Proteasome-Rich PaCS as an Oncofetal UPS Structure Handling Cytosolic Polyubiquitinated Proteins. In Vivo Occurrence, in Vitro Induction, and Biological Role

Abstract
:1. Introduction
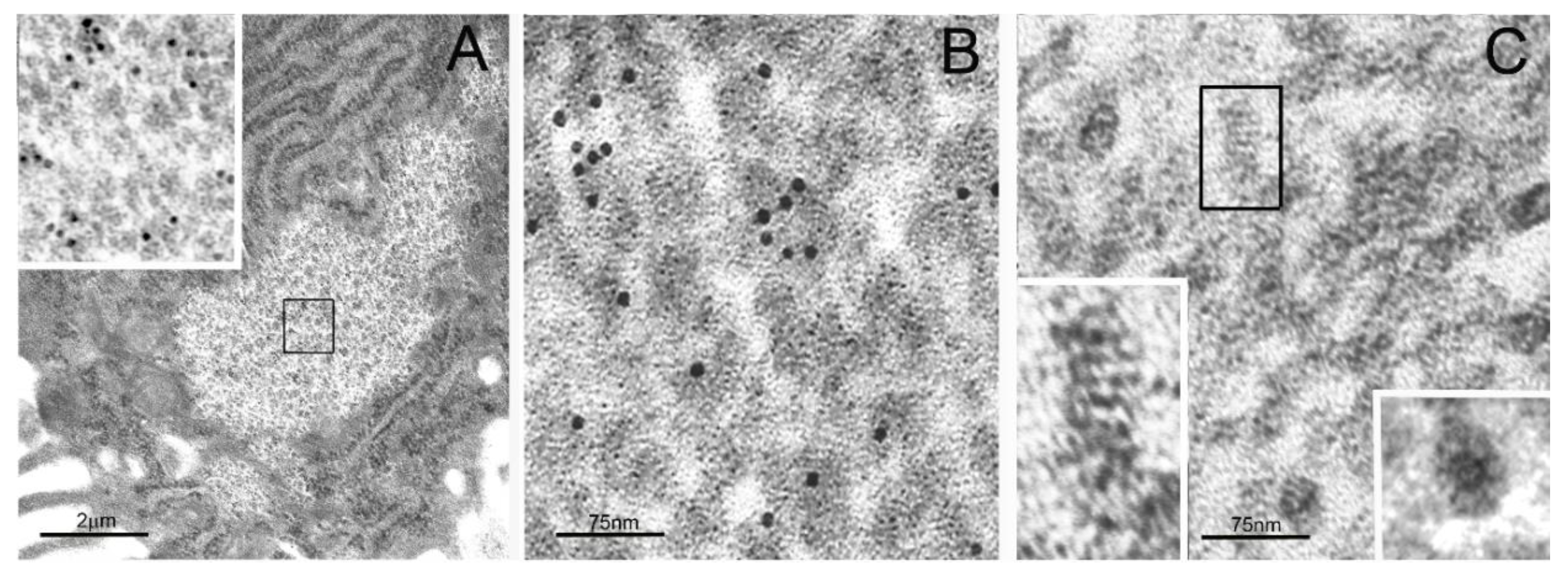
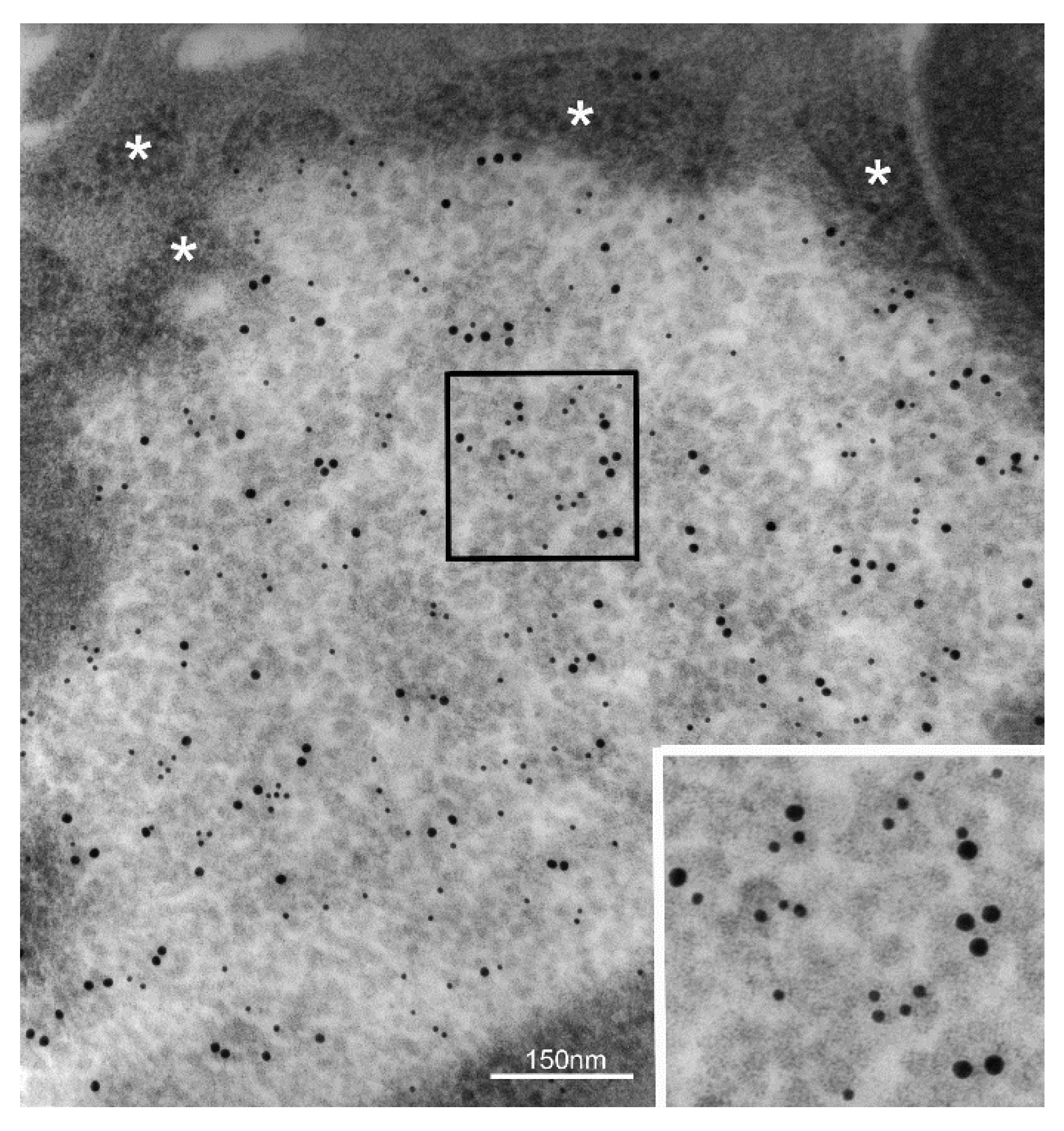
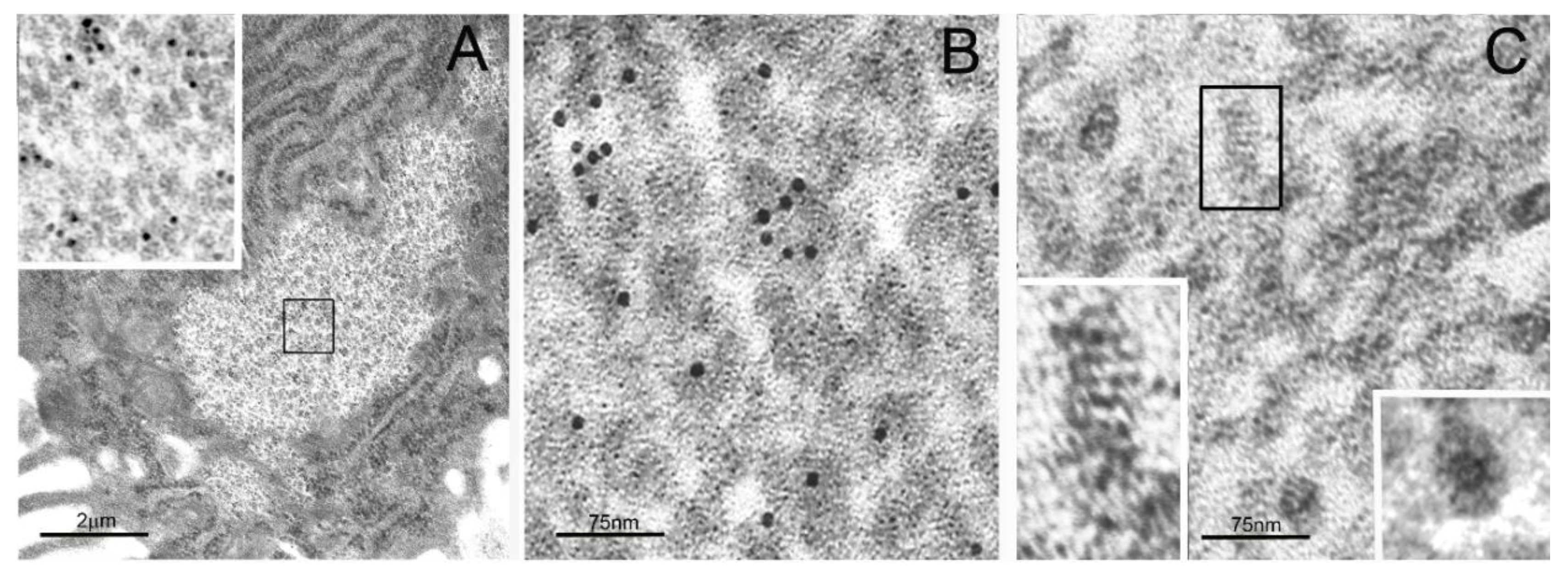
2. Particulate Cytoplasmic Structure (PaCS), an Oncofetal Cytoplasmic Structure Concentrating Proteasome Particles, PUbPs, and Heat-Shock Proteins
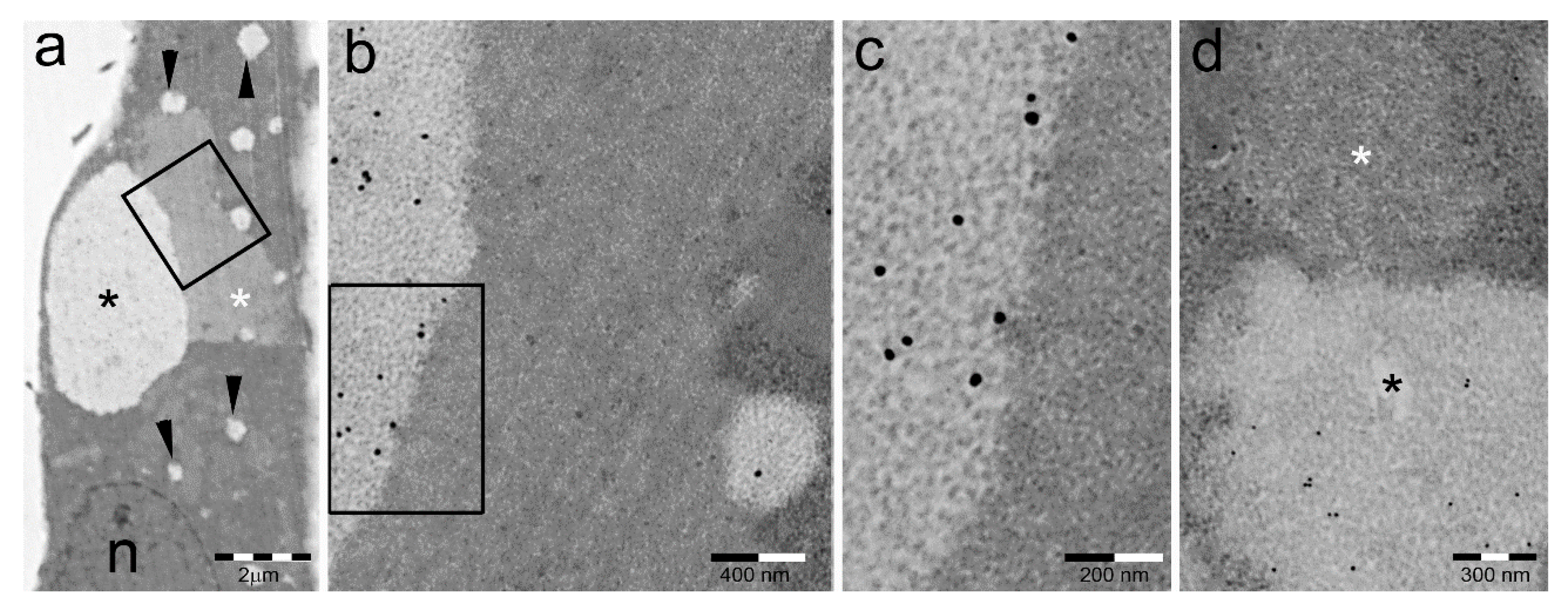
3. Distribution of PaCS in Fetal and Neoplastic Cells
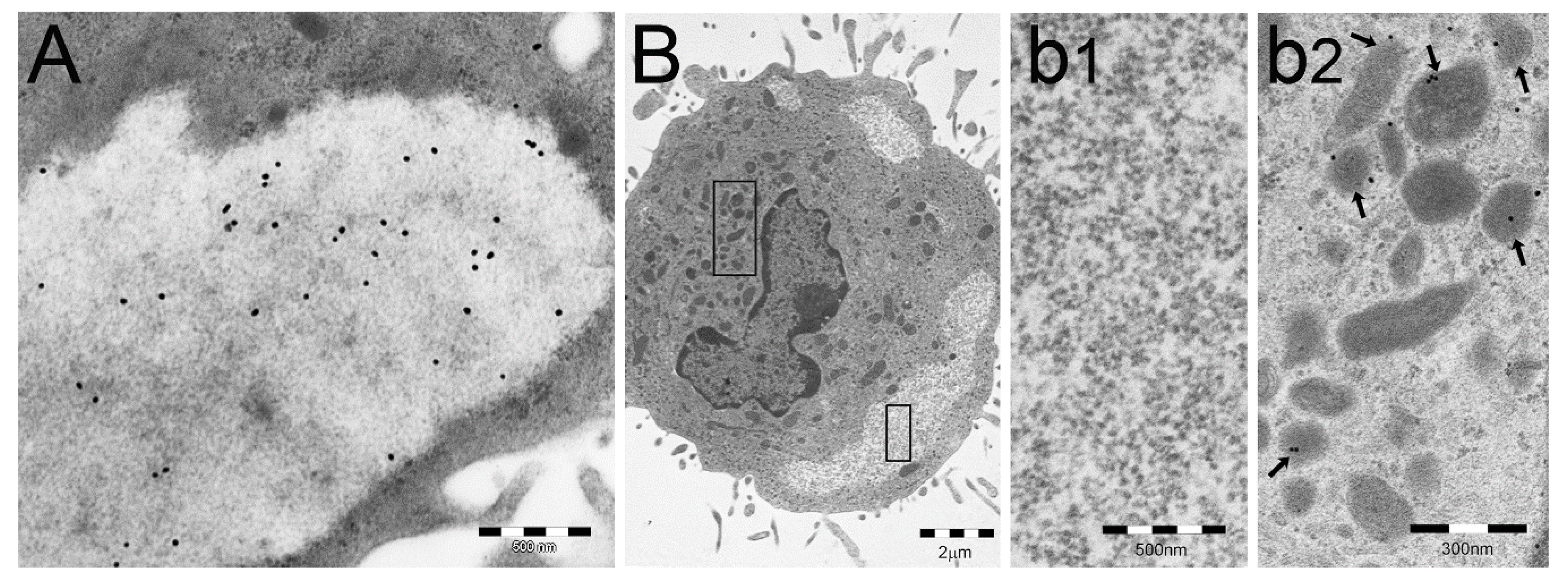
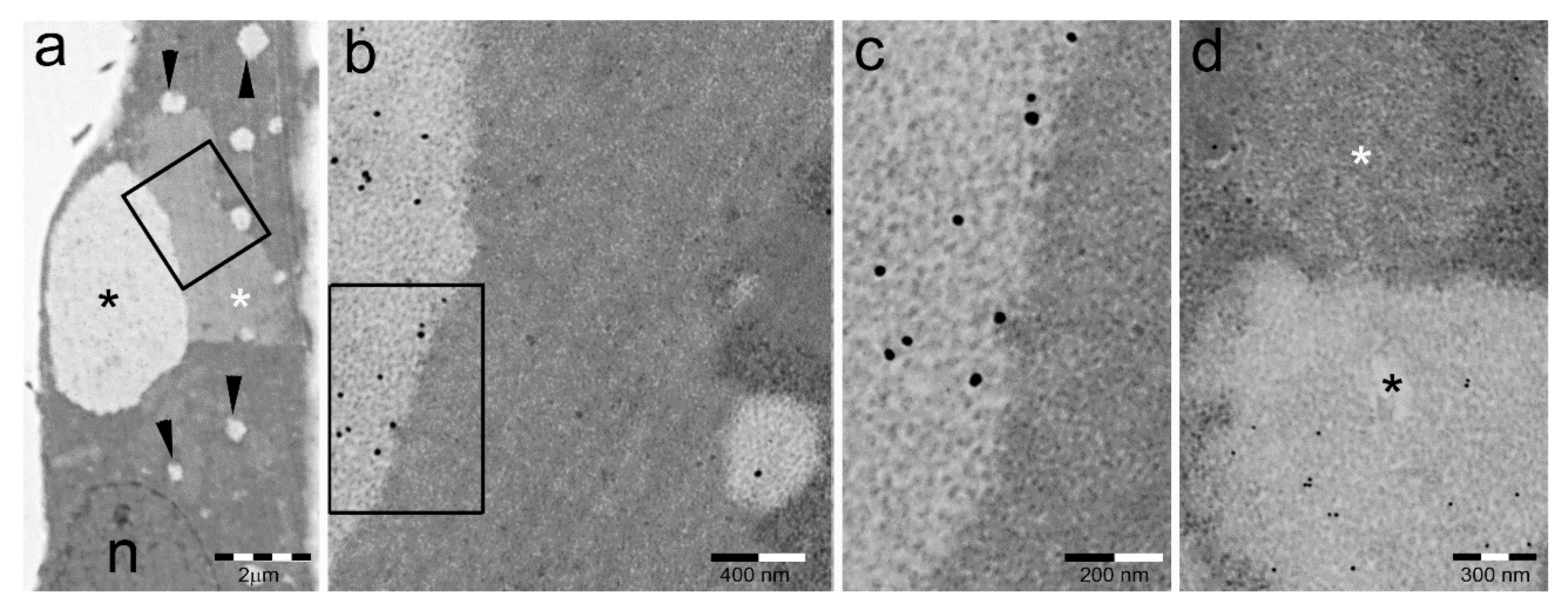
3.1. Fetal Tissues
3.2. In Neoplastic and Preneoplastic Cells
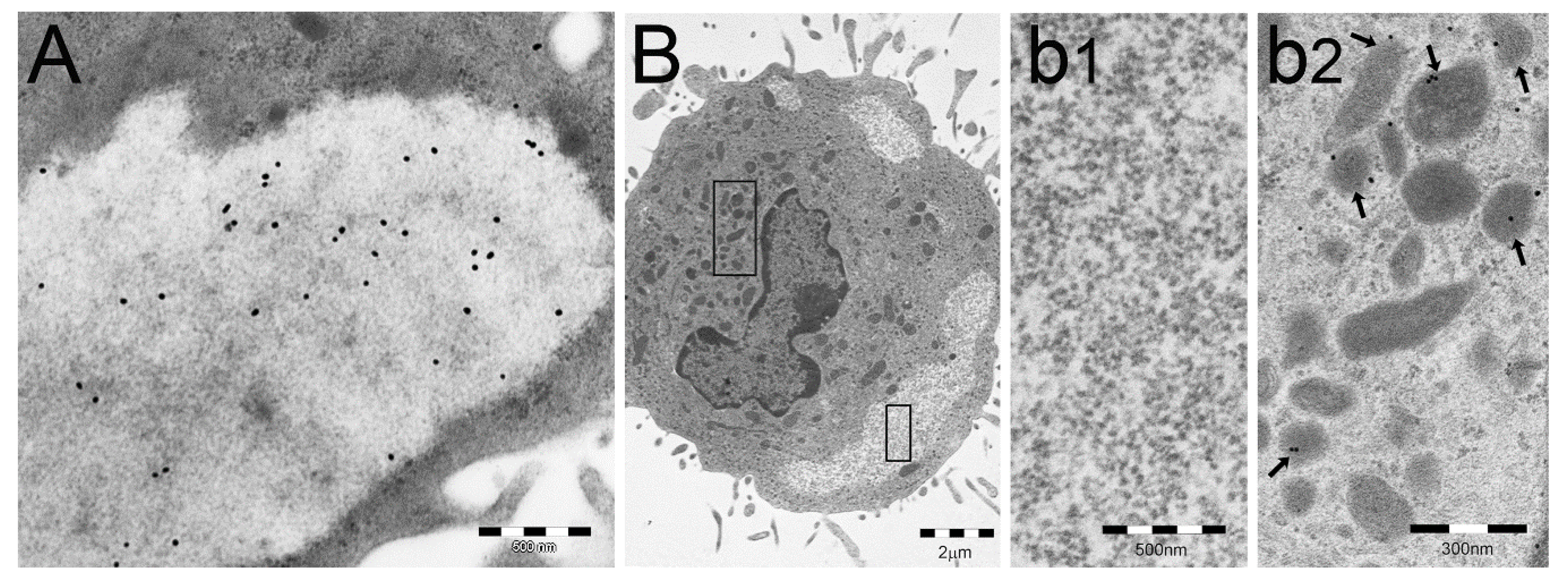
4. PaCS Induction in Cell Cultures under Trophic Factors/Interleukins Treatment
5. PaCS Intracellular and Extracellular Fate
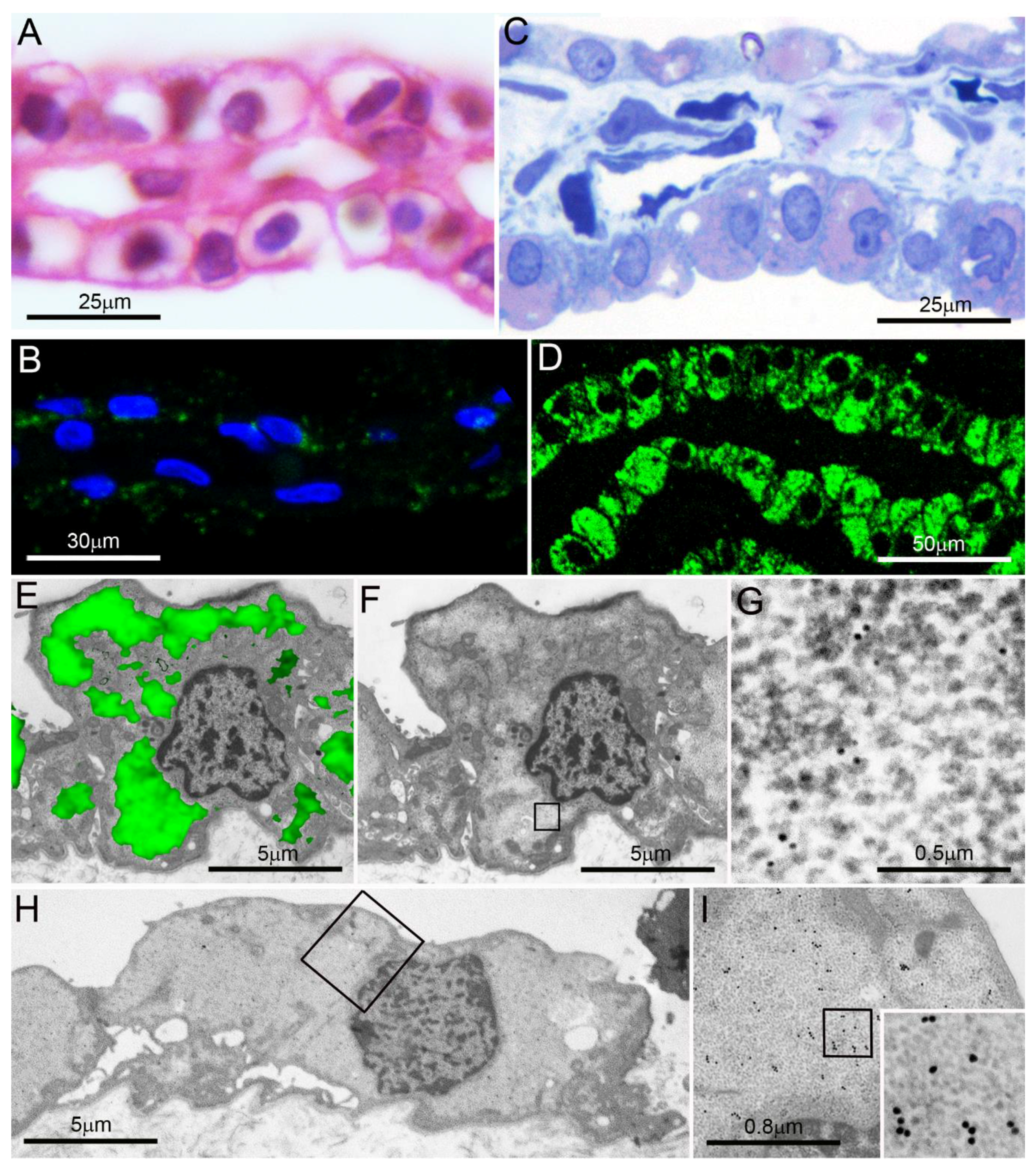
5.1. PaCS Intracellular Dissolution and Autophagy
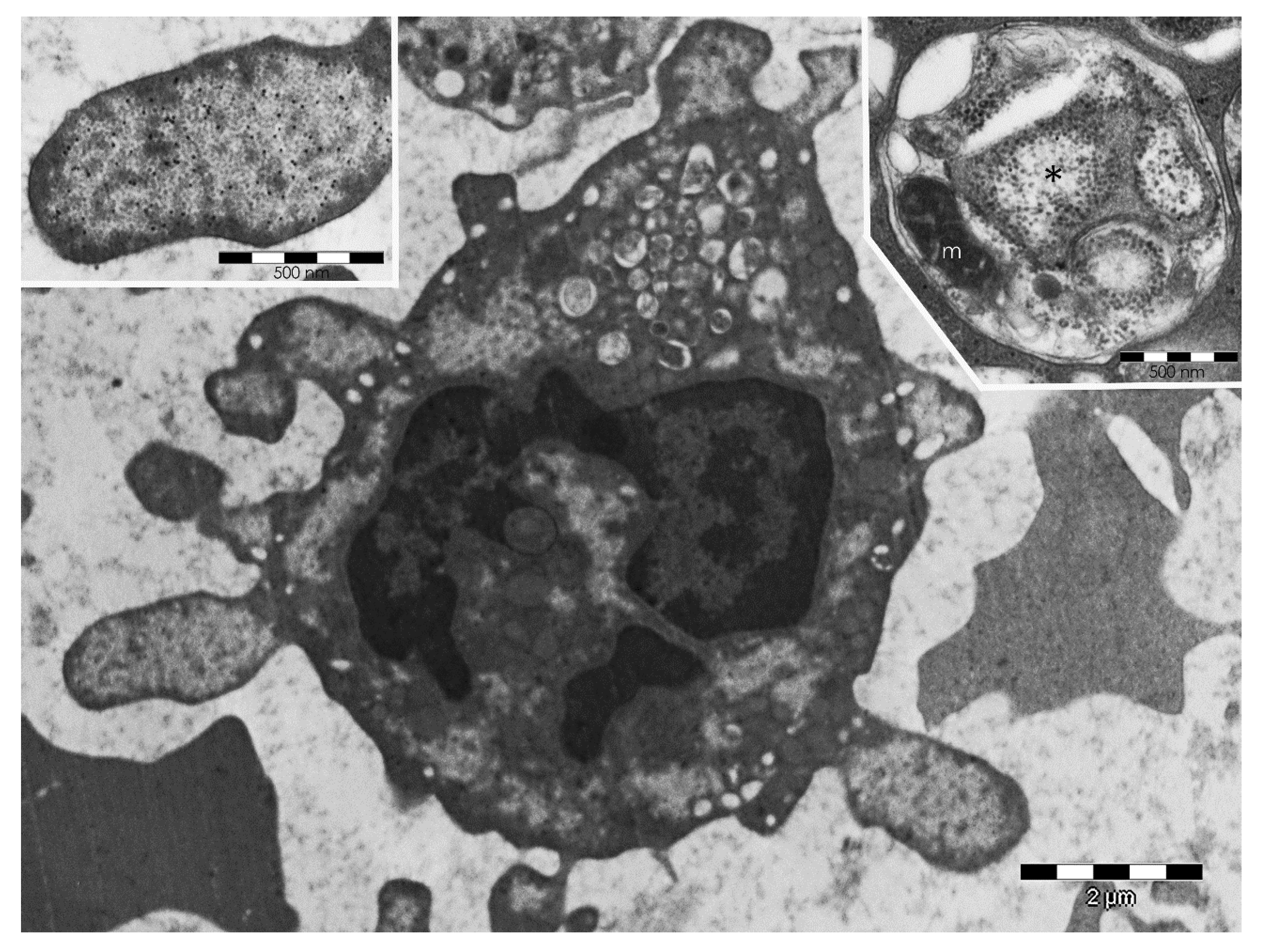
5.2. PaCS-Filled Cell Blebs
6. PaCS versus Sequestosomes, Aggresomes, and Inclusion Bodies of Degenerative Diseases
7. PaCS Biological and Pathological Role
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cell |
| DALIS | DC aggresome-like induced structures |
| DC | dendritic cell |
| EGFR | EGF receptor |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum-associated degradative |
| HIF | hypoxia-inducible factor |
| Hsp | heat-shock protein |
| IPOD | insoluble protein deposit |
| JUNQ | juxtanuclear quality-control compartment |
| NK | natural killer |
| PaCS | particulate cytoplasmic structure |
| PSCN | pancreatic serous cystic neoplasm |
| pUbPs | polyubiquitinated proteins |
| Rb | retinoblastoma tumor-suppressor protein |
| TAP | transporter for antigen processing |
| TEM | transmission electron microscopy |
| TPO | thrombopoietin |
| UPS | ubiquitin–proteasome system |
| VHL | Von Hippel–Lindau |
References
- McClellan, A.J.; Tam, S.; Kaganovich, D.; Frydman, J. Protein quality control: Chaperones culling corrupt conformations. Nat. Cell Biol. 2005, 7, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Rivett, A.J.; Palmer, A.; Knecht, E. Electron microscopy localization of the multicatalytic proteinase complex in rat liver and in cultured cells. J. Histochem. Cytochem. 1992, 40, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.; Murray, R.Z.; Mason, G.G.F.; Hendil, K.B.; Rivett, A.J. Association of immunoproteasomes with the endoplasmic reticulum. Biochem. J. 2000, 352, 611–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, A.L.; Kyritsis, C.; Tampé, R.; Cresswell, P. Access of soluble antigens to the endoplasmic reticulum can explain cross-presentation by dendritic cells. Nat. Immunol. 