Discovery of Potent Inhibitors for the Large Neutral Amino Acid Transporter 1 (LAT1) by Structure-Based Methods


 ,
,  , , ,
, , , 
Abstract
:
1. Introduction
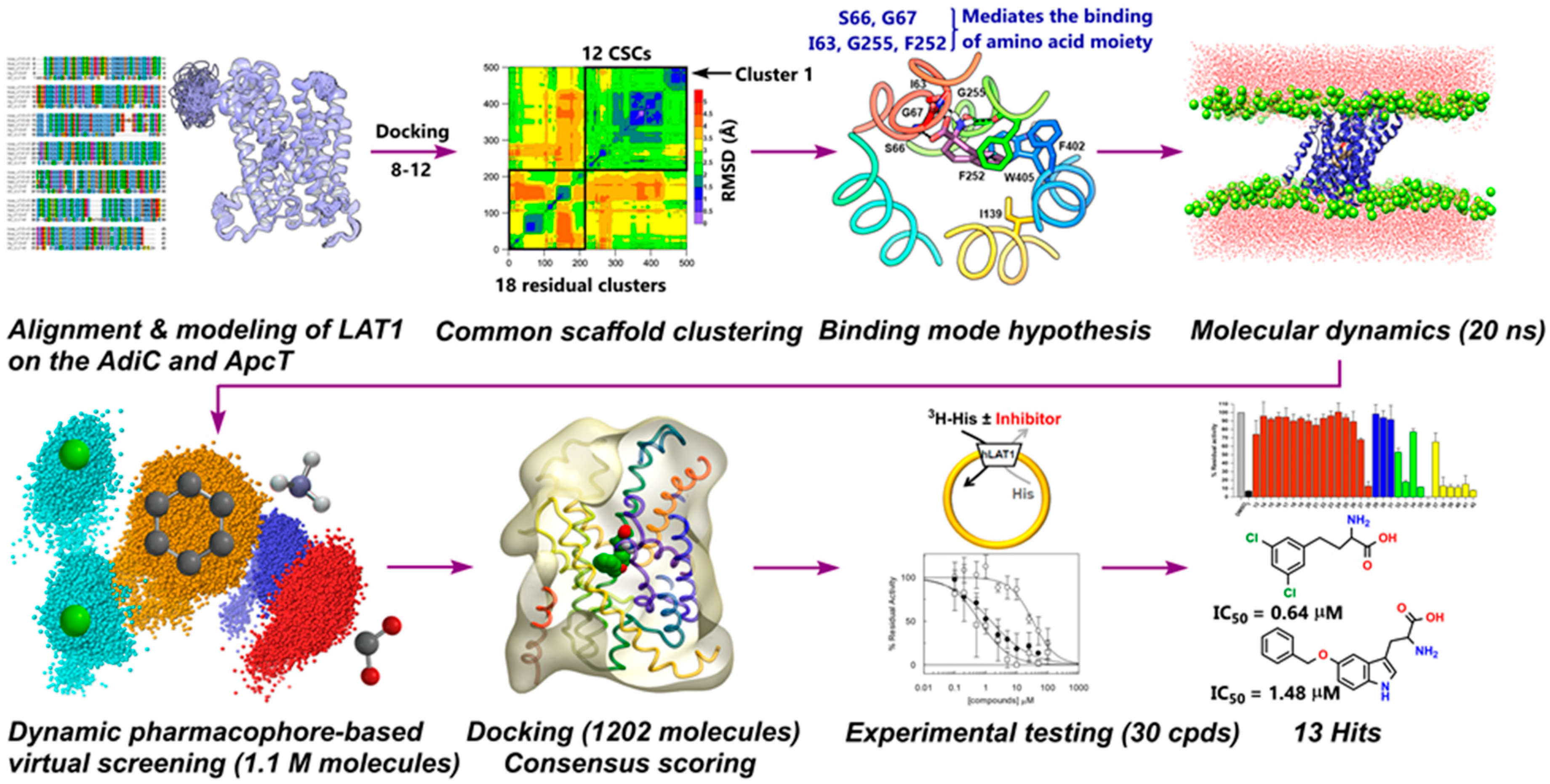
2. Results
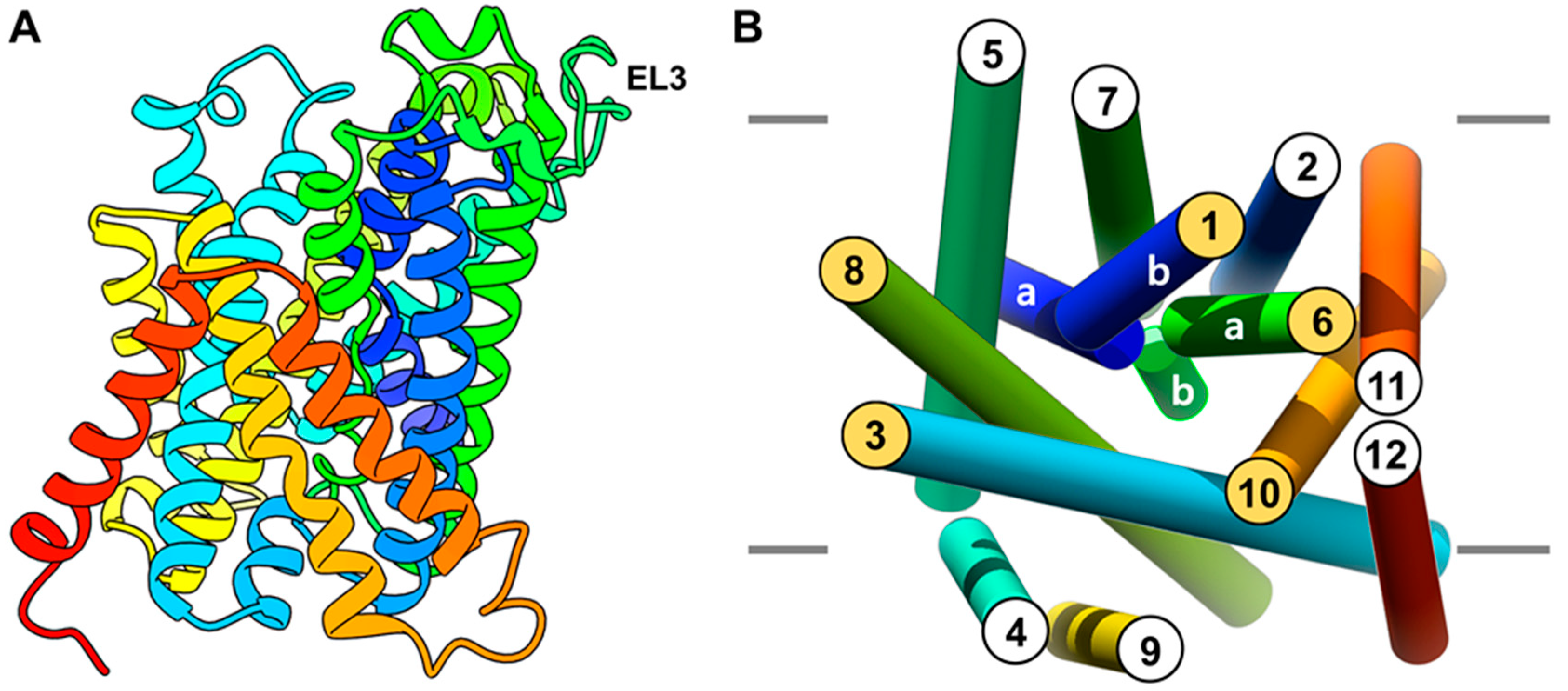
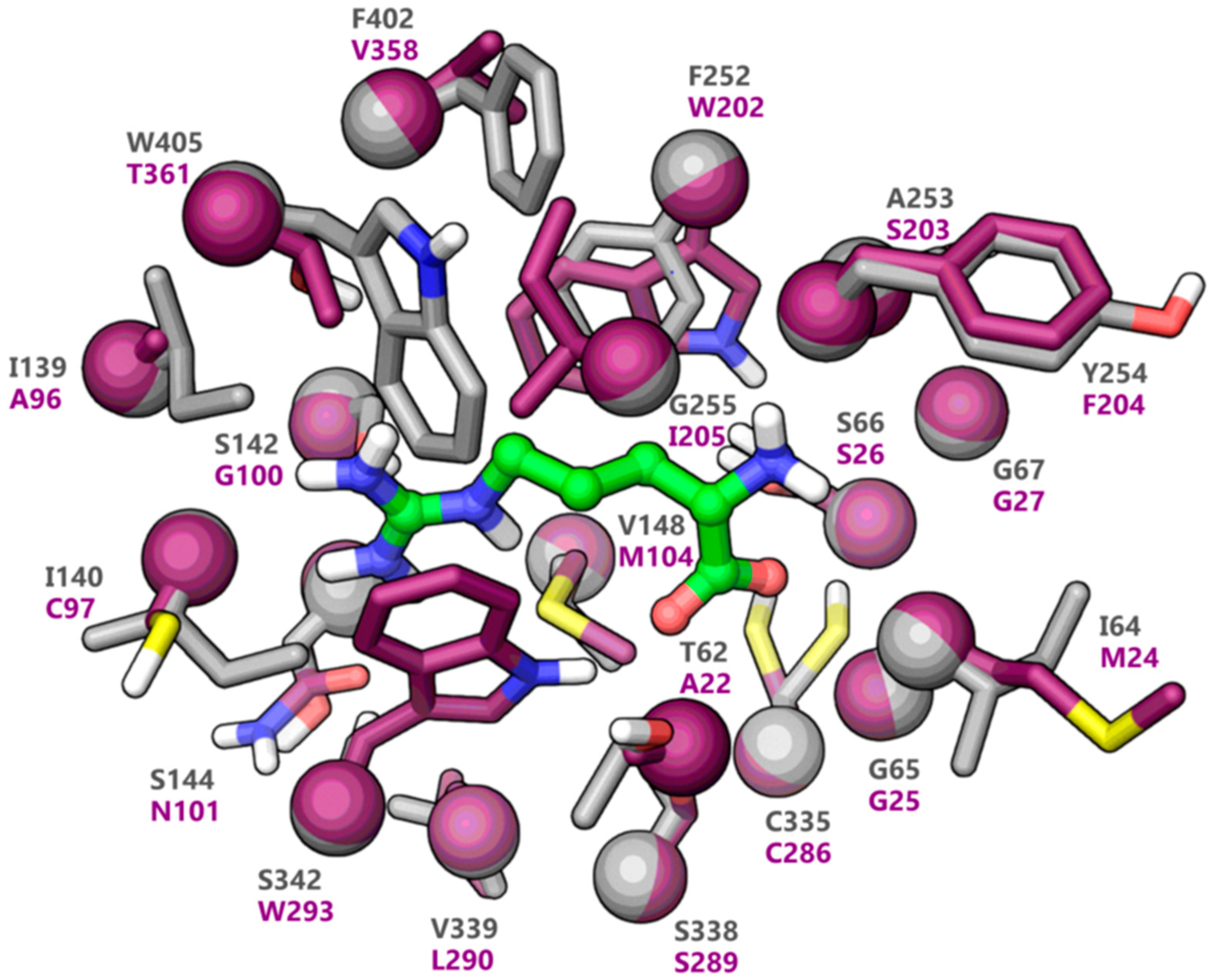
2.1. Homology Modeling of LAT1
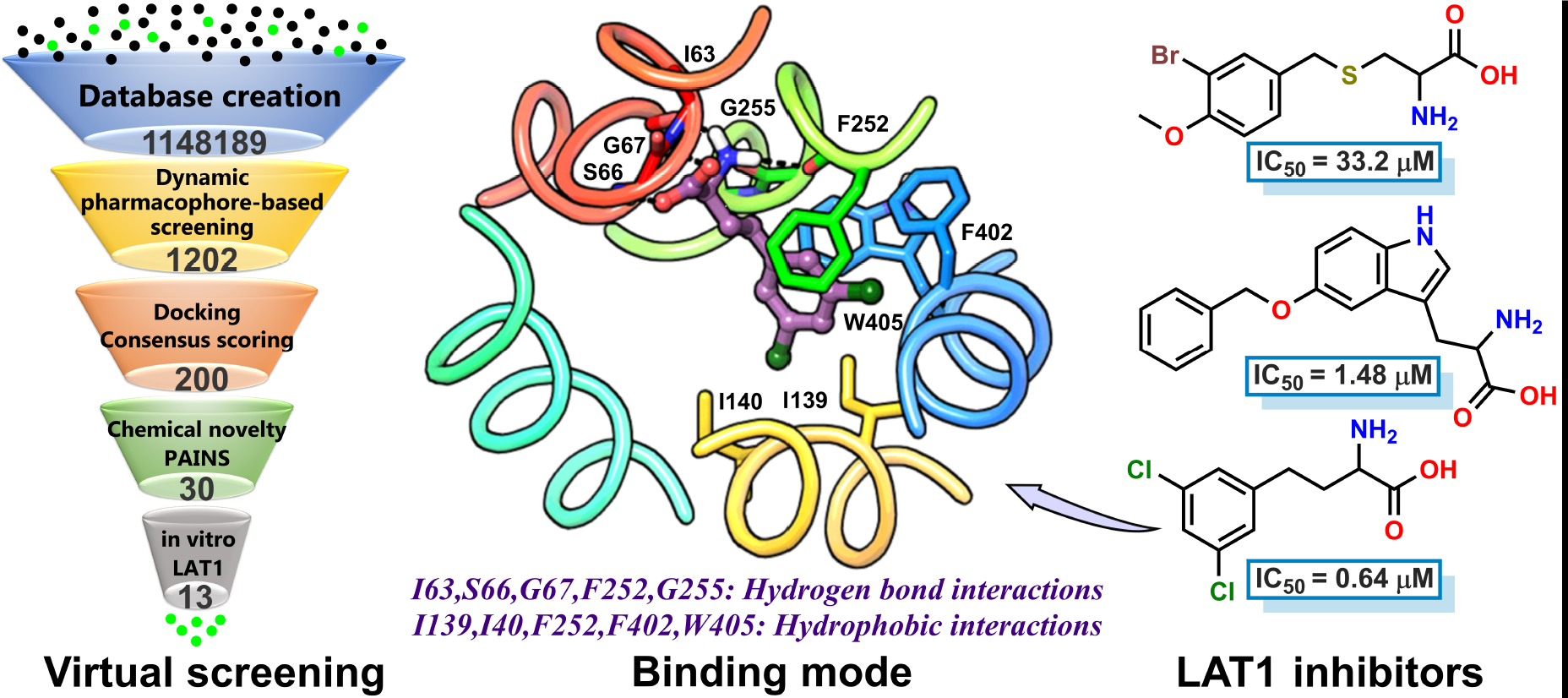
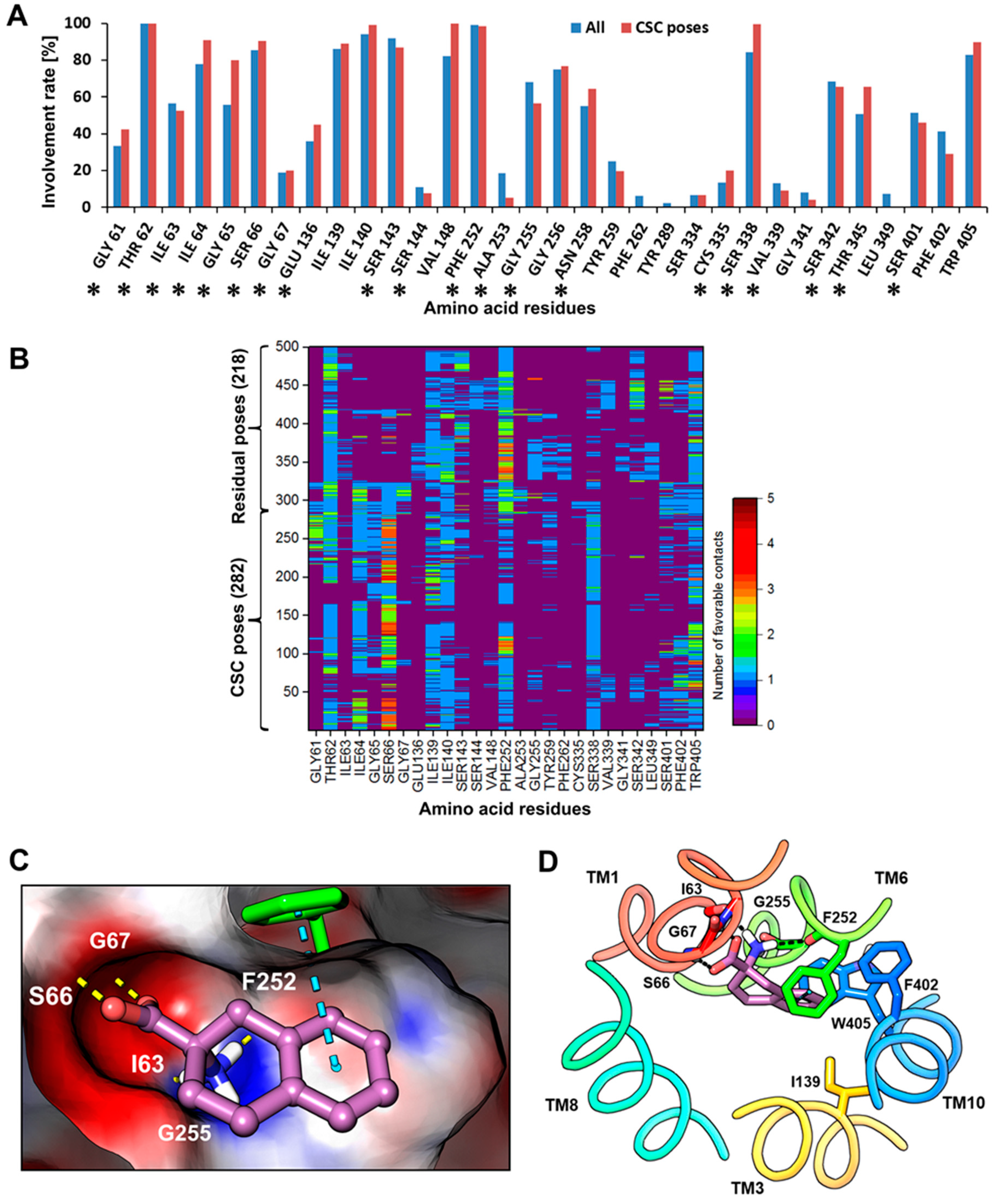
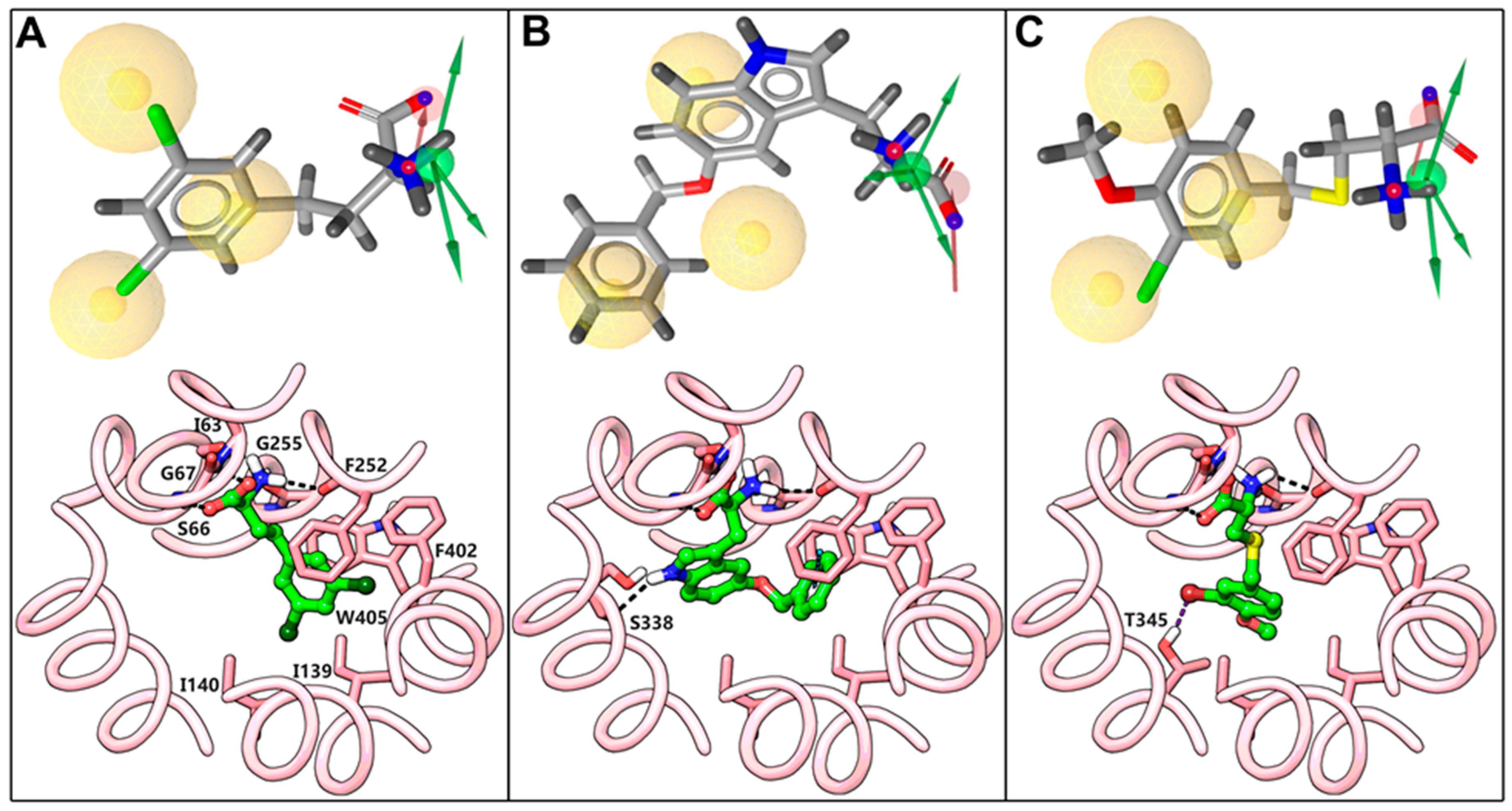
2.2. Docking and Binding Mode Hypothesis
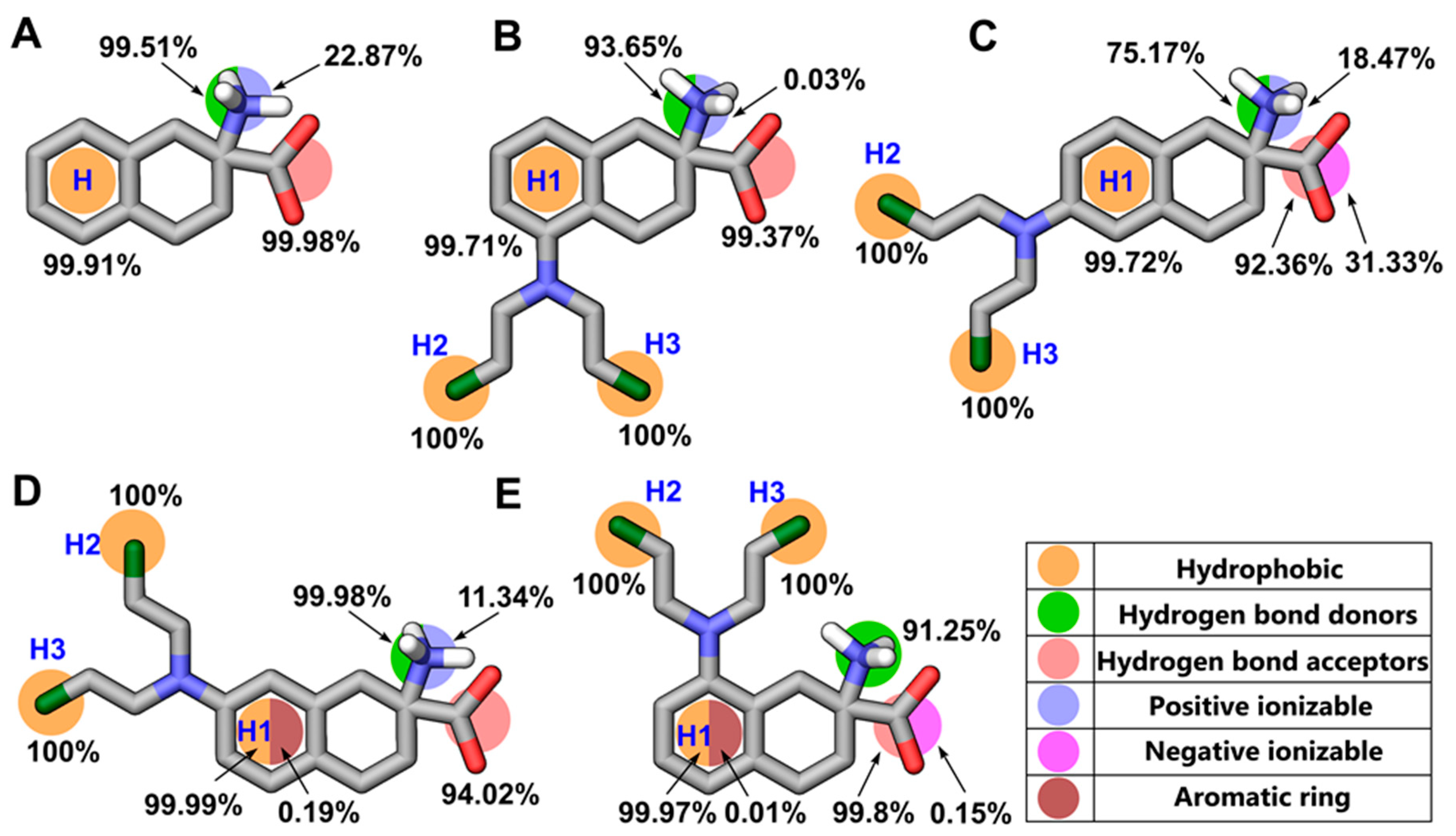
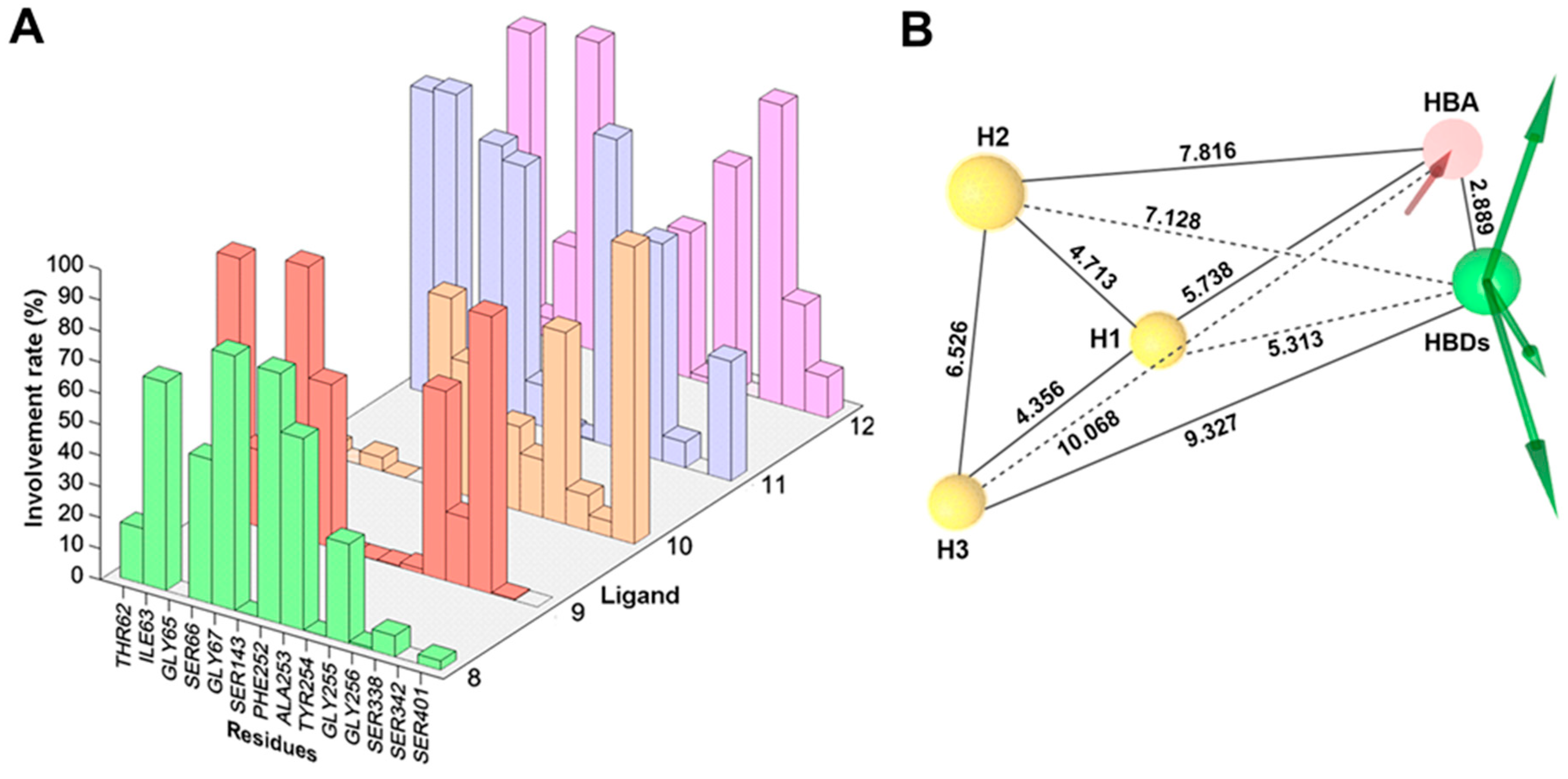
2.3. Dynamic Pharmacophore Modeling
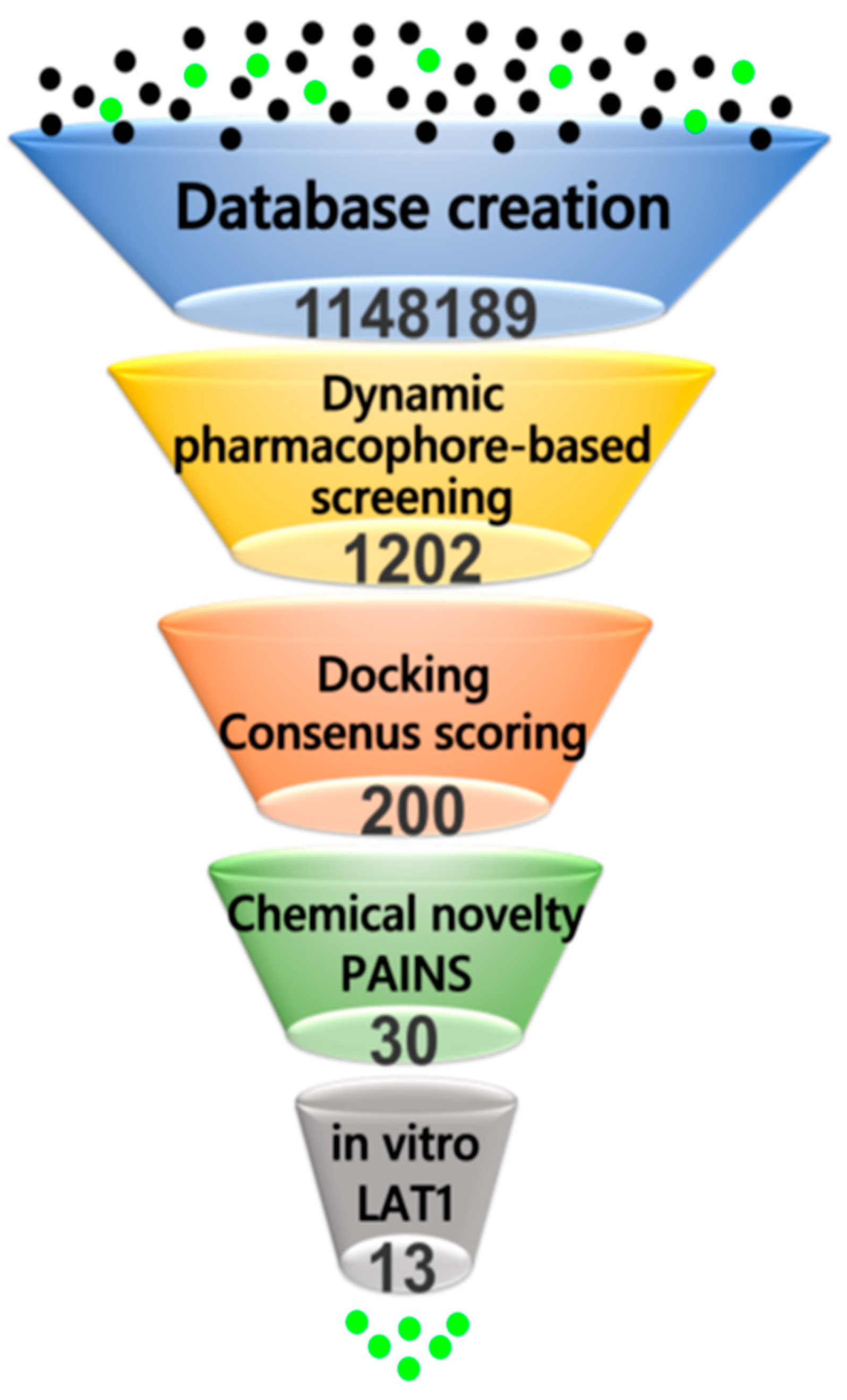
2.4. Virtual Screening
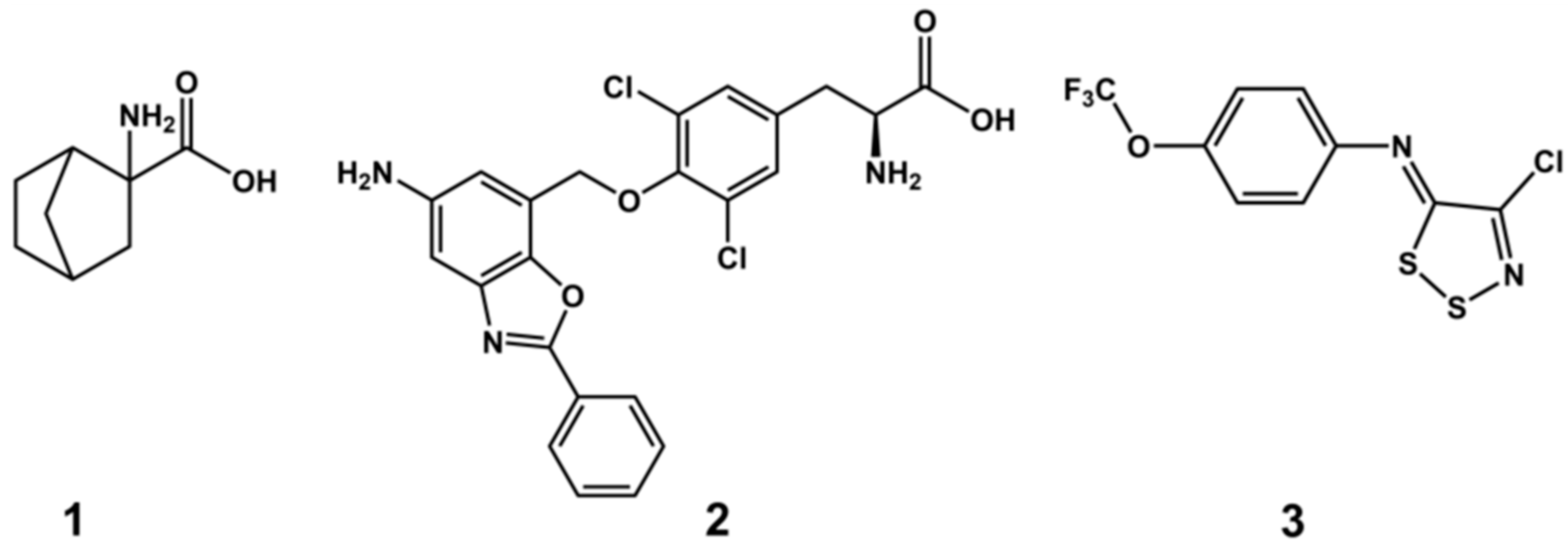
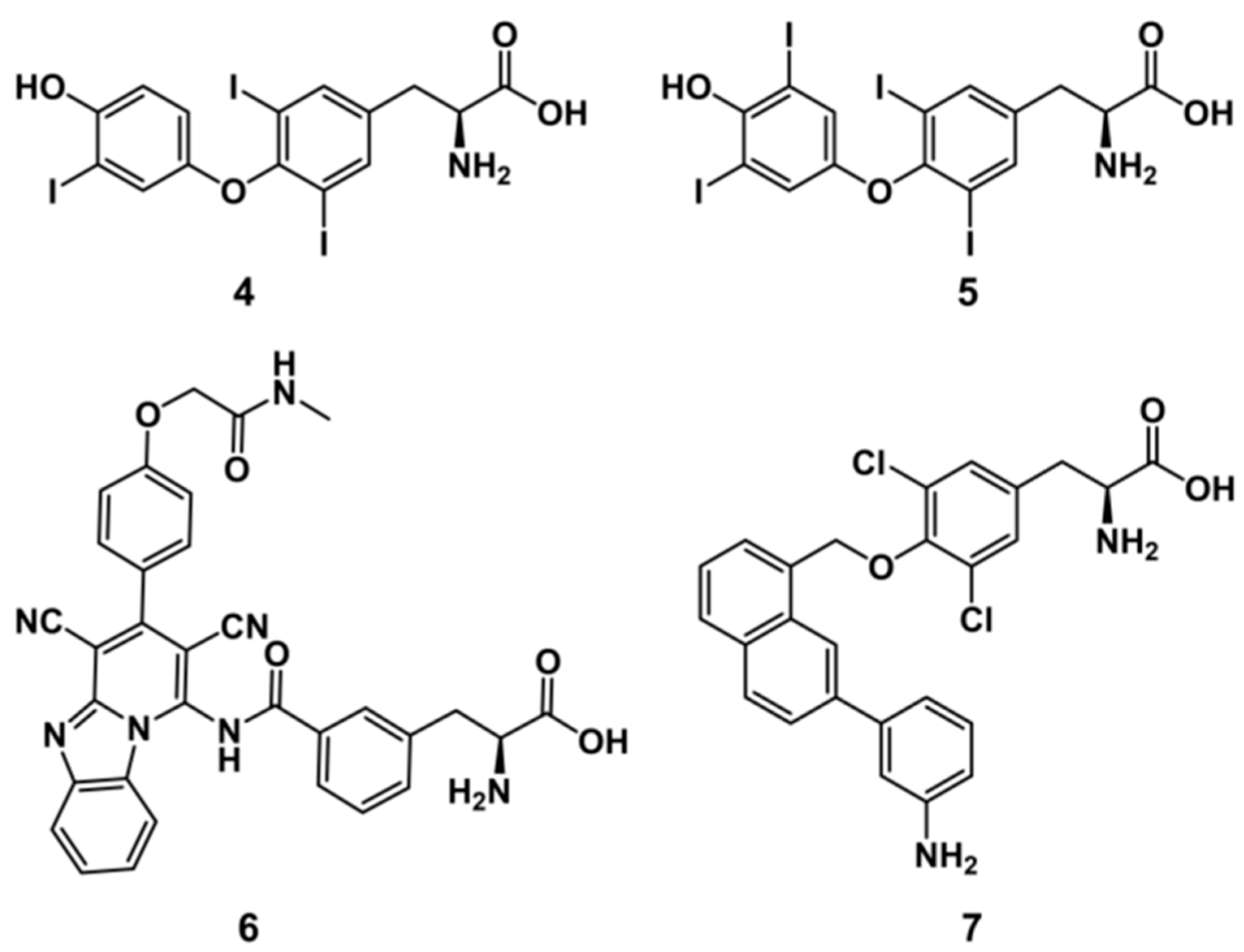
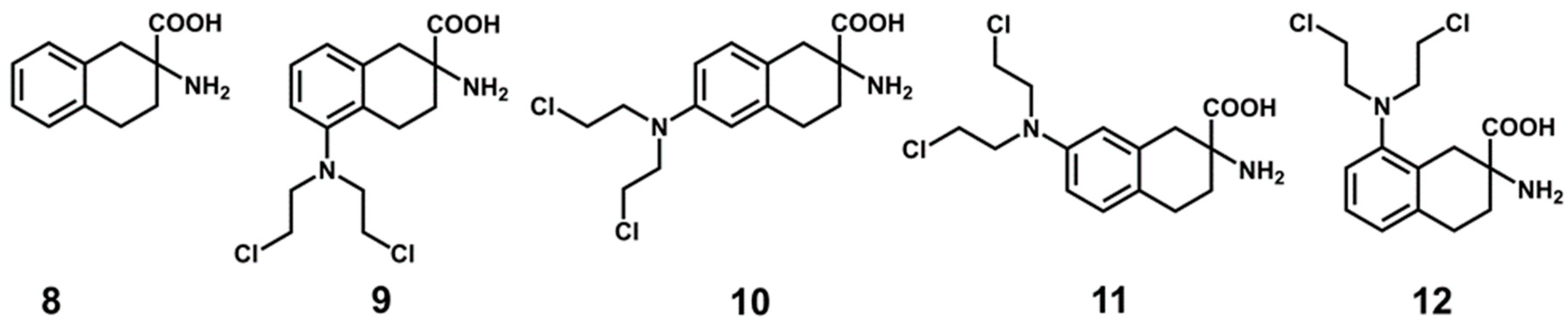
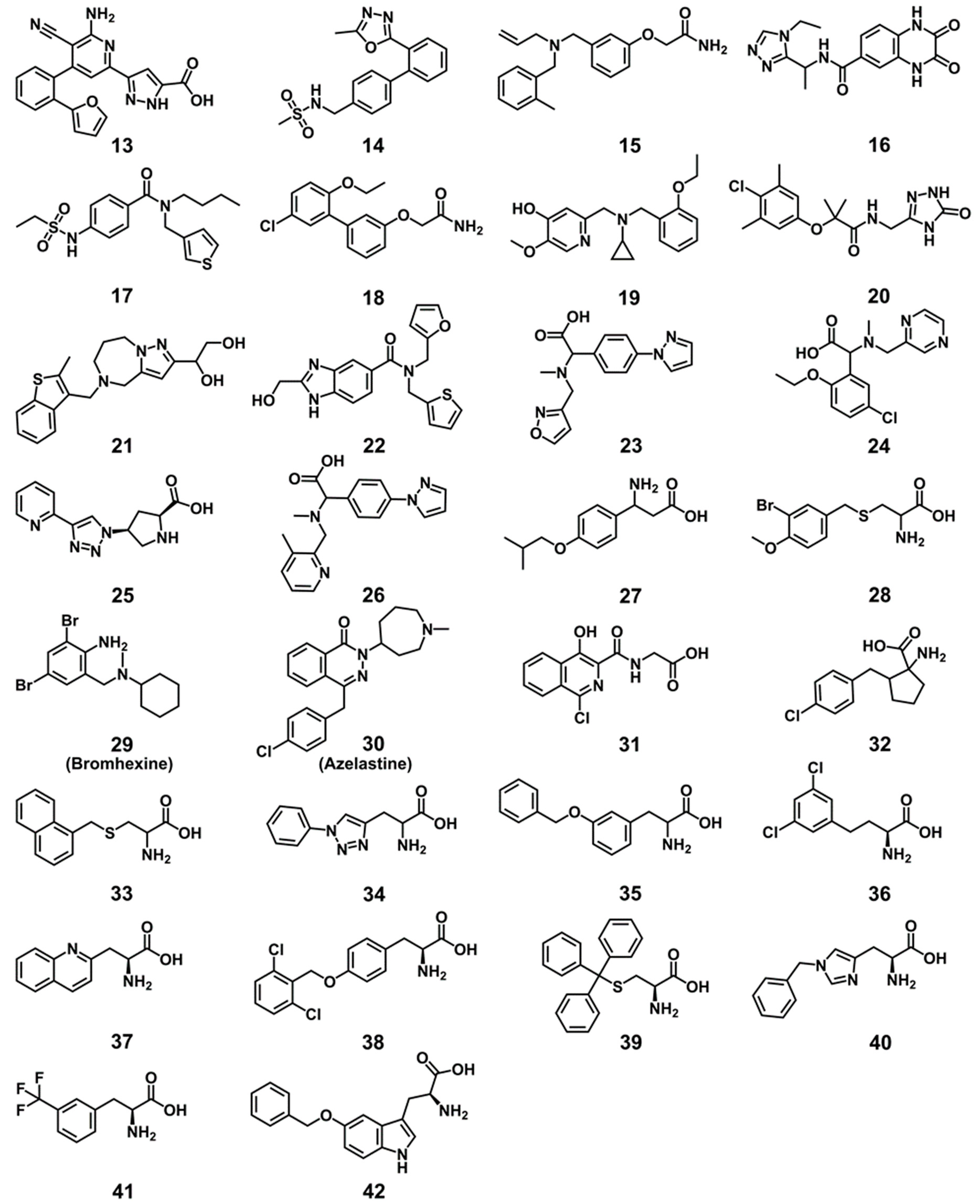
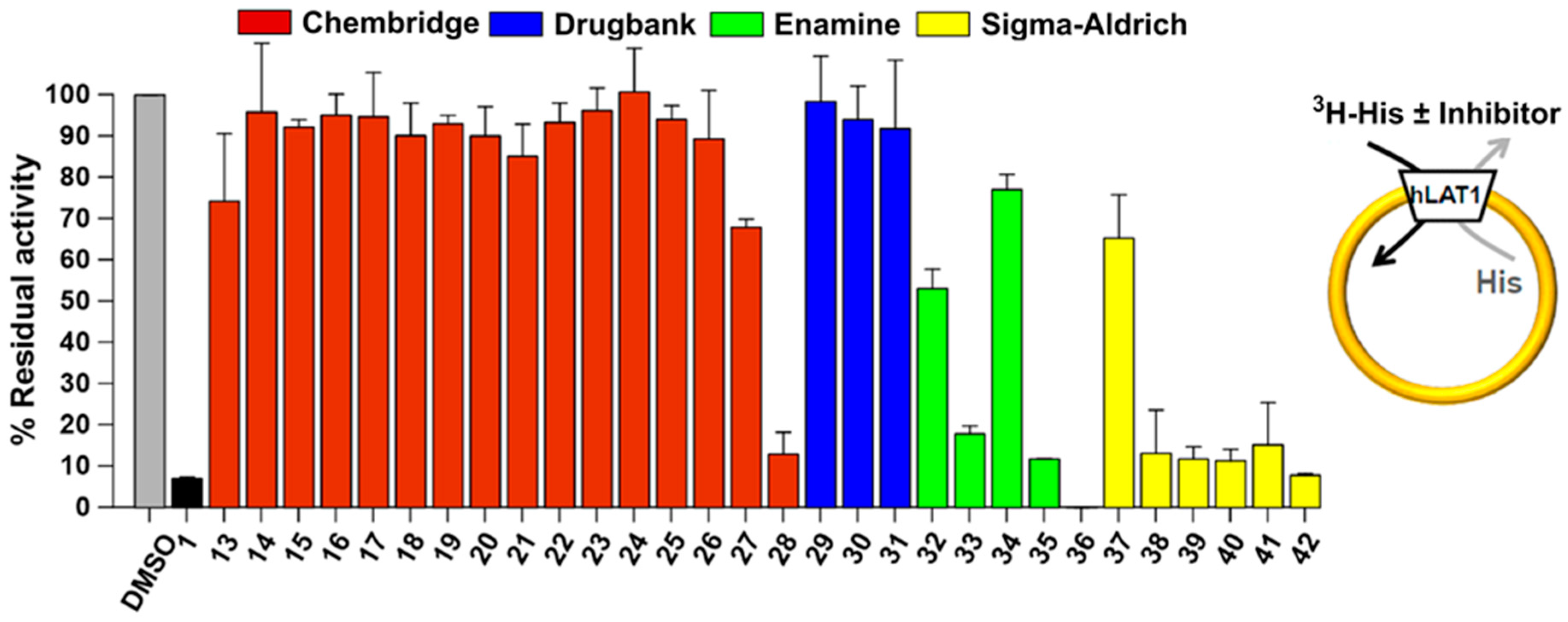
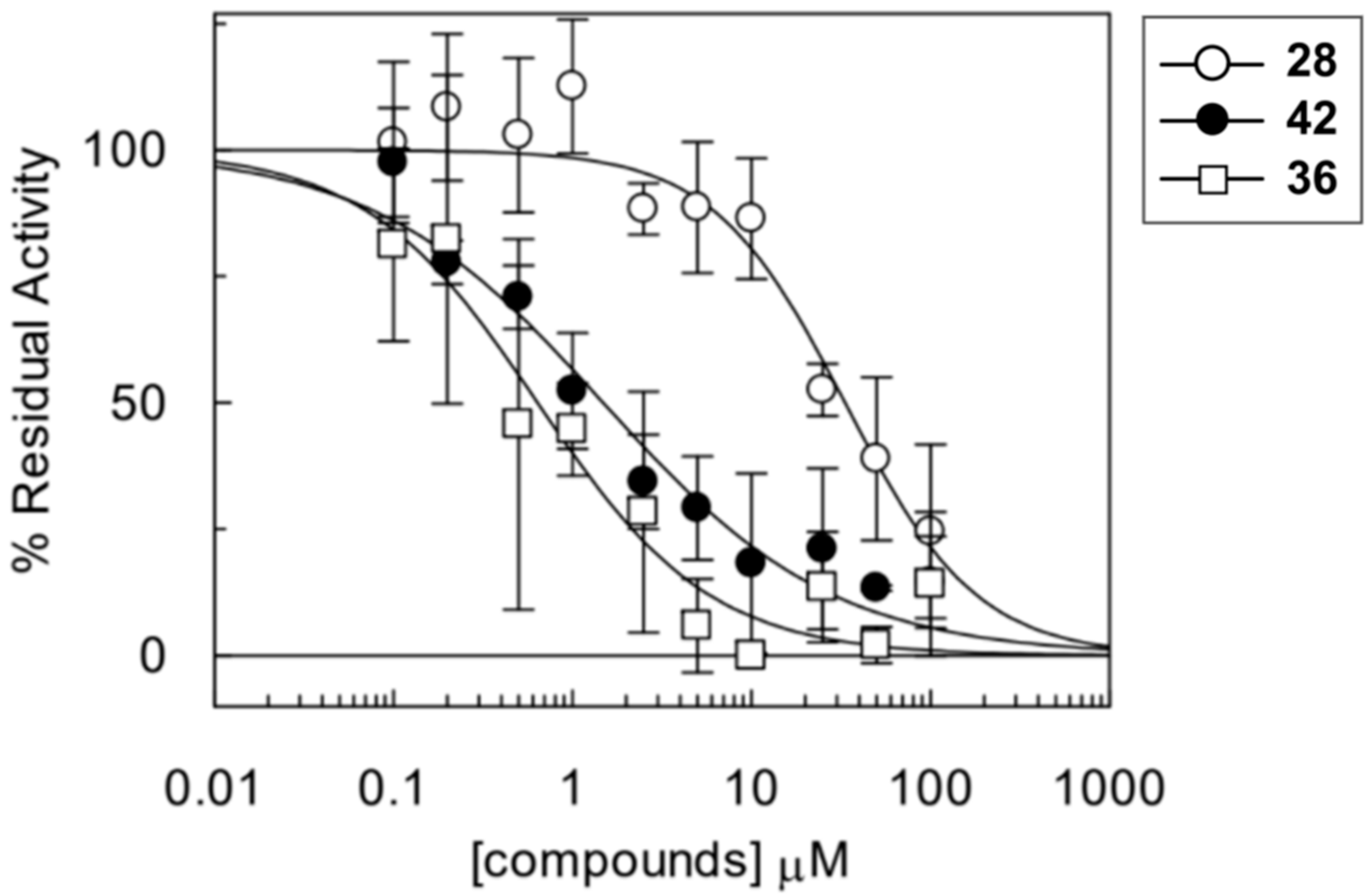
2.5. Experimental Testing
3. Discussion
4. Methods
4.1. Homology Modeling
4.2. Molecular Docking
4.3. Cluster Analysis
4.4. Molecular Dynamics Calculations
4.5. Structure-Based Pharmacophore Modeling
4.6. Pharmacophore Clustering and Validation
4.7. Virtual Screening
4.8. Compounds Filtering and Similarity Calculations
4.9. Chemicals
4.10. Pharmacological Testing
4.10.1. Purification and Reconstitution of Human LAT1
4.10.2. Transport Measurements
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of interest
Abbreviations
References
- Gomes, P.; Soares-da-Silva, P. L-DOPA transport properties in an immortalised cell line of rat capillary cerebral endothelial cells, RBE 4. Brain Res. 1999, 829, 143–150. [Google Scholar] [CrossRef]
