TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family
by

Nina Rol
1,2,†, 
Konda Babu Kurakula
3,†,
Chris Happé
1,2,
Harm Jan Bogaard
1 and
Marie-José Goumans
3,* 1
Department of Pulmonology, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Cardiovascular Sciences, 1081HV Amsterdam, The Netherlands
2
Department of Physiology, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Cardiovascular Sciences, 1081HV Amsterdam, The Netherlands
3
Department of Cell and Chemical Biology, Leiden University Medical Center, 2333ZA Leiden, The Netherlands
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2018, 19(9), 2585; https://doi.org/10.3390/ijms19092585
Submission received: 27 July 2018
/
Revised: 21 August 2018
/
Accepted: 23 August 2018
/
Published: 31 August 2018
(This article belongs to the Special Issue TGF-Beta Super Family Signaling)
Abstract
:Knowledge pertaining to the involvement of transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP) signaling in pulmonary arterial hypertension (PAH) is continuously increasing. There is a growing understanding of the function of individual components involved in the pathway, but a clear synthesis of how these interact in PAH is currently lacking. Most of the focus has been on signaling downstream of BMPR2, but it is imperative to include the role of TGF-β signaling in PAH. This review gives a state of the art overview of disturbed signaling through the receptors of the TGF-β family with respect to vascular remodeling and cardiac effects as observed in PAH. Recent (pre)-clinical studies in which these two pathways were targeted will be discussed with an extended view on cardiovascular research fields outside of PAH, indicating novel future perspectives.
1. Introduction
Pulmonary arterial hypertension (PAH) is a condition defined by an increase in mean pulmonary artery pressure and characterized by remodeling of the pulmonary vasculature [1]. Abnormalities in vessel functionality and responses to stressors culminate in aberrant growth of endothelial cells (ECs) and smooth muscle cells (SMCs), leading to vascular obstruction and the formation of plexiform lesions. The increased pulmonary vascular resistance enhances the load upon the right ventricle (RV). The RV will compensate with hypertrophy, which progresses to RV-failure and death. Current available therapies for PAH mainly target vasoconstriction to reduce pressures and relieve the load, with some showing anti-proliferative effects in vitro. These drugs decelerate, but do not stop disease progression [2,3].
The transforming growth factor-β (TGF-β) family plays a major role in the initiation and progression of PAH. TGF-β is not only an important regulator of vascular remodelling and inflammation in the lung, but also of hypertrophy and fibrosis in the heart [4,5,6,7,8]. Of all receptors belonging to the TGF-β family (Figure 1), the bone morphogenetic protein type 2 receptor (BMPR2) is the most relevant for PAH. Mutations in the BMPR2 gene were the first discovered and most studied mutations underlying hereditary PAH to date [9,10]. BMPR2 is closely entangled with other members of the TGF-β family, but the roles of many of the ligands and receptors in the TGF-β family are still underappreciated in PAH. Although bone morphogenetic protein (BMP) ligands and their receptors play an important role in disease progression and could function as therapeutic targets [11], agents effectively decreasing TGF-β1 activity, together with selective TGF-β ligand traps open up new treatment possibilities [12,13,14,15,16].
Here, we give a comprehensive update on TGF-β signaling in PAH, summarized in Table 1. Furthermore, we provide insights into current (pre)-clinical studies targeting the TGF-β pathway in other diseases that may be useful in designing therapeutic strategies for the deadly condition of PAH.
2. TGF-β Signaling
Members of the TGF-β family are widely expressed in diverse tissues and play an essential role throughout life, starting from gastrulation and the onset of body axis asymmetry to organ-specific morphogenesis and adult tissue homeostasis [55,56,57,58]. At the cellular level, TGF-β family members regulate fundamental processes important for tissue homeostasis and embryogenesis, such as cell proliferation, differentiation, apoptosis, migration, adhesion, cytoskeletal organization, extracellular matrix production, in a context- and cell type-dependent manner. Consistent with this pleiotropic activity, disrupted TGF-β signaling is associated with several developmental disorders, cancer, auto-immune, cardiovascular and fibrotic diseases [55,56,57,59,60].
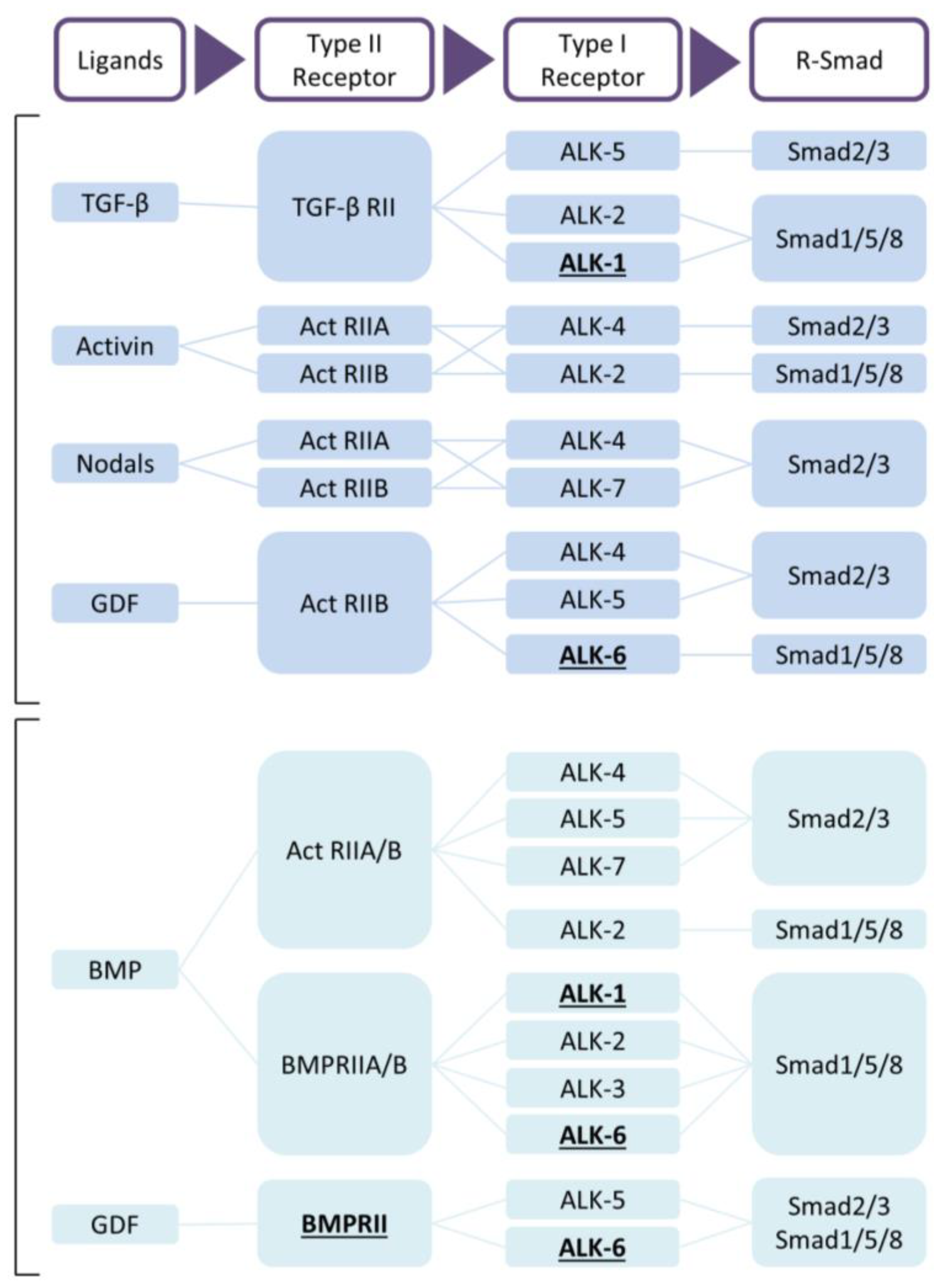
The TGF-β family members are subdivided into two functional groups: the TGF-β group that comprises the three mammalian TGF-β isoforms, activins, nodals and some growth and differentiation factors (GDFs) and the BMP group that includes all BMPs and most GDFs (Figure 1) [57,59]. TGF-β family members form functional dimers, bind to heterotetrameric complex of type I and type II serine/threonine kinase transmembrane receptors and signal through both Smad-dependent and Smad-independent pathways [57,58,61,62] (Figure 2). In mammals, seven type I receptors, also known as activin receptor-like kinases (ALKs), and five type II receptors have been reported so far. To control duration and intensity of TGF-β signaling, agonists, antagonists, co-receptors and intracellular signaling play key roles in ligand access and posttranslational modification of the receptors and downstream mediators in a cell- and context-dependent manner [60,61,63]. TGF-β is secreted in its latent form and needs to be proteolytically processed before being able to bind to signaling receptors [4]. This complex activation mechanism could open up new therapeutic targets. TGF-β signals in most cells by binding to TβRII forming a complex with TβRI (or ALK5). Activins bind to activin receptor type IIA (ActRIIA) or ActRIIB in a complex with ALK4, while BMPs signal via BMP type II receptor (BMPRII), ActRIIA or ActRIIB, in combination with ALK1, 2, 3 or 6. Although TβRII/TβRI is the preferable high affinity signaling complex, in endothelial cells, TGF-β can also signal through TβRII/ALK1/ALK5 [64,65].