2005, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Zamarchi, R.; Merlo, A.; Gorza, M.; Piovan, E.; Mandruzzato, S.; Bronte, V.; Wang, X.; Ferrone, S.; Amadori, A.; et al. Differential expression of constitutive and inducible proteasome subunits in human monocyte-derived DC differentiated in the presence of IFN-α or IL-4. Eur. J. Immunol. 2009, 39, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Amigorena, S.; Savina, A. Intracellular mechanisms of antigen cross-presentation in dendritic cells. Curr. Opin. Immunol. 2010, 22, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sijts, E.J.A.M.; Kloetzel, P.-M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. 2011, 68, 1491–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadaro, F.; Lapenta, C.; Donati, S.; Abalsamo, L.; Barnaba, V.; Belardelli, F.; Santini, S.M.; Ferrantini, M. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood 2012, 119, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.; Boehringer, D.; Ban, N.; Bukau, B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 2009, 16, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Matsumoto, M.; Yada, M.; Nakayama, K.I. Interaction of U-box-type ubiquitin-protein ligases (E3s) with molecular chaperones. Genes Cells 2004, 9, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClellan, A.J.; Scott, M.D.; Frydman, J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 2005, 121, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C. Mechanisms of the Hsp70 chaperone system. Biochem. Cell. Biol. 2010, 88, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.J.; Benham, A.M.; Plougastel, B.; Neefjes, J.; Trowsdale, J. Dynamics of proteasome distribution in living cells. EMBO J. 1997, 16, 6087–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, S.-M.; Chen, L.; Lambertson, D.; Anand, M.; Kinzy, T.G.; Madura, K. Proteasome-mediated degradation of cotranslationally damaged proteins involves translation elongation factor 1A. Mol. Cell. Biol. 2005, 25, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Sommi, P.; Ricci, V.; Solcia, E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS ONE 2010, 5, e9716. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Sommi, P.; Vanoli, A.; Manca, R.; Ricci, V.; Solcia, E. Proteasome particle-rich structures are widely present in human epithelial neoplasms: Correlative light, confocal and electron microscopy study. PLoS ONE 2011, 6, e21317. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W.; Walz, J.; Zühl, F.; Seemüller, E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998, 92, 367–380. [Google Scholar] [CrossRef]
