RSK2-Mediated ELK3 Activation Enhances Cell Transformation and Breast Cancer Cell Growth by Regulation of c-fos Promoter Activity

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
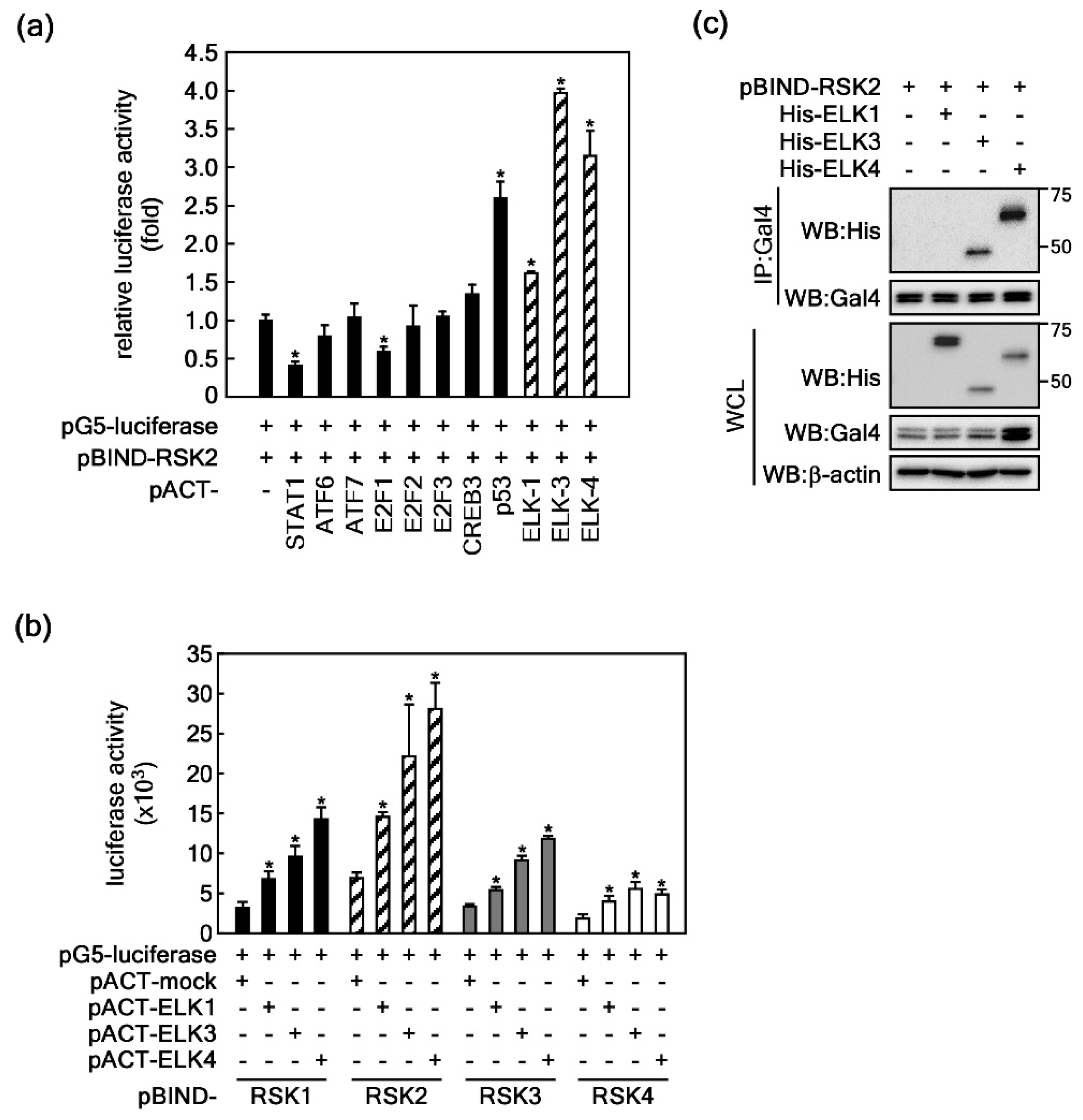
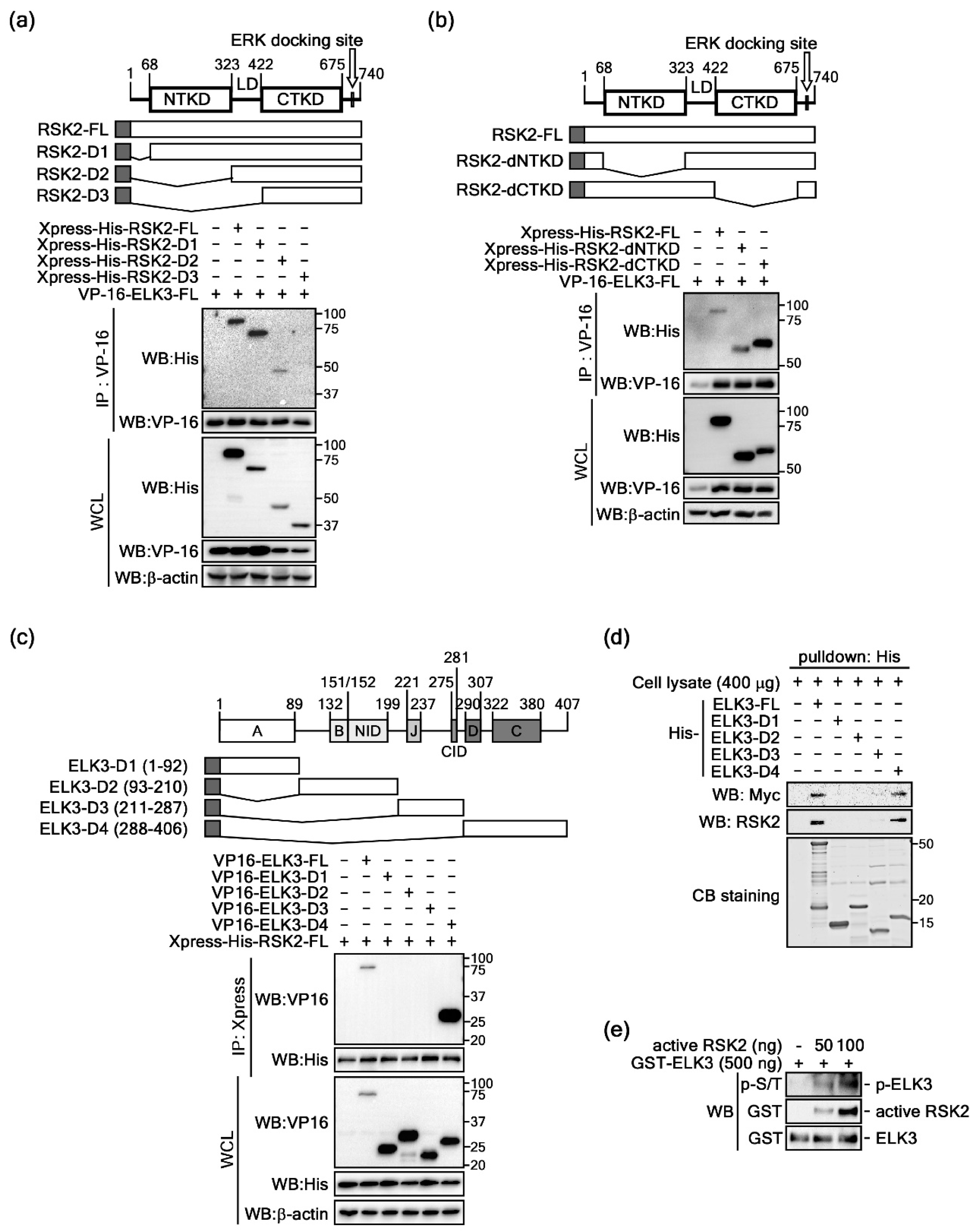
2.1. ELKs Are Novel Binding Partners with RSK2
2.2. ELK3 Is a Substrate of RSK2
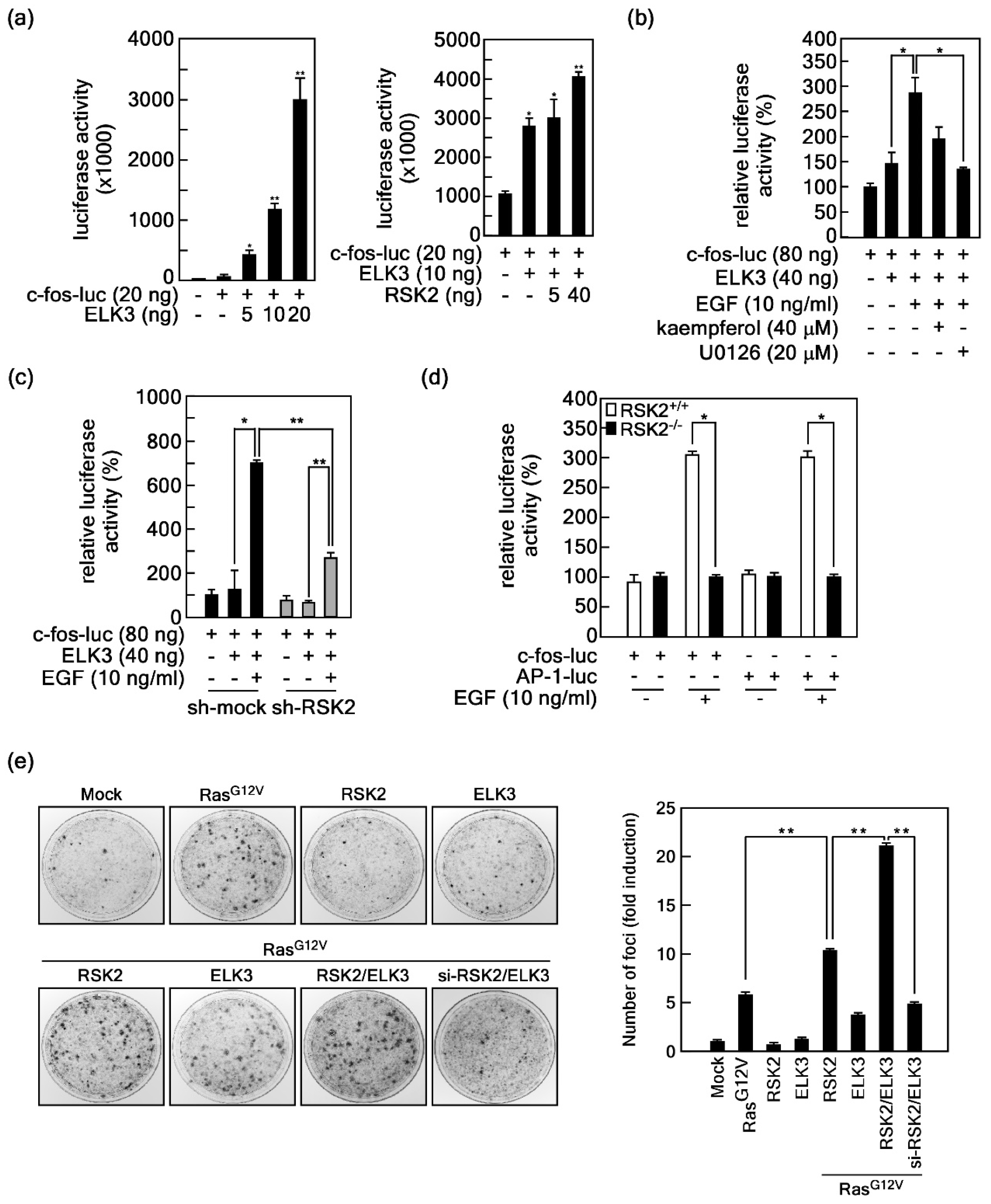
2.3. RSK2-Mediated ELK3 Transactivation Activity Regulates Ras-Mediated Cell Transformation
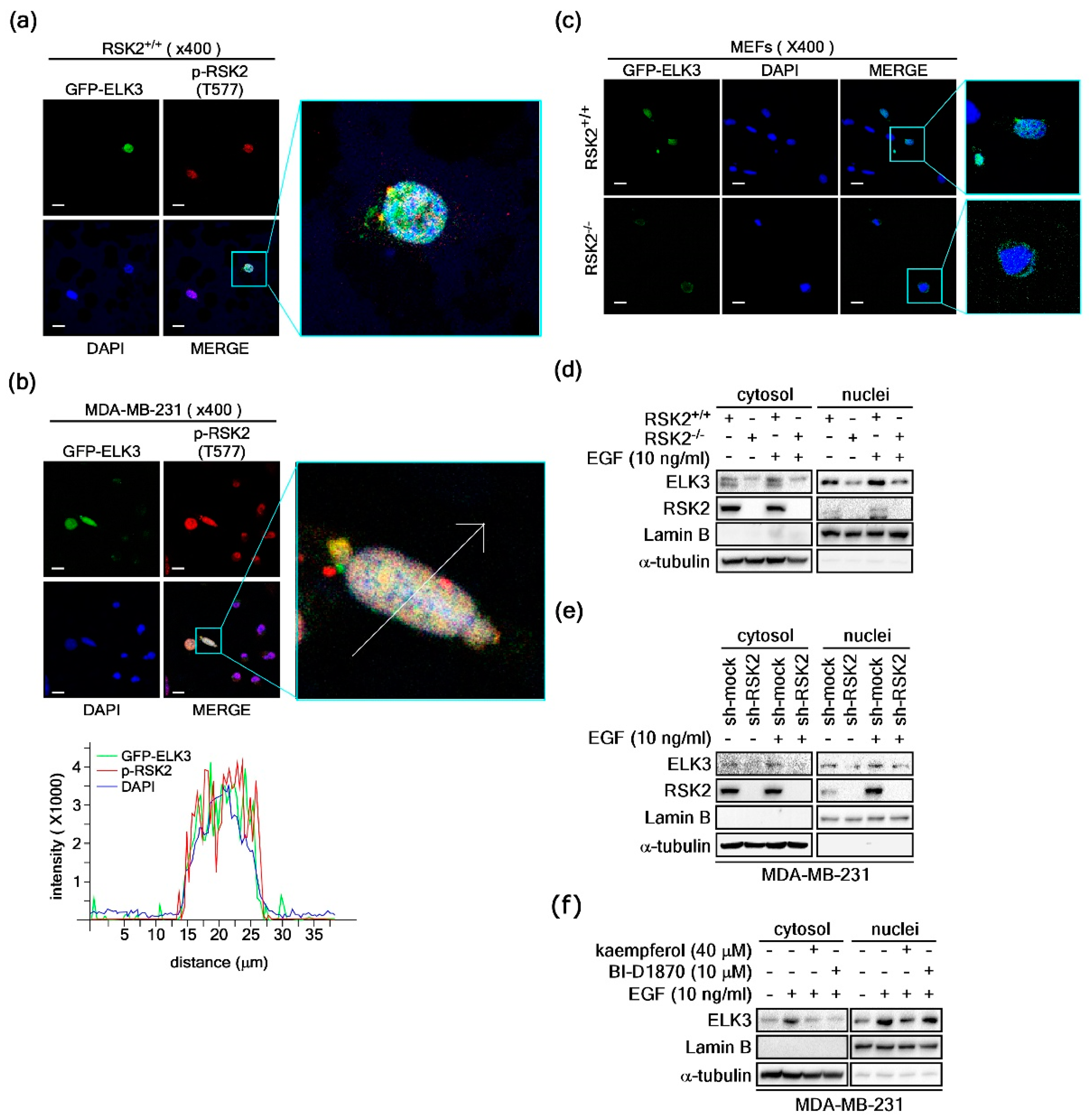
2.4. RSK2 Regulates ELK3 Nuclear Localization
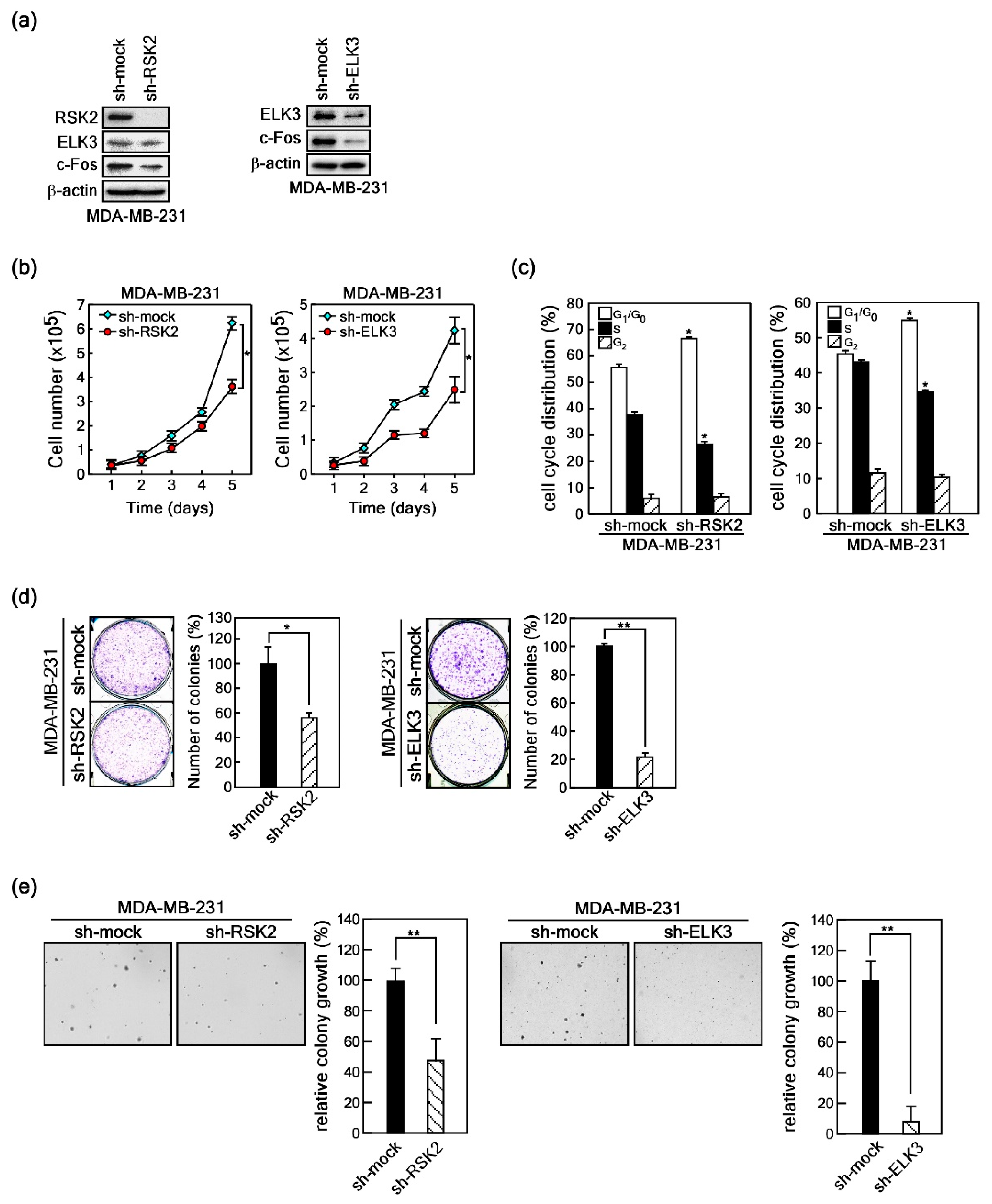
2.5. RSK2/ELK3 Signaling Axis Regulates MDA-MB-231 Breast Cancer Cell Growth
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture and Transfection
4.3. Mammalian Two-Hybrid Assay
4.4. Western Blotting
4.5. Immunoprecipitation
4.6. Gene Silencing
4.7. Cell Cycle Analysis
4.8. Reporter Gene Assay
4.9. In Vitro Kinase Assay
4.10. Vector Construction
4.11. Immunocytofluorescence Assay
4.12. Cell Count Assay
4.13. Anchorage-Independent Colony Growth Assay
4.14. Ni-IDA Pulldown Assay
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ducret, C.; Maira, S.M.; Dierich, A.; Wasylyk, B. The net repressor is regulated by nuclear export in response to anisomycin, UV, and heat shock. Mol. Cell. Biol. 1999, 19, 7076–7087. