Tanshinone IIA Attenuates Insulin Like Growth Factor 1 -Induced Cell Proliferation in PC12 Cells through the PI3K/Akt and MEK/ERK Pathways

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
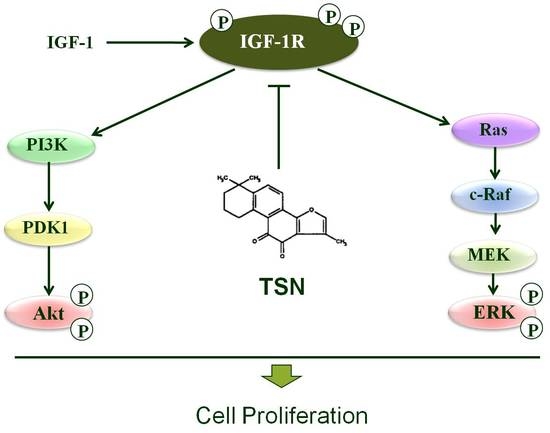
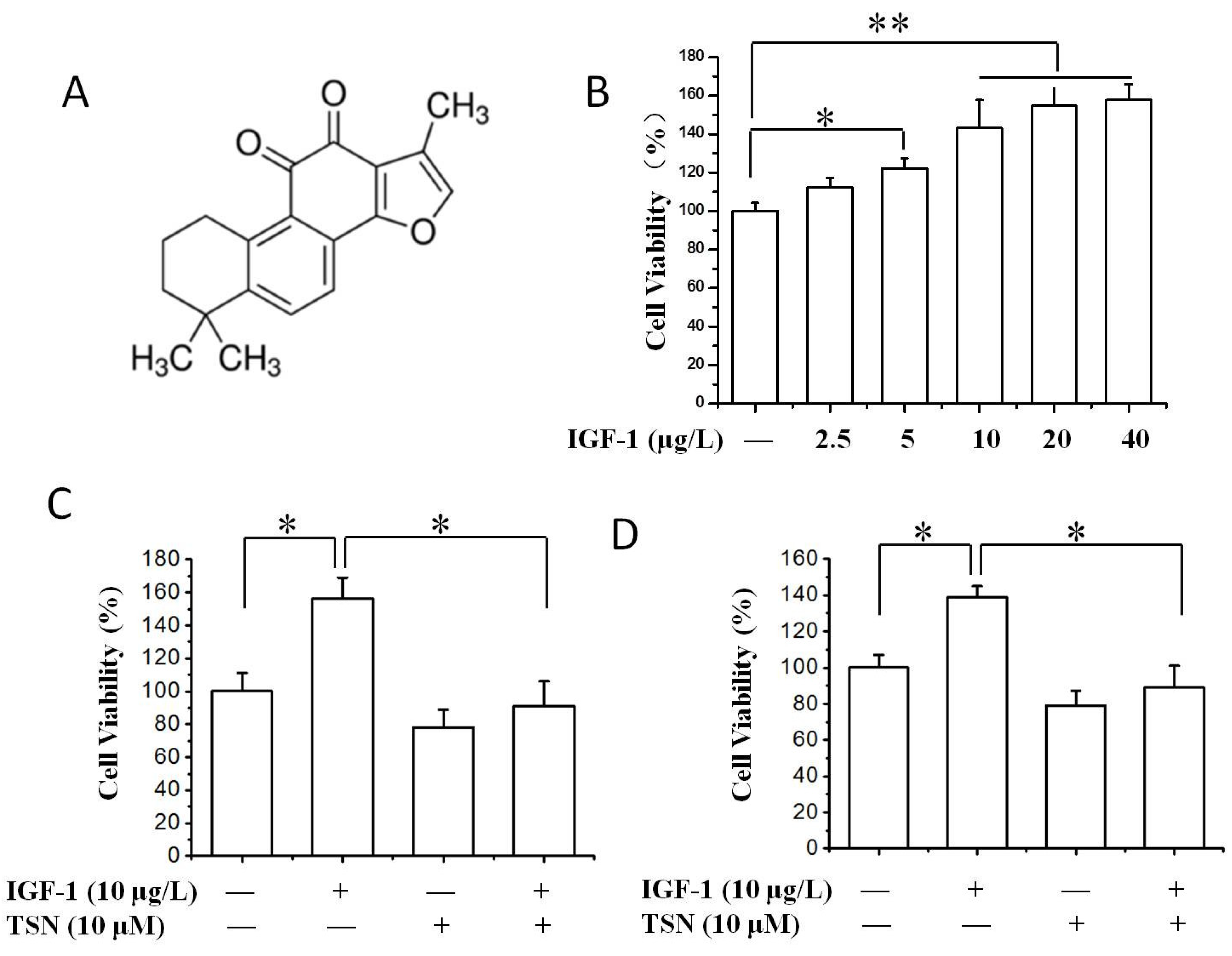
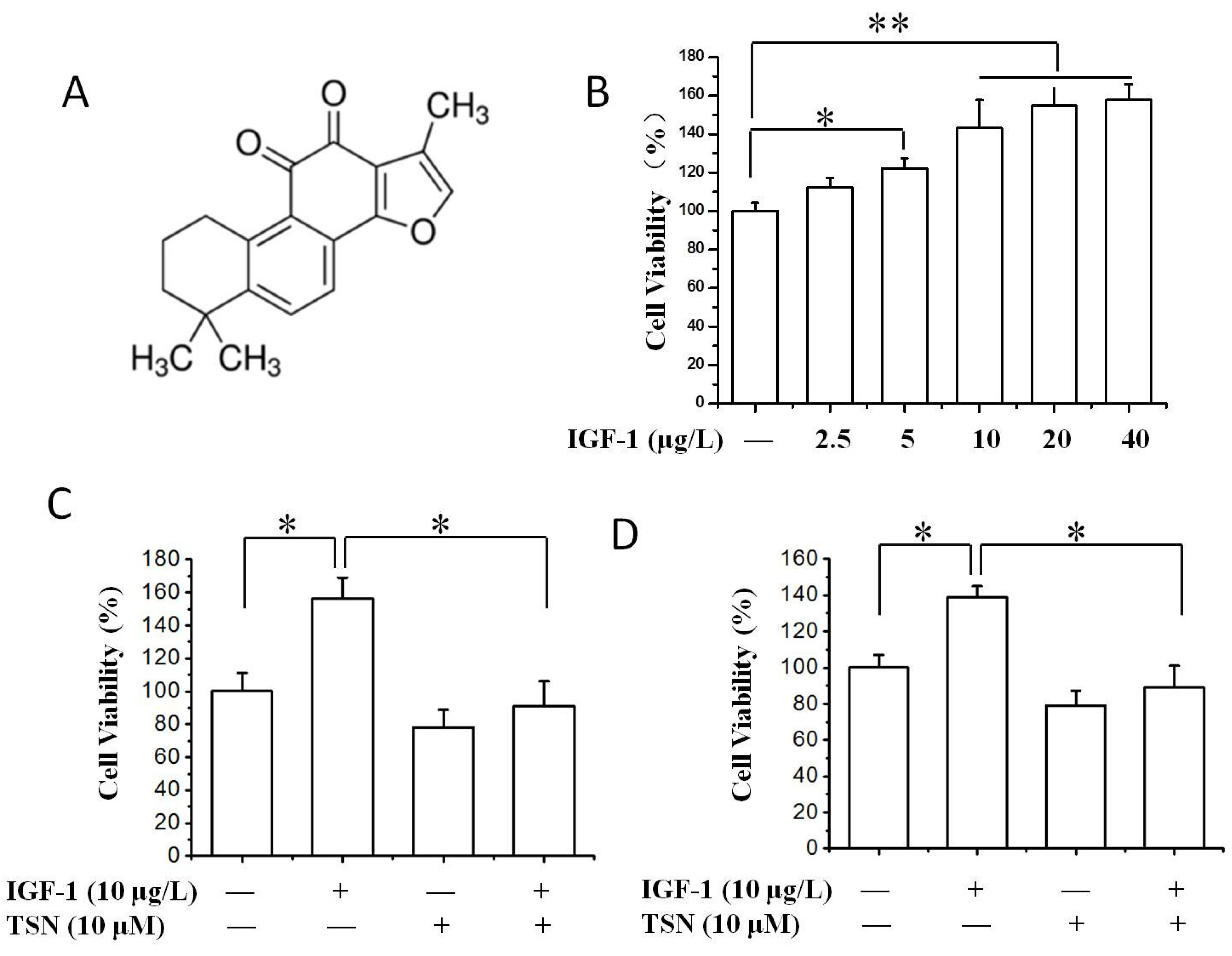
2.1. TSN Suppressed Cell Growth Induced by IGF-1 in PC12 Cells
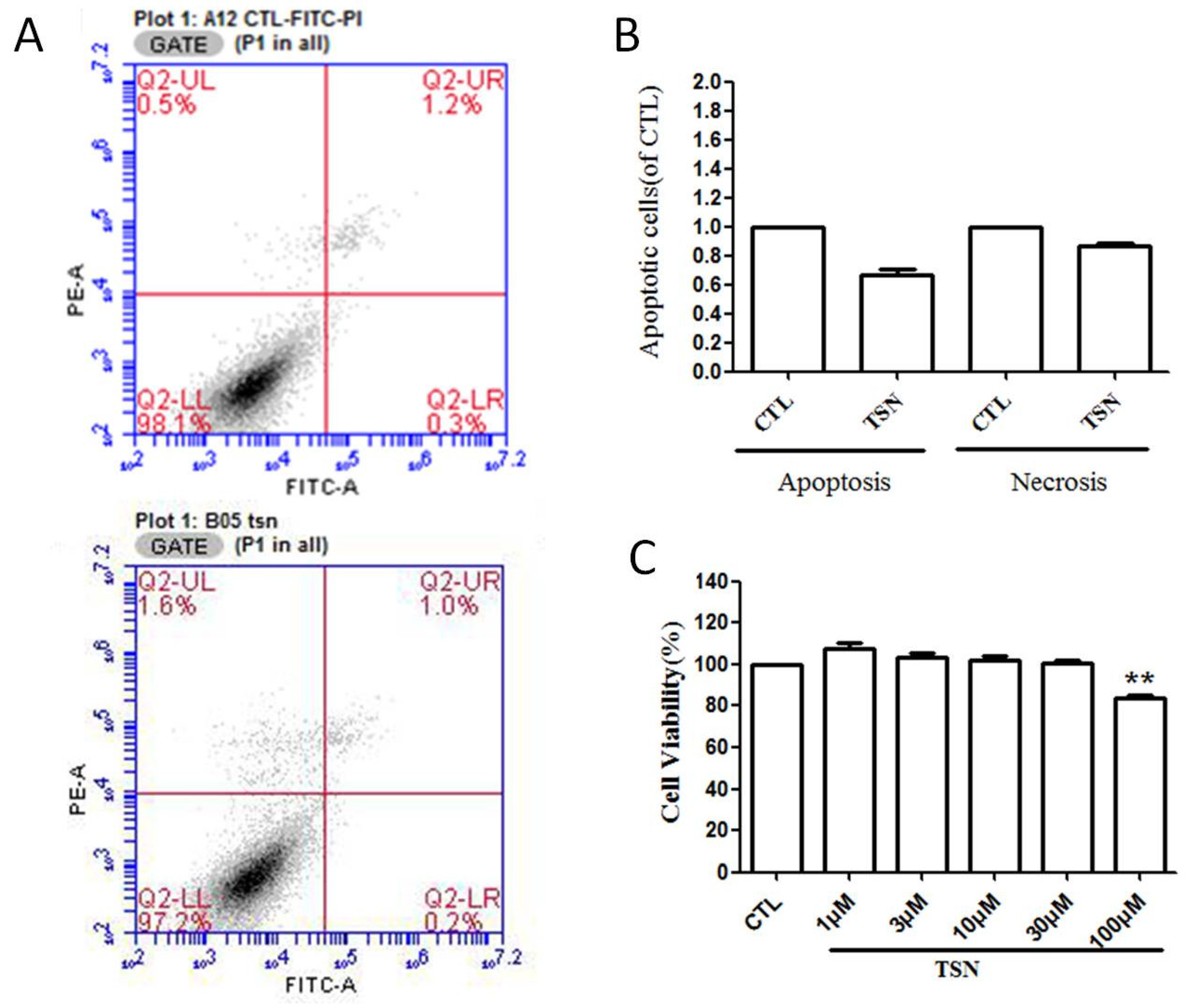
2.2. TSN had No Effect on the Apoptosis of PC12 Cells
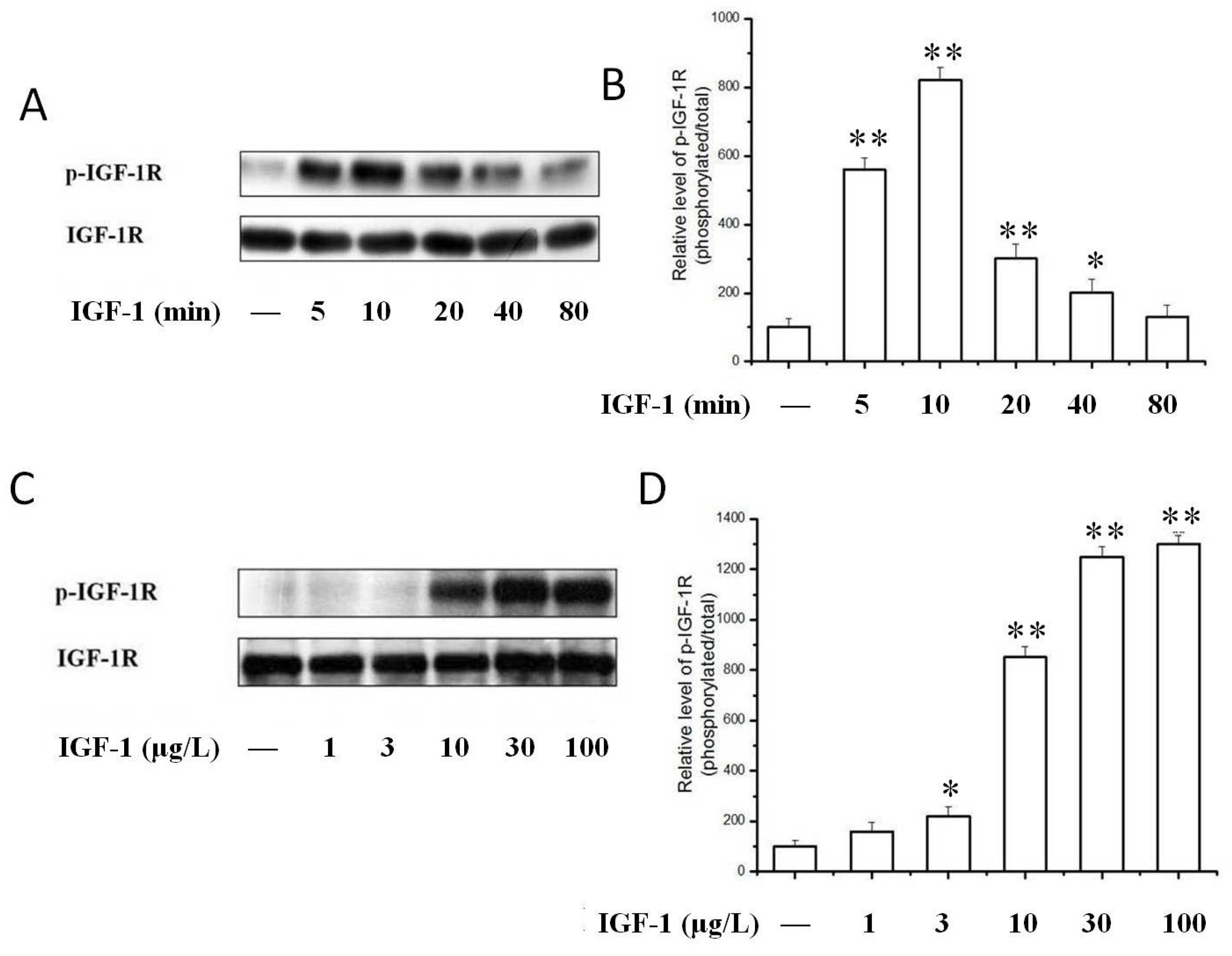
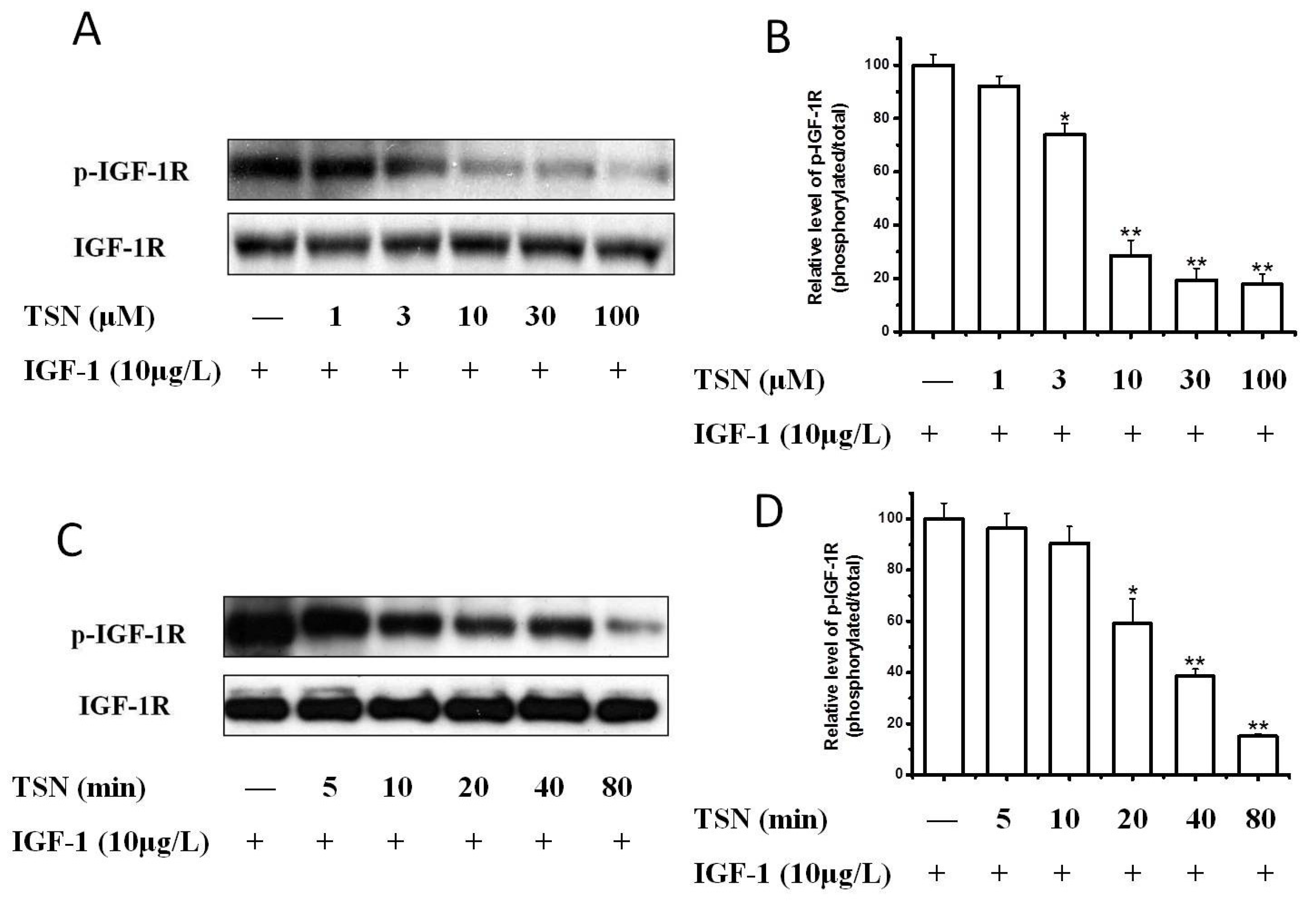
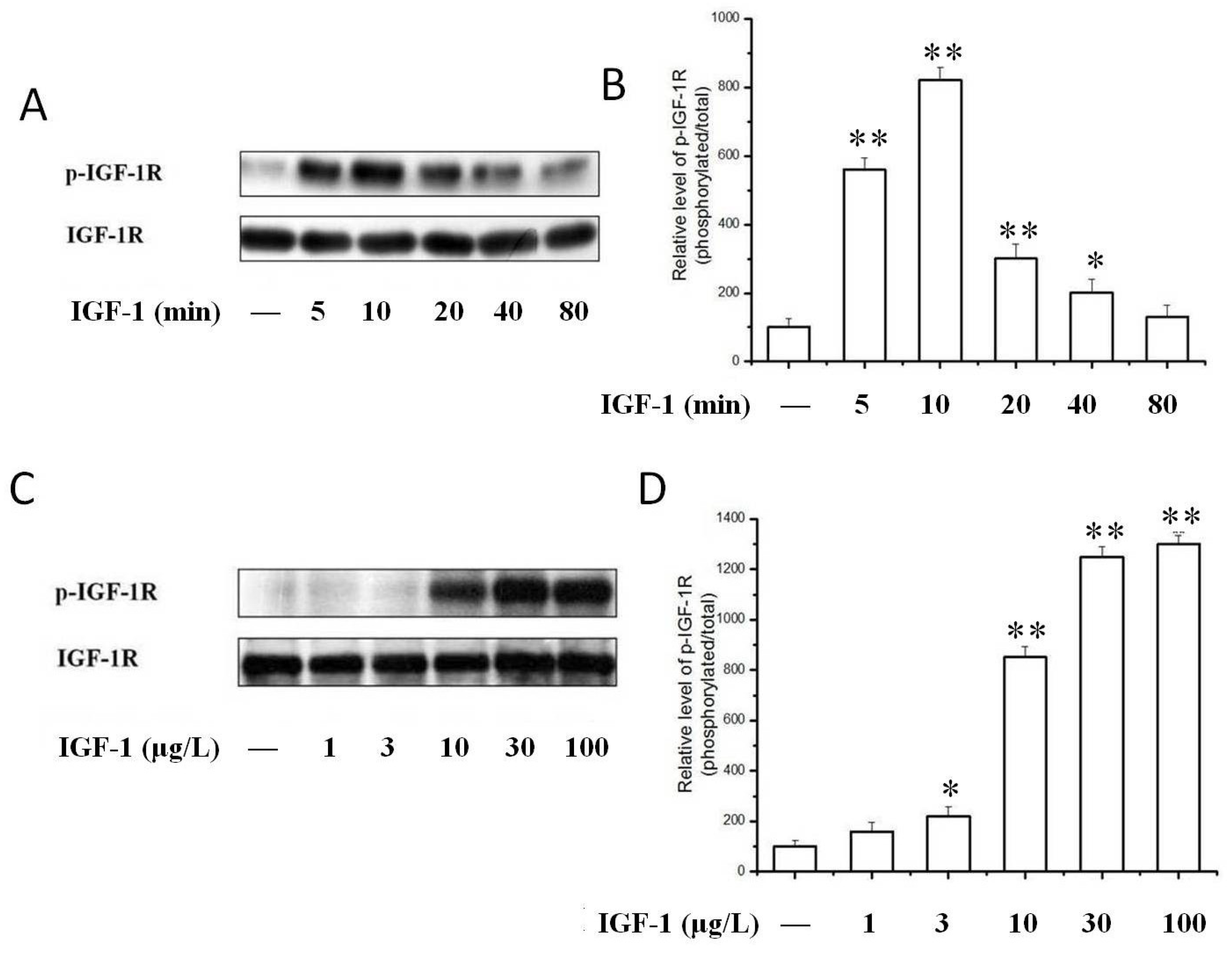
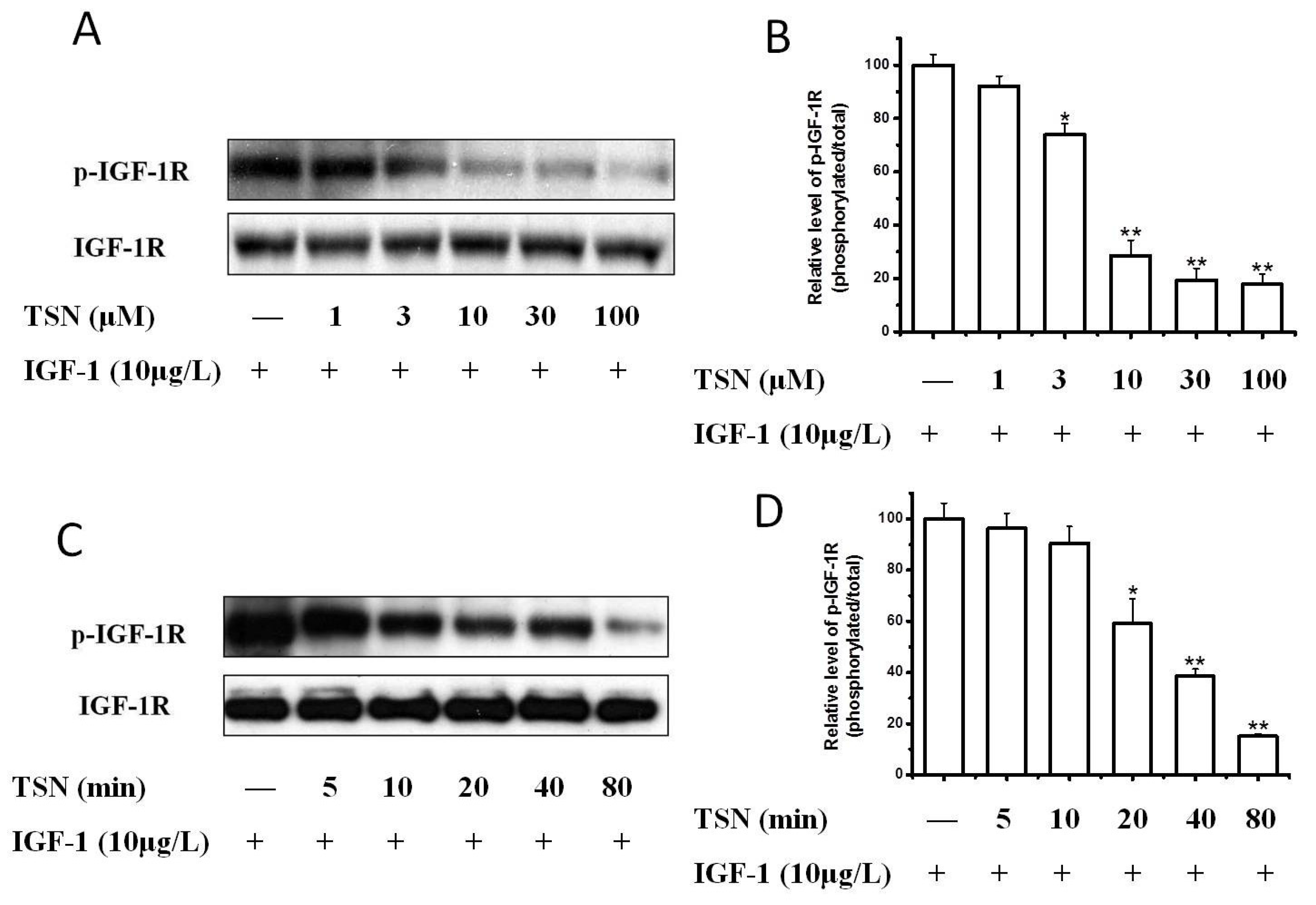
2.3. TSN Inhibited IGF-1-Induced Tyrosine Phosphorylation of IGF-1R in PC12 Cells
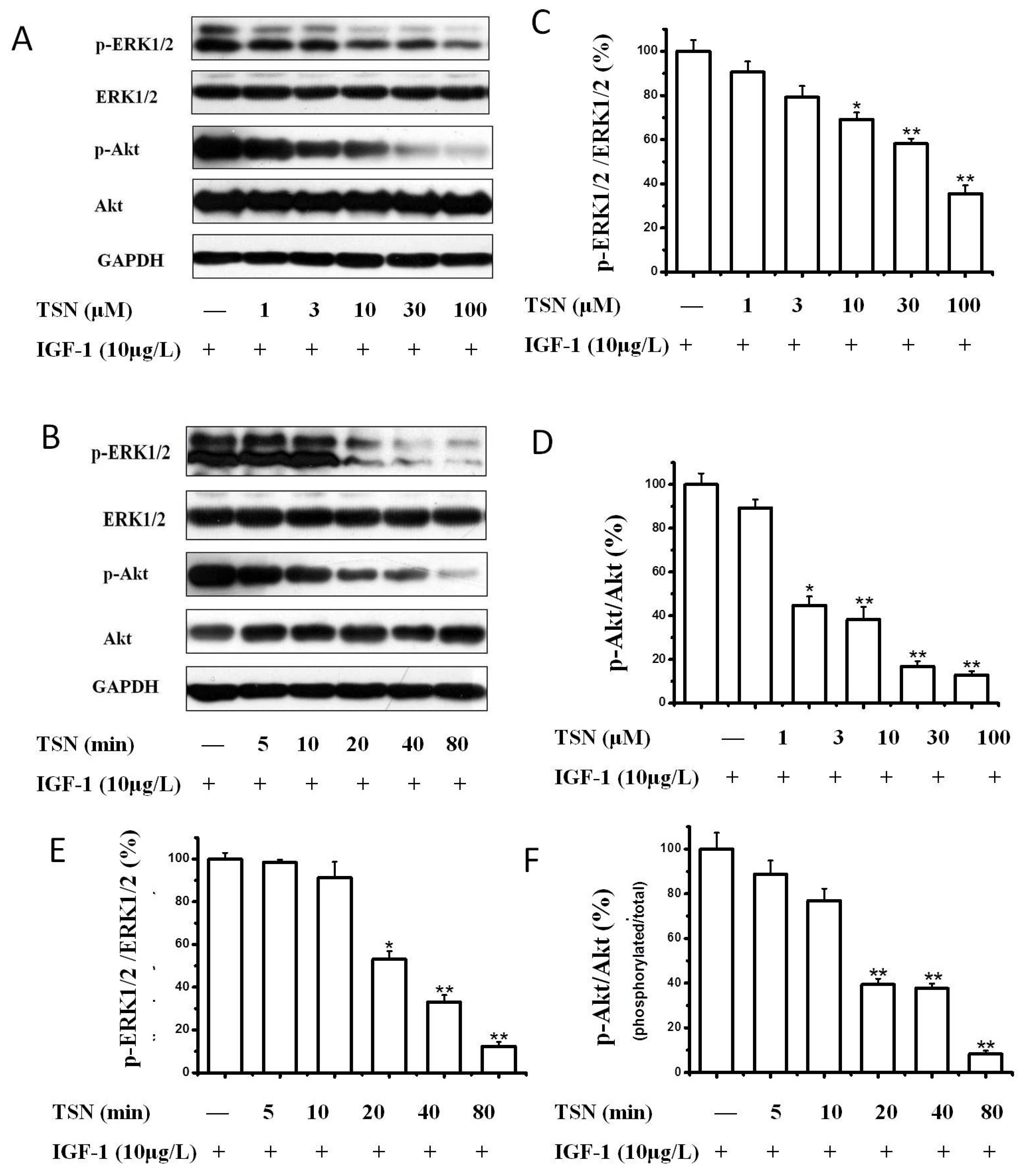
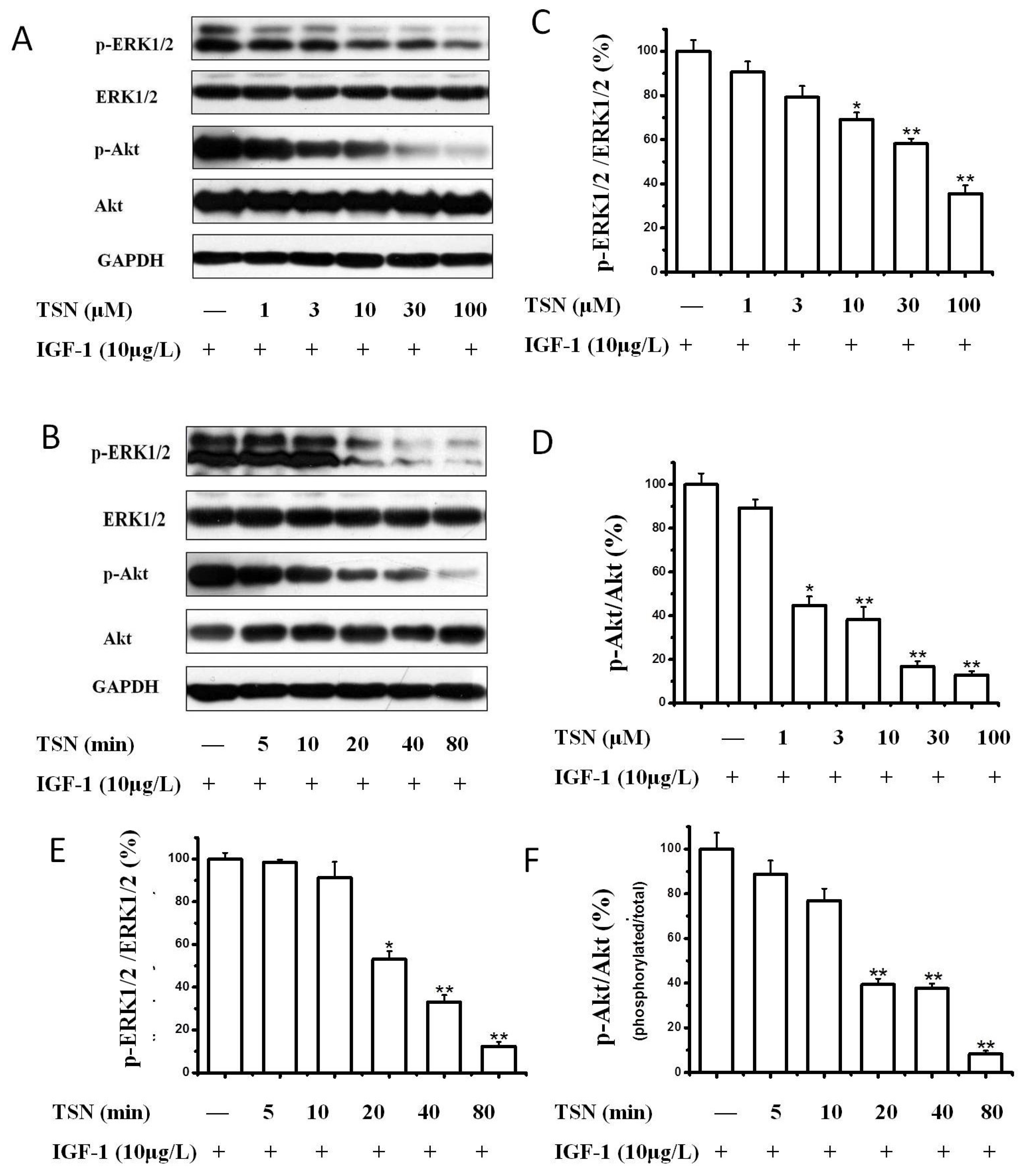
2.4. TSN Attenuated the Activation of Akt and MAPK Induced by IGF-1
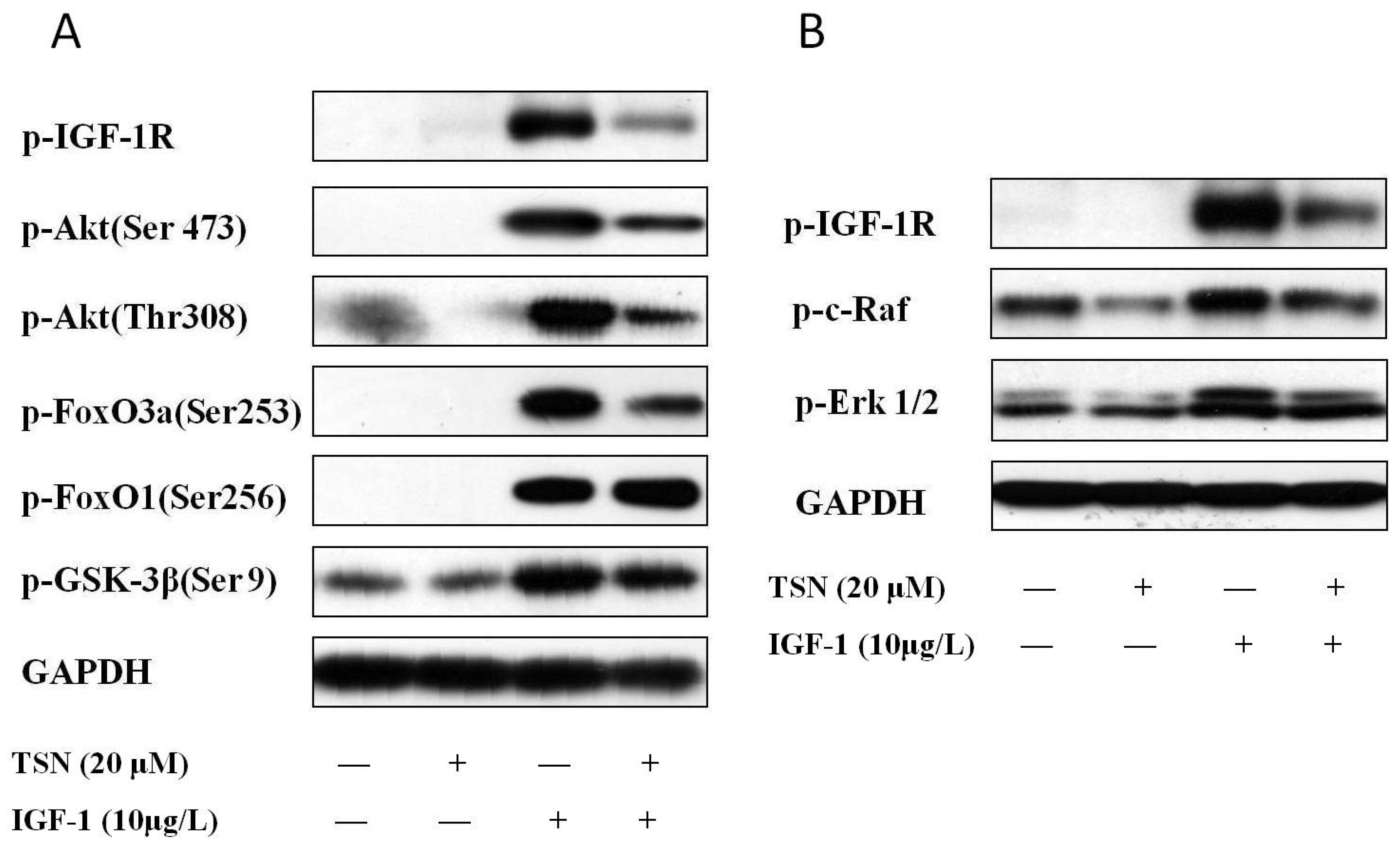
2.5. TSN Inhibited IGF-1R Mediated Akt and Mitogen-Activated Protein Kinase Kinase (MEK) Signaling Transduction
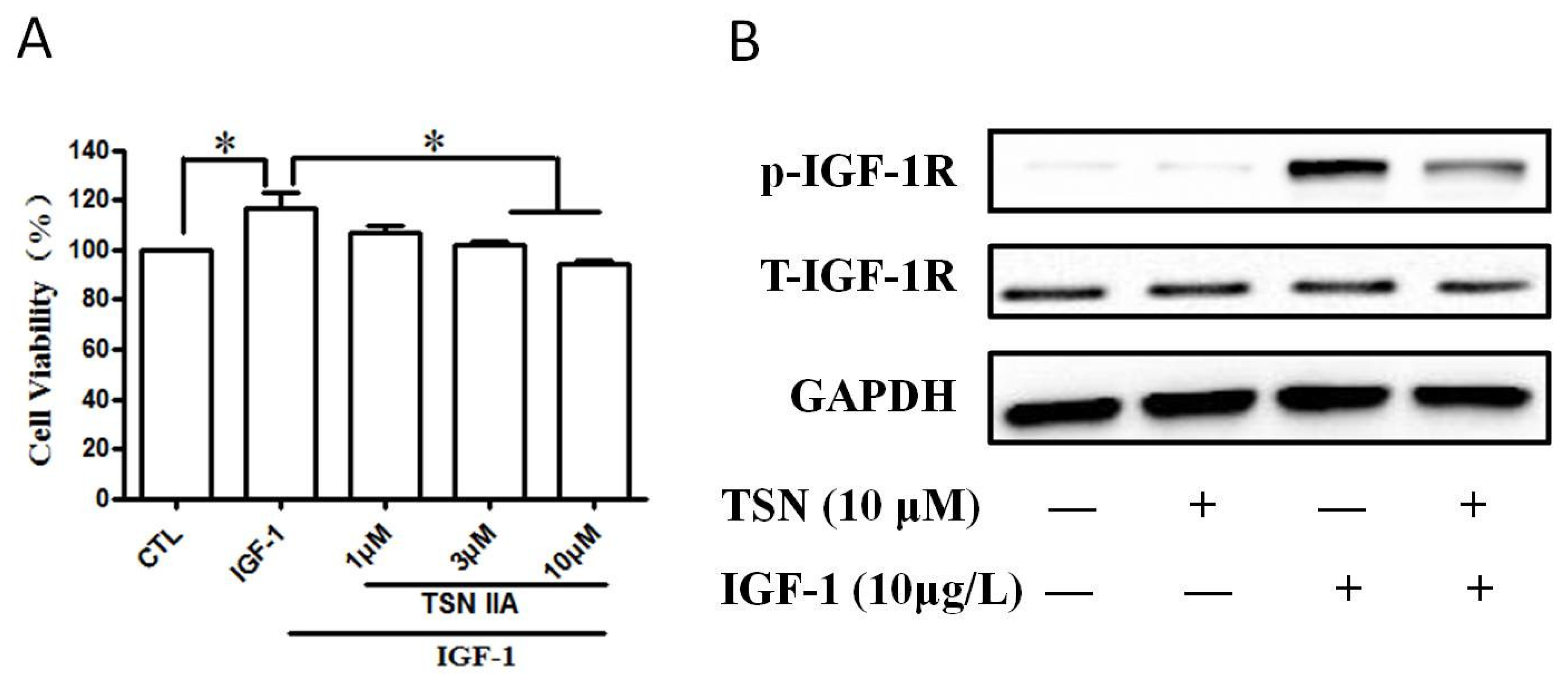
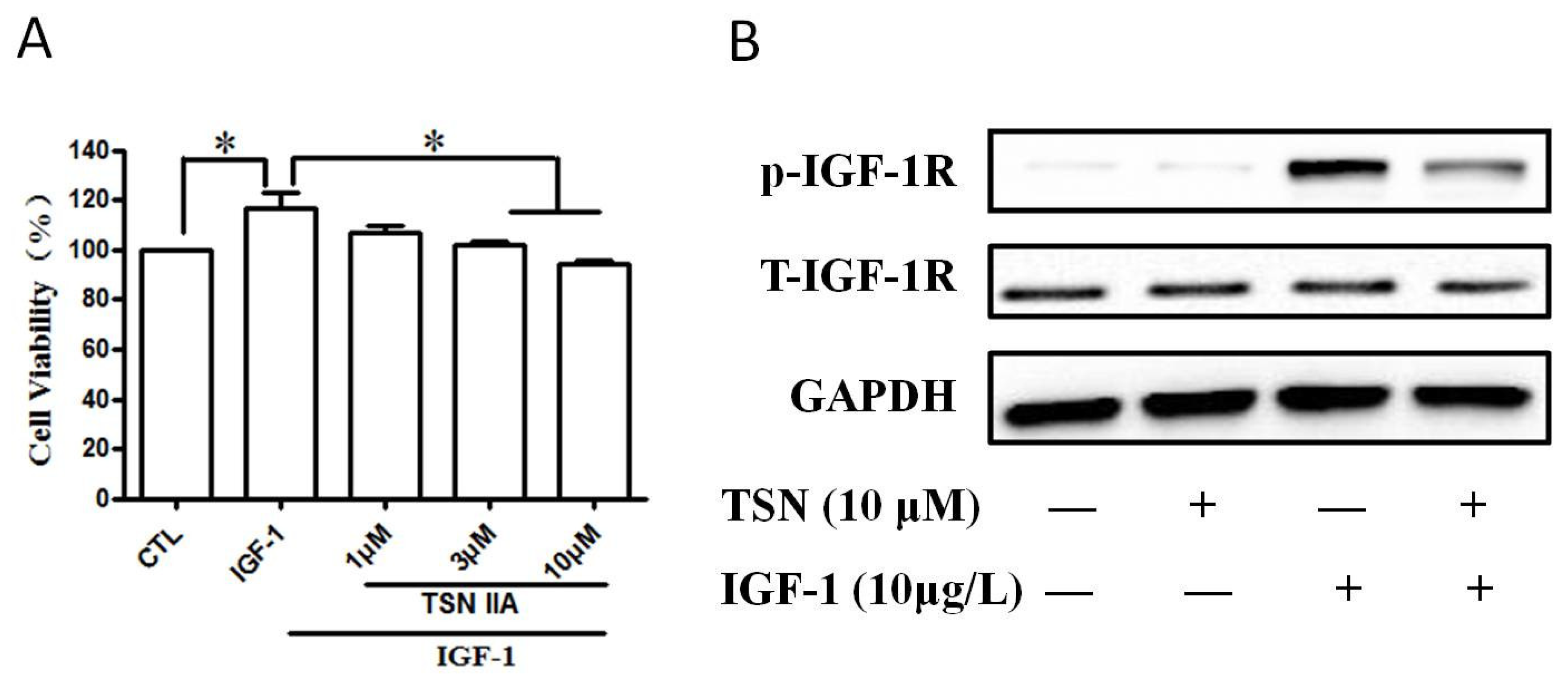
2.6. TSN Inhibited IGF-1-Induced Cell Growth and Tyrosine Phosphorylation of IGF-1R in SH-SY5Y Cells
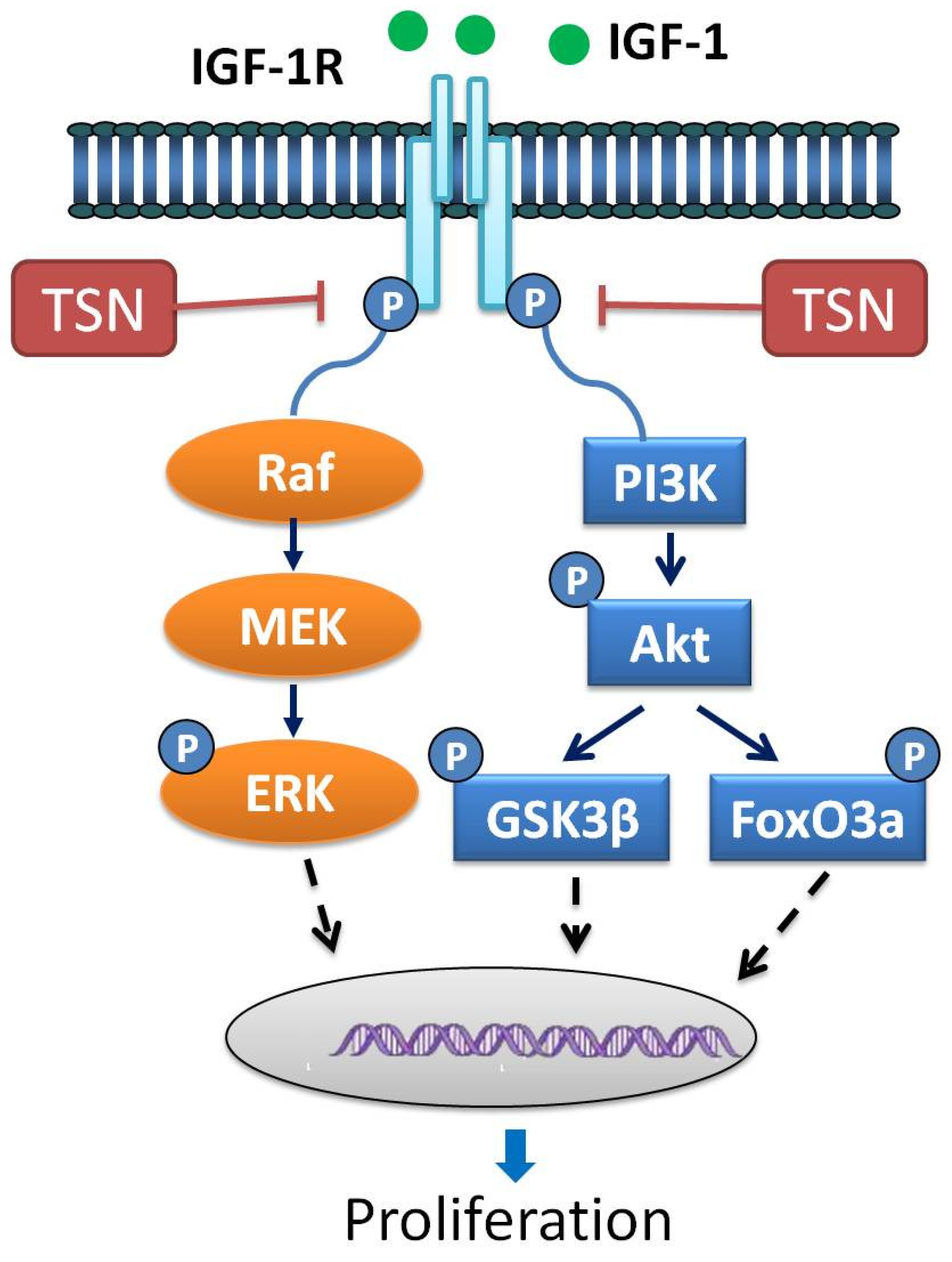
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Treatment
4.4. MTT Assay
4.5. CCK-8 Assay
4.6. Flow Cytometry Assay
4.7. Western Blotting
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| CCK-8 | cell counting kit-8 |
| CDKs | cyclin-dependent kinases |
| DMSO | dimethyl sulfoxide |
| ERK1/2 | extracellular-signal related kinase 1/2 |
