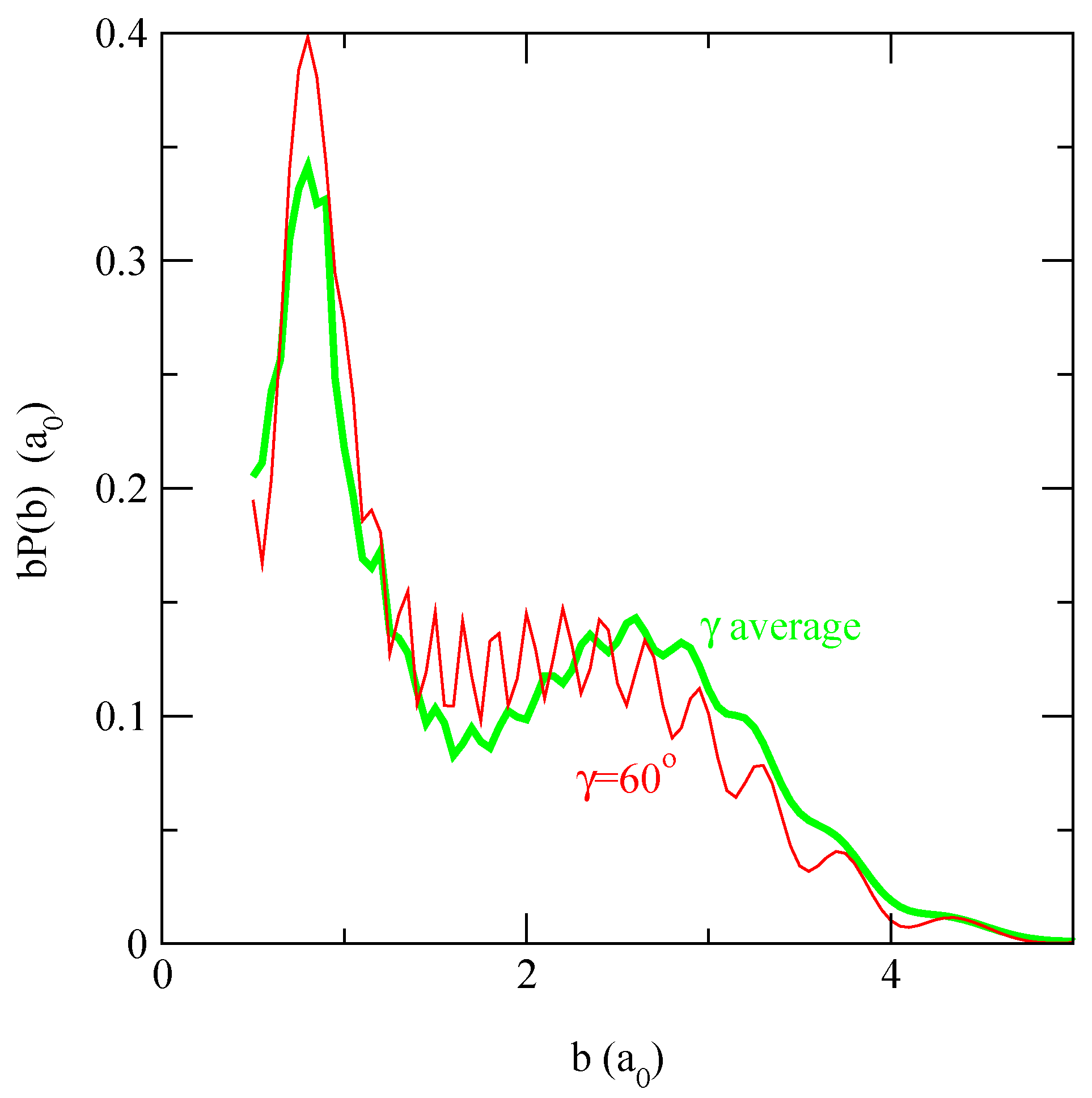
in fig. 5. We find that b(b; γ0 = 60°), which is very close to the probability obtained with the average interaction matrix, approximately reproduces the shape of the orientation averaged transition probabilities.