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.N.; Huebner, K.; Isobe, M.; ar-Rushdi, A.; Croce, C.M.; Reddy, E.S. Elk, tissue-specific ets-related genes on chromosomes X and 14 near translocation breakpoints. Science 1989, 244, 66–70. [Google Scholar] [CrossRef]
- Buchwalter, G.; Gross, C.; Wasylyk, B. Ets ternary complex transcription factors. Gene 2004, 324, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gille, H.; Strahl, T.; Shaw, P.E. Activation of ternary complex factor Elk-1 by stress-activated protein kinases. Curr. Biol. 1995, 5, 1191–1200. [Google Scholar] [CrossRef] [Green Version]
- Maira, S.M.; Wurtz, J.M.; Wasylyk, B. Net (ERP/SAP2) one of the Ras-inducible TCFs, has a novel inhibitory domain with resemblance to the helix-loop-helix motif. EMBO J. 1996, 15, 5849–5865. [Google Scholar] [CrossRef]
- Giovane, A.; Pintzas, A.; Maira, S.M.; Sobieszczuk, P.; Wasylyk, B. Net, a new ets transcription factor that is activated by Ras. Genes Dev. 1994, 8, 1502–1513. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene 2000, 19, 6594–6599. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Pharmacological inhibitors of the ERK signaling pathway: Application as anticancer drugs. Prog. Cell Cycle Res. 2003, 5, 219–224. [Google Scholar]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Price, M.A.; Cruzalegui, F.H.; Treisman, R. The p38 and ERK MAP kinase pathways cooperate to activate Ternary Complex Factors and c-fos transcription in response to UV light. EMBO J. 1996, 15, 6552–6563. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Rogers, A.E.; Treisman, R. Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2 (ERP/NET). EMBO J. 1995, 14, 2589–2601. [Google Scholar] [CrossRef]
- Cruzalegui, F.H.; Cano, E.; Treisman, R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 1999, 18, 7948–7957. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R.; Hunter, T. Activation of the Sap-1a transcription factor by the c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 4219–4224. [Google Scholar] [CrossRef]
- Janknecht, R.; Hunter, T. Convergence of MAP kinase pathways on the ternary complex factor Sap-1a. EMBO J. 1997, 16, 1620–1627. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Yang, S.H.; Su, M.S.; Sharrocks, A.D.; Davis, R.J. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol. Cell. Biol. 1997, 17, 2360–2371. [Google Scholar] [CrossRef]
- Yang, S.H.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998, 17, 1740–1749. [Google Scholar] [CrossRef]
- Gavin, A.C.; Nebreda, A.R. A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr. Biol. 1999, 9, 281–284. [Google Scholar] [CrossRef]
- Smith, J.A.; Poteet-Smith, C.E.; Malarkey, K.; Sturgill, T.W. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J. Biol. Chem. 1999, 274, 2893–2898. [Google Scholar] [CrossRef]
- Roux, P.P.; Richards, S.A.; Blenis, J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol. Cell. Biol. 2003, 23, 4796–4804. [Google Scholar] [CrossRef]
- Sutherland, C.; Campbell, D.G.; Cohen, P. Identification of insulin-stimulated protein kinase-1 as the rabbit equivalent of rskmo-2. Identification of two threonines phosphorylated during activation by mitogen-activated protein kinase. Eur. J. Biochem. 1993, 212, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.Y.; Yao, K.; Pugliese, A.; Malakhova, M.L.; Bode, A.M.; Dong, Z. A regulatory mechanism for RSK2 NH(2)-terminal kinase activity. Cancer Res. 2009, 69, 4398–4406. [Google Scholar] [CrossRef]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. A J. Virtual Libr. 2008, 13, 4258–4275. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Lee, M.H.; Lee, C.J.; Yao, K.; Lee, H.S.; Bode, A.M.; Dong, Z. RSK2 as a key regulator in human skin cancer. Carcinogenesis 2012, 33, 2529–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayadi, A.; Zheng, H.; Sobieszczuk, P.; Buchwalter, G.; Moerman, P.; Alitalo, K.; Wasylyk, B. Net-targeted mutant mice develop a vascular phenotype and up-regulate egr-1. EMBO J. 2001, 20, 5139–5152. [Google Scholar] [CrossRef] [Green Version]