- Cundy, K.C.; Annamalai, T.; Bu, L.; De Vera, J.; Estrela, J.; Luo, W.; Shirsat, P.; Torneros, A.; Yao, F.; Zou, J. XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, G.J.; Lam, H.Y.; Begleiter, A. Active carrier-mediated transport of melphalan by two separate amino acid transport systems in LPC-1 plasmacytoma cells in vitro. J. Biol. Chem. 1979, 254, 1057–1064. [Google Scholar] [PubMed]
- Smith, Q.R. Carrier-mediated transport to enhance drug delivery to brain. Int. Congr. Ser. 2005, 1277, 63–74. [Google Scholar] [CrossRef]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Holst, J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar]
- Wie, L.; Tominaga, H.; Ohgaki, R.; Wiriyasermkul, P.; Hagiwara, K.; Okuda, S.; Kaira, K.; Oriuchi, N.; Nagamori, S.; Kanai, Y. Specific transport of 3-fluoro-l-α-methyl-tyrosine by LAT1 explains its specificity to malignant tumors in imaging. Cancer Sci. 2016, 107, 347–352. [Google Scholar]
- Wie, L.; Tominaga, H.; Ohgaki, R.; Wiriyasermkul, P.; Hagiwara, K.; Okuda, S.; Kaira, K.; Kato, Y.; Oriuchi, N.; Nagamori, S.; et al. Transport of 3-Fluoro-l-A-Methyl-Tyrosine (famt) by Organic Ion Transporters Explains Renal Background in [18f]famt Positron Emission Tomography. J. Pharmacol. Sci. 2016, 130, 101–109. [Google Scholar]
- Wiriyasermkul, P.; Nagamori, S.; Tominaga, H.; Oriuchi, N.; Kaira, K.; Nakao, H.; Kitashoji, T.; Ohgaki, R.; Tanaka, H.; Endou, H. Transport of 3-fluoro-l-α-methyl-tyrosine by tumor-upregulated L-type amino acid transporter 1: A cause of the tumor uptake in PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2012, 53, 1253–1261. [Google Scholar] [CrossRef]