Upon complex formation, the activated type I receptor kinase will transduce the signal from the membrane to the nucleus by phosphorylating Smad transcription factors [61]. Smads are divided into three major classes: receptor-regulated Smads (R-Smads), common mediator Smad (co-Smad) and the inhibitory Smads (I-Smads). R-Smads (Smad1, Smad2, Smad3, Smad5 and Smad8) function as direct substrates for specific type I receptor kinases. ALK4, -5 and -7 phosphorylate Smad2 and Smad3, whereas Smad1, Smad5 and Smad8 become phosphorylated by the BMP type I receptors ALK1, -2, -3 and -6 [66]. Upon phosphorylation, R-Smads form a complex with the co-Smad, Smad4, and translocate to the nucleus. In the nucleus, Smad complexes engage in cooperative interactions with DNA and other DNA-binding proteins such as FAST1, FAST2, Fos/Jun and ATF2 to mediate the transcription of specific target genes [60,67]. The two I-Smads, Smad6 and Smad7, first identified in 1997 as vascular Smads, can compete with and inhibit R-Smads for type I interaction preventing phosphorylation [61,68]. Furthermore, they can induce proteasomal degradation of the type I receptor by recruiting Smurf1/2 E3 ubiquitin ligases [55,56,57,59,60]. For a more extensive description of TGF-β signaling, we refer to recent reviews [58,69,70,71,72,73,74,75,76,77].
3. Role of TGF-β Ligands in Pulmonary Arterial Hypertension
The presence of different TGF-β isoforms in the pulmonary vascular wall in the context of tissue remodeling in PAH was already described in 1994. Particularly, TGF-β3 is highly upregulated in both medial and intimal layers of remodeled pulmonary vessels [20]. More recent studies report that the increased presence of active TGF-β ligands co-localizes with SMCs in pulmonary arterioles and a strong expression of TGF-β1 in ECs and the interstitium of the plexiform lesions [46,78]. TGF-β signaling can directly inhibit BMP-Smad signaling in SMCs, and ligands from this side of the signaling balance can function as antagonists by competing for type II receptor binding [79,80]. Interestingly, pulmonary arterial ECs (PAECs) expressing a mutant BMPR2 release higher levels of TGF-β into the medium, thereby accelerating SMC growth [81]. As the quiescent effect that TGF-β typically has on SMC growth is impaired in PAH, the elevated TGF-β levels cause medial hypertrophy [82,83,84]. TGF-β-single nucleotide polymorphisms (SNP) on top of heterozygous BMPR2 mutation modulate the age of diagnosis and penetrance of familial PAH [45]. Other circulating ligands, such as activins and GDFs, are increased in PAH, as well, possibly stimulating cell growth and thereby contributing to pulmonary vascular remodeling [18,19,31,32,33,38]. The different animal models for pulmonary hypertension (PH) confirm the human pathology harboring more TGF-β and activins in the serum, pulmonary arteries and the RV in hypoxia or monocrotaline (MCT)-induced PH in rats [12,22,23,26]. The imperative role of TGF-β in PAH development is also illustrated by the dependency on this ligand in PAH associated with schistosomiasis in rats and required enhanced TGF-β signaling in a mouse model of scleroderma-related PH (SSc-PH) [24,85,86]. A recent study demonstrated that bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension via activation of TGF-β [87].
4. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension
ECs can change their endothelial cobblestone morphology to a mesenchymal phenotype, a process referred to as endothelial-to-mesenchymal transition (EndoMT). In this process, ECs progressively lose their characteristics, i.e., cell-cell junctions and specific markers such as CD31, VE-cadherin and CD34 and gain markers such as α-SMA, collagen-I and vimentin migrate and invade into the surrounding tissues [88,89]. Although EndoMT takes place during embryogenesis where the transition contributes to the development of the valves of the heart, it does not occur under normal physiological circumstances [90]. An imbalance in the TGF-β/BMP axis and disturbed inflammation contribute to the induction of EndoMT [91]. EndoMT is stimulated by increased TGF-β receptor signaling and attenuated by intact BMPR2 signaling [21,92]. This process has been reported in pathologies such as inflammatory bowel disease, chronic kidney disease, cardiac fibrosis and portal hypertension [88,93,94,95]. In vitro, TGF-β-induced EndoMT in PAECs leads to higher migration rates, lower proliferation rates and decreased barrier integrity [91].
Both pre-clinical and clinical studies demonstrate that EndoMT plays a role in the pathogenesis of PAH [89,96]. EndoMT is also detected in the pulmonary vasculature of systemic sclerosis-associated PAH patients [91]. This study additionally shows that in vitro-induced EndoMT leads to reduced barrier integrity of PAECs with the production of pro-inflammatory cytokines such as IL-6, IL-8 and TNF-α and high trans-endothelial migration of immune cells.
In the pulmonary vasculature of MCT rats, overexpression of Twist-1 and VE-cadherin and repression of p120-catenin indicate the induction of EndoMT [21]. Rapamycin, an immunosuppressive drug, reverses experimental PH by inhibiting the migration of PAECs and reducing EndoMT markers. Ponatinib, a multi-target tyrosine-kinase inhibitor, attenuates TGF-β-induced EndoMT in human pulmonary microvascular ECs [96].