- Walz, J.; Erdmann, A.; Kania, M.; Typke, D.; Koster, A.J.; Baumeister, W. 26S proteasome structure revealed by three-dimensional electron microscopy. J. Struct. Biol. 1998, 121, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Sommi, P.; Necchi, V.; Vitali, A.; Montagna, D.; De Luigi, A.; Salmona, M.; Ricci, V.; Solcia, E. PaCS is a novel cytoplasmic structure containing functional proteasome and inducible by cytokines/trophic factors. PLoS ONE 2013, 8, e82560. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Sommi, P.; Vitali, A.; Vanoli, A.; Savoia, A.; Ricci, V.; Solcia, E. Polyubiquitinated proteins, proteasome, and glycogen characterize the particle-rich cytoplasmic structure (PaCS) of neoplastic and fetal cells. Histochem. Cell Biol. 2014, 141, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Vanoli, A.; Necchi, V.; Barozzi, S.; Manca, R.; Pecci, A.; Solcia, E. Chaperone molecules concentrate together with the ubiquitin-proteasome system inside particulate cytoplasmic structure: Possible role in metabolism of misfolded proteins. Histochem. Cell Biol. 2015, 144, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Necchi, V.; Barozzi, S.; Vitali, A.; Boveri, E.; Elena, C.; Bernasconi, P.; Noris, P.; Solcia, E. Particulate cytoplasmic structures with high concentration of ubiquitin-proteasome accumulate in myeloid neoplasms. J. Hematol. Oncol. 2015, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimuro, M.; Sawada, H.; Yokosawa, H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett. 1994, 349, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Montagna, D.; Sommi, P.; Necchi, V.; Vitali, A.; Montini, E.; Turin, I.; Ferraro, D.; Ricci, V.; Solcia, E. Different polyubiquitinated bodies in human dendritic cells: IL-4 causes PaCS during differentiation while LPS or IFNα induces DALIS during maturation. Sci. Rep. 2017, 7, 1844. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Sommi, P.; Vanoli, A.; Fiocca, R.; Ricci, V.; Solcia, E. Natural history of Helicobacter pylori VacA toxin in human gastric epithelium in vivo: Vacuoles and beyond. Sci. Rep. 2017, 7, 14526. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.W.; Ali, M.; Wood, T.E.; Wong, D.; Maclean, N.; Wang, X.; Gronda, M.; Skrtic, M.; Li, X.; Hurren, R.; et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010, 115, 2251–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peth, A.; Besche, H.C.; Goldberg, A.L. Ubiquitinated proteins activate the proteasome by binding to USP14/UBP6 which causes 20S gate opening. Mol. Cell 2009, 36, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanayama, H.; Tanaka, K.; Aki, M.; Kagawa, S.; Miyaji, H.; Satoh, M.; Okada, F.; Sato, S.; Shimbara, N.; Ichihara, A. Changes in expressions of proteasome and ubiquitin genes in human renal cancer cells. Cancer Res. 1991, 51, 6677–6685. [Google Scholar] [PubMed]
