PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease

 , , ,
, , ,
Abstract
:
1. Introduction
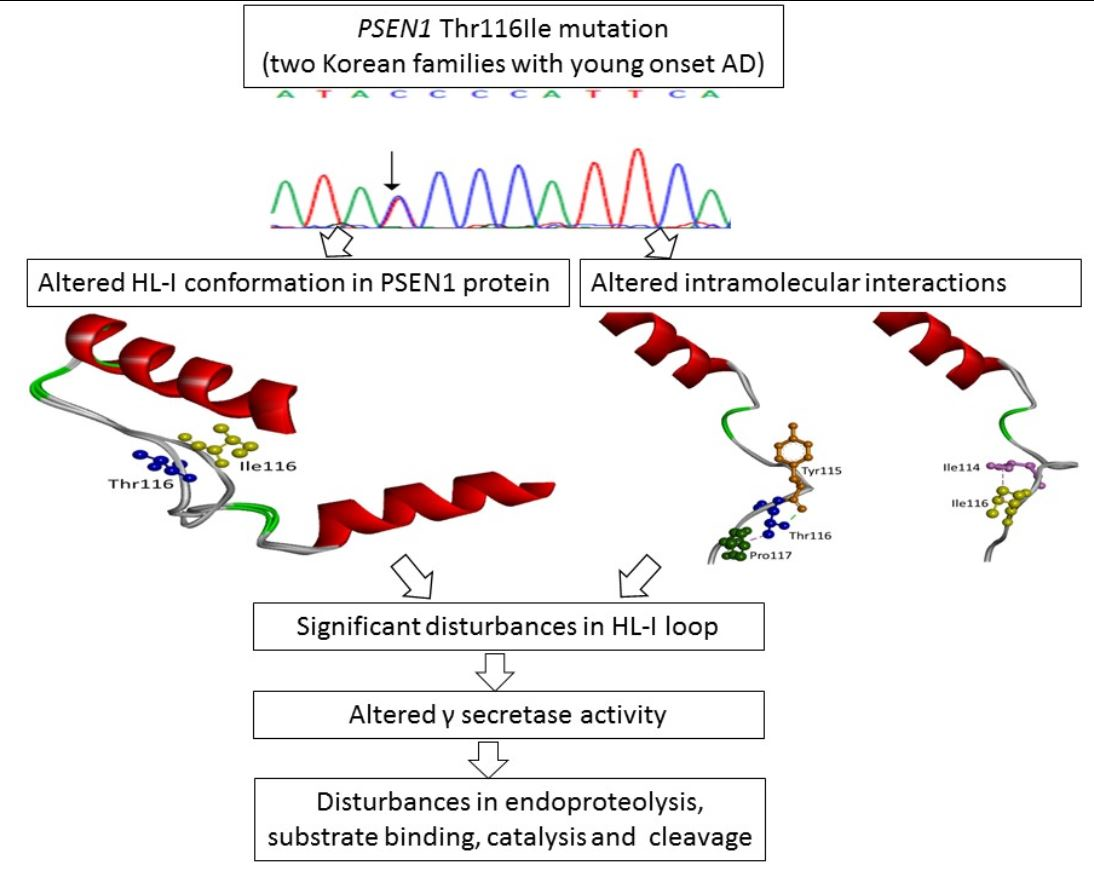
2. Results
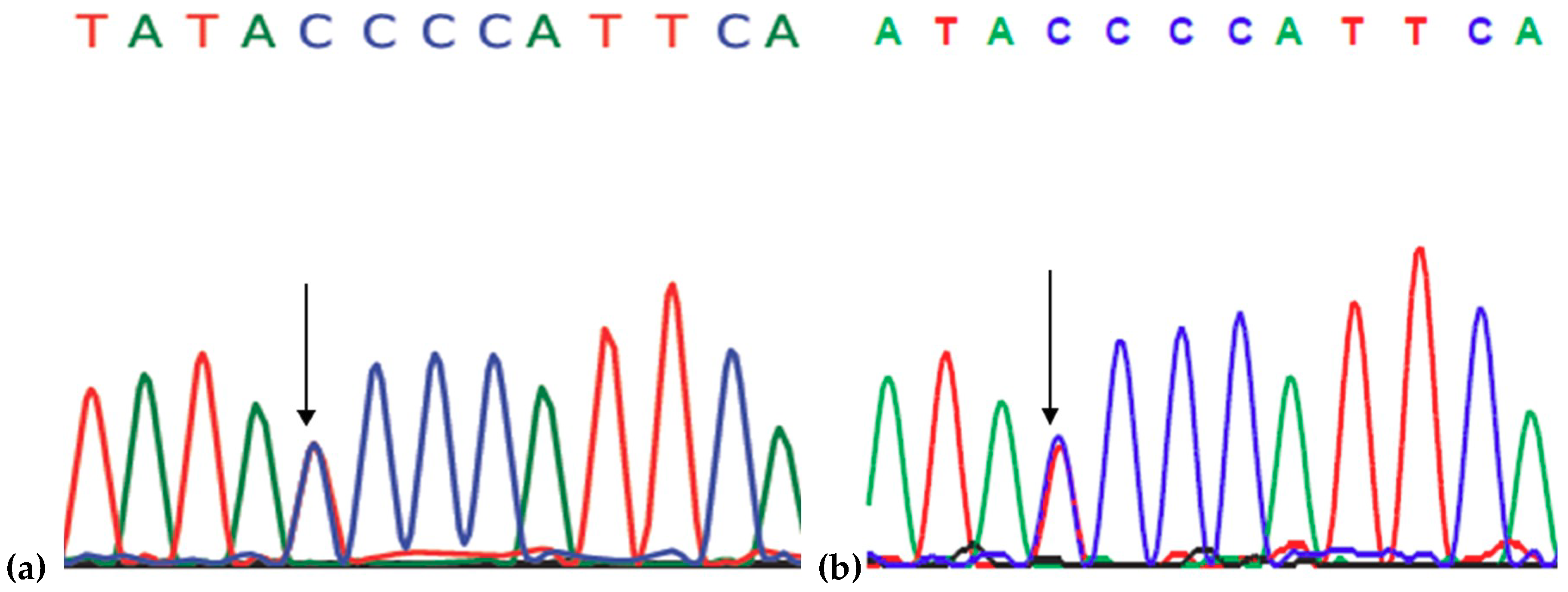
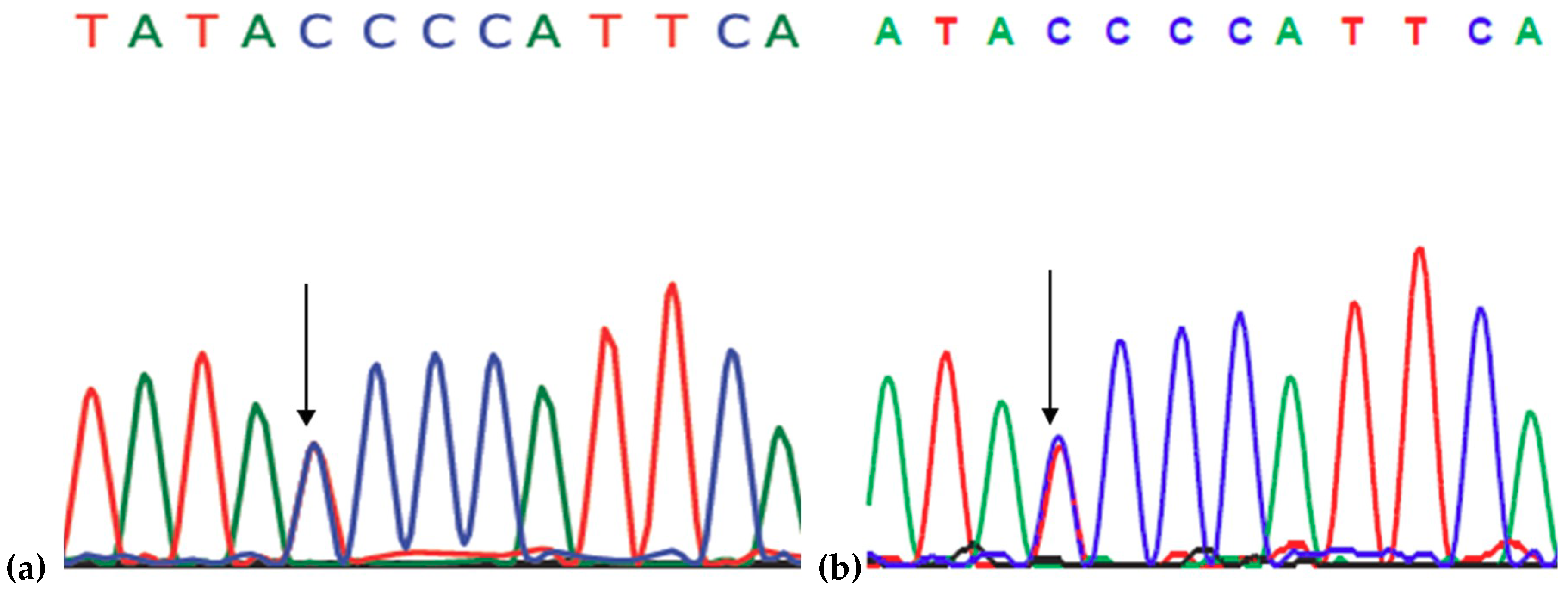
2.1. Genetic Analysis
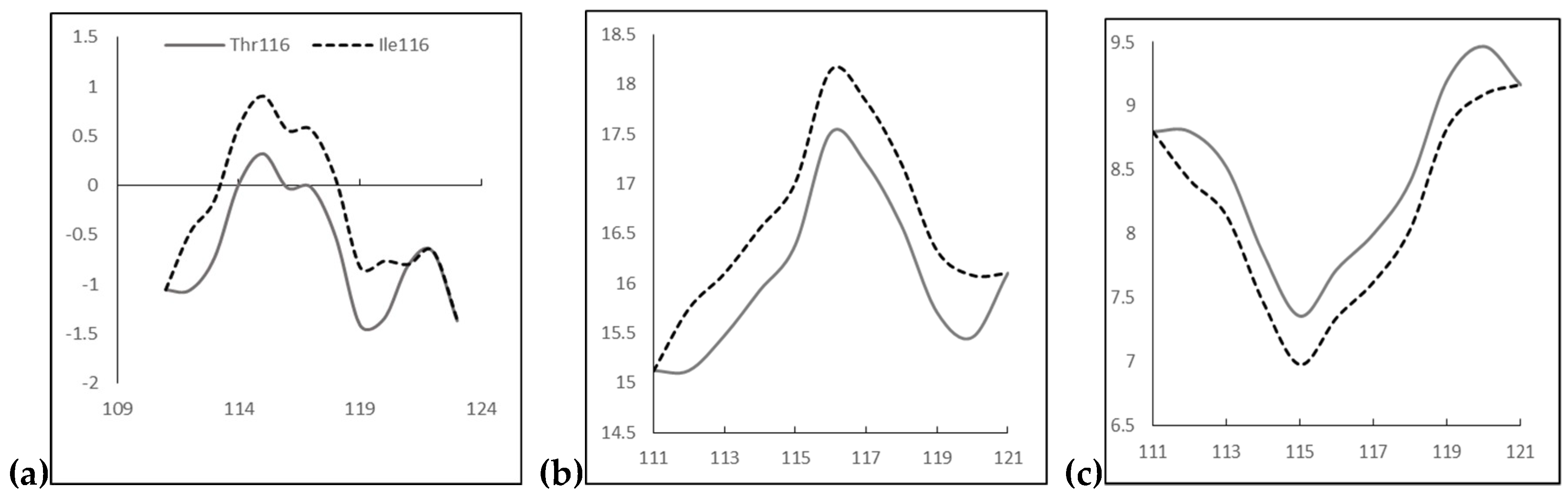
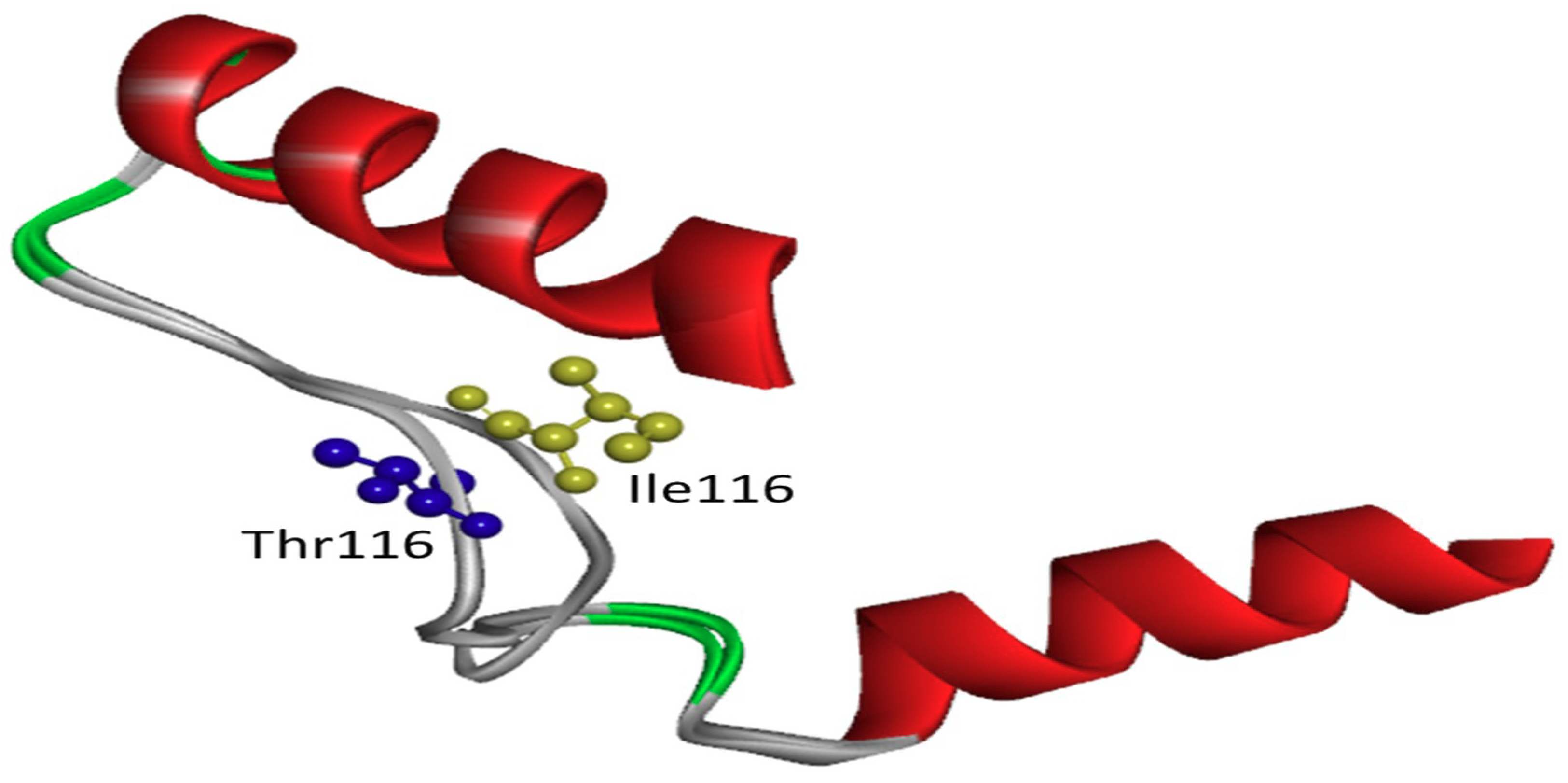
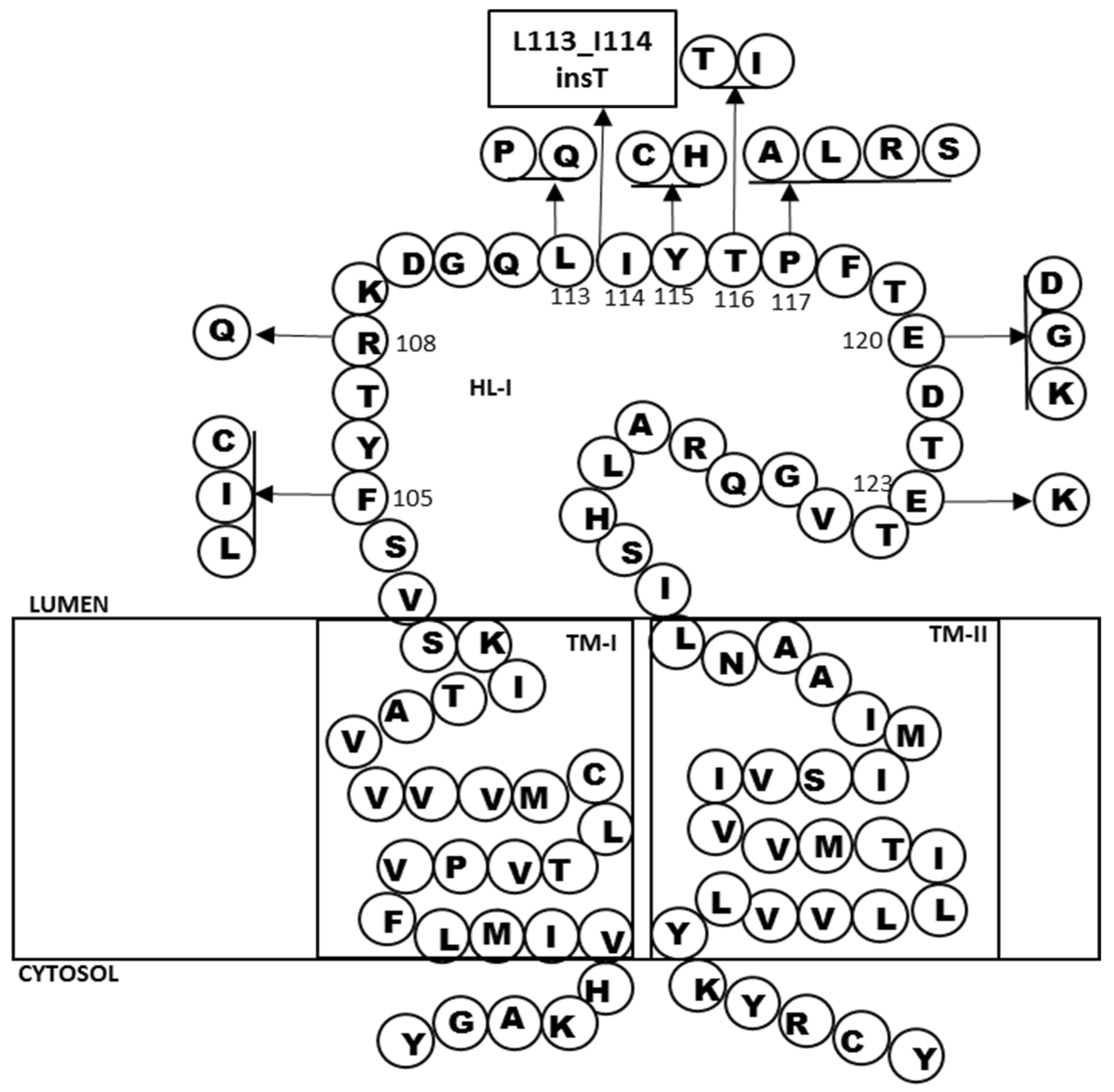
2.2. In Silico Predictions and 3D Modeling
3. Discussion
4. Materials and Methods
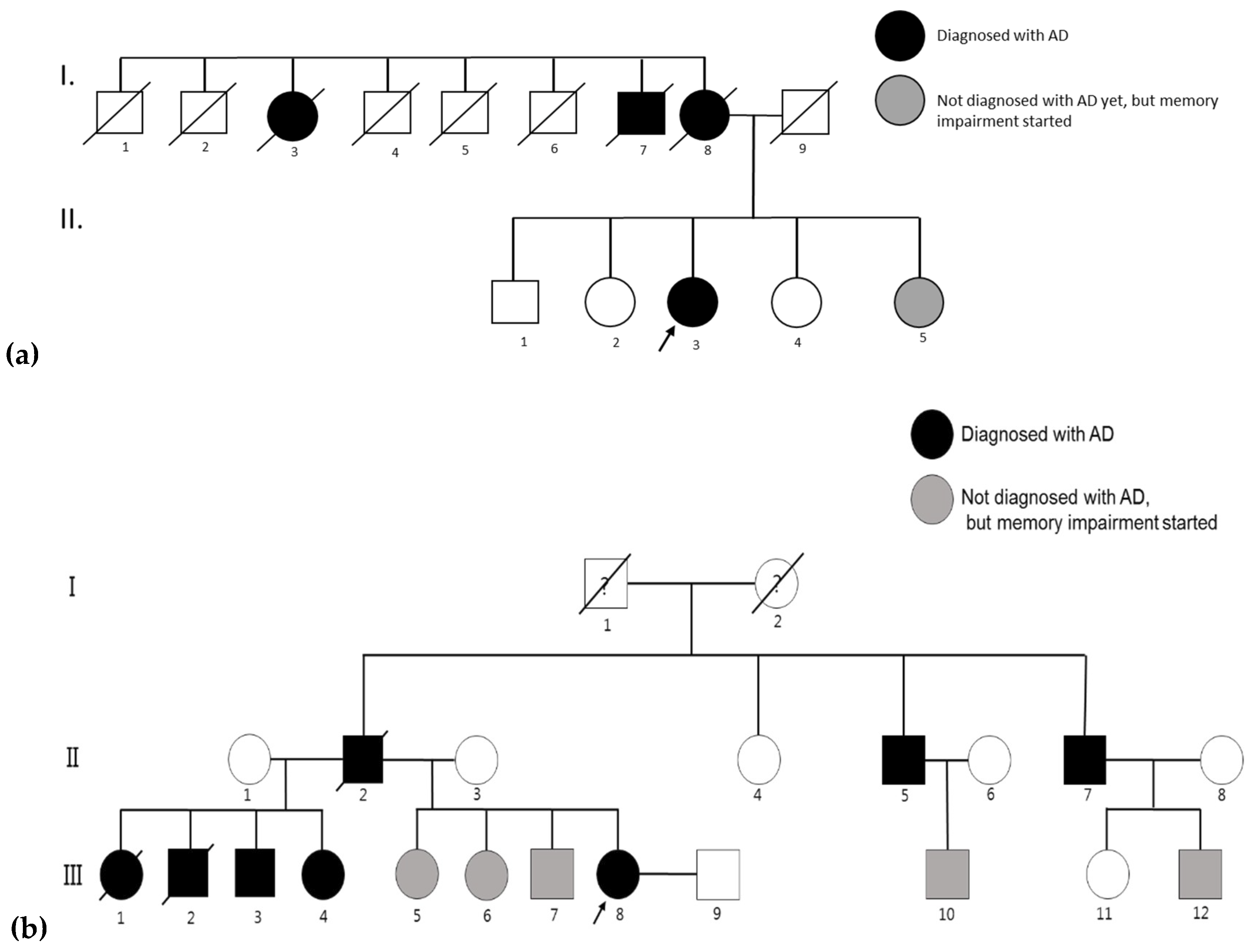
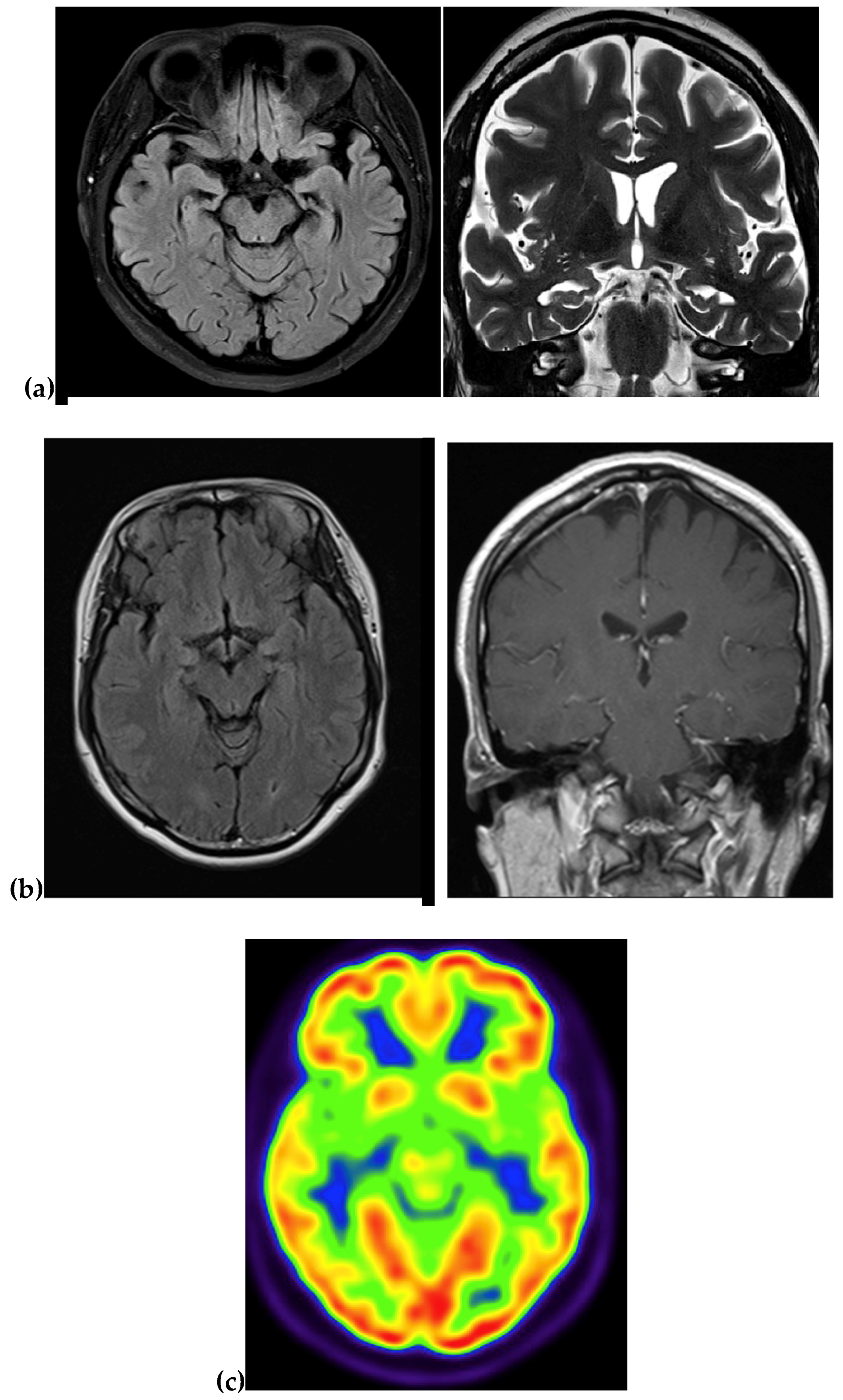
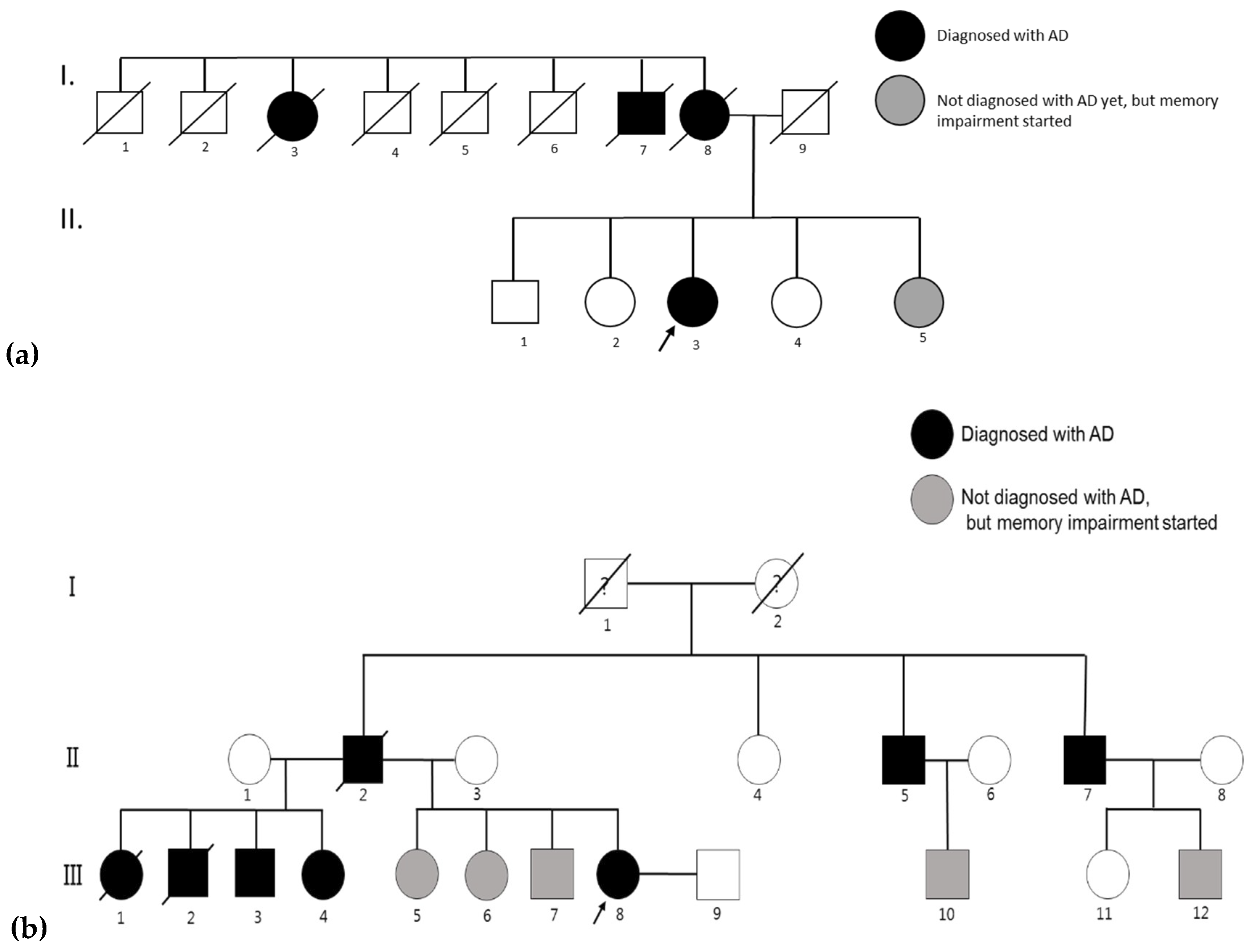
4.1. AD Patients and Their Families
4.1.1. Family 1
4.1.2. Family 2
4.2. Genetic Analysis
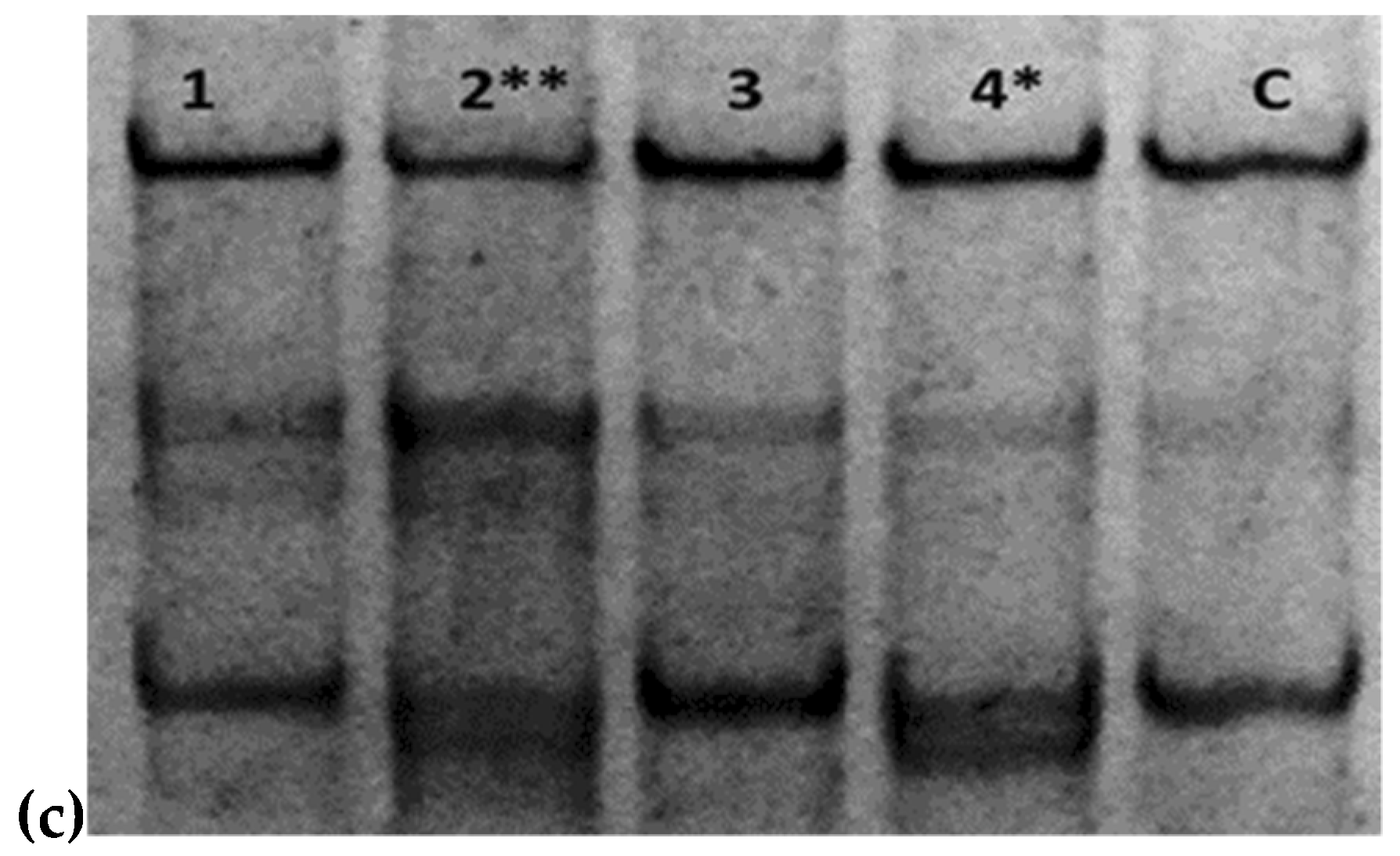
4.3. Single Strand Conformation Polymorphism (SSCP)
4.4. In Silico Screening and Structure Predictions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S.Y. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure, mechanism and inhibition of gamma-secretase and presenilin-like proteases. Biol. Chem. 2010, 391, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Liu, C.; Che, C.; Huang, H. Clinical genetics of Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 291862. [Google Scholar] [CrossRef] [PubMed]
- Ertekin-Taner, N. Genetics of Alzheimer’s disease: A centennial review. Neurol. Clin. 2007, 25, 611–667. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Flaman, J.M.; Brice, A.; Hannequin, D.; Dubois, B.; Martin, C.; Moreau, V.; Charbonnier, F.; Didierjean, O.; Tardieu, S.; et al. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum. Mol. Genet. 1995, 4, 2373–2377. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.; Park, S.A.; Bagyinszky, E.; Bae, S.O.; Kim, Y.J.; Im, J.Y.; Park, K.W.; Park, K.H.; Kim, E.J.; Jeong, J.H.; et al. A genetic screen of the mutations in the Korean patients with early-onset Alzheimer’s disease. Clin. Interv. Aging 2016, 11, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- La Bella, V.; Liguori, M.; Cittadella, R.; Settipani, N.; Piccoli, T.; Manna, I.; Quattrone, A.; Piccoli, F. A novel mutation (Thr116Ile) in the presenilin 1 gene in a patient with early-onset Alzheimer’s disease. Eur. J. Neurol. 2004, 11, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Raux, G.; Guyant-Marechal, L.; Martin, C.; Bou, J.; Penet, C.; Brice, A.; Hannequin, D.; Frebourg, T.; Campion, D. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: An update. J. Med. Genet. 2005, 42, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Quillard-Muraine, M.; Guyant-Maréchal, L.; Martinaud, O.; Pariente, J.; Puel, M.; Rollin-Sillaire, A.; Pasquier, F.; et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: Mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimers Dis. 2012, 30, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Jorgensen, P.; Bolwig, G.; Fraser, P.E.; Rogaeva, E.; Mann, D.; Havsager, A.M.; Jørgensen, A.L. A presenilin-1 Thr116Asn substitution in a family with early-onset Alzheimer’s disease. Neuroreport 1999, 10, 2255–2260. [Google Scholar] [CrossRef] [PubMed]
- Tysoe, C.; Whittaker, J.; Xuereb, J.; Cairns, N.J.; Cruts, M.; Van Broeckhoven, C.; Wilcock, G.; Rubinsztein, D.C. A presenilin-1 truncating mutation is present in two cases with autopsy-confirmed early-onset Alzheimer disease. Am. J. Hum. Genet. 1998, 62, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Dobricic, V.; Stefanova, E.; Jankovic, M.; Gurunlian, N.; Novakovic, I.; Hardy, J.; Kostic, V.; Guerreiro, R. Genetic testing in familial and young-onset Alzheimer’s disease: Mutation spectrum in a serbian cohort. Neurobiol. Aging 2012, 33, 1481.e7–1481.e12. [Google Scholar] [CrossRef] [PubMed]
