Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production

 , , ,
, , ,
Abstract
:1. Introduction
2. Differential Roles of PKA, CaMKIV and ERK/p38 MAPK Axis in CREB Activation in the Central Nervous System
3. Roles of ERK1/2 and p38 MAPK in Periostin Production in Cardiovascular Disease
3.1. Alcoholic Cardiomyopathy
3.2. Cardiac Remodeling
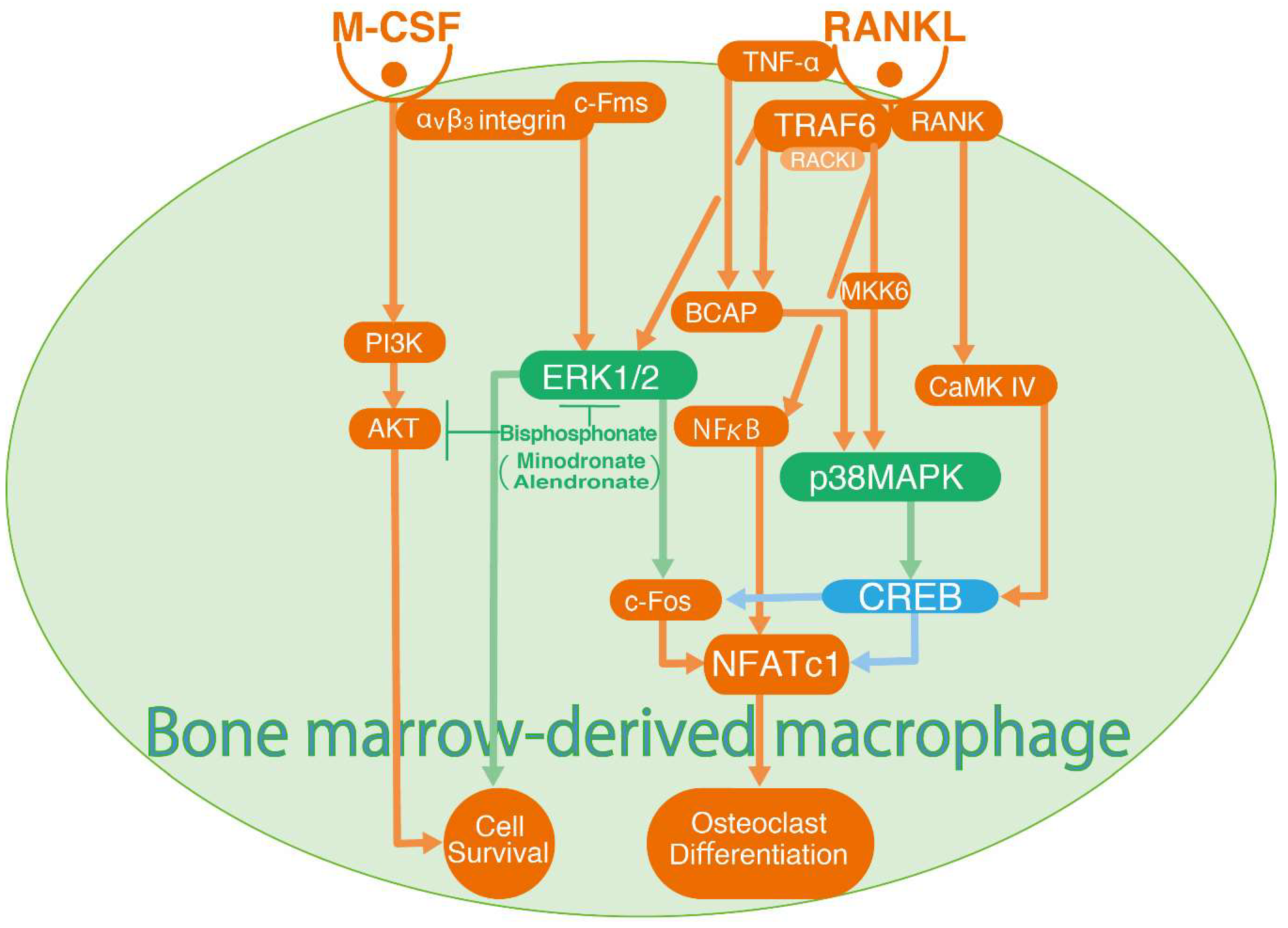
4. Crosstalk between ERK1/2 and CREB-p38 MAPK Signalling in Osteoclast Differentiation
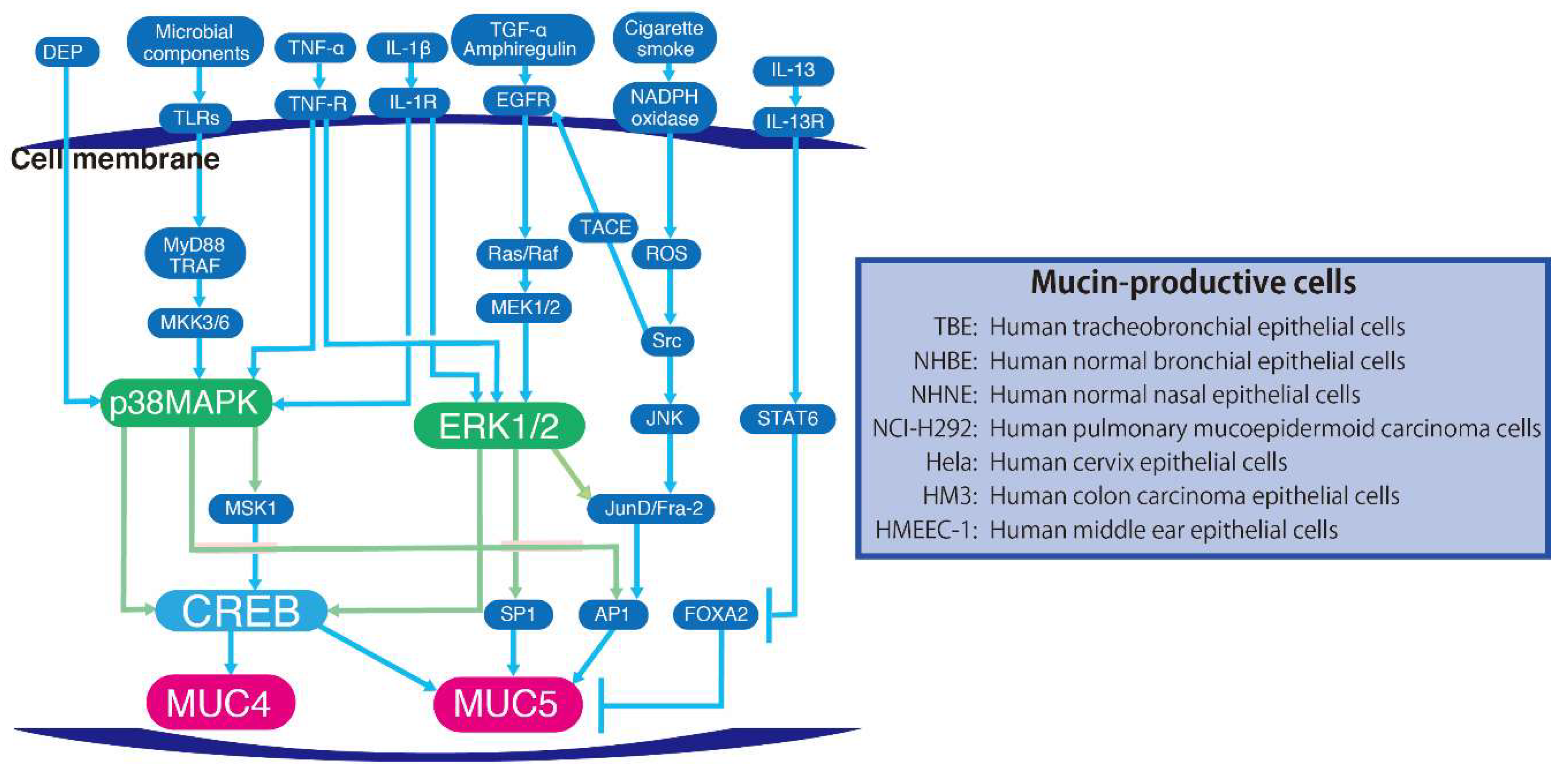
5. Mucin Production and MAPK Signalling
6. Roles of ERK1/2 and p38 MAPK-CREB Signalling in Vascular Smooth Muscle Cell Migration
7. Regulatory Mechanism of GM-CSF Secretion by cAMP and the EKR1/2-p38 MAPK Signalling Pathway
8. Roles of ERK1/2 and p38 MAPK Mediated CREB Activation in Cytokine and Steroid Synthesis
9. Roles of cAMP in CREB Activation
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CREB | cyclic AMP response element-binding protein |
| RSK | pp90 ribosomal S6 kinase |
| MSK1 | Mitogen- and stress-activated protein kinase 1 |
| CaMKs | Calcium/calmodulin-dependent protein kinases |
| EPAC | exchange protein directly activated by cAMP |
| NGF | nerve growth factor |
| MAPKAP kinase 2 | MAPK-activated protein kinase 2 |
| VSMC | Vascular smooth muscle cell |
| ACM | Alcoholic cardiomyopathy |
| ACA | Acetaldehyde |
| ADH | Alcohol dehydrogenase |
| ALDH | Acetaldehyde dehydrogenase |
| ECM | Extracellular cardiac matrix |
| RAS | Renin–angiotensin system |
| Ang II | Angiotensin II |
| DUSPs | Dual-specificity phosphatases |
| NF-κB | Nuclear factor kappa B |
| RANKL | Receptor activator of NF-κB, ligand |
| NFATc1 | Nuclear factor of activated T-cells, cytoplasmic 1 |
| RACK1 | Receptor for activated C kinase 1 |
| TRAF6 | TNF receptor-associated factor 6 |
| Pl3K | Phosphatidylinositol 3-kinase |
| BCAP | B-cell adaptor for Pl3K |
| Ambn | Ameloblastin |
| COPD | Chronic obstructive pulmonary disease |
| EGFR | Epidermal growth factor receptor |
| TLR | Toll like receptor |
| AP1 | Activator protein 1 |
| SP1 | Specificity protein 1 |
| TAK1 | transforming growth factor-activated kinase 1 |
| FOXA2 | Forkhead box protein A2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| ROS | Reactive oxygen species |
| Fra-2 | Fos-related antigen 2 |
| DEPs | Diesel exhaust particles |
| HETEs | Hydroxyeicosatetraenoic acids |
| ET-1 | Endothelin-1 |
| PDE4 | Phosphodiesterase 4 |
| StAR | Steroidogenic acute regulatory protein |
References
- Gonzalez, G.A.; Yamamoto, K.K.; Fischer, W.H.; Karr, D.; Menzel, P.; Biggs, W., 3rd; Vale, W.W.; Montminy, M.R. A cluster of phosphorylation sites on the cyclic AMP-regulated nuclear factor CREB predicted by its sequence. Nature 1989, 337, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB phosphorylation and dephosphorylation: A Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef]
- Xie, H.; Rothstein, T.L. Protein kinase C mediates activation of nuclear cAMP response element-binding protein (CREB) in B lymphocytes stimulated through surface Ig. J. Immunol. 1995, 154, 1717–1723. [Google Scholar] [PubMed]
- Ginty, D.D.; Bonni, A.; Greenberg, M.E. Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 1994, 77, 713–725. [Google Scholar] [CrossRef]
- Sheng, M.; Thompson, M.A.; Greenberg, M.E. CREB: A Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 1991, 252, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Rouse, J.; Zhang, A.; Cariati, S.; Cohen, P.; Comb, M.J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996, 15, 4629–4642. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ginty, D.D.; Greenberg, M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 1996, 273, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Cai, D.; Qiu, J.; Cao, Z.; McAtee, M.; Bregman, B.S.; Filbin, M.T. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J. Neurosci. 2001, 21, 4731–4739. [Google Scholar] [CrossRef]
- Batty, N.J.; Fenrich, K.K.; Fouad, K. The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci. Lett. 2017, 652, 56–63. [Google Scholar] [CrossRef]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Kiermayer, S.; Biondi, R.M.; Imig, J.; Plotz, G.; Haupenthal, J.; Zeuzem, S.; Piiper, A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol. Biol. Cell 2005, 16, 5639–5648. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Shewan, D.A. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol. Cell. Neurosci. 2008, 38, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Dash, P.K.; Karl, K.A.; Colicos, M.A.; Prywes, R.; Kandel, E.R. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 5061–5065. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Sun, L.D.; Atkins, C.M.; Soderling, T.R.; Wilson, M.A.; Tonegawa, S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 2001, 106, 771–783. [Google Scholar] [CrossRef]
- Ahn, S.; Ginty, D.D.; Linden, D.J. A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron 1999, 23, 559–568. [Google Scholar] [CrossRef]
