Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells

 , and
, and 
Abstract
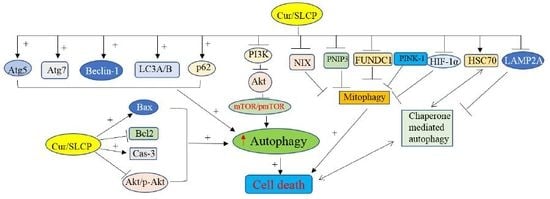
:
1. Introduction
2. Results
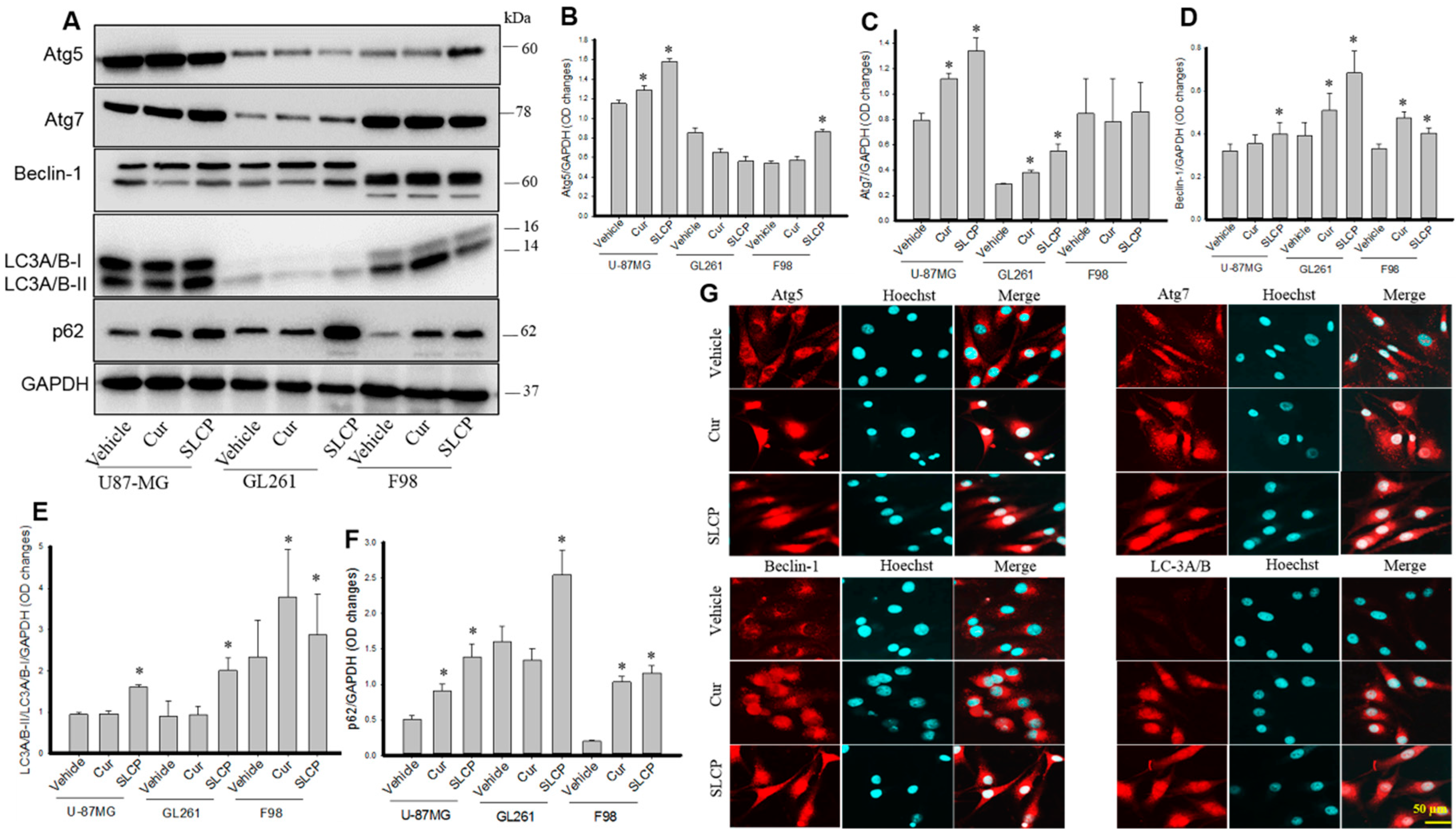
2.1. SLCP Induced Autophagy Greater