- Finckh, U.; Kuschel, C.; Anagnostouli, M.; Patsouris, E.; Pantes, G.V.; Gatzonis, S.; Kapaki, E.; Davaki, P.; Lamszus, K.; Stavrou, D.; et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics 2005, 6, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Raux, G.; Gantier, R.; Thomas-Anterion, C.; Boulliat, J.; Verpillat, P.; Hannequin, D.; Brice, A.; Frebourg, T.; Campion, D. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 2000, 55, 1577–1578. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Dowjat, W.K.; Buxbaum, J.D.; Khorkova, O.; Efthimiopoulos, S.; Kulczycki, J.; Lojkowska, W.; Wegiel, J.; Wisniewski, H.M.; Frangione, B. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport 2008, 9, 217–221. [Google Scholar] [CrossRef]
- Cruts, M.; van Duijn, C.M.; Backhovens, H.; Van den Broeck, M.; Wehnert, A.; Serneels, S.; Sherrington, R.; Hutton, M.; Hardy, J.; St George-Hyslop, P.H.; et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 1998, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Sharma, V.; Anderson, L.; Nemens, E.; Alonso, M.E.; Orr, H.; White, J.; Heston, L.; Bird, T.D.; Schellenberg, G.D. Missense mutations in the chromosome 14 familial Alzheimer’s disease presenilin 1 gene. Hum. Mutat. 1998, 11, 216–221. [Google Scholar] [CrossRef]