- Bazzaro, M.; Lee, M.K.; Zoso, A.; Stirling, W.L.; Santillan, A.; Shih, I.-M.; Roden, R.B. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006, 66, 3754–3763. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; De Luca, F. Inhibition of the proteasomal function in chondrocytes down-regulates growth plate chondrogenesis and longitudinal bone growth. Endocrinology 2006, 147, 3761–3768. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.-Y.; Maehr, R.; Gilchrist, C.A.; Long, M.A.; Bouley, D.M.; Mueller, B.; Ploegh, H.L.; Kopito, R.R. The mouse polyubiquitin gene UbC is essential for fetal liver development, cell-cycle progression and stress tolerance. EMBO J. 2007, 26, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumatori, A.; Tanaka, K.; Inamura, N.; Sone, S.; Ogura, T.; Matsumoto, T.; Tachikawa, T.; Shin, S.; Ichihara, A. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7071–7075. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, Y.; Shiratori, K.; Hatori, T.; Fujita, I.; Kimijima, A.; Yamamoto, M.; Kobayashi, M.; Furukawa, T. Association of epidermal growth factor receptor and mitogen-activated protein kinase with cystic neoplasms of the pancreas. Mod. Pathol. 2010, 23, 1127–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Périgny, M.; Hammel, P.; Corcos, O.; Larochelle, O.; Giraud, S.; Richard, S.; Sauvanet, A.; Belghiti, J.; Ruszniewski, P.; Bedossa, P.; et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: Characterization by VHL/HIF pathway proteins expression. Am. J. Surg. Pathol. 2009, 33, 739–748. [Google Scholar] [PubMed]
- Vortmeyer, A.O.; Lubensky, I.A.; Fogt, F.; Linehan, W.M.; Khettry, U.; Zhuang, Z. Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am. J. Pathol. 1997, 151, 951–956. [Google Scholar] [PubMed]
- Moore, P.S.; Zamboni, G.; Brighenti, A.; Lissandrini, D.; Antonello, D.; Capelli, P.; Rigaud, G.; Falconi, M.; Scarpa, A. Molecular characterization of pancreatic serous microcystic adenomas: Evidence for a tumor suppressor gene on chromosome 10q. Am. J. Pathol. 2001, 158, 317–321. [Google Scholar] [CrossRef]
- Wu, J.; Jiao, Y.; Dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Candusso, M.E.; Tava, F.; Luinetti, O.; Ventura, U.; Fiocca, R.; Ricci, V.; Solcia, E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 2007, 132, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Zabler, D.; Schmidt, D.; Hartig, R.; Brandt, S.; Backert, S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: Antagonistic effects of the vacuolating cytotoxin VacA. Cell. Microbiol. 2009, 11, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M., Jr.; Shaw, L.M.; Kelly, C.P. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 2001, 276, 48127–48134. [Google Scholar] [CrossRef] [PubMed]