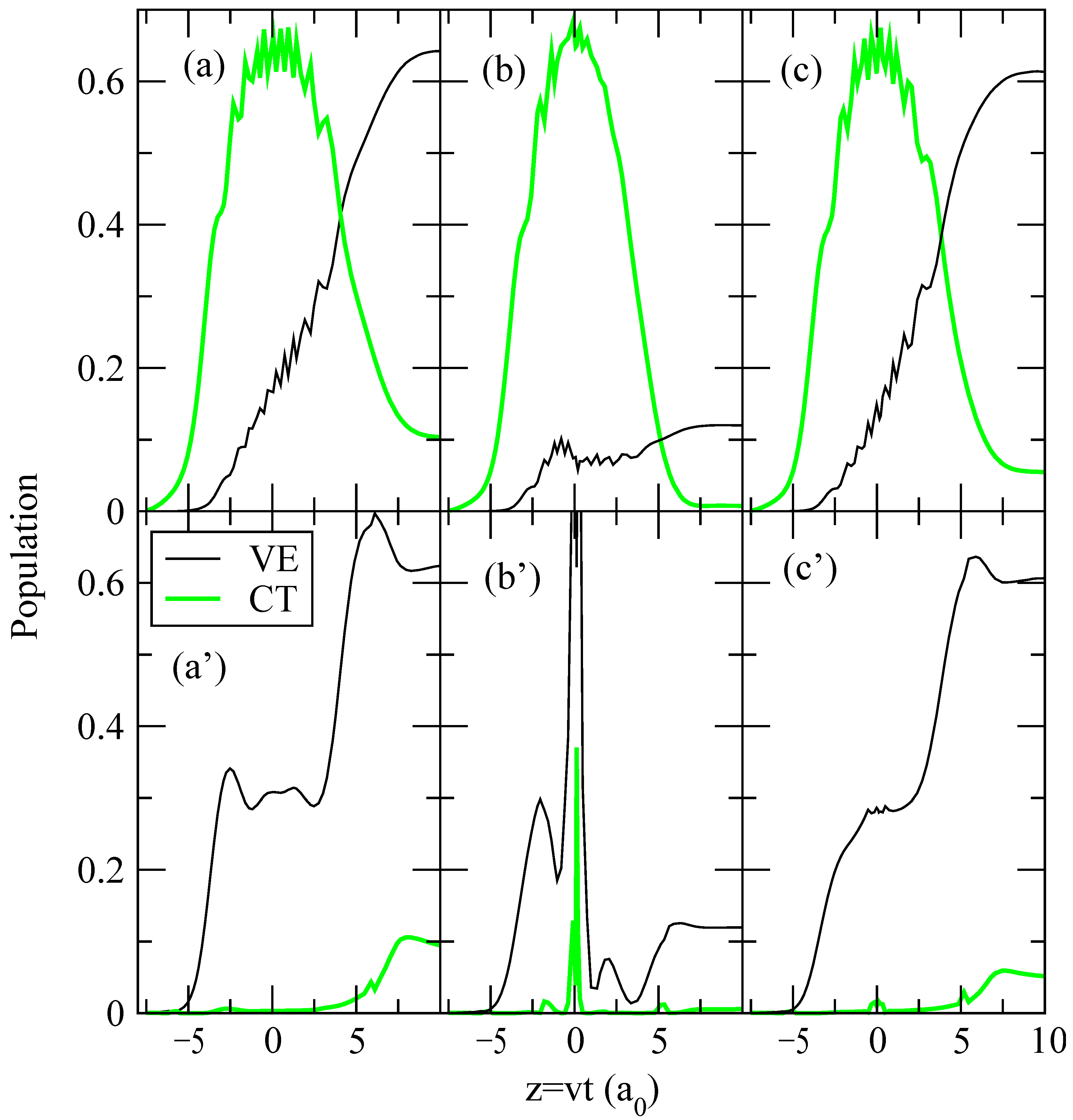
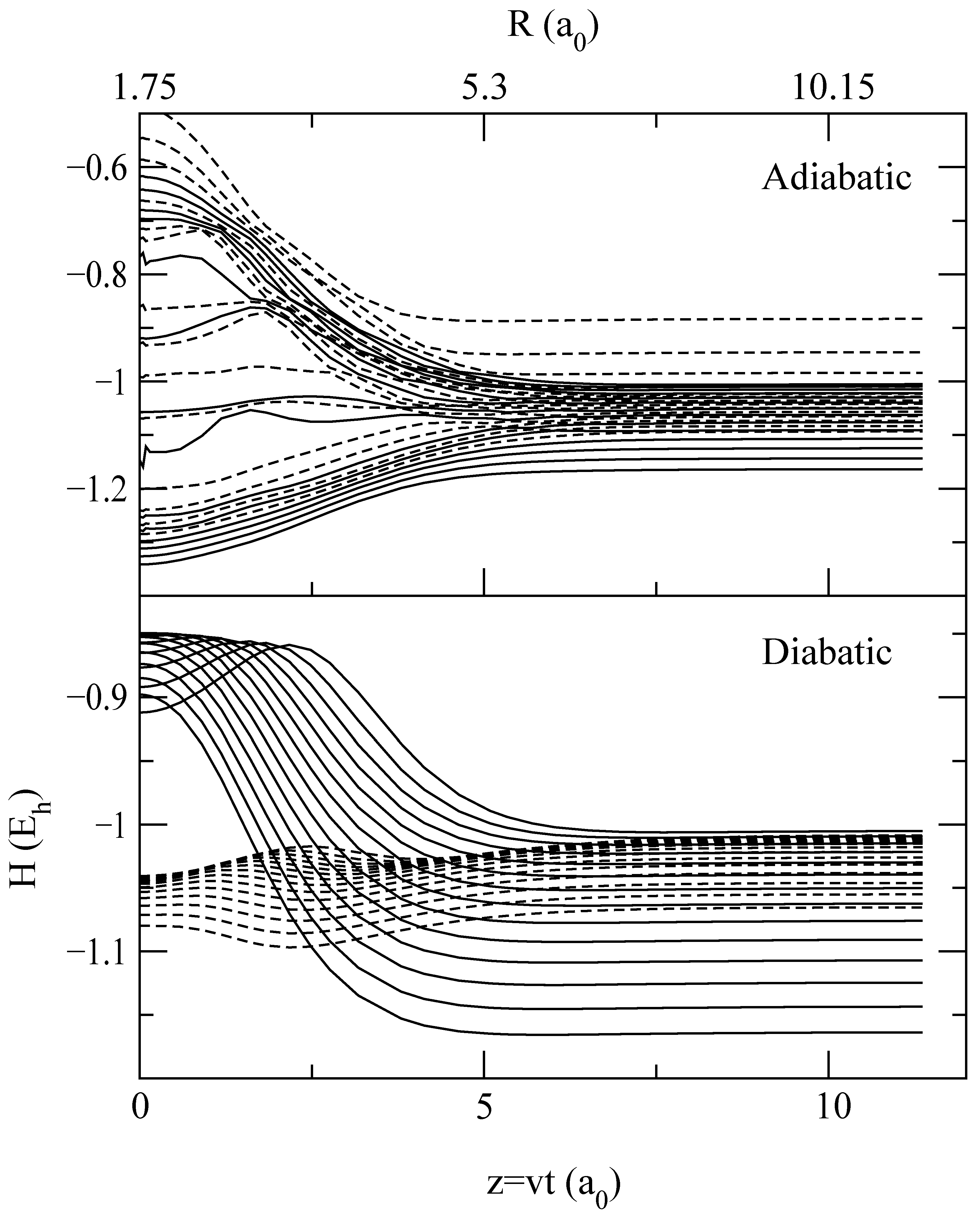
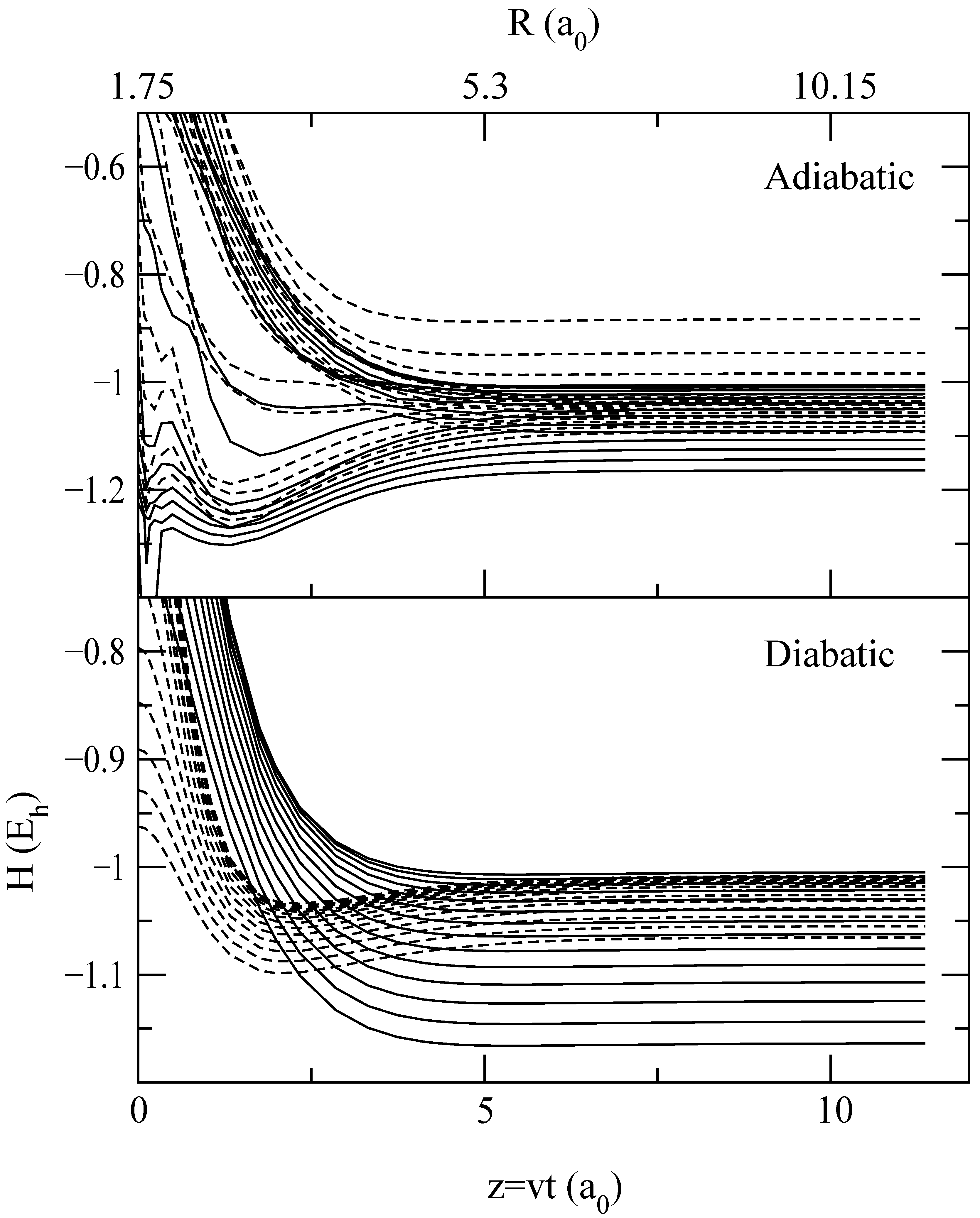
A more detailed information on the origin of the differences between transition probabilities for trajectories I, II and III is obtained from the “collision histories”, which are the time evolution of the populations |ajν|2 of the vibronic states (see eq. (12)). To relate these populations with the adiabatic CT mechanism at low v, described in [9], the collision histories for b = 1.75 a0 and E = 62.5 eV are plotted in fig. 6 in diabatic and adiabatic bases (see eq. (16)), and to help to understand them, the energy correlation diagrams in both bases are shown in fig. 7 and fig. 8 for trajectories I and II respectively. In this respect, it is interesting to note the large differences between adiabatic and diabatic correlation diagrams. This indicates that the interaction matrix elements in the latter are very strong, which makes difficult to understand the mechanism in this basis; e.g. the crossings are not traversed quasi diabatically.
The mechanism for trajectories I and III, in the diabatic representation, involves strong transitions from the entrance channel to the charge transfer ones for t < 0 through the series of crossings. This gives rise to the oscillations shown in the total charge transfer population of fig. 6. For t > 0, the population is transferred back from the charge transfer states to the entrance channel and the excited vibrational states of H2. The mechanism in the adiabatic basis is rather different: the first steps, at t < 0, are transitions from the entrance channel to the first excitation channels at R < 5 a0, where the relevant energy surfaces approach. In this basis, CT takes place at t > 0 through transitions from excited vibrational states of H2 to the charge transfer ones, because they are quasi degenerate; this is the mechanism described in [9] and first suggested in the trajectory surface hopping study of ref. [23].