- Canon, E.; Cosgaya, J.M.; Scsucova, S.; Aranda, A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol. Biol. Cell 2004, 15, 5583–5592. [Google Scholar] [CrossRef]
- Gao, Y.; Nikulina, E.; Mellado, W.; Filbin, M.T. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J. Neurosci. 2003, 23, 11770–11777. [Google Scholar] [CrossRef]
- Impey, S.; Smith, D.M.; Obrietan, K.; Donahue, R.; Wade, C.; Storm, D.R. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat. Neurosci. 1998, 1, 595–601. [Google Scholar] [CrossRef]
- Zanassi, P.; Paolillo, M.; Feliciello, A.; Avvedimento, E.V.; Gallo, V.; Schinelli, S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J. Biol. Chem. 2001, 276, 11487–11495. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Kohno, T.; Zhuang, Z.Y.; Brenner, G.J.; Wang, H.; Van Der Meer, C.; Befort, K.; Woolf, C.J.; Ji, R.R. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J. Neurosci. 2004, 24, 8310–8321. [Google Scholar] [CrossRef]
- Laonigro, I.; Correale, M.; Di Biase, M.; Altomare, E. Alcohol abuse and heart failure. Eur. J. Heart Fail. 2009, 11, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95. [Google Scholar] [CrossRef]
- Cheng, C.P.; Cheng, H.J.; Cunningham, C.; Shihabi, Z.K.; Sane, D.C.; Wannenburg, T.; Little, W.C. Angiotensin II type 1 receptor blockade prevents alcoholic cardiomyopathy. Circulation 2006, 114, 226–236. [Google Scholar] [CrossRef]
- Tan, Y.; Li, X.; Prabhu, S.D.; Brittian, K.R.; Chen, Q.; Yin, X.; McClain, C.J.; Zhou, Z.; Cai, L. Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. J. Am. Coll. Cardiol. 2012, 59, 1477–1486. [Google Scholar] [CrossRef]
- Mustroph, J.; Lebek, S.; Maier, L.S.; Neef, S. Mechanisms of cardiac ethanol toxicity and novel treatment options. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- Doser, T.A.; Turdi, S.; Thomas, D.P.; Epstein, P.N.; Li, S.Y.; Ren, J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation 2009, 119, 1941–1949. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, R.; Wei, S.; Yuan, Q.; Xue, M.; Hao, P.; Xu, F.; Wang, J.; Chen, Y. ALDH2 protects against alcoholic cardiomyopathy through a mechanism involving the p38 MAPK/CREB pathway and local renin-angiotensin system inhibition in cardiomyocytes. Int. J. Cardiol. 2018, 257, 150–159. [Google Scholar] [CrossRef]
- Brandt, M.; Wenzel, P. Alcohol puts the heart under pressure: Acetaldehyde activates a localized renin angiotensin aldosterone system within the myocardium in alcoholic cardiomyopathy. Int. J. Cardiol. 2018, 257, 220–221. [Google Scholar] [CrossRef]
- Levy, D.; Kenchaiah, S.; Larson, M.G.; Benjamin, E.J.; Kupka, M.J.; Ho, K.K.; Murabito, J.M.; Vasan, R.S. Long-term trends in the incidence of and survival with heart failure. N. Engl. J. Med. 2002, 347, 1397–1402. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Ohishi, M.; Katsuya, T.; Ito, N.; Ikushima, M.; Kaibe, M.; Tatara, Y.; Shiota, A.; Sugano, S.; Takeda, S.; et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Oparil, S.; Feng, W.; Chen, Y.F. Hypoxia-responsive growth factors upregulate periostin and osteopontin expression via distinct signaling pathways in rat pulmonary arterial smooth muscle cells. J. Appl. Physiol. 2004, 97, 1550–1558; discussion 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Amizuka, N.; Takeshita, S.; Takamatsu, H.; Katsuura, M.; Ozawa, H.; Toyama, Y.; Bonewald, L.F.; Kudo, A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone Miner. Res. 1999, 14, 1239–1249. [Google Scholar] [CrossRef]
- Wang, D.; Oparil, S.; Feng, J.A.; Li, P.; Perry, G.; Chen, L.B.; Dai, M.; John, S.W.; Chen, Y.F. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension 2003, 42, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Litvin, J.; Blagg, A.; Mu, A.; Matiwala, S.; Montgomery, M.; Berretta, R.; Houser, S.; Margulies, K. Periostin and periostin-like factor in the human heart: Possible therapeutic targets. Cardiovasc. Pathol. 2006, 15, 24–32. [Google Scholar] [CrossRef]
- Katsuragi, N.; Morishita, R.; Nakamura, N.; Ochiai, T.; Taniyama, Y.; Hasegawa, Y.; Kawashima, K.; Kaneda, Y.; Ogihara, T.; Sugimura, K. Periostin as a novel factor responsible for ventricular dilation. Circulation 2004, 110, 1806–1813. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W., 2nd; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fan, D.; Wang, C.; Wang, J.Y.; Cui, X.B.; Wu, D.; Zhou, Y.; Wu, L.L. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-beta1 pathways in cardiac fibroblasts. Cardiovasc. Res. 2011, 91, 80–89. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, V.P.; Baker, K.M. The intracellular renin-angiotensin system: Implications in cardiovascular remodeling. Curr. Opin. Nephrol. Hypertens. 2008, 17, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Hieu, T.B.; Ma, F.; Yu, Y.; Cao, Z.; Wang, M.; Wu, W.; Mao, Y.; Rose, P.; Law, B.Y.; et al. ZYZ-168 alleviates cardiac fibrosis after myocardial infarction through inhibition of ERK1/2-dependent ROCK1 activation. Sci. Rep. 2017, 7, 43242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.D.; Li, F.; Ma, D.B.; Deng, X.; Zhang, H.; Gao, J.; Hao, L.; Liu, D.D.; Wang, J. Periostin mediates cigarette smoke extract-induced proliferation and migration in pulmonary arterial smooth muscle cells. Biomed. Pharmacother. 2016, 83, 514–520. [Google Scholar] [CrossRef]