5. Receptors in Pulmonary Arterial Hypertension
Besides BMPR2 mutations, rare variants in other TGF-β receptor superfamily member genes are also associated with autosomal dominant familial PAH. Mutations in the type I receptor ALK1 and co-receptor endoglin are found in hereditary hemorrhagic telangiectasia (HHT)-associated PAH [35,36]. The increased prevalence of (h)PAH in HHT1 and HHT2 could be explained by the involvement of arteriovenous malformations, caused by ALK1 and ENG mutations, in the pathophysiology in both diseases [97,98,99]. Interestingly, in idiopathic PAH (iPAH) mRNA and protein levels of ALK1 and endoglin are specifically increased in ECs, leading to enhanced Smad1/5 phosphorylation (pSmad1/5) when stimulated with TGF-β, indicating a disturbed TGF-β/BMP balance [19]. In mice carrying a kinase-deficient TβRII in fibroblasts, the disturbed TGF-β signaling leads to pulmonary vasculopathy with medial thickening and mildly elevated pulmonary artery pressures [42]. TGF-β type III receptor (TβRIII) or β-glycan, a co-receptor acting as a reservoir of TGF-β2 for the type I and II receptors, is downregulated in familial PAH [28]. The functional consequences of these changes for the pathogenesis of PAH are yet unknown.
6. Canonical TGF-β Signaling in Pulmonary Arterial Hypertension
In the pulmonary vasculature, Smad2 phosphorylation after TGF-β receptor activation is increased, even though mRNA expression of Smad2 and Smad3 is decreased in whole lung lysates of PAH patients [27,39,45]. While pSmad2 (and not pSmad3) is likewise increased in the lungs of mice exposed to hypoxia, rats experimentally exposed to MCT develop PH 2–4 weeks after MCT injection, showing contrasting results with regards to canonical TGF-β signaling in the lung [25,31,46,100]. Some studies report increased pSmad2, while others show no change or even a decrease in Smad2 phosphorylation using Western blot analysis [12,37,43,46]. On the cellular, level increased Smad2 levels, driven by Activin A activation, are found in cultured SSc-PAH fibroblasts, responsible for collagen production [101]. Transgenic mice carrying an SMC specific dominant-negative BMPR2 gene do not show any alteration in Smad2 phosphorylation in lung tissue by Western blot analysis [102]. Although this could be due to technical differences between studies, it is possible that a BMPR2 mutation alone is not sufficient to regulate Smad2 phosphorylation.