- Betsunoh, H.; Fukuda, T.; Anzai, N.; Nishihara, D.; Mizuno, T.; Yuki, H.; Masuda, A.; Yamaguchi, Y.; Abe, H.; Yashi, M. Increased expression of system large amino acid transporter (LAT)-1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer 2013, 13, 509. [Google Scholar] [CrossRef]
- Ebara, T.; Kaira, K.; Saito, J.; Shioya, M.; Asao, T.; Takahashi, T.; Sakurai, H.; Kanai, Y.; Kuwano, H.; Nakano, T. L-type amino-acid transporter 1 expression predicts the response to preoperative hyperthermo-chemoradiotherapy for advanced rectal cancer. Anticancer Res. 2010, 30, 4223–4227. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2012, 103, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Otani, N.; Shinomiya, N.; Fukui, S.; Ooigawa, H.; Shima, K.; Matsuo, H.; Kanai, Y.; Endou, H. L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int. J. Cancer. 2006, 119, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, T.; Ferdous, G.; Tsuruta, T.; Satoh, T.; Baba, S.; Muto, T.; Ueno, A.; Kanai, Y.; Endou, H.; Okayasu, I. L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol. Int. 2009, 59, 7–18. [Google Scholar] [CrossRef]
- Takeuchi, K.; Ogata, S.; Nakanishi, K.; Ozeki, Y.; Hiroi, S.; Tominaga, S.; Aida, S.; Matsuo, H.; Sakata, T.; Kawai, T. LAT1 expression in non-small-cell lung carcinomas: Analyses by semiquantitative reverse transcription-PCR (237 cases) and immunohistochemistry (295 cases). Lung Cancer 2010, 68, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutté, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.N. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol. Rev. 1990, 70, 43–77. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Kaira, K.; Yamaguchi, A.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yokobori, T.; Miyazaki, T.; Asao, T. Efficacy of system l amino acid transporter 1 inhibition as a therapeutic target in esophageal squamous cell carcinoma. Cancer Sci. 2016, 107, 1499–1505. [Google Scholar] [CrossRef]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar]
- Kim, C.S.; Cho, S.H.; Chun, H.S.; Lee, S.Y.; Endou, H.; Kanai, Y.; Kim, D.K. BCH, an inhibitor of system L amino acid transporters, induces apoptosis in cancer cells. Biol. Pharm. Bull. 2008, 31, 1096–1100. [Google Scholar] [CrossRef]
- Shennan, D.B.; Thomson, J. Inhibition of system L (LAT1/CD98hc) reduces the growth of cultured human breast cancer cells. Oncol. Rep. 2008, 20, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; Endou, H. l-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Scalise, M.; Koyioni, M.; Koutentis, P.; Catto, M.; Eberini, I.; Parravicini, C.; Palazzolo, L.; Pisani, L.; Galluccio, M. Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem. Pharmacol. 2017, 143, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhou, L.; Jiao, X.; Lu, F.; Yan, C.; Zeng, X.; Wang, J.; Shi, Y. Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 2010, 463, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, F.; Zhou, L.; Dang, S.; Sun, L.; Li, X.; Wang, J.; Shi, Y. Structure and mechanism of an amino acid antiporter. Science 2009, 324, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, P.L.; Goehring, A.; Shankaranarayanan, A.; Gouaux, E. Structure