The mechanism for trajectory II (figure 6(b,b’)) looks very different of those for the other trajectories. However, the differences are mainly due to the shift effect to larger b values of bPct(b) mentioned above: for the particular value of b (1.75 a0) of fig. 6, and using the adiabatic representation, the transitions near Z = 0 in trajectory II take place in a series of narrow avoided crossings, which are reached in trajectories I and III at lower b. A common characteristic of the illustrations shown in fig. 6 is that the non-adiabatic transitions take place over a wide time interval, which implies a wide range of values γ, in trajectories I and II, leading to the inaccuracy of using a simplified treatment with couplings calculated for a single γ to evaluate the corresponding transition probabilities.
Figure 4.
Charge transfer transition probabilities for E = 4 keV (panels (a,b,c)) and E = 62.5 eV (panels (d,e,f)). Panels (a) and (d): bPct(b) for trajectories I, II y III, as indicated in the figure. Panels (b) and (e): b(b; γ0 = 60°) for γ0 = 0°, 45°, 90°, as indicated in the figure. Panels (c) and (f): Average of transition probabilities obtained with the isotropic approximation for several values of γ0 (dashed-thick lines) b(b) (eq. (25)); average of transition probabilities obtained for trajectories I, II and III (solid-thin line) bPct(b) (eq. (20)).
Figure 4.
Charge transfer transition probabilities for E = 4 keV (panels (a,b,c)) and E = 62.5 eV (panels (d,e,f)). Panels (a) and (d): bPct(b) for trajectories I, II y III, as indicated in the figure. Panels (b) and (e): b(b; γ0 = 60°) for γ0 = 0°, 45°, 90°, as indicated in the figure. Panels (c) and (f): Average of transition probabilities obtained with the isotropic approximation for several values of γ0 (dashed-thick lines) b(b) (eq. (25)); average of transition probabilities obtained for trajectories I, II and III (solid-thin line) bPct(b) (eq. (20)).

Figure 5.
Comparison between the charge transfer transition probabilities averaged over γ0 of Figure 4, panel (f), (grey thick line) with a single isotopic result at γ0 = 60° (thin line) at E = 62.5 eV.
Figure 5.
Comparison between the charge transfer transition probabilities averaged over γ0 of Figure 4, panel (f), (grey thick line) with a single isotopic result at γ0 = 60° (thin line) at E = 62.5 eV.
4 Conclusions
The calculations of the present work focus on the effect of the anisotropy of the interaction potential on charge transfer cross sections in H++H2(X1, ν = 0) collisions, as a benchmark case of ion-diatomic molecule collisions. In the calculation we have employed commonly-used DIM electronic wave functions. At high impact energies, E > 200 eV, this process takes place [9] through direct transitions from the entrance channel to the CT ones, which are accurately described by means of a sudden vibrational approach. We have shown that, at these energies, total CT cross sections arise as a consequence of transitions at relatively large impact parameters, and therefore large H++H2 distances, where coupling matrix elements are practically independent of the angle γ between vectors R and ρ. For E < 200 eV, a resonant mechanism becomes important; this involves transitions from excited H2 vibrational states, populated at short R, to quasi-degenerate CT channels. Since this latter mechanism involves transitions at short R, anisotropy effects become more important at low v. In order to describe the resonant mechanism, it is indispensable [9] to use a vibronic close-coupling basis, and our method is based on this formalism.
A first set of results of the present calculation includes cross sections calculated for trajectories
![Ijms 03 00142 g006]()
![Ijms 03 00142 g007]()
![Ijms 03 00142 g008]() where the H2 orientations with respect to the ion velocity are kept fixed , but γ changes during the collisions. In particular, we have considered the three trajectories of fig. 1; these calculations are directly relevant to possible orientation dependent experiments such as those reported in refs. [10, 11]. Our results at energies E < 200 eV show that, for this system, CT transition probabilities and cross sections cannot be obtained by assuming that transitions take place at a single value of γ, but an interval of angles are important, and this is the origin of the differences found between the calculated cross sections for the three trajectories. Besides, although this orientation dependence is a consequence of the anisotropy of the electronic Hamiltonian matrix elements, its influence on the final results is conditioned by the mechanism of the process in the vibronic basis. This is clear from the mechanisms discussed in fig. 6, with transitions taking place in the crossing regions of fig. 7, which do not have a counterpart in the electronic Hamiltonian matrix elements of fig. 2.