- Takemura, G.; Kanoh, M.; Minatoguchi, S.; Fujiwara, H. Cardiomyocyte apoptosis in the failing heart—A critical review from definition and classification of cell death. Int. J. Cardiol. 2013, 167, 2373–2386. [Google Scholar] [CrossRef]
- Huby, A.C.; Turdi, S.; James, J.; Towbin, J.A.; Purevjav, E. FasL expression in cardiomyocytes activates the ERK1/2 pathway, leading to dilated cardiomyopathy and advanced heart failure. Clin. Sci. 2016, 130, 289–299. [Google Scholar] [CrossRef]
- Liu, R.; van Berlo, J.H.; York, A.J.; Vagnozzi, R.J.; Maillet, M.; Molkentin, J.D. DUSP8 Regulates Cardiac Ventricular Remodeling by Altering ERK1/2 Signaling. Circ. Res. 2016, 119, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J. Mol. Med. 2005, 83, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Kim, N.; Kadono, Y.; Rho, J.; Lee, S.Y.; Lorenzo, J.; Choi, Y. Osteoimmunology: Interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 2006, 24, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Grigoriadis, A.E.; Wang, Z.Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Katagiri, H.; Kanegae, Y.; Takayanagi, H.; Sawada, Y.; Yamamoto, A.; Pando, M.P.; Asano, T.; Verma, I.M.; Oda, H.; et al. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J. Cell Biol. 2000, 148, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Nakamura, I.; Ikebe, T.; Akiyama, S.; Takahashi, N.; Suda, T. Activation of NF-kappaB is involved in the survival of osteoclasts promoted by interleukin-1. J. Biol. Chem. 1998, 273, 8799–8805. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Lee, K.B.; Kim, N. BCAP promotes osteoclast differentiation through regulation of the p38-dependent CREB signaling pathway. Bone 2018, 107, 188–195. [Google Scholar] [CrossRef]
- Chaweewannakorn, W.; Ariyoshi, W.; Okinaga, T.; Fujita, Y.; Maki, K.; Nishihara, T. Ameloblastin attenuates RANKL-mediated osteoclastogenesis by suppressing activation of nuclear factor of activated T-cell cytoplasmic 1 (NFATc1). J. Cell. Physiol. 2019, 234, 1745–1757. [Google Scholar] [CrossRef]
- Chaweewannakorn, W.; Ariyoshi, W.; Okinaga, T.; Morikawa, K.; Saeki, K.; Maki, K.; Nishihara, T. Ameloblastin and enamelin prevent osteoclast formation by suppressing RANKL expression via MAPK signaling pathway. Biochem. Biophys. Res. Commun. 2017, 485, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Nakashima, T.; Takemoto-Kimura, S.; Aoki, K.; Morishita, Y.; Asahara, H.; Ohya, K.; Yamaguchi, A.; Takai, T.; et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 2006, 12, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Deisseroth, K.; Tsien, R.W. Activity-dependent CREB phosphorylation: Convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 2808–2813. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Sudo, T.; Saito, T.; Osada, H.; Tsujimoto, M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL). J. Biol. Chem. 2000, 275, 31155–31161. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lee, D.; Choi, Y.; Lee, S.Y. The scaffold protein RACK1 mediates the RANKL-dependent activation of p38 MAPK in osteoclast precursors. Sci. Signal. 2015, 8, ra54. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; et al. Nitrogen-containing bisphosphonates inhibit RANKL- and M-CSF-induced osteoclast formation through the inhibition of ERK1/2 and Akt activation. J. Biomed. Sci. 2014, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Boucher, R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Investig. 2002, 109, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, C.L.; Khashayar, R.; Wong, H.H.; Ferrando, R.; Wu, R.; Hyde, D.M.; Hotchkiss, J.A.; Zhang, Y.; Novikov, A.; Dolganov, G.; et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am. J. Respir. Crit. Care Med. 2001, 163, 517–523. [Google Scholar] [CrossRef]
- Kirkham, S.; Kolsum, U.; Rousseau, K.; Singh, D.; Vestbo, J.; Thornton, D.J. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 178, 1033–1039. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Singh, A.P.; Batra, S.K. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008, 22, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Perrais, M.; Pigny, P.; Copin, M.C.; Aubert, J.P.; Van Seuningen, I. Induction of MUC2 and MUC5AC mucins by factors of the epidermal growth factor (EGF) family is mediated by EGF receptor/Ras/Raf/extracellular signal-regulated kinase cascade and Sp1. J. Biol. Chem. 2002, 277, 32258–32267. [Google Scholar] [CrossRef] [PubMed]