and mechanism of a Na+-independent amino acid transporter. Science 2009, 325, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Galluccio, M.; Scalise, M.; Parravicini, C.; Palazzolo, L.; Eberini, I.; Indiveri, C. Novel insights into the transport mechanism of the human amino acid transporter LAT1 (SLC7A5). Probing critical residues for substrate translocation. Biochim. Biophys. Acta 2017, 1861, 727–736. [Google Scholar] [CrossRef]
- Augustyn, E.; Finke, K.; Zur, A.A.; Hansen, L.; Heeren, N.; Chien, H.C.; Lin, L.; Giacomini, K.M.; Colas, C.; Schlessinger, A.; et al. LAT-1 activity of meta-substituted phenylalanine and tyrosine analogs. Bioorg. Med. Chem. Lett. 2016, 26, 2616–2621. [Google Scholar] [CrossRef]
- Tarlungeanu, D.C.; Deliu, E.; Dotter, C.P.; Kara, M.; Janiesch, P.C.; Scalise, M.; Galluccio, M.; Tesulov, M.; Morelli, E.; Sonmez, F.M. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 2016, 167, 1481–1494. [Google Scholar] [CrossRef]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M. Quantitative insight into the design of compounds recognized by the L-type amino acid transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Gynther, M.; Huttunen, J.; Puris, E.; Spicer, J.A.; Denny, W.A. A Selective and Slowly Reversible Inhibitor of l-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016, 59, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Peura, L.; Malmioja, K.; Leppänen, J.; Laine, K.; Poso, A.; Lahtela-Kakkonen, M.; Rautio, J. Structure-activity relationship study of compounds binding to large amino acid transporter 1 (LAT1) based on pharmacophore modeling and in situ rat brain perfusion. Eur. J. Pharm. Sci. 2013, 48, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Matharu, J.; Oki, J.; Worthen, D.R.; Smith, Q.R.; Crooks, P.A. Regiospecific and conformationally restrained analogs of melphalan and DL-2-NAM-7 and their affinities for the large neutral amino acid transporter (system LAT1) of the blood-brain barrier. Bioorg. Med. Chem. Lett. 2010, 20, 3688–3691. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Vistica, D.T.; Greig, N.H.; Purdon, D.; Rapoport, S.I.; Smith, Q.R. Rapid high-affinity transport of a chemotherapeutic amino acid across the blood-brain barrier. Cancer Res. 1992, 52, 2191–2196. [Google Scholar] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eramian, D.; Eswar, N.; Shen, M.Y.; Sali, A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. Publ. Protein Soc. 2008, 17, 1881–1893. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993, 231, 1049–1067. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinform. Oxf. Engl. 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Schrödinger Release 2015-1: SiteMap; Version 3.4; Schrödinger, LLC: New York, NY, USA, 2015.
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y. Human l-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef]
- Napolitano, L.; Scalise, M.; Galluccio, M.; Pochini, L.; Albanese, L.M.; Indiveri, C. LAT1 is the transport competent unit of the LAT1/CD98 heterodimeric amino acid transporter. Int. J. Biochem. Cell Biol. 2015, 67, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Krammer, E.