than Natural Cur in Different GBM Cells
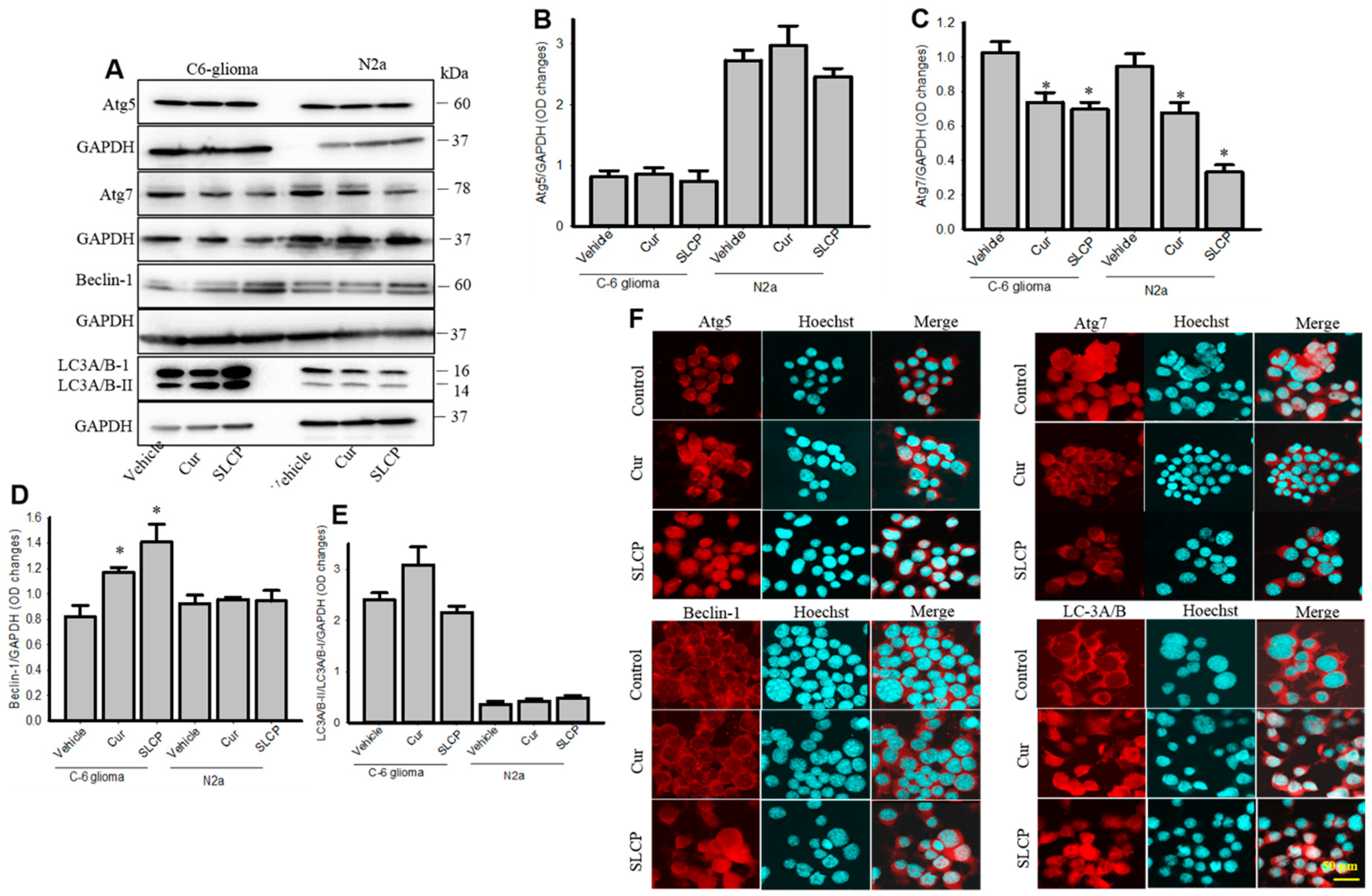
2.2. Cur and or SLCP Treatment Has Little Influence on Autophagy Pathways in Rat Glial Tumor Cells (C6-Glioma) and Mouse Neuroblatsoma (N2a) Cells