- Anheim, M.; Hannequin, D.; Boulay, C.; Martin, C.; Campion, D.; Tranchant, C. Ataxic variant of Alzheimer’s disease caused by Pro117Ala PSEN1 mutation. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1414–1415. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Maeda, K.; Hashimoto, M.; Yamashita, H.; Ikejiri, Y.; Bird, T.D.; Tanaka, C.; Schellenberg, G.D. A pedigree with a novel presenilin 1 mutation at a residue that is not conserved in presenilin 2. Arch. Neurol. 1999, 56, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Vetrivel, K.S.; Nguyen, P.D.; Meckler, X.; Cheng, H.; Kounnas, M.Z.; Wagner, S.L.; Parent, A.T.; Thinakaran, G. Mutation analysis of the presenilin 1 N-terminal domain reveals a broad spectrum of gamma-secretase activity toward amyloid precursor protein and other substrates. J. Biol. Chem. 2010, 285, 38042–38052. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Kosik, K.; Haltia, M.; Poyhonen, M.; Dickson, D.; Mann, D.; Neary, D.; Snowdon, J.; Lantos, P.; Lannfelt, L.; et al. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 1995, 11, 219–222. [Google Scholar]
- Takagi-Niidome, S.; Sasaki, T.; Osawa, S.; Sato, T.; Morishima, K.; Cai, T.; Iwatsubo, T.; Tomita, T. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of γ-secretase. J. Neurosci. 2015, 35, 2646–2656. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Tang, B.; Liu, X.; Xu, J.; Wang, Y.; Zhou, L.; Zhang, F.; Yan, X.; Zhou, Y.; Shen, L. Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol. Aging 2014, 35, 1957.e1–1957.e6. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Flaman, J.M.; Brice, A.; Hannequin, D.; Dubois, B.; Martin, C.; Moreau, V.; Charbonnier, F.; Didierjean, O.; Tardieu, S. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum. Mol. Genet. 1995, 4, 2373–2377. [Google Scholar] [CrossRef] [PubMed]