where the H2 orientations with respect to the ion velocity are kept fixed , but γ changes during the collisions. In particular, we have considered the three trajectories of fig. 1; these calculations are directly relevant to possible orientation dependent experiments such as those reported in refs. [10, 11]. Our results at energies E < 200 eV show that, for this system, CT transition probabilities and cross sections cannot be obtained by assuming that transitions take place at a single value of γ, but an interval of angles are important, and this is the origin of the differences found between the calculated cross sections for the three trajectories. Besides, although this orientation dependence is a consequence of the anisotropy of the electronic Hamiltonian matrix elements, its influence on the final results is conditioned by the mechanism of the process in the vibronic basis. This is clear from the mechanisms discussed in fig. 6, with transitions taking place in the crossing regions of fig. 7, which do not have a counterpart in the electronic Hamiltonian matrix elements of fig. 2.
Figure 6.
Collision histories of vibrational excitation (VE) and charge transfer (CT) channels for trajectories I (a,a’), II (b,b’) and III (c,c’). Collision energy is E = 62.5 eV and impact parameter b = 1.75 a0. Panels (a,b,c) contains results in the diabatic basis, while panels (a’,b’,c’) are in the adiabatic basis.
Figure 6.
Collision histories of vibrational excitation (VE) and charge transfer (CT) channels for trajectories I (a,a’), II (b,b’) and III (c,c’). Collision energy is E = 62.5 eV and impact parameter b = 1.75 a0. Panels (a,b,c) contains results in the diabatic basis, while panels (a’,b’,c’) are in the adiabatic basis.

Figure 7.
Energies of vibronic adiabatic (top panel) and diabatic (bottom panel) states as functions of the nuclear coordinate Z = vt for the trajectory I with b = 1.75 a0. Solid lines correspond to elastic and H2 vibrational excitation channels and dashed lines to charge transfer channels.
Figure 7.
Energies of vibronic adiabatic (top panel) and diabatic (bottom panel) states as functions of the nuclear coordinate Z = vt for the trajectory I with b = 1.75 a0. Solid lines correspond to elastic and H2 vibrational excitation channels and dashed lines to charge transfer channels.

Figure 8.
Same as fig. 7, but for trajectory II
Figure 8.
Same as fig. 7, but for trajectory II

The second point considered in this work refers to the calculation of OACS, which are the most common output of experiments. Since the evaluation of OACS requires the calculation of potential energy surfaces and couplings as functions of the three parameters R, ρ and γ, involving in general a considerable computational effort, it is useful to check on approximate methods to evaluate OACS. In this respect, we have considered an isotropic approximation where γ(t) is taken equal to a constant γ0. Such an isotropic calculation of CT cross sections has been carried out for γ0 = 45° with both DIM and ab initio couplings, yielding errors much smaller than the variation of DIM cross sections with γ0, which support the usefulness of the DIM method to study anisotropy effects. Our main conclusions on this second point are: 1) The CT cross sections averaged over trajectories are practically identical to the average over γ0 of cross sections from isotropic calculations. 2) The average of cross sections agrees with the cross section calculated with a Hamiltonian matrix averaged over γ that corresponds to a value of γ0 ≃ 60°. 3) Small differences are found between CT cross sections from isotropic calculations with γ0 in the range 4°–60°. These results are encouraging in order to simplify the calculation, but further work is required, since the agreement between cross sections seems to be due to cancellations in the average procedure of differences between transition probabilities for different γ0 or different trajectory orientations. However, it is expected that the CT reaction in H++H2 presents comparatively larger anisotropy than collisions involving multicharged ions, where transitions occur at large R, and because the special CT resonant mechanism at low v increases the importance of transitions at short R in CT cross sections.
Acknowledgements
This work has been partially supported by DGICYT projects BFM2000-0025 and FTN2000-0911. IR acknoledges the spanish MCyT for a “contrato de reincorporacioón”.
References
- Pauli, W. Handb. Phys. 1933, 24, 164.
- Baer, M. in State-Selected and State to State Ion-Molecule Reaction Dynamics. Advances in Chemical Physics Vol LXXXII; Baer, Michael, Ng, Cheuk-Yiu, John Wiley & Sons, Inc., Eds.; 1992. [Google Scholar]
- Sidis, V. Adv. At. Mol. Phys. 1990, 26, 161.