| FOXO1 | forkhead box O1 |
| FOXO3a | forkhead box O3a |
| GSK-3β | glycogen synthase kinase-3β |
| IGF-1 | insulin like growth factor 1 |
| IGF-1R | insulin like growth factor 1 receptor |
| JNK | Jun-amino-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| MTT | methyl thiazolytetrazolium |
| p70S6K | ribosomal protein S6 kinase |
| PC12 | pheochromocytoma |
| PI3K | phosphatidylinositol-3-kinase |
| TSN | Tanshinone IIA |
References
- Zhang, X.D.; He, C.X.; Cheng, J.; Wen, J.; Li, P.Y.; Wang, N.; Li, G.; Zeng, X.R.; Cao, J.M.; Yang, Y. Sodium Tanshinone II-A Sulfonate (DS-201) Induces Vasorelaxation of Rat Mesenteric Arteries via Inhibition of L-Type Ca(2+) Channel. Front. Pharmacol. 2018, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Xu, Q.; Xu, W.H.; Guo, X.H.; Zhang, S.; Chen, Y.D. Mechanisms of protection against diabetes-induced impairment of endothelium-dependent vasorelaxation by Tanshinone IIA. Biochim. Biophys. Acta 2015, 1850, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Li, W.W.; Ji, J.; Hu, Q.H.; Ji, H. The cardioprotective effect of sodium tanshinone IIA sulfonate and the optimizing of therapeutic time window in myocardial ischemia/reperfusion injury in rats. Atherosclerosis 2014, 235, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Yao, Y.; Wang, T.; Huang, H.; Xia, H. Tanshinone IIA ameliorates apoptosis of myocardiocytes by up-regulation of miR-133 and suppression of Caspase-9. Eur. J. Pharmacol. 