- Zekanowski, C.; Styczyńska, M.; Pepłońska, B.; Gabryelewicz, T.; Religa, D.; Ilkowski, J.; Kijanowska-Haładyna, B.; Kotapka-Minc, S.; Mikkelsen, S.; Pfeffer, A.; et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp. Neurol. 2003, 184, 991–996. [Google Scholar] [CrossRef]
- Dowjat, W.K.; Wisniewski, T.; Efthimiopoulos, S.; Wisniewski, H.M. Inhibition of neurite outgrowth by familial Alzheimer’s disease-linked presenilin-1 mutations. Neurosci. Lett. 1999, 267, 141–144. [Google Scholar] [CrossRef]
- Lladó, A.; Sánchez-Valle, R.; Rey, M.J.; Mercadal, P.; Almenar, C.; López-Villegas, D.; Fortea, J.; Molinuevo, J.L. New mutation in the PSEN1 (E120G) gene associated with early onset Alzheimer’s disease. Neurologia 2010, 25, 13–16. [Google Scholar] [CrossRef]
- Hutton, M.; Busfield, F.; Wragg, M.; Crook, R.; Perez-Tur, J.; Clark, R.F.; Prihar, G.; Talbot, C.; Phillips, H.; Wright, K.; et al. Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. Neuroreport 1996, 7, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: A case series. Lancet. Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Pericak-Vance, M.A.; Wijsman, E.M.; Moore, D.K.; Gaskell, P.C.; Yamaoka, L.A.; Bebout, J.L.; Anderson, L.; Welsh, K.A.; Clark, C.M.; et al. Linkage analysis of familial Alzheimer disease, using chromosome 21 markers. Am. J. Hum. Genet. 1991, 48, 563–583. [Google Scholar] [PubMed]
- Tanzi, R.E.; Vaula, G.; Romano, D.M.; Mortilla, M.; Huang, T.L.; Tupler, R.G.; Wasco, W.; Hyman, B.T.; Haines, J.L.; Jenkins, B.J.; et al. Assessment of amyloid β-protein precursor gene mutations in a large set of familial and sporadic alzheimer disease cases. Am. J. Hum. Genet. 1992, 51, 273–282. [Google Scholar] [PubMed]