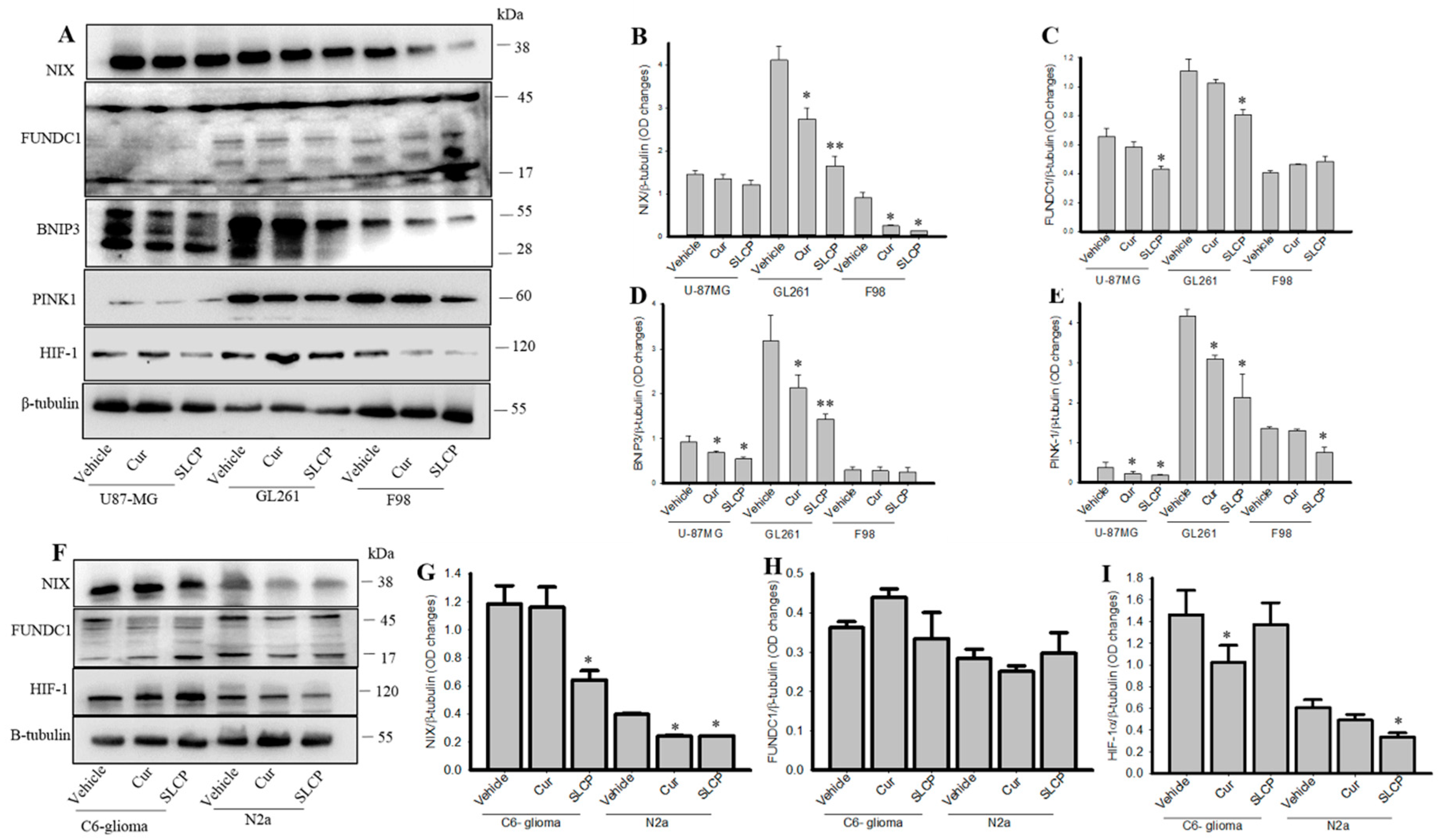
2.3. SLCP Inhibits Mitophagy Markers More than Cur in GBM Cells.
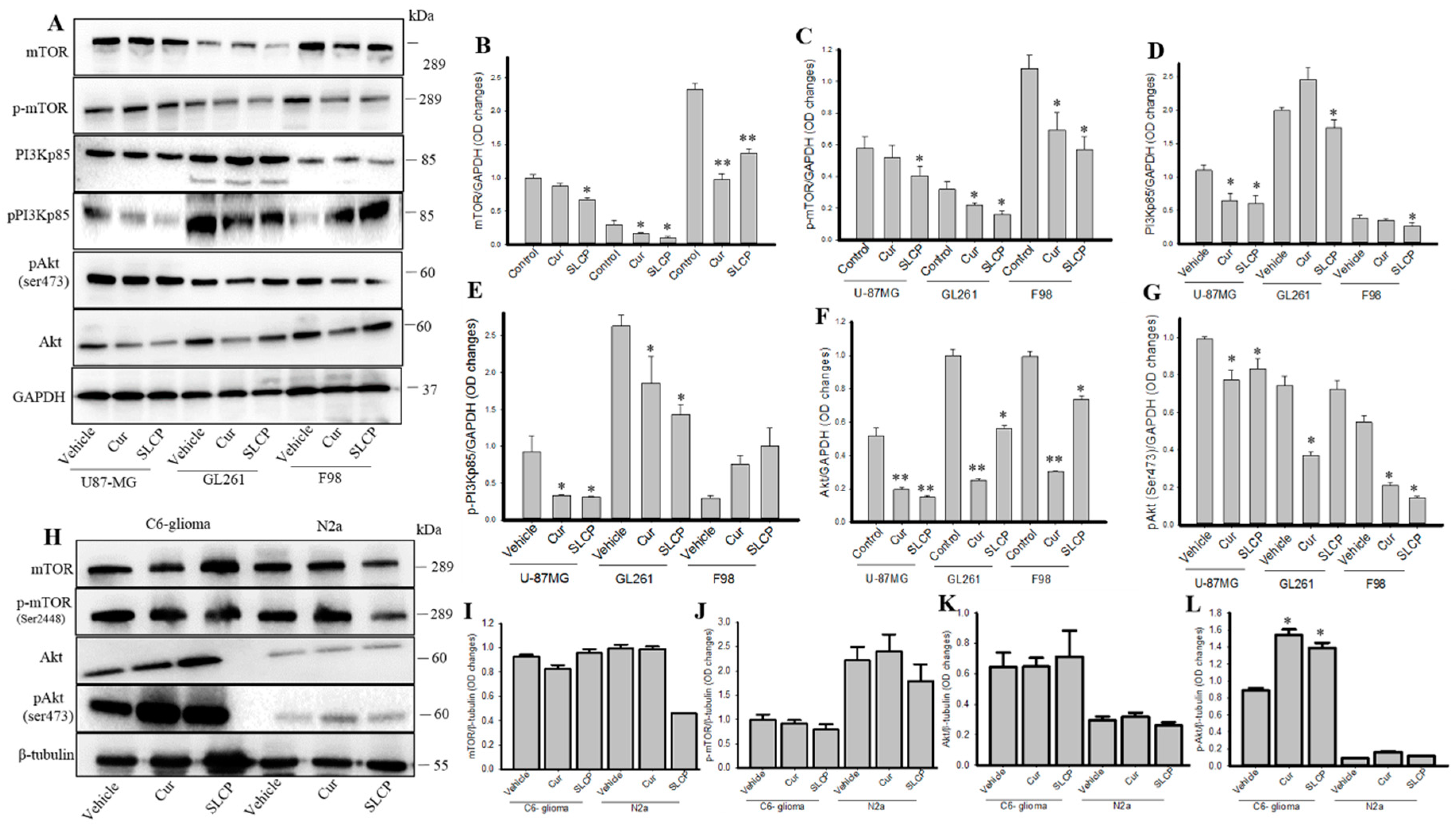
2.4. SLCP Inhibits PI3K-Akt/mTOR Pathway Activity in GBM Cells More than Cur does in GBM
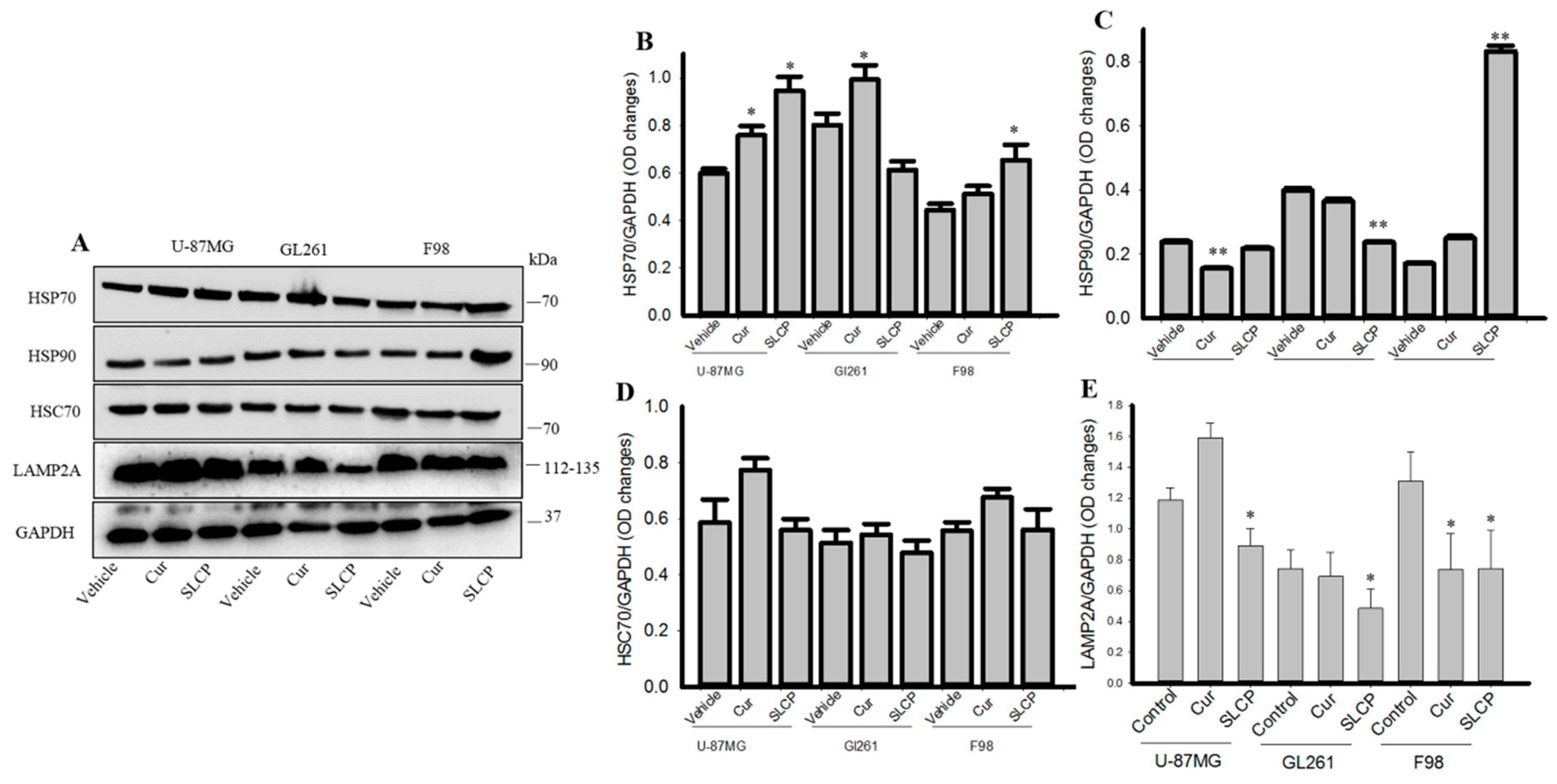
2.5. Cur and or SLCP Treatment Inhibited Chaperone-Mediated Autophagy (CMA) Markers in GBM Cells.
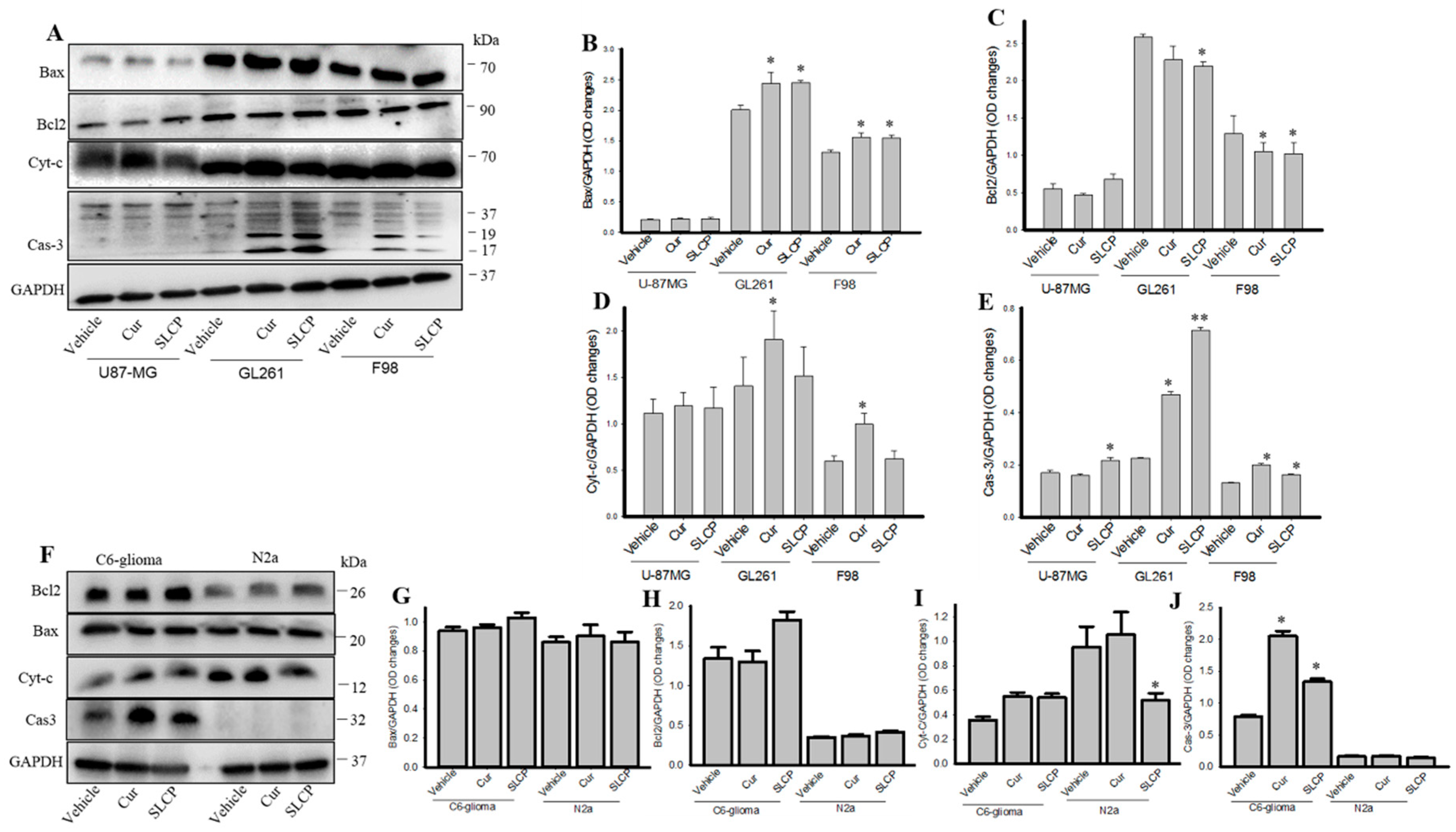
2.6. Cell Survival and Cell Death Markers Were also Modulated by SLCP More than Cur in GBM Cells.
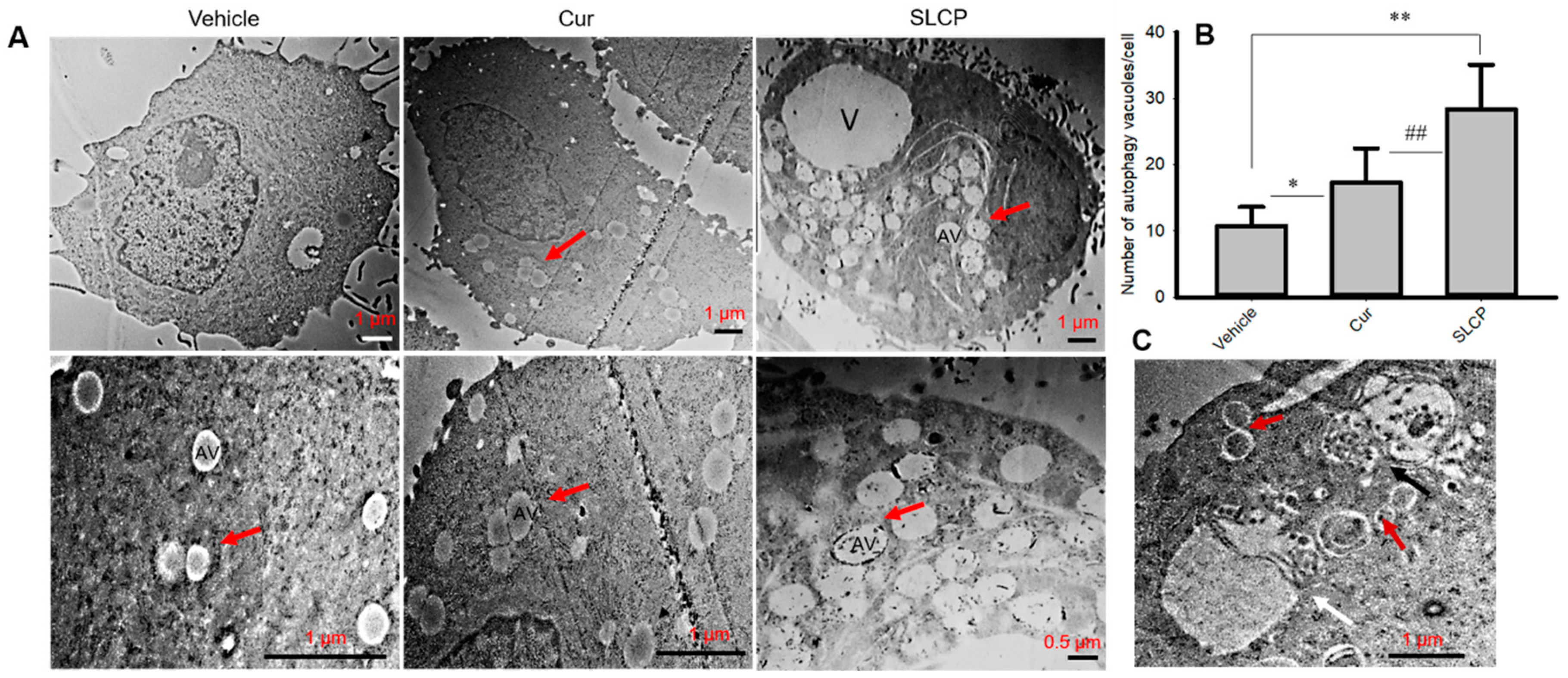
2.7. SLCP Treatment Produced Greater Increases in the Number of Autophagy Vacuoles in U-87MG Cells than Did Cur Treatments
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Curcumin and or SLCP Treatment
4.4. Immunocytochemistry and Confocal Imaging of Autophagy Markers.
4.5. Transmission Electron Microscopy (TEM)
4.6. Western Blot
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma |
| SLCP | Solid lipid curcumin particles |
| Cur | Curcumin |
| Atg | Autophagy-related protein |
| Akt | Protein kinase B |
| PI3K | Phosphatidyl inositol-3 kinase |
| mTOR | Mechanistic target of rapamycin |
| HSP | Heat shock protein |
| TMZ | Temozolomide |
| PCD | Program cell death |
| ATP | Adenosine triphosphate |
| SLPs | Solid lipid particles |
| AD | Alzheimer’s disease |
| PI | Propidium iodide |
| EMEM | Eagle’s Minimum Essential Medium, |
| FBS | Fetal bovine serum |
| HBSS | Hank’s balanced salt solution |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FITC | Fluorescent isothiocyanate |
| DPBS | Dulbecco’s phosphate buffer saline |
| ROS | Reactive oxygen species |
| BNIP3 | Bcl-2/adenovirus E1B 19 kDa-interacting protein |
| Bax | Bcl2-associated X protein |
| Bcl2 | B-cell lymphoma 2 |
| NF-kB | Nuclear factor kappa beta |
| ATCC | American type cell culture |
| SEM | Standard error of mean |
| PBS | Phosphate buffer saline |
| EDTA | Ethylene-di-amino-tetra-acetic-acid |
| RPM | Revolution per minute |
| mM | Millimolar |