2017, 815, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, X.; Patal, K.; Hu, R.; Chuang, S.; Zhang, G.; Zheng, J. Tanshinones inhibit amyloid aggregation by amyloid-beta peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 2013, 4, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Liu, Q.; Xu, G.; Huang, Z.; Luo, N.; Huang, Y.; Cai, K. Synergistic effect of tanshinone IIA and mesenchymal stem cells on preventing learning and memory deficits via anti-apoptosis, attenuating tau phosphorylation and enhancing the activity of central cholinergic system in vascular dementia. Neurosci. Lett. 2017, 637, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.; Qian, S.S.; Zhang, Y.J.; Wang, R.Q. Salvia miltiorrhiza: A source for anti-Alzheimer’s disease drugs. Pharm. Biol. 2016, 54, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, C.; Xiang, Z.; Jiao, B. Tanshinone IIA reduces the risk of Alzheimer’s disease by inhibiting iNOS, MMP2 and NFkappaBp65 transcription and translation in the temporal lobes of rat models of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Wenping, H.; Yuan, Z.; Jie, S.; Lijun, Z.; Zhezhi, W. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, P. Tanshinone II-A: New perspectives for old remedies. Expert Opin. Ther. Pat. 2013, 23, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, C.; Wu, Q.; Zheng, K.; Chen, J.; Lan, Y.; Qin, Y.; Mei, W.; Wang, B. Evaluation of Tanshinone IIA Developmental Toxicity in Zebrafish Embryos. Molecules 2017, 22, 660. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.Q.; Cheng, X.L.; Yang, Y.; Yan, L.; Gu, J.L.; Li, H.; Zeng, X.R.; Cao, J.M. Tanshinone II-A sodium sulfonate (DS-201) enhances human BKCa channel activity by selectively targeting the pore-forming alpha subunit. Acta Pharmacol. Sin. 2014, 35, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, W.; Zhao, J.; Wu, X.; Lu, J.J.; Chen, X.; Xu, Q.M.; Khan, I.A.; Yang, S. Tanshinones and diethyl blechnics with anti-inflammatory and anti-cancer activities from Salvia miltiorrhiza Bunge (Danshen). Sci. Rep. 2016, 6, 33720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.O.; Kang, S.E.; Im, C.R.; Lee, J.H.; Ahn, K.S.; Yang, W.M.; Um, J.Y.; Lee, S.G.; Yun, M. Tanshinone IIA induces TRAIL sensitization of human lung cancer cells through selective ER stress induction. Int. J. Oncol. 2016, 48, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Q.; Liu, F.; Cheng, X.; Wu, X.; Wang, H.; Wu, M.; Ma, Y.; Wang, G.; Hao, H. UDP-glucuronosyltransferase 1A compromises intracellular accumulation and anti-cancer effect of tanshinone IIA in human colon cancer cells. PLoS ONE 2013, 8, e79172. [Google Scholar] [CrossRef] [PubMed]
- Guerram, M.; Jiang, Z.Z.; Yousef, B.A.; Hamdi, A.M.; Hassan, H.M.; Yuan, Z.Q.; Luo, H.W.; Zhu, X.; Zhang, L.Y. The potential utility of acetyltanshinone IIA in the treatment of HER2-overexpressed breast cancer: Induction of cancer cell death by targeting apoptotic and metabolic signaling pathways. Oncotarget 2015, 6, 21865–21877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhou, J.; Geng, G.; Shi, Q.; Sauriol, F.; Wu, J.H. Antiandrogenic, maspin induction, and antiprostate cancer activities of tanshinone IIA and its novel derivatives with modification in ring A. J. Med. Chem. 2012, 55, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C. Tanshinone IIA inhibits gastric carcinoma AGS cells through increasing p-p38, p-JNK and p53 but reducing p-ERK, CDC2 and cyclin B1 expression. Anticancer Res. 2014, 34, 7097–7110. [Google Scholar] [PubMed]