Upon translocation into the nucleus, pSmad2 binds to the promoter of specific target genes like plasminogen activator inhibitor (PAI)-1, a well-acknowledged TGF-β target gene [103]. Interestingly, mRNA and protein expression of PAI-1 are decreased in iPAH, while circulating levels of PAI-1 are increased in both primary (idiopathic) and secondary PAH [49,50]. The latter is likely linked to the widespread development of thrombosis with intraluminal thrombin deposition [104]. The two widely-used experimental PH rat models, MCT and SuHx (VEGF receptor inhibitor Sugen combined with hypoxia), show conflicting results compared to the human situation, with increased mRNA expression of PAI-1 [12].
The co-Smad, Smad4, forms an intracellular complex with the TGF-β and BMP-mediated phosphorylated R-Smads and is needed for nuclear translocation [60]. Mutations in Smad4, together with ACVRL1 (ALK1) and ENG are causative of the vascular disorder HHT [105]. Nasim et al. report two independent iPAH cases with a missense and splice site mutation in Smad4, but no differential protein expression was found in PAECs and SMCs of iPAH patients [39,47]. In contrast with these human findings, Smad4 is reduced in MCT-induced PH on both the mRNA and protein level [43,48]. Transcription of the I-Smads, Smad6 and Smad7, is also reduced in lung tissue of these animals [48]. Differences in expression of I-Smads in human PAH tissue have not been reported to date. However, it has been shown that Smad6 is suppressed by the prostanoid Iloprost, thereby enhancing the intensity and duration of the TGF-β/Smad responses [106].
7. Non-Canonical TGF-β Signaling
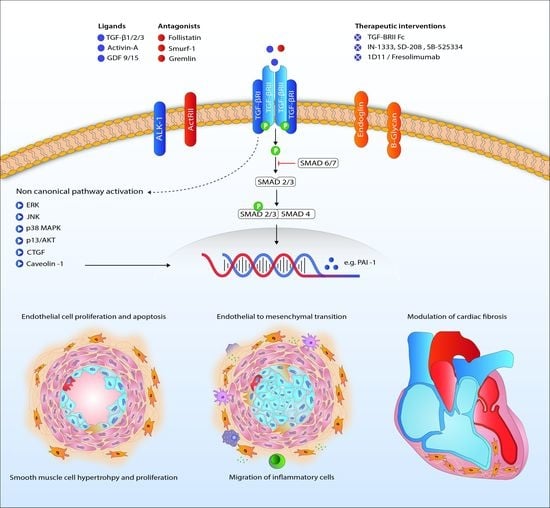
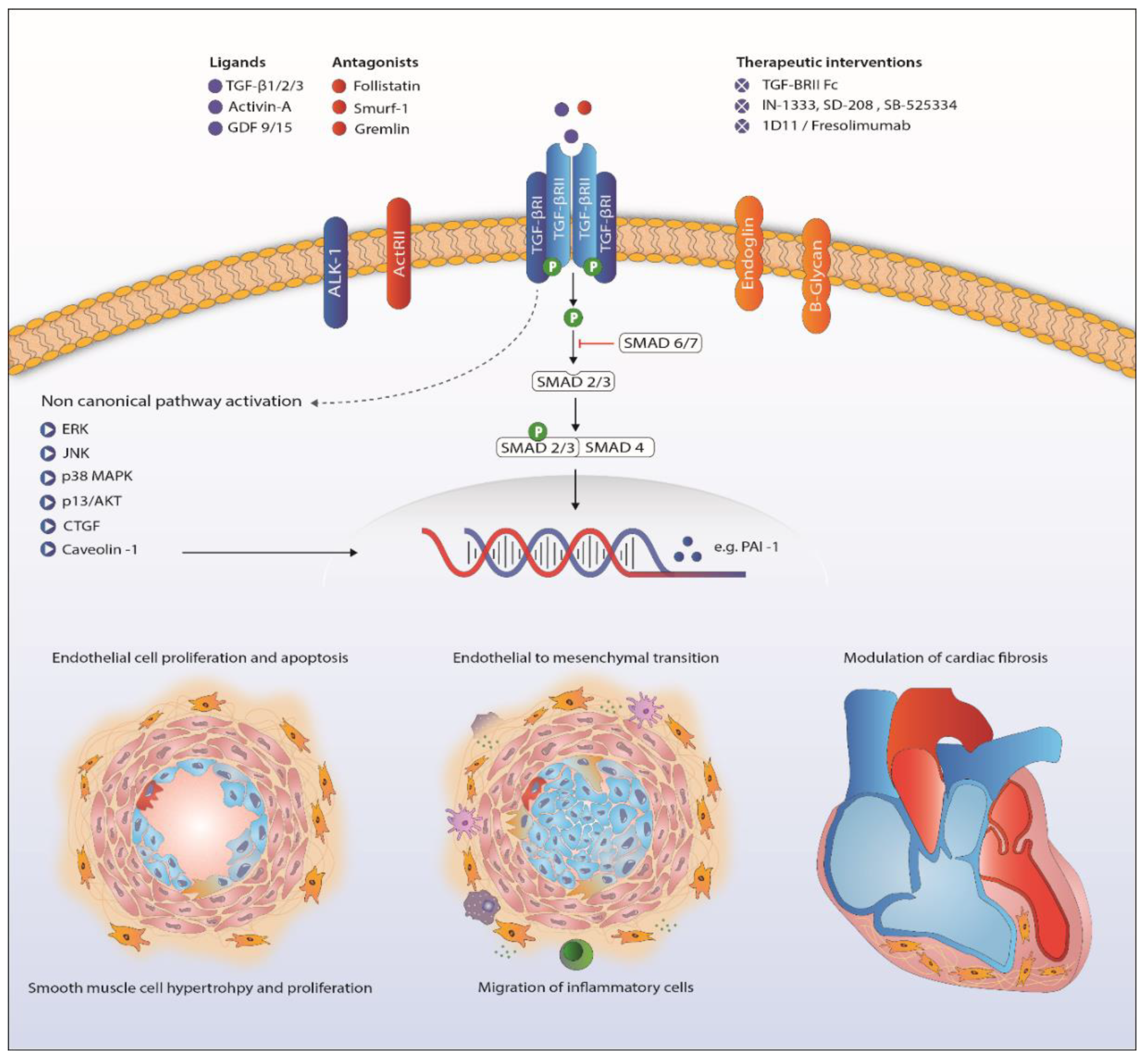
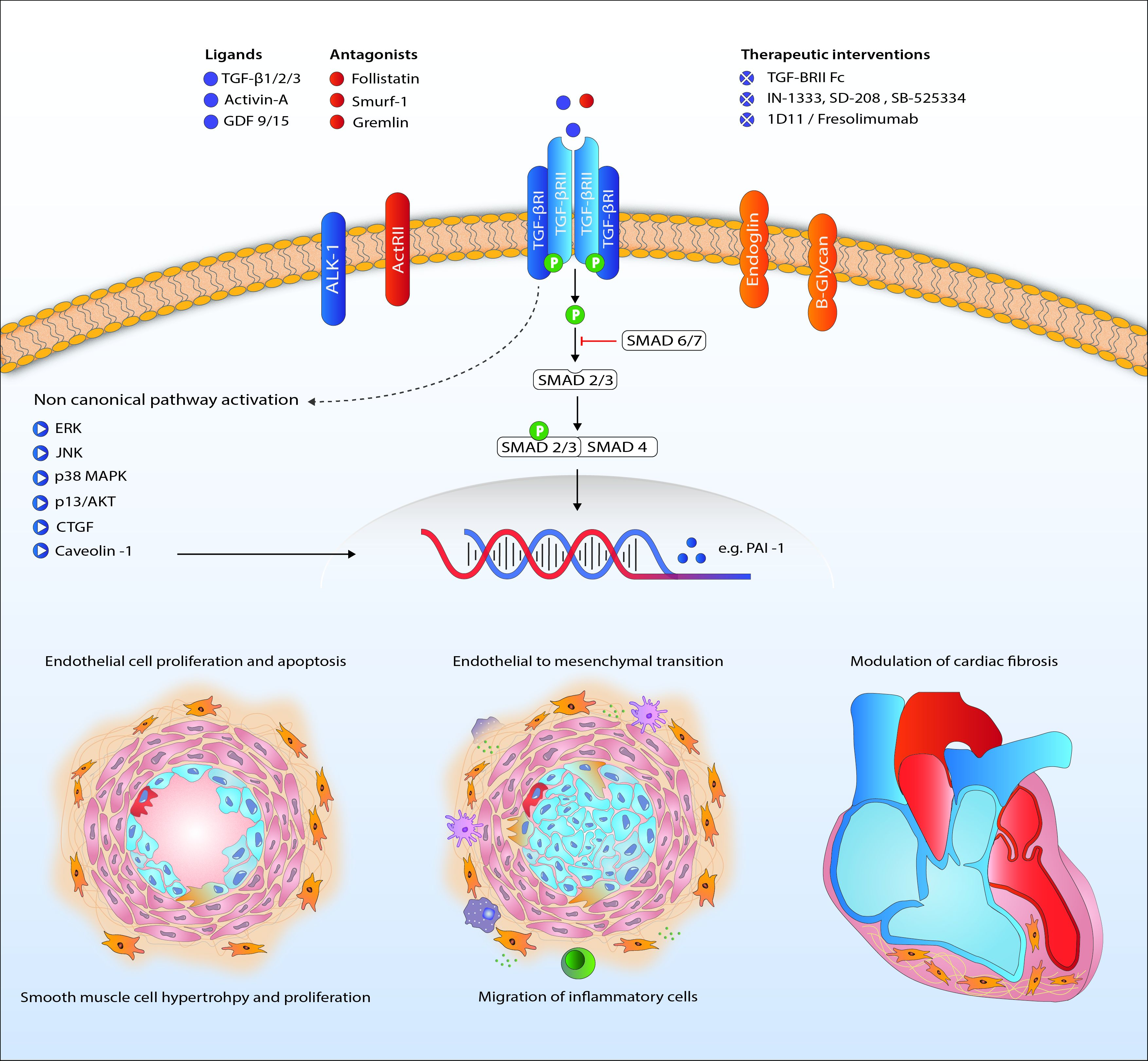
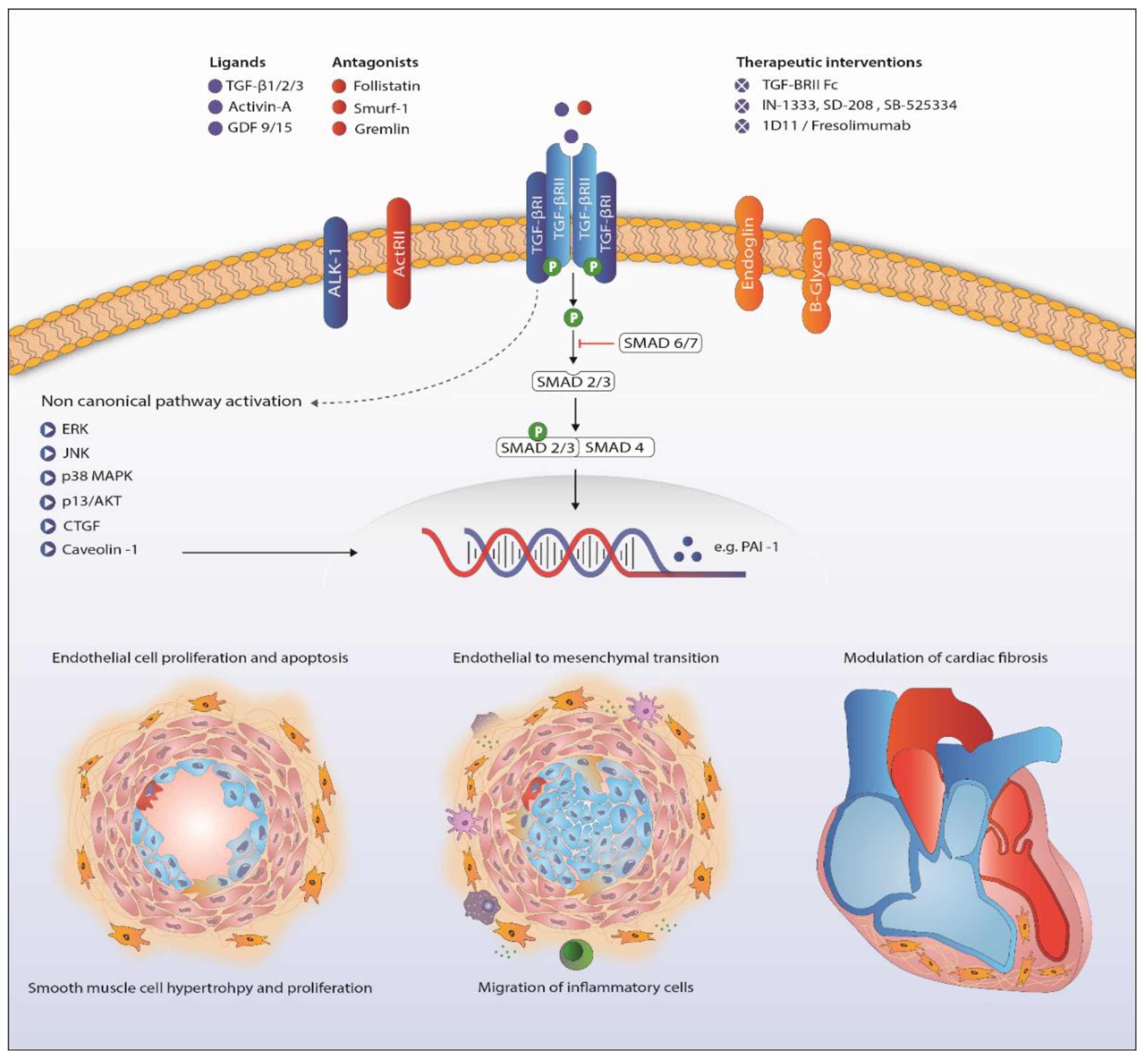
Downstream signaling of TGF-β goes beyond phosphorylation of the Smad proteins. Activation of ERK, JNK/p38, Rho-like GTPases and PI3K/Akt is involved in the non-Smad pathway and also familiar in PAH research (Figure 2) [107,108,109,110]. Upregulation of these proteins in PAH has been shown before, although only a few in the context of disturbed TGF-β signaling [13,51,111]. Besides BMPR2 mutations, caveolin 1 (CAV1) mutations are a rare cause of PAH, influencing both canonical and non-canonical TGF-β/BMP signaling [36,52,112].