- Tsang, Y.H.; Lamb, A.; Romero-Gallo, J.; Huang, B.; Ito, K.; Peek, R.M., Jr.; Ito, Y.; Chen, L.F. Helicobacter pylori CagA targets gastric tumor suppressor RUNX3 for proteasome-mediated degradation. Oncogene 2010, 29, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Buti, L.; Spooner, E.; Van der Veen, A.G.; Rappuoli, R.; Covacci, A.; Ploegh, H.L. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. USA 2011, 108, 9238–9243. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Noto, J.M.; Zaika, E.; Romero-Gallo, J.; Piazuelo, M.B.; Schneider, B.; El-Rifai, W.; Correa, P.; Peek, R.M.; Zaika, A.I. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut 2015, 64, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Nakahara, T.; Ueno, T.; Sasaki, K.; Yoshida, S.; Kyo, S.; Howley, P.M.; Sakai, H. Requirement of E7 oncoprotein for viability of HeLa cells. Microbes Infect. 2006, 8, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: P53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S. proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Minelli, A.; Sommi, P.; Vitali, A.; Caruso, R.; Longoni, D.; Frau, M.R.; Nasi, C.; De Gregorio, F.; Zecca, M.; et al. Ubiquitin-proteasome-rich cytoplasmic structures in neutrophils of patients with Shwachman-Diamond syndrome. Haematologica 2012, 97, 1057–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menne, T.F.; Goyenechea, B.; Sanchez-Puig, N.; Wong, C.C.; Tonkin, L.M.; Ancliff, P.J.; Brost, R.L.; Costanzo, M.; Boone, C.; Warren, A.J. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 2007, 39, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, K.A.; Austin, K.M.; Lee, C.S.; Dias, A.; Malsch, M.M.; Reed, R.; Shimamura, A. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood 2007, 110, 1458–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambekar, C.; Das, B.; Yeger, H.; Dror, Y. SBDS-deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr. Blood Cancer 2010, 55, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Krüger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic is maintained by granulocyte/macrophage colony stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of dendritic cell development by GM-CSF: Molecular control and implications for immune homeostasis and therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.H.; Wientjens, G.J.; Fibbe, W.E.; Willemze, R.; Kluin-Nelemans, H.C. Inhibition of human macrophage colony formation by interleukin 4. J. Exp. Med. 1989, 170, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, A.D.; Zamorano, J. Regulation of gene expression, growth, and cell survival by IL-4: Contribution of multiple signaling pathways. Cell Res. 1998, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.W.; Darling, D.; Farzaneh, F.; Galea-Lauri, J. Influence of interleukin-4 on the phenotype and function of bone marrow-derived murine dendritic cells generated under serum-free conditions. Scand. J. Immunol. 