- Takeyama, K.; Dabbagh, K.; Jeong Shim, J.; Dao-Pick, T.; Ueki, I.F.; Nadel, J.A. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: Role of neutrophils. J. Immunol. 2000, 164, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Thai, P.; Zhao, Y.H.; Ho, Y.S.; DeSouza, M.M.; Wu, R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 2003, 278, 17036–17043. [Google Scholar] [CrossRef] [PubMed]
- Song, K.S.; Lee, W.J.; Chung, K.C.; Koo, J.S.; Yang, E.J.; Choi, J.Y.; Yoon, J.H. Interleukin-1 beta and tumor necrosis factor-alpha induce MUC5AC overexpression through a mechanism involving ERK/p38 mitogen-activated protein kinases-MSK1-CREB activation in human airway epithelial cells. J. Biol. Chem. 2003, 278, 23243–23250. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.Z.; Simard, F.A.; Cloutier, A.; Vardhan, H.; Dubois, C.M.; McDonald, P.P. The p38-MSK1 signaling cascade influences cytokine production through CREB and C/EBP factors in human neutrophils. J. Immunol. 2013, 191, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Luyimbazi, J.; Xu, X.; Schofield, B.; Neben, T.Y.; Karp, C.L.; Donaldson, D.D. Interleukin-13: Central mediator of allergic asthma. Science 1998, 282, 2258–2261. [Google Scholar] [CrossRef]
- Zhen, G.; Park, S.W.; Nguyenvu, L.T.; Rodriguez, M.W.; Barbeau, R.; Paquet, A.C.; Erle, D.J. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am. J. Respir. Cell Mol. Biol. 2007, 36, 244–253. [Google Scholar] [CrossRef]
- Chen, G.; Wan, H.; Luo, F.; Zhang, L.; Xu, Y.; Lewkowich, I.; Wills-Karp, M.; Whitsett, J.A. Foxa2 programs Th2 cell-mediated innate immunity in the developing lung. J. Immunol. 2010, 184, 6133–6141. [Google Scholar] [CrossRef]
- Rose, M.C.; Voynow, J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 2006, 86, 245–278. [Google Scholar] [CrossRef]
- Li, D.; Gallup, M.; Fan, N.; Szymkowski, D.E.; Basbaum, C.B. Cloning of the amino-terminal and 5’-flanking region of the human MUC5AC mucin gene and transcriptional up-regulation by bacterial exoproducts. J. Biol. Chem. 1998, 273, 6812–6820. [Google Scholar] [CrossRef] [PubMed]
- Ha, U.; Lim, J.H.; Jono, H.; Koga, T.; Srivastava, A.; Malley, R.; Pages, G.; Pouyssegur, J.; Li, J.D. A novel role for IkappaB kinase (IKK) alpha and IKKbeta in ERK-dependent up-regulation of MUC5AC mucin transcription by Streptococcus pneumoniae. J. Immunol. 2007, 178, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Lim, D.J.; Han, J.; Kim, Y.S.; Basbaum, C.B.; Li, J.D. Novel cytoplasmic proteins of nontypeable Haemophilus influenzae up-regulate human MUC5AC mucin transcription via a positive p38 mitogen-activated protein kinase pathway and a negative phosphoinositide 3-kinase-Akt pathway. J. Biol. Chem. 2002, 277, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lim, J.H.; Jono, H.; Gu, X.X.; Kim, Y.S.; Basbaum, C.B.; Murphy, T.F.; Li, J.D. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 2004, 324, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar, H.; Li, D.; Gallup, M.; Sidhu, S.; Drori, E.; Basbaum, C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulin. J. Biol. Chem. 2003, 278, 26202–26207. [Google Scholar] [CrossRef] [PubMed]
- Gensch, E.; Gallup, M.; Sucher, A.; Li, D.; Gebremichael, A.; Lemjabbar, H.; Mengistab, A.; Dasari, V.; Hotchkiss, J.; Harkema, J.; et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J. Biol. Chem. 2004, 279, 39085–39093. [Google Scholar] [CrossRef] [PubMed]
- Rokutan-Kurata, M.; Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Menju, T.; Momose, M.; Koyama, M.; Shigeto, S.; Fujimoto, M.; Zhang, M.; et al. Lung Adenocarcinoma With MUC4 Expression Is Associated With Smoking Status, HER2 Protein Expression, and Poor Prognosis: Clinicopathologic Analysis of 338 Cases. Clin. Lung Cancer 2017, 18, e273–e281. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumida, H.; Goto, M.; Kitajima, S.; Kubota, I.; Hirotsu, Y.; Wakimoto, J.; Batra, S.K.; Imai, K.; Yonezawa, S. MUC4 expression correlates with poor prognosis in small-sized lung adenocarcinoma. Lung Cancer 2007, 55, 195–203. [Google Scholar] [CrossRef]
- Park, I.H.; Kang, J.H.; Kim, J.A.; Shin, J.M.; Lee, H.M. Diesel Exhaust Particles Enhance MUC4 Expression in NCI-H292 Cells and Nasal Epithelial Cells via the p38/CREB Pathway. Int. Arch. Allergy Immunol. 2016, 171, 209–216. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Yeh, E.T.; Zhang, S.; Wu, H.D.; Korbling, M.; Willerson, J.T.; Estrov, Z. Transdifferentiation of human peripheral blood CD34+-enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation 2003, 108, 2070–2073. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, N.; Koga, Y.; Ikebe, M. Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem. Biophys. Res. Commun. 2002, 297, 773–778. [Google Scholar] [CrossRef]
- Niiro, N.; Koga, Y.; Ikebe, M. Agonist-induced changes in the phosphorylation of the myosin- binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem. J. 2003, 369 Pt 1, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Koga, Y.; Ikebe, M. p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J. Biol. Chem. 2005, 280, 4983–4991. [Google Scholar] [CrossRef] [PubMed]