8. Downstream Targets of TGF-β in the Lung
Several reports demonstrated that vascular thrombosis plays an essential role in the pathophysiology of iPAH. Indeed, anticoagulation treatment confers a survival benefit in iPAH patients [113,114], perhaps because in situ thrombosis of pulmonary vessels may contribute to the pathogenesis of this disease [115,116]. Transcriptional activity of PAI-1 is elevated in patients with PAH along with other coagulation-associated genes. This increase in PAI-1 activity may explain impaired fibrinolysis in iPAH patients [116,117]. In contrast, two other studies demonstrated no change in PAI-1 activity in the serum of iPAH and chronic thrombo-embolic pulmonary hypertension (CTEPH) patients at rest or after venous occlusion [118,119]. However, the same group reported later that there is an increase in PAI-1 activity in female iPAH patients before and after venous occlusion [120]. The discrepancies between these studies may be caused by the use of different assays, gender differences and a low sample size per group. Given the heterogeneity in PAH patients, more studies are warranted to unravel the true function of PAI-1 in the pulmonary vasculature in PAH.
The inhibitor of DNA binding family of proteins (ID proteins) is a major downstream transcriptional target of BMP signaling [121]. In mammalian cells, four members of the Id family, Id1–4, have been identified so far. It has been reported that ID1, ID2 and ID3 are induced by BMPs in PAECs and SMCs through a canonical Smad-dependent pathway [11,106,121,122]. In adult organs, ID4 is mostly expressed in testis, brain and kidney and left out of the scope of this review [123]. Interestingly, BMP9 strongly induces the expression of ID proteins in PAECs, while BMP4 and BMP6 increase the expression of ID proteins in SMCs [121,124]. Mutations in BMPR2 strongly reduced the expression of ID proteins in both PAECs and SMCs. Unexpectedly, the expression of ID proteins along with BMPR2 expression are also reduced in PAECs and SMCs of some iPAH patients. In line with these observations in cultured cells, the expression of ID proteins is attenuated in experimental MCT-PH lungs, as well as in human lungs [48]. ID proteins also regulate the cell cycle and proliferation of SMCs in a BMP4-dependent manner [106,121]. Recently, BMP9 has been shown to selectively increase BMPR2, ID1 and ID3 proteins in endothelial cells in vitro and thereby decrease experimental PH in vivo, suggesting the potential involvement of these genes in PAH [11].
9. TGF-β Signaling in the Heart in Pulmonary Arterial Hypertension
Survival of PAH patients is determined by the ability of the RV to adapt to the increased pressures in the pulmonary vasculature [125]. The challenged RV suffers from neurohormonal activation, capillary loss inflammation, apoptosis, oxidative stress and metabolic shifts leading to hypertrophy and fibrosis [126]. Cardiac fibrosis is related to increased TGF-β signaling [127,128]. In rat, cardiac fibrosis induced by increased RV afterload, as seen in PH, is likely to be mediated through TGF-β-induced connective tissue growth factor signaling (Figure 2) [53,54]. The beneficial effects of carvedilol (β-blocker), iloprost (prostacyclin) and losartan (angiotensin receptor blocker) on RV function in animals are partly ascribed to attenuated TGF-β-mediated fibrosis [53,54,129]. Furthermore, nintedanib, a tyrosine kinase inhibitor known to inhibit TGF-β-mediated fibrosis, attenuated cardiac fibrosis in experimental pulmonary hypertension [130,131]
Hemnes et al. showed an upregulation of the TGF-β pathway in the RV of PAH patients by increased transcription of TGF-β3 [29]. TGF-β inhibition by either pan-TGFβ antibodies or specifically binding to TGF-β1 and TGF-β3 showed lowering of RV systolic pressures and attenuated RV hypertrophy in MCT and SuHx rat models [12,132]. One of the few studies investigating downstream TGF-β signaling in the heart in the context of PAH showed decreased phosphorylation of Smad2 in both RV and LV. This observation was independent of the presence of a BMPR2 mutation [40].
10. Therapeutic Interventions Relevant in PAH
In 13 preclinical and nearly twenty phase I–III clinical trials, TGF-β signaling is targeted to treat cancer and fibrotic diseases [72,77,133]. TGF-β signaling can be targeted mainly in three different ways in clinical trials: specific antibodies, antisense oligonucleotides and receptor kinase inhibitors. As ECs and SMCs in PAH produce excessive amounts of TGF-β, the use of these targets may decrease vascular remodeling through their inhibitory effect on these cells. In contrast to the clear role of TGF-β signaling in tumorigenesis, vascular diseases are more complex with simultaneous up- and down-regulation of the pathway and interactions with the BMP pathway [72].
Beneficial effects of inhibiting TGF-β ligands on pulmonary vascular and cardiac remodeling have previously been shown in experimental MCT- and hypoxia-induced rat PH models [12]. Targeting the ALK5 kinase with SD208, a drug known to suppress tumor metastasis in rodent models, ameliorated MCT-induced PH [134]. Beneficial anti-remodeling effects of prostacyclin analogues, used in PAH treatment strategies, can partly be explained by TGF-β inhibition [13,135]. How these effects in rat models can provide implications for the human disease are uncertain; besides increased availability of TGF-β ligands, different regulation patterns are observed. As TGF-β signaling is crucial for many physiological functions, prolonged inhibition of this signaling might lead to harmful side effects. Preclinical studies in PAH patient-derived cells could give valuable information about expected responses, illustrated by different effects in ECs and SMCs upon TGF-β stimulation [19,82,83].