- Buchwalter, G.; Gross, C.; Wasylyk, B. The ternary complex factor Net regulates cell migration through inhibition of PAI-1 expression. Mol. Cell. Biol. 2005, 25, 10853–10862. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Buchwalter, G.; Dubois-Pot, H.; Cler, E.; Zheng, H.; Wasylyk, B. The ternary complex factor net is downregulated by hypoxia and regulates hypoxia-responsive genes. Mol. Cell. Biol. 2007, 27, 4133–4141. [Google Scholar] [CrossRef]
- Gross, C.; Dubois-Pot, H.; Wasylyk, B. The ternary complex factor Net/Elk-3 participates in the transcriptional response to hypoxia and regulates HIF-1 alpha. Oncogene 2008, 27, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Xia, L.; Mo, P.; Huang, W.; Zhang, L.; Wang, Y.; Zhu, H.; Tian, D.; Liu, J.; Chen, Z.; Zhang, Y.; et al. The TNF-alpha/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012, 33, 2250–2259. [Google Scholar] [CrossRef]
- Choi, S.H.; Shin, H.W.; Park, J.Y.; Yoo, J.Y.; Kim, D.Y.; Ro, W.S.; Yun, C.O.; Han, K.H. Effects of the knockdown of hypoxia inducible factor-1alpha expression by adenovirus-mediated shRNA on angiogenesis and tumor growth in hepatocellular carcinoma cell lines. Korean J. Hepatol. 2010, 16, 280–287. [Google Scholar] [CrossRef]