- Ihara, E.; Yu, Q.; Chappellaz, M.; MacDonald, J.A. ERK and p38MAPK pathways regulate myosin light chain phosphatase and contribute to Ca2+ sensitization of intestinal smooth muscle contraction. Neurogastroenterol. Motil. 2015, 27, 135–146. [Google Scholar] [CrossRef]
- Schmitz, U.; Berk, B.C. Angiotensin II signal transduction: Stimulation of multiple mitogen-activated protein kinase pathways. Trends Endocrinol. Metab. 1997, 8, 261–266. [Google Scholar] [CrossRef]
- Nagayama, K.; Kyotani, Y.; Zhao, J.; Ito, S.; Ozawa, K.; Bolstad, F.A.; Yoshizumi, M. Exendin-4 Prevents Vascular Smooth Muscle Cell Proliferation and Migration by Angiotensin II via the Inhibition of ERK1/2 and JNK Signaling Pathways. PLoS ONE 2015, 10, e0137960. [Google Scholar] [CrossRef]
- Shi, L.; Ji, Y.; Liu, D.; Liu, Y.; Xu, Y.; Cao, Y.; Jiang, X.; Xu, C. Sitagliptin attenuates high glucose-induced alterations in migration, proliferation, calcification and apoptosis of vascular smooth muscle cells through ERK1/2 signal pathway. Oncotarget 2017, 8, 77168–77180. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Luo, Y.; Hu, P.; Dou, L.; Huang, S. Tanshinone IIA inhibits AGEs-induced proliferation and migration of cultured vascular smooth muscle cells by suppressing ERK1/2 MAPK signaling. Iran. J. Basic Med Sci. 2018, 21, 83–88. [Google Scholar]
- Chen, C.; Du, P.; Wang, J. Paeoniflorin ameliorates acute myocardial infarction of rats by inhibiting inflammation and inducible nitric oxide synthase signaling pathways. Mol. Med. Rep. 2015, 12, 3937–3943. [Google Scholar] [CrossRef]
- Fan, X.; Wu, J.; Yang, H.; Yan, L.; Wang, S. Paeoniflorin blocks the proliferation of vascular smooth muscle cells induced by plateletderived growth factorBB through ROS mediated ERK1/2 and p38 signaling pathways. Mol. Med. Rep. 2018, 17, 1676–1682. [Google Scholar]
- Li, Y.X.; Run, L.; Shi, T.; Zhang, Y.J. CTRP9 regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation, apoptosis and migration via TGF-beta1/ERK1/2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 490, 1319–1325. [Google Scholar] [CrossRef]
- Ono, H.; Ichiki, T.; Fukuyama, K.; Iino, N.; Masuda, S.; Egashira, K.; Takeshita, A. cAMP-response element-binding protein mediates tumor necrosis factor-alpha-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1634–1639. [Google Scholar] [CrossRef]
- Wihlborg, A.K.; Balogh, J.; Wang, L.; Borna, C.; Dou, Y.; Joshi, B.V.; Lazarowski, E.; Jacobson, K.A.; Arner, A.; Erlinge, D. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of UTP in man during myocardial infarction. Circ. Res. 2006, 98, 970–976. [Google Scholar] [CrossRef]
- Jalvy, S.; Renault, M.A.; Lam Shang Leen, L.; Belloc, I.; Reynaud, A.; Gadeau, A.P.; Desgranges, C. CREB mediates UTP-directed arterial smooth muscle cell migration and expression of the chemotactic protein osteopontin via its interaction with activator protein-1 sites. Circ. Res. 2007, 100, 1292–1299. [Google Scholar] [CrossRef]
- Reddy, M.A.; Sahar, S.; Villeneuve, L.M.; Lanting, L.; Natarajan, R. Role of Src tyrosine kinase in the atherogenic effects of the 12/15-lipoxygenase pathway in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 387–393. [Google Scholar] [CrossRef]
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Rao, G.N. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 809–815. [Google Scholar] [CrossRef]
- Lopez, A.F.; Williamson, D.J.; Gamble, J.R.; Begley, C.G.; Harlan, J.M.; Klebanoff, S.J.; Waltersdorph, A.; Wong, G.; Clark, S.C.; Vadas, M.A. Recombinant human granulocyte-macrophage colony-stimulating factor stimulates in vitro mature human neutrophil and eosinophil function, surface receptor expression, and survival. J. Clin. Investig. 1986, 78, 1220–1228. [Google Scholar] [CrossRef]
- Metcalf, D. The molecular biology and functions of the granulocyte-macrophage colony-stimulating factors. Blood 1986, 67, 257–267. [Google Scholar]
- Takizawa, H. Airway epithelial cells as regulators of airway inflammation (Review). Int. J. Mol. Med. 1998, 1, 367–378. [Google Scholar] [CrossRef]
- Xing, Z.; Ohkawara, Y.; Jordana, M.; Graham, F.; Gauldie, J. Transfer of granulocyte-macrophage colony-stimulating factor gene to rat lung induces eosinophilia, monocytosis, and fibrotic reactions. J. Clin. Investig. 1996, 97, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Braciak, T.; Ohkawara, Y.; Sallenave, J.M.; Foley, R.; Sime, P.J.; Jordana, M.; Graham, F.L.; Gauldie, J. Gene transfer for cytokine functional studies in the lung: The multifunctional role of GM-CSF in pulmonary inflammation. J. Leukoc. Biol. 1996, 59, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Colony stimulating factors, cytokines and monocyte-macrophages—Some controversies. Immunol. Today 1993, 14, 18–24. [Google Scholar] [CrossRef]
- Sato, E.; Camhi, S.L.; Koyama, S.; Robbins, R.A. Methotrexate stimulates lung fibroblasts and epithelial cells to release eosinophil chemotactic activity. J. Rheumatol. 2001, 28, 502–508. [Google Scholar]