| RIPA | Radio immunoprecipitation assay |
| SDS | Sodium dodecyl sulfate |
| BCA | Bicinchoninic acid assay |
| PVDF | Polyvinylidene fluoride |
| ANOVA | One-way analysis of variance |
| HSD | Honestly significant difference |
| µM | Micromolar |
| AU | Arbitrary unit |
| OD | Optical density |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBS | Tris buffer saline |
| CMA | Chaperone-mediated autophagy |
| HSC70 | Heat shock cognate 70 |
| LAMP-2A | Lysosome-associated membrane protein type 2A |
| G2/M | Gap2/mitosis |
| VEGF | Vascular endothelial growth factor |
References
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. 5), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.A.; Sordillo, P.P.; Helson, L. Curcumin for the Treatment of Glioblastoma. Anticancer Res. 2015, 35, 6373–6378. [Google Scholar] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, C.; Matteoni, S.; Signore, M.; Cardone, L.; Nath, K.; Glickson, J.D.; Paggi, M.G. Drug repurposing for the treatment of glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2017, 36, 169. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Song, X.; Lee, D.H.; Dilly, A.K.; Lee, Y.S.; Choudry, H.A.; Kwon, Y.T.; Bartlett, D.L.; Lee, Y.J. Crosstalk Between Apoptosis and Autophagy Is Regulated by the Arginylated BiP/Beclin-1/p62 Complex. Mol. Cancer Res. 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Aggarwal, B.B. Turmeric, the Golden Spice: From Traditional Medicine to Modern Medicine. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Zhao, J.; Zhu, J.; Lv, X.; Xing, J.; Liu, S.; Chen, C.; Xu, Y. Curcumin potentiates the potent antitumor activity of ACNU against glioblastoma by suppressing the PI3K/AKT and NF-kappaB/COX-2 signaling pathways. OncoTargets Ther. 2017, 10, 5471–5482. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Paladugu, L.; Dunbar, G.L. Solid lipid curcumin particles provide greater anti-amyloid, anti-inflammatory and neuroprotective effects than curcumin in the 5xFAD mouse model of Alzheimer’s disease. BMC Neurosci. 