- Xiang, Z.L.; Zeng, Z.C.; Fan, J.; Tang, Z.Y.; Zeng, H.Y.; Gao, D.M. Gene expression profiling of fixed tissues identified hypoxia-inducible factor-1alpha, VEGF, and matrix metalloproteinase-2 as biomarkers of lymph node metastasis in hepatocellular carcinoma. Clin. Cancer Res. 2011, 17, 5463–5472. [Google Scholar] [CrossRef]
- Zheng, H.; Wasylyk, C.; Ayadi, A.; Abecassis, J.; Schalken, J.A.; Rogatsch, H.; Wernert, N.; Maira, S.M.; Multon, M.C.; Wasylyk, B. The transcription factor Net regulates the angiogenic switch. Genes Dev. 2003, 17, 2283–2297. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Lee, M.H.; Yoo, S.M.; Choi, K.I.; Song, J.H.; Jang, J.H.; Oh, S.R.; Ryu, H.W.; Lee, H.S.; Surh, Y.J.; et al. Magnolin inhibits cell migration and invasion by targeting the ERKs/RSK2 signaling pathway. BMC Cancer 2015, 15, 576. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Yao, K.; Kim, H.G.; Kang, B.S.; Zheng, D.; Bode, A.M.; Dong, Z. Ribosomal S6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Res. 2007, 67, 8104–8112. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Lee, M.H.; Lee, J.Y.; Song, J.H.; Lee, H.S.; Cho, Y.Y. RSK2-induced stress tolerance enhances cell survival signals mediated by inhibition of GSK3beta activity. Biochem. Biophys. Res. Commun. 2013, 440, 112–118. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef]