- Gislason, E. A.; Parlant, G.; Sizun, M. in State-Selected and State to State Ion-Molecule Reaction Dynamics. Advances in Chemical Physics Vol LXXXII; Baer, Michael, Ng, Cheuk-Yiu, John Wiley & Sons, Inc., Eds.; 1992. [Google Scholar]
- Errea, L. F.; Gorfinkiel, J. D.; Macías, A.; Méndez, L.; Riera, A. J. Phys. B: At. Mol. Opt. Phys. 1997, 30, 3855.
- Errea, L. F.; Gorfinkiel, J. D.; Macías, A.; Méndez, L.; Riera, A. J. Phys. B: At. Mol. Opt. Phys. 1999, 32, 1705.
- D. Elizaga, Errea, L. F.; Gorfinkiel, J. D.; Macías, A.; Méndez, L.; Riera, A.; Rojas, A. J. Phys. B: At. Mol. Opt. Phys. 2000, 33, 2037.
- Errea, L. F.; Macías, A.; Méndez, L.; Rabadán, I.; Riera, A. J. Phys. B: At. Mol. Opt. Phys. 2000, 33, L615.
- Errea, L. F.; Mac½as, A.; Méndez, L.; Rabadán, I.; Riera, A. Phys. Rev. A. 2001, 65, 010701(R).
- Frémont, F.; Bedouet, C.; Tarisien, M.; Adoui, A.; Cassimi, A.; Dubois, A.; Chesnel, J.-Y.; Husson, X. J. Phys. B: At. Mol. Opt. Phys.. 2000, 33, L249.
- Bräauning, H.; Reiser, I.; Diehl, A.; Theiß, A.; Sidkey, A.; Cocke, C. L.; Salzborn, E. J. Phys. B: At. Mol. Opt. Phys. 2001, 34, L321.
- Ichihara, A.; Iwamoto, O.; Janev, R. K. J. Phys. B: At. Mol. Opt. Phys. 2000, 33, 4747.
- Krstić, P. S.; Schultz, D. R.; Janev, R. K. Physica Scripta 2002, in press.
- Kimura, M. Phys. Rev. A. 1985, 32, 802. [PubMed]
- Baer, B.; Niedner-Schatteburg, G.; Toennies, J. P. J. Chem. Phys. 1989, 91, 4169.
- Ellison, F. O.; Huff, N. T.; Patel, J. C. J. Am. Chem. Soc. 1963, 85, 3544.
- Tully, J. C.; Preston, R. K. J. Chem. Phys. 1971, 55, 562.
- Florescu, A.; Sizun, M.; Sidis, V. J. Chem. Phys. 1993, 99, 7277.
- Bates, D. R.; McCarroll, R. Proc. Roy. Soc. A. 1958, 245, 175.
- Macías, A.; Errea, L. F.; Gorfinkiel, J. D.; Méndez, L.; A., Riera. J. Mol. Struct. (Theochem) 1998, 433, 169.
- Gealy, M. W.; Van Zyl, B. I. Phys. Rev. A. 1987, 36, 3091. [PubMed]
- Illescas, C.; Riera, A. J. Phys. B: At. Mol. Opt. Phys. 1998, 31, 2777.
- Niedner, G.; Noll, M.; Toennies, J. P.; Schlier, Ch. J. Chem. Phys. 1987, 87, 2685.
© 2002 by MDPI. Reproduction for noncommercial purposes permitted.
Share and Cite
MDPI and ACS Style
Errea, L.F.; Macías, A.; Méndez, L.; Rabadán, I.; Riera, A. Anisotropy effects in H++H2 collisions. Int. J. Mol. Sci. 2002, 3, 142-161. https://doi.org/10.3390/i3030142
AMA Style
Errea LF, Macías A, Méndez L, Rabadán I, Riera A. Anisotropy effects in H++H2 collisions. International Journal of Molecular Sciences. 2002; 3(3):142-161. https://doi.org/10.3390/i3030142
Chicago/Turabian StyleErrea, L. F., A. Macías, L. Méndez, I. Rabadán, and A. Riera. 2002. "Anisotropy effects in H++H2 collisions" International Journal of Molecular Sciences 3, no. 3: 142-161. https://doi.org/10.3390/i3030142