-M.; Ghaddar, K.; André, B.; Prévost, M. Unveiling the Mechanism of Arginine Transport through AdiC with Molecular Dynamics Simulations: The Guiding Role of Aromatic Residues. PLoS ONE 2016, 11, e0160219. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of amino acid-related compounds mediated by l-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Docter, R.; Moerings, E.P.; Verrey, F.; Krenning, E.P.; Hennemann, G.; Visser, T.J. Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology 2001, 142, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of a novel series of inhibitors for cancer type transporter l-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H.; Bembenek, S.D.; Tounge, B.A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Klepsch, F.; Chiba, P.; Ecker, G.F. Exhaustive Sampling of Docking Poses Reveals Binding Hypotheses for Propafenone Type Inhibitors of P-Glycoprotein. PLoS Comput. Biol. 2011, 7, e1002036. [Google Scholar] [CrossRef]
- Richter, L.; de Graaf, C.; Sieghart, W.; Varagic, Z.; Mörzinger, M.; de Esch, I.J.; Ecker, G.F.; Ernst, M. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 2012, 8, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Weissensteiner, R.; Steiner, I.; Sitte, H.H.; Ecker, G.F.; Freissmuth, M.; Sucic, S. The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin transporter. Mol. Pharmacol. 2010, 78, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural interaction fingerprint (SIFt): A novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- ChemBridge Online Chemical Store. Available online: http://www.hit2lead.com (accessed on 7 September 2016).