- Cho, Y.Y.; He, Z.; Zhang, Y.; Choi, H.S.; Zhu, F.; Choi, B.Y.; Kang, B.S.; Ma, W.Y.; Bode, A.M.; Dong, Z. The p53 protein is a novel substrate of ribosomal S6 kinase 2 and a critical intermediary for ribosomal S6 kinase 2 and histone H3 interaction. Cancer Res. 2005, 65, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Yao, K.; Bode, A.M.; Bergen, H.R., 3rd; Madden, B.J.; Oh, S.M.; Ermakova, S.; Kang, B.S.; Choi, H.S.; Shim, J.H.; et al. RSK2 mediates muscle cell differentiation through regulation of NFAT3. J. Biol. Chem. 2007, 282, 8380–8392. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Fey, D.; Matallanas, D.; Rauch, J.; Rukhlenko, O.S.; Kholodenko, B.N. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin. Cell Dev. Biol. 2016, 58, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Arul, N.; Cho, Y.Y. A rising cancer prevention target of RSK2 in human skin cancer. Front. Oncol. 2013, 3, 201. [Google Scholar] [CrossRef]
- Cho, Y.Y. RSK2 and its binding partners in cell proliferation, transformation and cancer development. Arch. Pharm. Res. 2017, 40, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y. Molecular targeting of ERKs/RSK2 signaling in cancers. Curr. Pharm. Des. 2017, 23, 4247–4258. [Google Scholar] [CrossRef] [PubMed]
- McCandless, S.E.; Schwartz, S.; Morrison, S.; Garlapati, K.; Robin, N.H. Adult with an interstitial deletion of chromosome 10 [del(10)(q25. 1q25.3)]: Overlap with Coffin-Lowry syndrome. Am. J. Med. Genet. 2000, 95, 93–98. [Google Scholar] [CrossRef]