- Kamimura, K.; Tanahashi, H.; Yamanaka, H.; Takahashi, K.; Asada, T.; Tabira, T. Familial Alzheimer’s disease genes in Japanese. J. Neurol. Sci. 1998, 160, 76–81. [Google Scholar] [CrossRef]
- Jeong, B.H.; Ju, W.K.; Huh, K.; Lee, E.A.; Choi, I.S.; Im, J.H.; Choi, E.K.; Kim, Y.S. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J. Korean Med. Sci. 1998, 13, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Van Swieten, J.C.; Joosse, M.; Hasegawa, M.; Stevens, M.; Tibben, A.; Niermeijer, M.F.; Hillebrand, M.; Ravid, R.; Oostra, B.A.; et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 1999, 64, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K. PCR-SSCP: A simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991, 1, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | First Case | Second Case | Third Case | Korean-1 | Korean-2 |
|---|---|---|---|---|---|
| Country | Italy | France | France | Korea | Korea |
| Age of onset (years) | 45 years | 40–47 years | 38–44 years | 41 years | 38 years |
| Disease | EOAD | EOAD | EOAD | EOAD | EOAD |
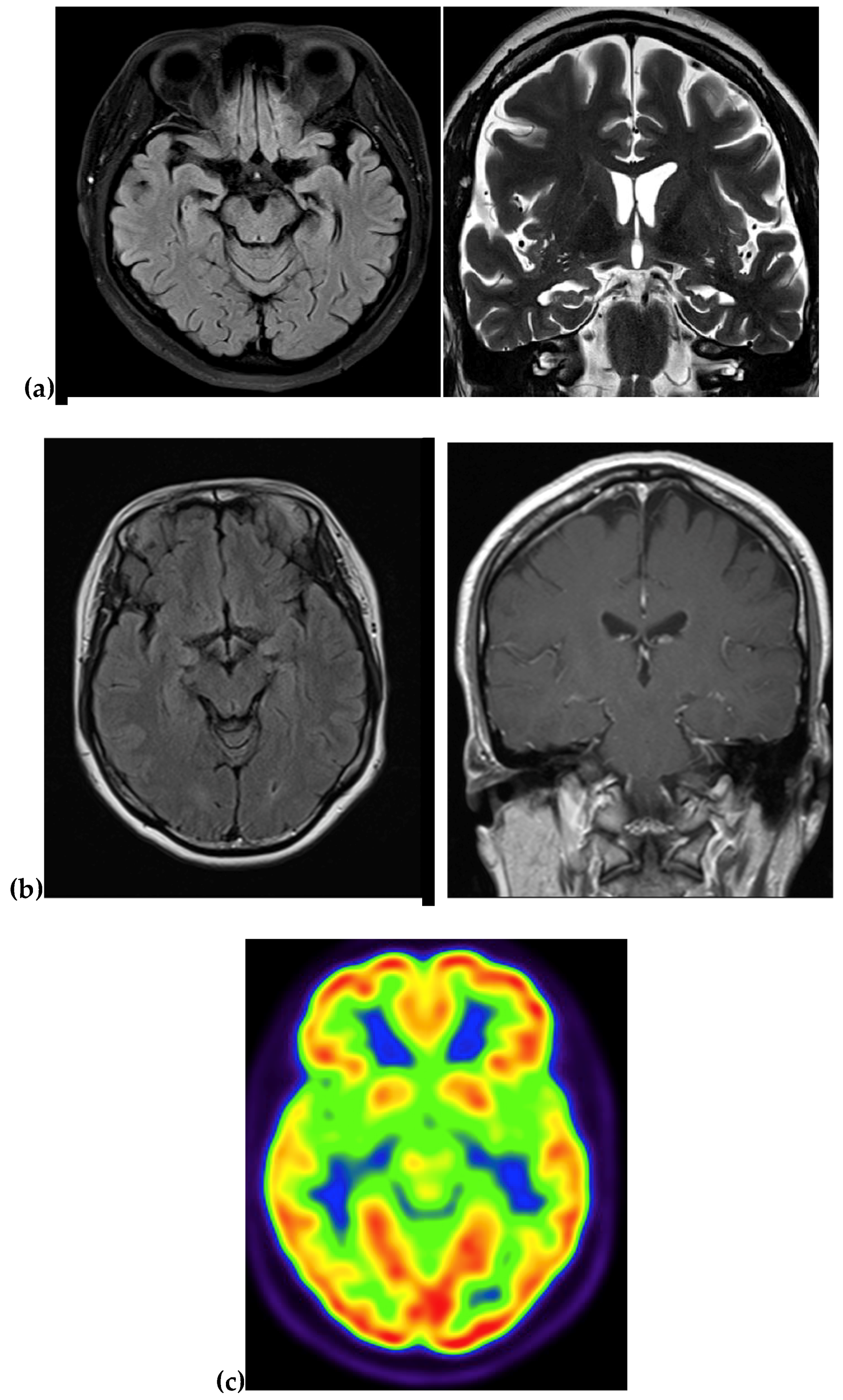
| Imaging | Atrophy and fronto-parieto-temporal enlargement of cortical sulci | NA | NA | MRI: atrophy in the right temporal and parietal regions PET: NA | MRI: Atrophy in the medial region PET: reduced metabolism in temporal and parietal region |
| MMSE | 14/30 | NA | NA | Initially 24/30, later 18/30 | 23/30 |
| Clinical phenotype | Memory loss, confusion and disorientation, followed by progressive memory loss | No detailed information, but patients fulfilled the NINCDS-ADRDA criteria | No detailed information | Memory impairment, confusion, visuospatial dysfunction, speech disturbances. | Impairment in memory and mood, marked ideomotor apraxia |