- Numanami, H.; Koyama, S.; Sato, E.; Haniuda, M.; Nelson, D.K.; Hoyt, J.C.; Freels, J.L.; Habib, M.P.; Robbins, R.A. Serine protease inhibitors modulate chemotactic cytokine production by human lung fibroblasts in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L882–L890. [Google Scholar] [CrossRef]
- Koga, Y.; Hisada, T.; Ishizuka, T.; Utsugi, M.; Ono, A.; Yatomi, M.; Kamide, Y.; Aoki-Saito, H.; Tsurumaki, H.; Dobashi, K.; et al. CREB regulates TNF-alpha-induced GM-CSF secretion via p38 MAPK in human lung fibroblasts. Allergol. Int. 2016, 65, 406–413. [Google Scholar] [CrossRef]
- Cromwell, O.; Hamid, Q.; Corrigan, C.J.; Barkans, J.; Meng, Q.; Collins, P.D.; Kay, A.B. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology 1992, 77, 330–337. [Google Scholar]
- Nakamura, Y.; Azuma, M.; Okano, Y.; Sano, T.; Takahashi, T.; Ohmoto, Y.; Sone, S. Upregulatory effects of interleukin-4 and interleukin-13 but not interleukin-10 on granulocyte/macrophage colony-stimulating factor production by human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1996, 15, 680–687. [Google Scholar] [CrossRef]
- Noah, T.L.; Becker, S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am. J. Physiol. 1993, 265, L472–L478. [Google Scholar] [CrossRef]
- Hashimoto, S.; Matsumoto, K.; Gon, Y.; Maruoka, S.; Kujime, K.; Hayashi, S.; Takeshita, I.; Horie, T. p38 MAP kinase regulates TNF alpha-, IL-1 alpha- and PAF-induced RANTES and GM-CSF production by human bronchial epithelial cells. Clin. Exp. Allergy 2000, 30, 48–55. [Google Scholar] [CrossRef]
- Krull, M.; Bockstaller, P.; Wuppermann, F.N.; Klucken, A.C.; Muhling, J.; Schmeck, B.; Seybold, J.; Walter, C.; Maass, M.; Rosseau, S.; et al. Mechanisms of Chlamydophila pneumoniae-mediated GM-CSF release in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2006, 34, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Meja, K.K.; Seldon, P.M.; Nasuhara, Y.; Ito, K.; Barnes, P.J.; Lindsay, M.A.; Giembycz, M.A. p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NF-kappa B-independent mechanism. Br. J. Pharmacol. 2000, 131, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Esnault, S.; Malter, J.S. Extracellular signal-regulated kinase mediates granulocyte-macrophage colony-stimulating factor messenger RNA stabilization in tumor necrosis factor-alpha plus fibronectin-activated peripheral blood eosinophils. Blood 2002, 99, 4048–4052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, S.M.; Chi, D.S.; Hall, H.K.; Reynolds, S.A.; Aramide, O.; Lee, S.A.; Krishnaswamy, G. GM-CSF induction in human lung fibroblasts by IL-1beta, TNF-alpha, and macrophage contact. J. Interferon Cytokine Res. 2003, 23, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.M.; Chi, D.S.; Lee, S.A.; Hall, K.; Krishnaswamy, G. Inhibition of GM-CSF production in fibroblast-monocyte coculture by prednisone and effects of rhGM-CSF on human lung fibroblasts. Front. Biosci. 2004, 9, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Tannheimer, S.L.; Wright, C.D.; Salmon, M. Combination of roflumilast with a beta-2 adrenergic receptor agonist inhibits proinflammatory and profibrotic mediator release from human lung fibroblasts. Respir. Res. 2012, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Gorbacheva, A.M.; Korneev, K.V.; Kuprash, D.V.; Mitkin, N.A. The Risk G Allele of the Single-Nucleotide Polymorphism rs928413 Creates a CREB1-Binding Site That Activates IL33 Promoter in Lung Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 2911. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Totiger, T.; Shi, C.; Castellanos, J.; Lamichhane, P.; Dosch, A.R.; Messaggio, F.; Kashikar, N.; Honnenahally, K.; Ban, Y.; et al. Tobacco Carcinogen-Induced Production of GM-CSF Activates CREB to Promote Pancreatic Cancer. Cancer Res. 2018, 78, 6146–6158. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Peters, H.; Jungck, D.; Muller, K.; Strauch, J.; Koch, A. TNFalpha-induced GM-CSF release from human airway smooth muscle cells depends on activation of an ET-1 autoregulatory positive feedback mechanism. Thorax 2009, 64, 1044–1052. [Google Scholar] [CrossRef]
- McDonald, P.P. Transcriptional regulation in neutrophils: Teaching old cells new tricks. Adv. Immunol. 2004, 82, 1–48. [Google Scholar]
- Cloutier, A.; Ear, T.; Borissevitch, O.; Larivee, P.; McDonald, P.P. Inflammatory cytokine expression is independent of the c-Jun N-terminal kinase/AP-1 signaling cascade in human neutrophils. J. Immunol. 2003, 171, 3751–3761. [Google Scholar] [CrossRef]
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef]