2018, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ahuja, A.; Ali, J.; Baboota, S. Conundrum and therapeutic potential of curcumin in drug delivery. Crit. Rev. Ther. Drug Carrier Syst. 2010, 27, 279–312. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Hall, T.C.; Paladugu, L.; Kolli, N.; Learman, C.; Rossignol, J.; Dunbar, G.L. A comparative study of dietary curcumin, nanocurcumin, and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5x-familial Alzheimer’s disease mice. Histochem. Cell Biol. 2016, 146, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Al-Gharaibeh, A.; Kolli, N.; Dunbar, G.L. Solid Lipid Curcumin Particles Induce More DNA Fragmentation and Cell Death in Cultured Human Glioblastoma Cells than Does Natural Curcumin. Oxid. Med. Cell. Longev. 2017, 2017, 9656719. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.P.; Maiti, P.; et al. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef]
- Frautschy, S.A.; Cole, G.M. Why pleiotropic interventions are needed for Alzheimer’s disease. Mol. Neurobiol. 2010, 41, 392–409. [Google Scholar] [CrossRef]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Dunbar, G.L. Comparative Neuroprotective Effects of Dietary Curcumin and Solid Lipid Curcumin Particles in Cultured Mouse Neuroblastoma Cells after Exposure to Abeta42. Int. J. Alzheimers Dis. 2017, 2017, 4164872. [Google Scholar] [PubMed]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed. Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, 93621. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Salumbides, B.C.; Black, K.L.; Koronyo-Hamaoui, M. Alzheimer’s disease in the retina: Imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 2012, 10, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Molczyk, A. Side effects of temozolomide treatment in patient with glioblastoma multiforme—Case study. Prz. Lek. 2010, 67, 445–446. [Google Scholar]