- Huang, S.T.; Huang, C.C.; Huang, W.L.; Lin, T.K.; Liao, P.L.; Wang, P.W.; Liou, C.W.; Chuang, J.H. Tanshinone IIA induces intrinsic apoptosis in osteosarcoma cells both in vivo and in vitro associated with mitochondrial dysfunction. Sci. Rep. 2017, 7, 40382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Shan, C.; Liu, L.; Zhou, T.; Zhou, J.; Hu, X.; Chen, Y.; Cui, H.; Gao, N. Tanshinone IIA inhibits HIF-1alpha and VEGF expression in breast cancer cells via mTOR/p70S6K/RPS6/4E-BP1 signaling pathway. PLoS ONE 2015, 10, e0117440. [Google Scholar]
- Tsai, M.Y.; Yang, R.C.; Wu, H.T.; Pang, J.H.; Huang, S.T. Anti-angiogenic effect of Tanshinone IIA involves inhibition of matrix invasion and modification of MMP-2/TIMP-2 secretion in vascular endothelial cells. Cancer Lett. 2011, 310, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Fernagut, P.O.; Bezard, E.; Meissner, W.G. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: Targets for disease modification? Prog. Neurobiol. 2014, 118, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bruchim, I.; Werner, H. Targeting IGF-1 signaling pathways in gynecologic malignancies. Expert Opin. Ther. Targets 2013, 17, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Worrall, C.; Takahashi, S.