| Family history | Unknown | Familial | Probable familial | Familial | Familial |
| References | [12] | [13] | [14] | Our data | Our data [11] |
| Mutation | Thr116Asn | Thr116Ile |
|---|---|---|
| Pathogenicity | Pathogenic | Pathogenic |
| Age of onset | 35–41 years (Familial) | 40–47 years (Familial and de novo) |
| Clinical phenotype | EOAD (no clinical data) | EOAD |
| PolyPhen2 scores (HumDiv) | 1.00 (probably damaging) | 1.00 (probably damaging) |
| SIFT scores | 0 (damaging) | 0.07 (tolerated) |
| References | [12,13,14] | [15] |
| Mutation | Dx | Clinical Symptoms/Pathology | Age of Onset | Family History | Functional Studies | References |
|---|---|---|---|---|---|---|
| Phe105Cys | AD | Memory impairment and behavioral changes | 45–60 years | Positive | NA | [28] |
| Phe105Ile | AD | NA | 53–58 years | Positive | NA | [19] |
| Ple105Leu | AD | Parkinson’s like symptoms, severe dementia | 52 years | Probable positive | NA | [18] |
| Arg108Gln | AD | Progressive cognitive decline, myoclonic jerks | 45 years | Segregation could not proven | NA | [17] |
| L113_I114insT | AD | Postmortem studies confirmed the AD | 34–45 years | Familial | 3.4-fold higher Aβ42 levels | [16] |
| Leu113Pro | FTD | Behavioral impairments, myoclonic jerks, seizures | 38–50 years | Familial | NA | [19] |
| Leu113Gln | AD | Rapid progressive dementia, drop attacks, myoclonic seizures, and bilateral spasticity. | 33–36 years | Familial | NA | [18] |
| Tyr115Cys | AD | Postmortem studies confirmed the AD | 39–45 years | Familial | 5.4-fold higher Aβ42 levels | [21] |
| Tyr115His | AD | Epileptic seizure, myoclonus | 35–40 years | Familial | Increased levels of Aβ42, lower Aβ40 | [29] |