- Colburn, N.H.; Wendel, E.J.; Abruzzo, G. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: Mitogen-resistant variants are sensitive to promotion. Proc. Natl. Acad. Sci. USA 1981, 78, 6912–6916. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, S.-M.; Lee, C.-J.; An, H.-J.; Lee, J.Y.; Lee, H.S.; Kang, H.C.; Cho, S.-J.; Kim, S.-M.; Park, J.; Kim, D.J.; et al. RSK2-Mediated ELK3 Activation Enhances Cell Transformation and Breast Cancer Cell Growth by Regulation of c-fos Promoter Activity. Int. J. Mol. Sci. 2019, 20, 1994. https://doi.org/10.3390/ijms20081994
Yoo S-M, Lee C-J, An H-J, Lee JY, Lee HS, Kang HC, Cho S-J, Kim S-M, Park J, Kim DJ, et al. RSK2-Mediated ELK3 Activation Enhances Cell Transformation and Breast Cancer Cell Growth by Regulation of c-fos Promoter Activity. International Journal of Molecular Sciences. 2019; 20(8):1994. https://doi.org/10.3390/ijms20081994
Chicago/Turabian StyleYoo, Sun-Mi, Cheol-Jung Lee, Hyun-Jung An, Joo Young Lee, Hye Suk Lee, Han Chang Kang, Sung-Jun Cho, Seung-Min Kim, Juhee Park, Dae Joon Kim, and et al. 2019. "RSK2-Mediated ELK3 Activation Enhances Cell Transformation and Breast Cancer Cell Growth by Regulation of c-fos Promoter Activity" International Journal of Molecular Sciences 20, no. 8: 1994. https://doi.org/10.3390/ijms20081994