2005, 61, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Necchi, V.; Balduini, A.; Noris, P.; Barozzi, S.; Sommi, P.; di Buduo, C.; Balduini, C.L.; Solcia, E.; Pecci, A. Ubiquitin/proteasome-rich particulate cytoplasmic structures (PaCSs) in the platelets and megakaryocytes of ANKRD26-related thrombo-cytopenia. Thromb. Haemost. 2013, 109, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Balduini, A.; Balayn, N.; Currao, M.; Nurden, P.; Deswarte, C.; Leverger, G.; Noris, P.; Perrotta, S.; Solary, E.; et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J. Clin. Investig. 2014, 124, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Gao, Y.S.; Sztul, E. Hassles with taking out the garbage: Aggravating aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Wigley, W.C.; Fabunmi, R.P.; Lee, M.G.; Marino, C.R.; Muallem, S.; DeMartino, G.N.; Thomas, P.J. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 1999, 145, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Lelouard, H.; Gatti, E.; Cappello, F.; Gresser, O.; Camosseto, V.; Pierre, P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 2002, 417, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lelouard, H.; Ferrand, V.; Marguet, D.; Bania, J.; Camossetto, V.; David, A.; Gatti, E.; Pierre, P. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J. Cell Biol. 2004, 164, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondylis, V.; van Nispen Tot Pannerden, H.E.; van Dijk, S.; Ten Broeke, T.; Wubbolts, R.; Geerts, W.J.; Seinen, C.; Mutis, T.; Heijnen, H.F. Endosome-mediated autophagy: An unconventional MIIC-driven autophagic pathway operational in dendritic cells. Autophagy 2013, 9, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Lavabre-Bertrand, T.; Henry, L.; Carillo, S.; Guiraud, I.; Ouali, A.; Dutaud, D.; Aubry, L.; Rossi, J.F.; Bureau, J.P. Plasma proteasome level is a potential marker in patients with solid tumors and hemopoietic malignancies. Cancer 2001, 92, 2493–2500. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Chen, C.C.; Chen, L.L.; Lee, C.C.; Huang, T.S. Secreted heat shock protein 90α (HSP90α) induces nuclear factor-κB-mediated TCF12 protein expression to down-regulate E-cadherin and to enhance colorectal cancer cell migration and invasion. J. Biol. Chem. 2013, 288, 9001–9010. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solcia, E.; Sommi, P.; Necchi, V.; Vitali, A.; Manca, R.; Ricci, V. Particle-rich cytoplasmic structure (PaCS): Identification, natural history, role in cell biology and pathology. Biomolecules 2014, 4, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.G.; García-Alai, M.M.; Smal, C.; Centeno, J.M.; Iacono, R.; Castaño, E.; Gualfetti, P.; de Prat-Gay, G. The HPV16 E7 viral oncoprotein self-assembles into defined spherical oligomers. Biochemistry 2004, 43, 3310–3317. [Google Scholar] [CrossRef] [PubMed]