11. Conclusions
In PAH, several mutations in components of the TGF-β/BMP signaling pathway have been identified. However, most research over the years has focused on BMP signaling, in particular BMPR2. Enhanced expression of TGF-β has been found systemically (i.e., in serum) and locally (i.e., in ECs and SMCs of the pulmonary vasculature) in PH patients and animal models. Furthermore, TGF-β has been shown to be involved in proliferation, inflammation, angiogenesis and fibrosis in lungs in PAH. In addition, TGF-β induces EndoMT, which is also involved in PAH, and as such, TGF-β could be interesting as a treatment target. Inhibition of TGF-β signaling, either directly or through targeting intermediates, may be a novel therapeutic strategy in PAH.
TGF-β signaling plays an essential role in vascular cells, immune cells and other cells such as epithelial cells in lungs. However, TGF-β signaling is very complex, as there are numerous ligands and diverse receptors that exhibit distinct functions in a cell- and context-dependent manner through interaction with other proteins, thereby affecting multiple signaling cascades. This intricate pathway is crucial for vessel wall homeostasis in many diseases, including PAH. Therefore, a deeper understanding of this pathway is necessary for the development of safer and efficient therapies for PAH.
In conclusion, overactive TGF-β signaling is an important regulator of pulmonary vascular remodelling in PAH, e.g., by balancing BMP signaling. TGF-β inhibitors have entered clinical trials for treatment of cancer and fibrotic diseases with encouraging first clinical results. The future of specific TGF-β inhibitors are promising and open new challenges in PAH research.
Author Contributions
Drafted or substantively revised the manuscript: N.R., K.B.K., C.H., H.J.B., M.J.G.; Has approved the final version of the manuscript: N.R., K.B.K., C.H., H.J.B., M.J.G.
Funding
We gratefully acknowledge the support from the Dutch Lung Foundation (5.2.17.198JO) and the Netherlands CardioVascular Research Initiative: The Dutch Heart Foundation, the Dutch Federation of University Medical Centers, the Netherlands Organisation for Health Research and Development and the Royal Netherlands Academy of Sciences (2012-08).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk, N.A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Rich, S.; Kaufmann, E.; Levy, P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Tantini, B.; Manes, A.; Fiumana, E.; Pignatti, C.; Guarnieri, C.; Zannoli, R.; Branzi, A.; Galie, N. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic. Res. Cardiol. 2005, 100, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ten, D.P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 857–869. [Google Scholar]
- Ten, D.P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar]
- Liang, H.; Zhang, C.; Ban, T.; Liu, Y.; Mei, L.; Piao, X.; Zhao, D.; Lu, Y.; Chu, W.; Yang, B. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int. J. Biochem. Cell. Biol. 2012, 44, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Ten Dijke, P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., III; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective TGFβ Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 134, A19307. [Google Scholar]
- Ogo, T.; Chowdhury, H.M.; Yang, J.; Long, L.; Li, X.; Torres Cleuren, Y.N.; Morrell, N.W.; Schermuly, R.T.; Trembath, R.C.; Nasim, M.T. Inhibition of overactive transforming growth factor-β signaling by prostacyclin analogs in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Granton, E.; Hu, Y.; Miranda, M.Z.; Weichelt, U.; Breuils Bonnet, S.; Bonnet, S.; Morrell, N.W.; Connelly, K.A.; Provencher, S.; et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. Am. J. Respir Crit. Care Med. 2018, 197, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.S.; Yanagihara, T.; Granton, E.; Sato, S.; Shimbori, C.; Upagupta, C.; Imani, J.; Hambly, N.; Ask, K.; Gauldie, J.; et al. Macitentan reduces progression of TGF-β1-induced pulmonary fibrosis and pulmonary hypertension. Eur. Respir. J. 2018, 2018, 1701857. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Zuo, C.; He, Y.; Chen, G.; Piao, L.; Zhang, J.; Xiao, B.; Shen, Y.; Tang, J.; Kong, D.; et al. EP3 receptor deficiency attenuates pulmonary hypertension through suppression of Rho/TGF-β1 signaling. J. Clin. Investing. 2015, 125, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rivas, G.; Jerjes-Sanchez, C.; Rodriguez, D.; Garcia-Pelaez, J.; Trevino, V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med. Genet. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, N.; Bergh, C.H.; Andersson, B.; Sakiniene, E.; Carlsten, H.; Rundqvist, B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur. Respir. J. 2009, 34, 662–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef] [PubMed]
- Botney, M.D.; Bahadori, L.; Gold, L.I. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-β. Am. J. Pathol. 1994, 144, 286–295. [Google Scholar] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Wang, W.; Zhu, X.C.; Ye, W.J.; Cai, H.; Wu, P.L.; Huang, X.Y.; Wang, L.X. The potential of asiaticoside for TGF-β1/Smad signaling inhibition in prevention and progression of hypoxia-induced pulmonary hypertension. Life Sci. 2015, 137, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.; Hu, H.; Shi, L.; Chen, J.; Wang, B.; Chen, C.; Zhu, H.; Li, Y.; Li, Q.; et al. Potential therapeutic targets for hypoxia-induced pulmonary artery hypertension. J. Transl. Med. 2014, 12, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, B.B.; Chabon, J.; Gebreab, L.; Poole, J.; Debella, E.; Davis, L.; Tanaka, T.; Sanders, L.; Dropcho, N.; Bandeira, A.; et al. Transforming growth factor-β signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation 2013, 128, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-β signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.A.; Obaid, A.A.; Zaki, H.F.; Agha, A.M. Role of oxidative stress, inflammation, nitric oxide and transforming growth factor-β in the protective effect of diosgenin in monocrotaline-induced pulmonary hypertension in rats. Eur. J. Pharmacol. 2014, 740, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1235–H1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.E.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: A gene microarray analysis. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vinasco, L.; Gomberg-Maitland, M.; Maitland, M.L.; Desai, A.A.; Singleton, P.A.; Sammani, S.; Sam, L.; Liu, Y.; Husain, A.N.; Lang, R.M.; et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genom. 2008, 33, 278–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yndestad, A.; Larsen, K.O.; Oie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjonsberg, O.H.; Pedersen, T.M.; Anfinsen, O.G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, N.; Kempf, T.; Tapken, H.; Tongers, J.; Laenger, F.; Lehmann, U.; Golpon, H.; Olsson, K.; Wilkins, M.R.; Gibbs, J.S.; et al. Growth differentiation factor-15 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Wharton, J.; Howard, L.S.; Gibbs, J.S.; Wilkins, M.R. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart 2011, 97, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Nickel, N.; Jonigk, D.; Kempf, T.; Bockmeyer, C.L.; Maegel, L.; Rische, J.; Laenger, F.; Lehmann, U.; Sauer, C.; Greer, M.; et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir. Res. 2011, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Coulet, F.; Girerd, B.; Montani, D.; Humbert, M.; Lacombe, P.; Chinet, T.; Gouya, L.; Roume, J.; Axford, M.M.; et al. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin. Genet. 2012, 82, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.F.; Lame, M.W.; Segall, H.J.; Wilson, D.W. Smad signaling in the rat model of monocrotaline pulmonary hypertension. Toxicol. Pathol. 2008, 36, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Jachec, W.; Foremny, A.; Domal-Kwiatkowska, D.; Smolik, S.; Tomasik, A.; Mazurek, U.; Wodniecki, J. Expression of TGF-β1 and its receptor genes (TβR I, TβR II, and TβR III-βglycan) in peripheral blood leucocytes in patients with idiopathic pulmonary arterial hypertension and Eisenmenger’s syndrome. Int. J. Mol. Med. 2008, 21, 99–107. [Google Scholar] [PubMed]