- Funding, A.T.; Johansen, C.; Kragballe, K.; Otkjaer, K.; Jensen, U.B.; Madsen, M.W.; Fjording, M.S.; Finnemann, J.; Skak-Nielsen, T.; Paludan, S.R.; et al. Mitogen- and stress-activated protein kinase 1 is activated in lesional psoriatic epidermis and regulates the expression of pro-inflammatory cytokines. J. Investig. Dermatol. 2006, 126, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, J.; Nakayama, M.; Isomoto, H.; Kurazono, H.; Mukaida, N.; Mukhopadhyay, A.K.; Azuma, T.; Yamaoka, Y.; Sap, J.; Yamasaki, E.; et al. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-kappaB activation. J. Immunol. 2008, 180, 5017–5027. [Google Scholar] [CrossRef]
- Gray, J.G.; Chandra, G.; Clay, W.C.; Stinnett, S.W.; Haneline, S.A.; Lorenz, J.J.; Patel, I.R.; Wisely, G.B.; Furdon, P.J.; Taylor, J.D.; et al. A CRE/ATF-like site in the upstream regulatory sequence of the human interleukin 1 beta gene is necessary for induction in U937 and THP-1 monocytic cell lines. Mol. Cell. Biol. 1993, 13, 6678–6689. [Google Scholar] [CrossRef]
- Barabitskaja, O.; Foulke, J.S., Jr.; Pati, S.; Bodor, J.; Reitz, M.S., Jr. Suppression of MIP-1beta transcription in human T cells is regulated by inducible cAMP early repressor (ICER). J. Leukoc. Biol. 2006, 79, 378–387. [Google Scholar] [CrossRef]
- Proffitt, J.; Crabtree, G.; Grove, M.; Daubersies, P.; Bailleul, B.; Wright, E.; Plumb, M. An ATF/CREB-binding site is essential for cell-specific and inducible transcription of the murine MIP-1 beta cytokine gene. Gene 1995, 152, 173–179. [Google Scholar] [CrossRef]
- Khuu, C.H.; Barrozo, R.M.; Hai, T.; Weinstein, S.L. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol. Immunol. 2007, 44, 1598–1605. [Google Scholar] [CrossRef]
- Zhao, C.; Hui, W.; Fernandes, M.J.; Poubelle, P.E.; Bourgoin, S.G. Lysophosphatidic acid-induced IL-8 secretion involves MSK1 and MSK2 mediated activation of CREB1 in human fibroblast-like synoviocytes. Biochem. Pharmacol. 2014, 90, 62–72. [Google Scholar] [CrossRef]
- Choi, E.Y.; Park, Z.Y.; Choi, E.J.; Oh, H.M.; Lee, S.; Choi, S.C.; Lee, K.M.; Im, S.H.; Chun, J.S.; Jun, C.D. Transcriptional regulation of IL-8 by iron chelator in human epithelial cells is independent from NF-kappaB but involves ERK1/2- and p38 kinase-dependent activation of AP-1. J. Cell. Biochem. 2007, 102, 1442–1457. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y. Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal. 2007, 19, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Che, W.; Manetsch, M.; Quante, T.; Rahman, M.M.; Patel, B.S.; Ge, Q.; Ammit, A.J. Sphingosine 1-phosphate induces MKP-1 expression via p38 MAPK- and CREB-mediated pathways in airway smooth muscle cells. Biochim. Biophys. Acta 2012, 1823, 1658–1665. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.X.; Cai, W.; Andres, D.A. Rit-mediated stress resistance involves a p38-mitogen- and stress-activated protein kinase 1 (MSK1)-dependent cAMP response element-binding protein (CREB) activation cascade. J. Biol. Chem. 2012, 287, 39859–39868. [Google Scholar] [CrossRef]
- Dang, X.; Zhu, Q.; He, Y.; Wang, Y.; Lu, Y.; Li, X.; Qi, J.; Wu, H.; Sun, Y. IL-1beta Upregulates StAR and Progesterone Production Through the ERK1/2- and p38-Mediated CREB Signaling Pathways in Human Granulosa-Lutein Cells. Endocrinology 2017, 158, 3281–3291. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Stocco, D.M. The role of specific mitogen-activated protein kinase signaling cascades in the regulation of steroidogenesis. J. Signal Transduct. 2011, 2011, 821615. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Q.; Ma, Z.; Wang, M.; Shen, W.J.; Azhar, S.; Guo, Z.; Hu, Z. Feedback inhibition of CREB signaling by p38 MAPK contributes to the negative regulation of steroidogenesis. Reprod. Biol. Endocrinol. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delghandi, M.P.; Johannessen, M.; Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005, 17, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Tang, X.Q.; Liu, H.; Mao, X.F.; Wang, Y.X. Both classic Gs-cAMP/PKA/CREB and alternative Gs-cAMP/PKA/p38beta/CREB signal pathways mediate exenatide-stimulated expression of M2 microglial markers. J. Neuroimmunol. 2018, 316, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.; Verrecchia, F.; Mauviel, A. Cyclic adenosine 3’,5’-monophosphate-elevating agents inhibit transforming growth factor-beta-induced SMAD3/4-dependent transcription via a protein kinase A-dependent mechanism. Oncogene 2003, 22, 8881–8890. [Google Scholar] [CrossRef]
- Swaney, J.S.; Roth, D.M.; Olson, E.R.; Naugle, J.E.; Meszaros, J.G.; Insel, P.A. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. USA 2005, 102, 437–442. [Google Scholar] [CrossRef]
- Liu, X.; Sun, S.Q.; Hassid, A.; Ostrom, R.S. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol. Pharmacol. 2006, 70, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Patel, H.H.; Lai, N.C.; Aroonsakool, N.; Roth, D.M.; Insel, P.A. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. USA 2008, 105, 6386–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surinkaew, S.; Aflaki, M.; Takawale, A.; Chen, Y.; Qi, X.Y.; Gillis, M.A.; Shi, Y.F.; Tardif, J.C.; Chattipakorn, N.; Nattel, S. Exchange protein activated by cyclic-adenosine monophosphate (Epac) regulates atrial fibroblast function and controls cardiac remodelling. Cardiovasc. Res. 2019, 115, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ryu, J.; Lee, Y.; Lee, Z.H.; Kim, H.H. Adenylate cyclase and calmodulin-dependent kinase have opposite effects on osteoclastogenesis by regulating the PKA-NFATc1 pathway. J. Bone Miner. Res. 2011, 26, 1217–1229. [Google Scholar] [CrossRef] [Green Version]