- Dinnes, J.; Cave, C.; Huang, S.; Milne, R. A rapid and systematic review of the effectiveness of temozolomide for the treatment of recurrent malignant glioma. Br. J. Cancer 2002, 86, 501–505. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Shah, A.H.; Gersey, Z.C.; Shah, S.S.; Bregy, A.; Komotar, R.J.; Graham, R.M. Investigating the therapeutic role and molecular biology of curcumin as a treatment for glioblastoma. Ther. Adv. Med. Oncol. 2016, 8, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘menage-a-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutouja, F.; Brinkmeier, R.; Mastalski, T.; El Magraoui, F.; Platta, H.W. Regulation of the Tumor-Suppressor BECLIN 1 by Distinct Ubiquitination Cascades. Int. J. Mol. Sci. 2017, 18, 2541. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome Proteins LC3A, LC3B and LC3C Have Distinct Subcellular Distribution Kinetics and Expression in Cancer Cell Lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Long, M.; Li, X.; Zhu, S.; Zhang, M.; Yang, Z. Curcumin activates autophagy and attenuates oxidative damage in EA. hy926 cells via the Akt/mTOR pathway. Mol. Med. Rep. 2016, 13, 2187–2193. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Braganhol, E.; Klafke, K.; Figueiró, F.; Terra, SR.; Paludo, F.J.; Morrone, M.; Bristot, I.J.; Battastini, A.M.; Forcelini, C.M.; et al. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett. 2015, 358, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duzgun, Z.; Eroglu, Z.; Biray Avci, C. Role of mTOR in glioblastoma. Gene 2016, 575 Pt 1, 187–190. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neurooncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Landen, C.N. The importance of the PI3K/AKT/MTOR pathway in the progression of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 8213–8227. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Shen, G.; Khor, T.O.; Kim, J.H.; Kong, A.N. Curcumin inhibits Akt/mammalian target of rapamycin signaling through protein phosphatase-dependent mechanism. Mol. Cancer Ther. 2008, 7, 2609–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapuy, O.; Vinod, P.K.; Banhegyi, G. mTOR inhibition increases cell viability via autophagy induction during endoplasmic reticulum stress—An experimental and modeling study. FEBS Open Bio 2014, 4, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Mendez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1alpha in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Liu, Y.; Zhang, L.; Cai, F.; Zhu, L.; Xu, J. C0818, a novel curcumin derivative, interacts with Hsp90 and inhibits Hsp90 ATPase activity. Acta Pharm. Sin. B 2017, 7, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Klionsky, D.J. Autophagy in the eukaryotic cell. Eukaryot. Cell 2002, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Thorburn, A. Autophagy and cell death. Essays Biochem. 2013, 55, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Schrand, A.M.; Schlager, J.J.; Dai, L.; Hussain, S.M. Preparation of cells for assessing ultrastructural localization of nanoparticles with transmission electron microscopy. Nat. Protoc. 2010, 5, 744–757. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Source | Type | Company | Catalog No. | Address |
| Atg5 | Rabbit | Monoclonal | Cell signaling Technology | 12994S | Danvers, MA, USA |
| Atg7 | Rabbit | Monoclonal | Cell signaling Technology | 8558S | Danvers, MA, USA |
| Beclin-1 | Rabbit | Polyclonal | Cell signaling Technology | 3738S | Danvers, MA, USA |
| LC3A/B | Rabbit | Polyclonal | Cell signaling Technology | 4108S | Danvers, MA, USA |
| p62 | Rabbit | Polyclonal | Cell signaling Technology | 5114S | Danvers, MA, USA |
| mTOR | Rabbit | Polyclonal | Cell signaling Technology | 2972S | Danvers, MA, USA |
| p-mTOR | Rabbit | Monoclonal | Cell signaling Technology | 2971S | Danvers, MA, USA |
| PI3Kp85 | Rabbit | Monoclonal | Cell signaling Technology | 4292S | Danvers, MA, USA |
| BNIP3L/NIX | Rabbit | Monoclonal | Cell signaling Technology | 12396S | Danvers, MA, USA |