- Richter, A.; Yeager, M.E.; Zaiman, A.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Impaired transforming growth factor-β signaling in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit Care Med. 2004, 170, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, C.E.; Happe, C.M.; Dorfmuller, P.; Trip, P.; Spruijt, O.A.; Rol, N.; Hoevenaars, F.P.; Houweling, A.C.; Girerd, B.; Marcus, J.T.; et al. Bone Morphogenetic Protein Receptor Type 2 Mutation in Pulmonary Arterial Hypertension: A View on the Right Ventricle. Circulation 2016, 133, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Docx, C.; Holmes, A.M.; Beach, S.; Duggan, N.; England, K.; Leblanc, C.; Lebret, C.; Schindler, F.; Raza, F.; et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am. J. Pathol. 2009, 174, 380–389. [Google Scholar] [PubMed]
- Derrett-Smith, E.C.; Dooley, A.; Gilbane, A.J.; Trinder, S.L.; Khan, K.; Baliga, R.; Holmes, A.M.; Hobbs, A.J.; Abraham, D.; Denton, C.P. Endothelial injury in a transforming growth factor β-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013, 65, 2928–2939. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Kouri, F.M.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Sandu, R.; Dony, E.; Seeger, W.; Schermuly, R.T.; Eickelberg, O.; et al. The transforming growth factor-β/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xu, M.; Dong, Y.; Liu, J.; Li, Y.; Mao, W.; Wang, J.; Wang, L. 1,25(OH)2D3 attenuates pulmonary arterial hypertension via microRNA-204 mediated Tgfbr2/Smad signaling. Exp. Cell Res. 2018, 362, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A., III; Poling, J.S.; Phillips, C.A.; Stanton, K.C.; Austin, E.D.; Cogan, J.D.; Wheeler, L.; Yu, C.; Newman, J.H.; Dietz, H.C.; et al. Synergistic heterozygosity for TGFβ1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet. Med. 2008, 10, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Han, W.; Greer, P.A.; Tuder, R.M.; Toque, H.A.; Wang, K.K.; Caldwell, R.W.; Su, Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J. Clin. Investig. 2011, 121, 4548–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morty, R.E.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Zakrzewicz, A.; Kouri, F.M.; Peters, D.M.; Dumitrascu, R.; Seeger, W.; Knaus, P.; et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arteriosc. Thromb. Vasc. Biol. 2007, 27, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Kouri, F.M.; Queisser, M.A.; Konigshoff, M.; Chrobak, I.; Preissner, K.T.; Seeger, W.; Eickelberg, O. Plasminogen activator inhibitor type 1 inhibits smooth muscle cell proliferation in pulmonary arterial hypertension. Int. J. Biochem. Cell Biol. 2008, 40, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.H.; Hassell, K.L.; Badesch, D.B.; Kressin, D.C.; Marlar, R.A. Coagulation and fibrinolytic profiles in patients with severe pulmonary hypertension. Chest 1996, 110, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Happe, C.M.; Rol, N.; Fontijn, R.D.; Dickhoff, C.; Hartemink, K.J.; Grunberg, K.; Tu, L.; Timens, W.; Nossent, G.D.; et al. Delayed Microvascular Shear-adaptation in Pulmonary Arterial Hypertension: Role of PECAM-1 Cleavage. Am. J. Respir. Crit. Care Med. 2016, 193, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. The role of genetics in pulmonary arterial hypertension. J. Pathol. 2017, 241, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Cho, M.Y.; Li, J.; Assad, R.S.; Sun, M.; Rohailla, S.; Honjo, O.; Apitz, C.; Redington, A.N. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor-β1 signaling and is abrogated by angiotensin receptor blockade. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.; Sakagami, M.; Syed, A.A.; Farkas, L.; Van, T.B.; Kraskauskas, D.; Mizuno, S.; Abbate, A.; Bogaard, H.J.; Byron, P.R.; et al. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur. Respir. J. 2015, 45, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; Ten, D.P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFβ signaling. Cell. Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; Ten, D.P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Goumans, M.J.; Ten, D.P. Regulatory RNAs controlling vascular (dys)function by affecting TGF-β family signalling. EXCLI J. 2015, 14, 832–850. [Google Scholar] [PubMed]
- Schmierer, B.; Hill, C.S. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.H.; Hamel, L.; Chevalier, S.; Philip, A. Endoglin expression on human microvascular endothelial cells association with βglycan and formation of higher order complexes with TGF-β signalling receptors. Eur. J. Biochem. 2000, 267, 5550–5560. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten, D.P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; Ten, D.P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Massague, J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [PubMed]
- Labbe, E.; Silvestri, C.; Hoodless, P.A.; Wrana, J.L.; Attisano, L. Smad2 and Smad3 positively and negatively regulate TGF β-dependent transcription through the forkhead DNA-binding protein FAST2. Mol. Cell 1998, 2, 109–120. [Google Scholar] [CrossRef]
- Topper, J.N.; Cai, J.; Qiu, Y.; Anderson, K.R.; Xu, Y.Y.; Deeds, J.D.; Feeley, R.; Gimeno, C.J.; Woolf, E.A.; Tayber, O.; et al. Vascular MADs: Two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. USA 1997, 94, 9314–9319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Euler, G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Pardali, E.; Sanchez-Duffhues, G.; Ten, D.P. BMP signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Tajsic, T.; Morrell, N.W. Transforming growth factor-β(1) represses bone morphogenetic protein-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-β Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Long, L.; Reynolds, P.N.; Morrell, N.W. Expression of mutant BMPR-II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm. Circ. 2011, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β(1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.J.; Holmes, A.M.; Deighton, J.; Long, L.; Yang, X.; Barker, L.; Walker, C.; Budd, D.C.; Upton, P.D.; Morrell, N.W. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: Role of proinflammatory cytokines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L604–L615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Sun, S.; Zhu, J.; Gao, S.; Pang, J.; Zhu, D.; Sun, Z. Transforming growth factor-β1 upregulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension via the PTEN/AKT pathways. Int J. Biochem. Cell Biol. 2016, 77, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.B.; Kumar, R. Schistosomiasis and the pulmonary vasculature (2013 Grover Conference series). Pulm. Circ. 2014, 4, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am. J. Respir. Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Kassa, B.; Gebreab, L.; Robinson, J.C.; Koyanagi, D.E.; Sanders, L.; Barthel, L.; Meadows, C.; Fox, D.; et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat. Commun. 