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, S.; Geng, R.; Zheng, Y.; Liao, R.; Yan, F.; Thrimawithana, T.; Little, P.J.; Feng, Z.P.; Lazarovici, P.; et al. IGF-1 signaling via the PI3K/Akt pathway confers neuroprotection in human retinal pigment epithelial cells exposed to sodium nitroprusside insult. J. Mol. Neurosci. 2015, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Q.; Zhang, L.; Little, P.J.; Xie, X.; Meng, Q.; Ren, Y.; Zhou, L.; Gao, G.; Quirion, R.; et al. Insulin-like growth factor-1 induces the phosphorylation of PRAS40 via the PI3K/Akt signaling pathway in PC12 cells. Neurosci. Lett. 2012, 516, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultured hippocampal neurons. Mol. Pharmacol. 2002, 62, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.C.; Venara, M.; Nowicki, S.; Chemes, H.E.; Barontini, M.; Pennisi, P.A. Igf-I regulates pheochromocytoma cell proliferation and survival in vitro and in vivo. Endocrinology 2012, 153, 3724–3734. [Google Scholar] [CrossRef] [PubMed]
- Lonn, S.; Inskip, P.D.; Pollak, M.N.; Weinstein, S.J.; Virtamo, J.; Albanes, D. Glioma risk in relation to serum levels of insulin-like growth factors. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Brahmkhatri, V.P.; Prasanna, C.; Atreya, H.S. Insulin-like growth factor system in cancer: Novel targeted therapies. Biomed Res. Int. 2015, 2015, 538019. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ito, F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol. Pharm. Bull. 2011, 34, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Quirion, R. Insulin-like growth factor-1 (IGF-1) induces the activation/phosphorylation of Akt kinase and cAMP response element-binding protein (CREB) by activating different signaling pathways in PC12 cells. BMC Neurosci. 