- Weivoda, M.M.; Ruan, M.; Hachfeld, C.M.; Pederson, L.; Howe, A.; Davey, R.A.; Zajac, J.D.; Kobayashi, Y.; Williams, B.O.; Westendorf, J.J.; et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. J. Bone Miner. Res. 2016, 31, 65–75. [Google Scholar] [CrossRef]
- Mediero, A.; Perez-Aso, M.; Cronstein, B.N. Activation of adenosine A(2A) receptor reduces osteoclast formation via PKA- and ERK1/2-mediated suppression of NFkappaB nuclear translocation. Br. J. Pharmacol. 2013, 169, 1372–1388. [Google Scholar] [CrossRef]
- Tintut, Y.; Parhami, F.; Tsingotjidou, A.; Tetradis, S.; Territo, M.; Demer, L.L. 8-Isoprostaglandin E2 enhances receptor-activated NFkappa B ligand (RANKL)-dependent osteoclastic potential of marrow hematopoietic precursors via the cAMP pathway. J. Biol. Chem. 2002, 277, 14221–14226. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mizoguchi, T.; Take, I.; Kurihara, S.; Udagawa, N.; Takahashi, N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J. Biol. Chem. 2005, 280, 11395–11403. [Google Scholar] [CrossRef]
- Gray, T.; Nettesheim, P.; Loftin, C.; Koo, J.S.; Bonner, J.; Peddada, S.; Langenbach, R. Interleukin-1beta-induced mucin production in human airway epithelium is mediated by cyclooxygenase-2, prostaglandin E2 receptors, and cyclic AMP-protein kinase A signaling. Mol. Pharmacol. 2004, 66, 337–346. [Google Scholar] [CrossRef]
- Zhu, T.; Wu, X.L.; Zhang, W.; Xiao, M. Glucagon Like Peptide-1 (GLP-1) Modulates OVA-Induced Airway Inflammation and Mucus Secretion Involving a Protein Kinase A (PKA)-Dependent Nuclear Factor-kappaB (NF-kappaB) Signaling Pathway in Mice. Int. J. Mol. Sci. 2015, 16, 20195–20211. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Ghatak, S.; Akaike, T.; Segi-Nishida, E.; Iwasaki, S.; Iwamoto, M.; Misra, S.; Tamura, K.; et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J. Clin. Investig. 2006, 116, 3026–3034. [Google Scholar] [CrossRef] [Green Version]
- Indolfi, C.; Avvedimento, E.V.; Di Lorenzo, E.; Esposito, G.; Rapacciuolo, A.; Giuliano, P.; Grieco, D.; Cavuto, L.; Stingone, A.M.; Ciullo, I.; et al. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat. Med. 1997, 3, 775–779. [Google Scholar] [CrossRef]
- Bos, J.L. Epac: A new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003, 4, 733–738. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Jin, M.; Otsu, K.; Ulucan, C.; Wang, X.; Baljinnyam, E.; Takaoka, M.; et al. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1547–H1555. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Yokoyama, U.; Yanai, C.; Ishige, R.; Kurotaki, D.; Umemura, M.; Fujita, T.; Kubota, T.; Okumura, S.; Sata, M.; et al. Epac1 Deficiency Attenuated Vascular Smooth Muscle Cell Migration and Neointimal Formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2617–2625. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Kornhauser, J.M.; Xia, Z.; Thiele, E.A.; Greenberg, M.E. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol. Cell. Biol. 1998, 18, 1946–1955. [Google Scholar] [CrossRef]
- Xiao, X.; Li, B.X.; Mitton, B.; Ikeda, A.; Sakamoto, K.M. Targeting CREB for cancer therapy: Friend or foe. Curr. Cancer Drug Targets 2010, 10, 384–391. [Google Scholar] [CrossRef]
- Li, B.X.; Gardner, R.; Xue, C.; Qian, D.Z.; Xie, F.; Thomas, G.; Kazmierczak, S.C.; Habecker, B.A.; Xiao, X. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci. Rep. 2016, 6, 34513. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cAMP Pathway | ERK/p38MAPK-CREB Pathway | cAMP-ERK/p38MAPK Pathway | |
|---|---|---|---|
| Neuronal system | ↑ | ↑ | ↑ |
| Cardiac fibrosis | ↓ | ↑ | N.R. |
| Osteoclast differentiation | ↓ | ↑ | ↓ or ↑ |
| Mucin production | ↑ | ↑ | N.R. |
| VSMC migration | ↓(cAMP ) ↑(Epac) | ↑ | N.R. |
| GM-CSF production | ↓ | ↑ | N.R. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koga, Y.; Tsurumaki, H.; Aoki-Saito, H.; Sato, M.; Yatomi, M.; Takehara, K.; Hisada, T. Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int. J. Mol. Sci. 2019, 20, 1346. https://doi.org/10.3390/ijms20061346
Koga Y, Tsurumaki H, Aoki-Saito H, Sato M, Yatomi M, Takehara K, Hisada T. Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. International Journal of Molecular Sciences. 2019; 20(6):1346. https://doi.org/10.3390/ijms20061346
Chicago/Turabian StyleKoga, Yasuhiko, Hiroaki Tsurumaki, Haruka Aoki-Saito, Makiko Sato, Masakiyo Yatomi, Kazutaka Takehara, and Takeshi Hisada. 2019. "Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production" International Journal of Molecular Sciences 20, no. 6: 1346. https://doi.org/10.3390/ijms20061346