- Enamine Ltd. Enamine Building Blocks; Enamine Ltd.: Riga, Latvia, 2017. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large neutral amino acid transporter enables brain drug delivery via prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef]
- Nagamori, S.; Wiriyasermkul, P.; Okuda, S.; Kojima, N.; Hari, Y.; Kiyonaka, S.; Mori, Y.; Tominaga, H.; Ohgaki, R.; Kanai, Y. Structure-activity relations of leucine derivatives reveal critical moieties for cellular uptake and activation of mTORC1-mediated signaling. Amino Acids 2016, 48, 1045–1058. [Google Scholar] [CrossRef]
- Zur, A.A.; Chien, H.C.; Augustyn, E.; Flint, A.; Heeren, N.; Finke, K.; Hernandez, C.; Hansen, L.; Miller, S.; Lin, L. LAT1 activity of carboxylic acid bioisosteres: Evaluation of hydroxamic acids as substrates. Bioorg. Med. Chem. Lett. 2016, 26, 5000–5006. [Google Scholar] [CrossRef] [Green Version]
- Chien, H.C.; Colas, C.; Finke, K.; Springer, S.; Stoner, L.; Zur, A.A.; Venteicher, B.; Campbell, J.; Hall, C.; Flint, A. Reevaluating the Substrate Specificity of the l-Type Amino Acid Transporter (LAT1). J. Med. Chem. 2018, 61, 7358–7373. [Google Scholar] [CrossRef]
- Yunger, L.M.; Cramer, R.D. Measurement of correlation of partition coefficients of polar amino acids. Mol. Pharmacol. 1981, 20, 602–608. [Google Scholar] [PubMed]
- Chollet, J.F.; Delétage, C.; Faucher, M.; Miginiac, L.; Bonnemain, J.L. Synthesis and structure-activity relationships of some pesticides with an alpha-amino acid function. Biochim. Biophys. Acta 1997, 1336, 331–341. [Google Scholar] [CrossRef]
- Colas, C.; Grewer, C.; Otte, N.J.; Gameiro, A.; Albers, T.; Singh, K.; Shere, H.; Bonomi, M.; Holst, J.; Schlessinger, A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015, 11, e1004477. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Geier, E.; Fan, H.; Irwin, J.J.; Shoichet, B.K.; Giacomini, K.M.; Sali, A. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl. Acad. Sci. USA 2011, 108, 15810–15815. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Pajor, A.M.; Schlessinger, A. Structure-Based Identification of Inhibitors for the SLC13 Family of Na+/Dicarboxylate Cotransporters. Biochemistry 2015, 54, 4900–4908. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Sun, N.N.; Colas, C.; Pajor, A.M. Determinants of Substrate and Cation Transport in the Human Na+/Dicarboxylate Cotransporter NaDC3. J. Biol. Chem. 2014, 289, 16998–17008. [Google Scholar] [CrossRef]
- Jurik, A.; Zdrazil, B.; Holy, M.; Stockner, T.; Sitte, H.H.; Ecker, G.F. A Binding Mode Hypothesis of Tiagabine Confirms Liothyronine Effect on γ-Aminobutyric Acid Transporter 1 (GAT1). J. Med. Chem. 2015, 58, 2149–2158. [Google Scholar] [CrossRef]
- Garibsingh RA, A.; Otte, N.J.; Ndaru, E.; Colas, C.; Grewer, C.; Holst, J.; Schlessinger, A. Homology Modeling Informs Ligand Discovery for the Glutamine Transporter ASCT2. Front. Chem. 2018, 6, 279. [Google Scholar] [CrossRef]
- Jain, S.; Grandits, M.; Richter, L.; Ecker, G.F. Structure based classification for bile salt export pump (BSEP) inhibitors using comparative structural modeling of human BSEP. J. Comput. Aided Mol. Des. 2017, 31, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Krause, G.; Hinz, K.M. Thyroid hormone transport across L-type amino acid transporters: What can molecular modelling tell us? Mol. Cell. Endocrinol. 2017, 458, 68–75. [Google Scholar] [CrossRef]
- Nizami, B.; Sydow, D.; Wolber, G.; Honarparvar, B. Molecular insight on the binding of NNRTI to K103N mutated HIV-1 RT: Molecular dynamics simulations and dynamic pharmacophore analysis. Mol. Biosyst. 2016, 12, 3385–3395. [Google Scholar] [CrossRef]
- Peura, L.; Malmioja, K.; Laine, K.; Leppänen, J.; Gynther, M.; Isotalo, A.; Rautio, J. Large Amino Acid Transporter 1 (LAT1) Prodrugs of Valproic Acid: New Prodrug Design Ideas for Central Nervous System Delivery. Mol. Pharm. 2011, 8, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Grewer, C.; Grabsch, E. New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Meck, R.; Yunis, A. The inhibition of gamma-glutamyl transpeptidase from human pancreatic carcinoma cells by (alpha S,5S)-alpha-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125; NSC-163501). Res. Commun. Chem. Pathol. Pharmacol. 1980, 27, 175–182. [Google Scholar] [PubMed]
- Kreuzer, J.; Bach, N.C.; Forler, D.; Sieber, S.A. Target discovery of acivicin in cancer cells elucidates its mechanism of growth inhibition. Chem. Sci. 2015, 6, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: Proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Rosell, A.; Meury, M.; Álvarez-Marimon, E.; Costa, M.; Pérez-Cano, L.; Zorzano, A.; Fernández-Recio, M.; Palacín, M.; Fotiadis, D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc. Natl. Acad. Sci. USA 2014, 111, 2966–2971. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [Green Version]
- LigPrep; Version 3.3; Schrodinger, LLC: New York, NY, USA, 2015.
- MarvinSketch 16.6.27, 2016, ChemAxon. Available online: http://www.chemaxon.com (accessed on 10 April 2015).
- Schrodinger, LLC. Schrodinger Suite 2015 Protein Preparation Wizard; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nosé, S.A. Molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, N. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Wieder, M.; Perricone, U.; Boresch, S.; Seidel, T.; Langer, T. Evaluating the stability of pharmacophore features using molecular dynamics simulations. Biochem. Biophys. Res. Commun. 2016, 470, 685–689. [Google Scholar] [CrossRef]
- Gynther, M.; Jalkanen, A.; Lehtonen, M.; Forsberg, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Knuuti, J.; Rautio, J. Brain uptake of ketoprofen-lysine prodrug in rats. Int. J. Pharm. 2010, 399, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Wieder, M.; Garon, A.; Perricone, U.; Boresch, S.; Seidel, T.; Almerico, A.M.; Langer, T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Schrödinger Release 2015-1: Canvas; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Jasial, S.; Hu, Y.; Vogt, M.; Bajorath, J. Activity-relevant similarity values for fingerprints and implications for similarity searching. F1000Research 2016, 5, 591. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Lorusso, I.; De Palma, A.; Indiveri, C. The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim. Biophys. Acta. 2016, 1860, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Torchetti, E.M.; Brizio, C.; Colella, M.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Roberti, M.; Barile, M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 2010, 10, 263–273. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | pIC50 | LogP o/w | HAC | LE | LLE | FQ |
|---|---|---|---|---|---|---|
| 8 | 5.11 | −0.78 | 14 | 0.37 | 5.89 | 0.60 |
| 9 | 5.07 | 1.30 | 21 | 0.24 | 3.76 | 0.53 |
| 10 | 4.17 | 1.18 | 21 | 0.20 | 2.98 | 0.44 |
| 11 | 7.10 | 1.40 | 21 | 0.34 | 5.70 | 0.75 |
| 12 | 3.60 | 1.42 | 21 | 0.17 | 2.17 | 0.37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Scalise, M.; Galluccio, M.; Wieder, M.; Seidel, T.; Langer, T.; Indiveri, C.; Ecker, G.F. Discovery of Potent Inhibitors for the Large Neutral Amino Acid Transporter 1 (LAT1) by Structure-Based Methods. Int. J. Mol. Sci. 2019, 20, 27. https://doi.org/10.3390/ijms20010027
Singh N, Scalise M, Galluccio M, Wieder M, Seidel T, Langer T, Indiveri C, Ecker GF. Discovery of Potent Inhibitors for the Large Neutral Amino Acid Transporter 1 (LAT1) by Structure-Based Methods. International Journal of Molecular Sciences. 2019; 20(1):27. https://doi.org/10.3390/ijms20010027
Chicago/Turabian StyleSingh, Natesh, Mariafrancesca Scalise, Michele Galluccio, Marcus Wieder, Thomas Seidel, Thierry Langer, Cesare Indiveri, and Gerhard F. Ecker. 2019. "Discovery of Potent Inhibitors for the Large Neutral Amino Acid Transporter 1 (LAT1) by Structure-Based Methods" International Journal of Molecular Sciences 20, no. 1: 27. https://doi.org/10.3390/ijms20010027