2006, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Liu, S.; Shao, T.; Hua, H.; Kong, Q.; Wang, J.; Luo, T.; Jiang, Y. GSK3 protein positively regulates type I insulin-like growth factor receptor through forkhead transcription factors FOXO1/3/4. J. Biol. Chem. 2014, 289, 24759–24770. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Arachchige-Don, A.P.; Donaldson, M.S.; Patriarchi, T.; Horne, M.C. Cyclin G2 promotes cell cycle arrest in breast cancer cells responding to fulvestrant and metformin and correlates with patient survival. Cell Cycle 2016, 15, 3278–3295. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Zhu, Y. FOXO3a involvement in the release of TNF-alpha stimulated by ATP in spinal cord astrocytes. J. Mol. Neurosci. 2013, 51, 792–804. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelder, R.E.; Yoon, S.O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Olmos, Y. The complex biology of FOXO. Curr. Drug Targets 2011, 12, 1322–1350. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Fang, Y.; Dong, W.; Mu, X.; Liu, Q.; Du, J. IGF-1 receptor is down-regulated by sunitinib induces MDM2-dependent ubiquitination. FEBS Open Bio 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhu, J.; Yu, Y.; Zhang, Z.; Chen, G.; Zhou, X.; Qiao, C.; Hou, T.; Mao, X. A virtual screen identified C96 as a novel inhibitor of phosphatidylinositol 3-kinase that displays potent preclinical activity against multiple myeloma in vitro and in vivo. Oncotarget 2014, 5, 