- Dantur, K.; Alonso, L.; Castaño, E.; Morelli, L.; Centeno-Crowley, J.M.; Vighi, S.; de Prat-Gay, G. Cytosolic accumulation of HPV16 E7 oligomers supports different transformation routes for the prototypic viral oncoprotein: The amyloid-cancer connection. Int. J. Cancer 2009, 125, 1902–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T., Jr. Assembly of A beta amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 1999, 38, 8972–8980. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Sittler, A.; Schweiger, K.; Heiser, V.; Lurz, R.; Hasenbank, R.; Bates, G.P.; Lehrach, H.; Wanker, E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. USA 1999, 96, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Denk, H.; Stumptner, C.; Fuchsbichler, A.; Müller, T.; Farr, G.; Müller, W.; Terracciano, L.; Zatloukal, K. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J. Pathol. 2006, 208, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, E.; Salminen, A.; Alafuzoff, I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001, 12, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. p62 is a common component of cytoplasmic inclusion in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef]
- Waelter, S.; Boeddrich, A.; Lurz, R.; Scherzinger, E.; Lueder, G.; Lehrach, H.; Wanker, E.E. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 2001, 12, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Shashidharan, P.; Perl, D.P.; Jenner, P.; Olanow, C.W. Aggresome-related biogenesis of Lewy bodies. Eur. J. Neurosci. 2002, 16, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaarur, N.; Meriin, A.B.; Bejarano, E.; Xu, X.; Gabai, V.L.; Cuervo, A.M.; Sherman, M.Y. Proteasome failure promotes positioning of lysosomes around the aggresome via local block of microtubule-dependent transport. Mol. Cell. Biol. 2014, 34, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.J.; Lyakhovetsky, R.; Werdiger, A.C.; Gitler, A.D.; Soen, Y.; Kaganovich, D. Compartmentalization of superoxide dismutase 1 (SOD1G93A) aggregates determines their toxicity. Proc. Natl. Acad. Sci. USA 2012, 109, 15811–15816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.; Björklund, L.M.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Proteasome inhibition causes nigral degeneration with inclusion bodies in rats. Neuroreport 2002, 13, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Zaarur, N.; Meriin, A.B.; Gabai, V.L.; Sherman, M.Y. Triggering aggresome formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J. Biol. Chem. 2008, 283, 27575–27584. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Dubey, D.; Yamakawa, K.; Ganesh, S. Lafora disease proteins malin and laforin are recruited to aggresomes in response to proteasomal impairment. Hum. Mol. Genet. 