| FUNDC1 | Rabbit | Monoclonal | EMD Millipore | ABC506 | Burlington, MA, USA |
| HIF-1α | Rabbit | Monoclonal | Cell signaling Technology | 14179S | Danvers, MA, USA |
| PINK-1 | Rabbit | Monoclonal | Cell signaling Technology | 6946S | Danvers, MA, USA |
| Cyt-c | Rabbit | Monoclonal | Cell Signaling Technology | 4272S | Danvers, MA, USA |
| Caspase-3 | Rabbit | Monoclonal | Cell Signaling Technology | 9661S | Danvers, MA, USA |
| Bax | Rabbit | Polyclonal | Cell signaling Technology | 2772S | Danvers, MA, USA |
| Bcl-2 | Mouse | Monoclonal | Santa Cruz Biotech | Sc-7382 | Santa Cruz, CA, USA |
| Akt | Rabbit | Monoclonal | Cell signaling Technology | 9272S | Danvers, MA, USA |
| pAkt (Ser473) | Rabbit | Monoclonal | Cell signaling Technology | 4060S | Danvers, MA, USA |
| HSP70 | Rabbit | Polyclonal | Cell signaling Technology | 4872S | Danvers, MA, USA |
| HSP90 | Rabbit | Polyclonal | Cell signaling Technology | 4877S | Danvers, MA, USA |
| HSC70 | Rabbit | Polyclonal | Cell signaling Technology | 8444S | Danvers, MA, USA |
| LAMP2 | Mouse | Polyclonal | Santa Cruz Biotech | sc-20004 | Santa Cruz, CA, USA |
| GAPDH | Rabbit | Monoclonal | Cell signaling Technology | 2118S | Danvers, MA, USA |
| β-tubulin | Rabbit | Monoclonal | Cell signaling Technology | 2146S | Danvers, MA, USA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, P.; Scott, J.; Sengupta, D.; Al-Gharaibeh, A.; Dunbar, G.L. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 399. https://doi.org/10.3390/ijms20020399
Maiti P, Scott J, Sengupta D, Al-Gharaibeh A, Dunbar GL. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. International Journal of Molecular Sciences. 2019; 20(2):399. https://doi.org/10.3390/ijms20020399
Chicago/Turabian StyleMaiti, Panchanan, Jason Scott, Dipanwita Sengupta, Abeer Al-Gharaibeh, and Gary L. Dunbar. 2019. "Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells" International Journal of Molecular Sciences 20, no. 2: 399. https://doi.org/10.3390/ijms20020399