3836–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.Y.; Ahn, J.W.; Kim, H.S.; Lee, J.; Yoon, J.H. Apicidin inhibits cell growth by downregulating IGF-1R in salivary mucoepidermoid carcinoma cells. Oncol. Rep. 2015, 33, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, B.; Li, Y.; Niu, B.; Wang, X.; Cheng, Y.; Zhou, Z.; You, T.; Liu, Y.; Wang, H.; Xu, J. Involvement of PI3K/Akt/FoxO3a and PKA/CREB Signaling Pathways in the Protective Effect of Fluoxetine Against Corticosterone-Induced Cytotoxicity in PC12 Cells. J. Mol. Neurosci. 2016, 59, 567–578. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Su, X.; Fang, J.; Xin, X.; Zhao, X.; Gaur, U.; Wen, Q.; Xu, J.; Little, P.J.; Zheng, W. Tanshinone IIA Attenuates Insulin Like Growth Factor 1 -Induced Cell Proliferation in PC12 Cells through the PI3K/Akt and MEK/ERK Pathways. Int. J. Mol. Sci. 2018, 19, 2719. https://doi.org/10.3390/ijms19092719
Wang H, Su X, Fang J, Xin X, Zhao X, Gaur U, Wen Q, Xu J, Little PJ, Zheng W. Tanshinone IIA Attenuates Insulin Like Growth Factor 1 -Induced Cell Proliferation in PC12 Cells through the PI3K/Akt and MEK/ERK Pathways. International Journal of Molecular Sciences. 2018; 19(9):2719. https://doi.org/10.3390/ijms19092719
Chicago/Turabian StyleWang, Haitao, Xiaoying Su, Jiankang Fang, Xingan Xin, Xia Zhao, Uma Gaur, Qiang Wen, Jiangping Xu, Peter J. Little, and Wenhua Zheng. 2018. "Tanshinone IIA Attenuates Insulin Like Growth Factor 1 -Induced Cell Proliferation in PC12 Cells through the PI3K/Akt and MEK/ERK Pathways" International Journal of Molecular Sciences 19, no. 9: 2719. https://doi.org/10.3390/ijms19092719