| Thr116Ile | Was discussed in details in Table 1 and Table 2 | |||||
| Thr116Asn | ||||||
| Pro117Ala | AD | Issues with balance, tremor, ataxia | 29–35 years | Familial | Elevated Aβ42/40 ratio | [23] |
| Pro117Leu | AD | Progressive memory impairment, mood swings, seizure, myoclonus | 24–33 years | Familial/de novo | Elevated Aβ42, inhibited neural overgrowth | [20] |
| Pro117Arg | AD | Apathy, behavioral changes, seizures, myoclonus, gait impairment | 33–36 years | Unknown/ familial | Elevated total Aβ | [30] |
| Pro117Ser | AD | Personality changes, severe dementia, seizures, myoclonus, tremor | 29–33 years | Familial | Elevated Aβ42 levels | [31] |
| Glu120Asp | AD | Language impairment, seizures, disorientation | 34–53 years | Familial | NA | [22] |
| Glu120Gly | AD | Memory and cognitive decline, seizure, gait disturbances | 30–39 years | Familial | NA | [32] |
| Glu120Lys | AD | Spastic paraparesis | 43–45 years | Familial/ unknown | Elevated Aβ42/40 ratio | [33,34] |
| Glu123Lys | AD | Progressive aphasia, reduced visuospatial activity | 56–62 years | Familial | NA | [24] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagyinszky, E.; Lee, H.-M.; Van Giau, V.; Koh, S.-B.; Jeong, J.H.; An, S.S.A.; Kim, S. PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 2604. https://doi.org/10.3390/ijms19092604
Bagyinszky E, Lee H-M, Van Giau V, Koh S-B, Jeong JH, An SSA, Kim S. PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(9):2604. https://doi.org/10.3390/ijms19092604
Chicago/Turabian StyleBagyinszky, Eva, Hye-Mi Lee, Vo Van Giau, Seong-Beom Koh, Jee Hyang Jeong, Seong Soo A. An, and SangYun Kim. 2018. "PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 9: 2604. https://doi.org/10.3390/ijms19092604