2007, 16, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, J.; Gruart, A.; García-Rocha, M.; Delgado-García, J.M.; Guinovart, J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 2014, 23, 3147–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, J.; Zheng, H.; Su, H.; Powell, S.R. Proteasome functional insufficiency in cardiac pathogenesis. Am. J. Physiol. Heart. Circ. Physiol. 2011, 301, H2207–H2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, C.; Alberti, S.; Höhfeld, J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta 2004, 1695, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Divald, A.; Kivity, S.; Wang, P.; Hochhauser, E.; Roberts, B.; Teichberg, S.; Gomes, A.V.; Powell, S.R. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ. Res. 2010, 106, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.; Menéndez-Benito, V.; Dantuma, N.P.; et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Zemskov, E.A.; Wang, G.h.; Nukina, N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 2001, 10, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanbe, A.; Osinska, H.; Saffitz, J.E.; Glabe, C.G.; Kayed, R.; Maloyan, A.; Robbins, J. Desmin-related cardiomyopathy in transgenic mice: A cardiac amyloidosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10132–10136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PaCS | Sequestosome | |
|---|---|---|
| Ultrastructure * | Collection of barrel-like particles | Granulofibrillar arrays |
| Content | ||
| (a) Proteasome | Yes | No |
| (b) Polyubiquitinated proteins | Yes (likely K48-linked) | No |
| (c) Hsp 70 and 90 | Yes | No |
| (d) P62/SQSTM1 | No | Yes |
| Degradation by autophagy | Possible | Possible |
| Entering cell blebs and ectosomes | Frequent | Not found |
| Associated pathology | Clear cell neoplasms | Hepatocellular cancer |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solcia, E.; Necchi, V.; Sommi, P.; Ricci, V. Proteasome-Rich PaCS as an Oncofetal UPS Structure Handling Cytosolic Polyubiquitinated Proteins. In Vivo Occurrence, in Vitro Induction, and Biological Role. Int. J. Mol. Sci. 2018, 19, 2767. https://doi.org/10.3390/ijms19092767
Solcia E, Necchi V, Sommi P, Ricci V. Proteasome-Rich PaCS as an Oncofetal UPS Structure Handling Cytosolic Polyubiquitinated Proteins. In Vivo Occurrence, in Vitro Induction, and Biological Role. International Journal of Molecular Sciences. 2018; 19(9):2767. https://doi.org/10.3390/ijms19092767
Chicago/Turabian StyleSolcia, Enrico, Vittorio Necchi, Patrizia Sommi, and Vittorio Ricci. 2018. "Proteasome-Rich PaCS as an Oncofetal UPS Structure Handling Cytosolic Polyubiquitinated Proteins. In Vivo Occurrence, in Vitro Induction, and Biological Role" International Journal of Molecular Sciences 19, no. 9: 2767. https://doi.org/10.3390/ijms19092767