2017, 8, 15494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.; Duim, S.N.; Moerkamp, A.T.; Goumans, M.J. TGFβ and BMP signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation 2012, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Diez, M.; Musri, M.M.; Ferrer, E.; Barbera, J.A.; Peinado, V.I. Endothelial progenitor cells undergo an endothelial-to-mesenchymal transition-like process mediated by TGFβRI. Cardiovasc. Res. 2010, 88, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Kitao, A.; Sato, Y.; Sawada-Kitamura, S.; Harada, K.; Sasaki, M.; Morikawa, H.; Shiomi, S.; Honda, M.; Matsui, O.; Nakanuma, Y. Endothelial to mesenchymal transition via transforming growth factor-β1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 2009, 175, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Ji, Y.; Zhang, G.; Qu, Y.; Zhang, L.; Jiang, W. Ponatinib attenuates experimental pulmonary arterial hypertension by modulating Wnt signaling and vasohibin-2/vasohibin-1. Life Sci. 2016, 148, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.; Velthuis, S.; van Gent, M.; Westermann, C.; Snijder, R.; Mager, J.; Post, M. Pulmonary Hypertension in a Large Cohort with Hereditary Hemorrhagic Telangiectasia. Respiration 2017, 94, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Borthwick, G.M.; Hislop, A.A.; Arthur, H.M. Endoglin and activin receptor-like-kinase 1 are co-expressed in the distal vessels of the lung: Implications for two familial vascular dysplasias, HHT and PAH. Lab. Investig. 2009, 89, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, P.D.; Morrell, N.W. The transforming growth factor-β-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp. Physiol. 2013, 98, 1262–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, K.; Kawaguchi, Y.; Kawamoto, M.; Ota, Y.; Tochimoto, A.; Gono, T.; Katsumata, Y.; Takagi, M.; Hara, M.; Yamanaka, H. Activation of the activin A-ALK-Smad pathway in systemic sclerosis. J. Autoimmun. 2011, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; West, J. Rho-kinase inhibition alleviates pulmonary hypertension in transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor gene. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L667–L674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.; Oh, E.Y.; Lee, H.B. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat. Rev. Nephrol. 2009, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hassell, K.L. Altered hemostasis in pulmonary hypertension. Blood Coagul. Fibrinolysis 1998, 9, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Richards, J.A.; Letteboer, T.G.; Rushlow, D.; Prigoda, N.L.; Leedom, T.P.; Ganguly, A.; Castells, A.; Ploos van Amstel, J.K.; Westermann, C.J.; et al. SMAD4 mutations found in unselected HHT patients. J. Med. Genet. 2006, 43, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, X.; Al-Lamki, R.S.; Southwood, M.; Zhao, J.; Lever, A.M.; Grimminger, F.; Schermuly, R.T.; Morrell, N.W. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ. Res. 2010, 107, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell. Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol Lung Cell. Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef] [PubMed]
- Lambers, C.; Roth, M.; Zhong, J.; Campregher, C.; Binder, P.; Burian, B.; Petkov, V.; Block, L.H. The interaction of endothelin-1 and TGF-β1 mediates vascular cell remodeling. PLoS ONE 2013, 8, e73399. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Bottinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-β/SMAD signaling through an interaction with the TGF-β type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Preston, I.R.; Roberts, K.E.; Miller, D.P.; Sen, G.P.; Selej, M.; Benton, W.W.; Hill, N.S.; Farber, H.W. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015, 132, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Sosada, M.; Fabel, H. Plasma coagulation profiles in patients with severe primary pulmonary hypertension. Eur. Respir. J. 1998, 12, 1446–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.R.; Granton, J.T.; Mehta, S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest 2006, 130, 545–552. [Google Scholar] [CrossRef]
- Eisenberg, P.R.; Lucore, C.; Kaufman, L.; Sobel, B.E.; Jaffe, A.S.; Rich, S. Fibrinopeptide A levels indicative of pulmonary vascular thrombosis in patients with primary pulmonary hypertension. Circulation 1990, 82, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Beckmann, R.; Frank, H.; Kneussl, M.; Mlczoch, J.; Binder, B.R. Fibrinogen, t-PA, and PAI-1 plasma levels in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1994, 150, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Olman, M.A.; Marsh, J.J.; Lang, I.M.; Moser, K.M.; Binder, B.R.; Schleef, R.R. Endogenous fibrinolytic system in chronic large-vessel thromboembolic pulmonary hypertension. Circulation 1992, 86, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Christ, G.; Graf, S.; Huber-Beckmann, R.; Zorn, G.; Lang, I.; Kneussi, M.; Binder, B.R.; Huber, K. Impairment of the plasmin activation system in primary pulmonary hypertension: Evidence for gender differences. Thromb. Haemost. 2001, 86, 557–562. [Google Scholar] [PubMed]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id proteins are critical downstream effectors of BMP signaling in human pulmonary arterial smooth muscle cells. Am. J. Physiol Lung Cell. Mol. Physiol. 2013, 305, L312–L321. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Davies, R.J.; Southwood, M.; Long, L.; Yang, X.; Sobolewski, A.; Upton, P.D.; Trembath, R.C.; Morrell, N.W. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: Implications for familial pulmonary arterial hypertension. Circ. Res. 2008, 102, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, V.; van Cruchten, I.; Sablitzky, F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994, 22, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, M.C.; Kind, T.; Marcus, J.T.; Mauritz, G.J.; Heymans, M.W.; Bogaard, H.J.; Boonstra, A.; Marques, K.M.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 2011, 58, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Manes, A. The right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 2014, 23, 476–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaard, H.J.; Abe, K.; Vonk, N.A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Kato, H.; Honjo, O.; Breitling, S.; Kuebler, W.M.; Sun, M.; Friedberg, M.K. Carvedilol improves biventricular fibrosis and function in experimental pulmonary hypertension. J. Mol. Med. 2015, 93, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rol, N.; de Raaf, M.A.; Sun, X.; Kuiper, V.P.; da Silva Goncalves Bos, D.; Happe, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib improves cardiac fibrosis but leaves pulmonary vascular remodeling unaltered in experimental pulmonary hypertension. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Megalou, A.J.; Glava, C.; Oikonomidis, D.L.; Vilaeti, A.; Agelaki, M.G.; Baltogiannis, G.G.; Papalois, A.; Vlahos, A.P.; Kolettis, T.M. Transforming growth factor-β inhibition attenuates pulmonary arterial hypertension in rats. Int. J. Clin. Exp. Med. 2010, 3, 332–340. [Google Scholar] [PubMed]
- De Gramont, A.; Faivre, S.; Raymond, E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 2017, 6, e1257453. [Google Scholar] [CrossRef] [PubMed]
- Zaiman, A.L.; Podowski, M.; Medicherla, S.; Gordy, K.; Xu, F.; Zhen, L.; Shimoda, L.A.; Neptune, E.; Higgins, L.; Murphy, A.; et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Yilmaz, H.; Ghofrani, H.A.; Woyda, K.; Pullamsetti, S.; Schulz, A.; Gessler, T.; Dumitrascu, R.; Weissmann, N.; Grimminger, F.; et al. Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 358–363. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
TGF-β and BMP signaling. Receptors with evidence of mutations in pulmonary arterial hypertension (PAH) are underlined [17]. Abbreviations: ActRII, activin receptor type II; ALK, activing receptor-like kinase; BMP, bone morphogenetic protein; GDF, growth/differentiation factor; TGF, transforming growth factor.
Figure 1.
TGF-β and BMP signaling. Receptors with evidence of mutations in pulmonary arterial hypertension (PAH) are underlined [17]. Abbreviations: ActRII, activin receptor type II; ALK, activing receptor-like kinase; BMP, bone morphogenetic protein; GDF, growth/differentiation factor; TGF, transforming growth factor.
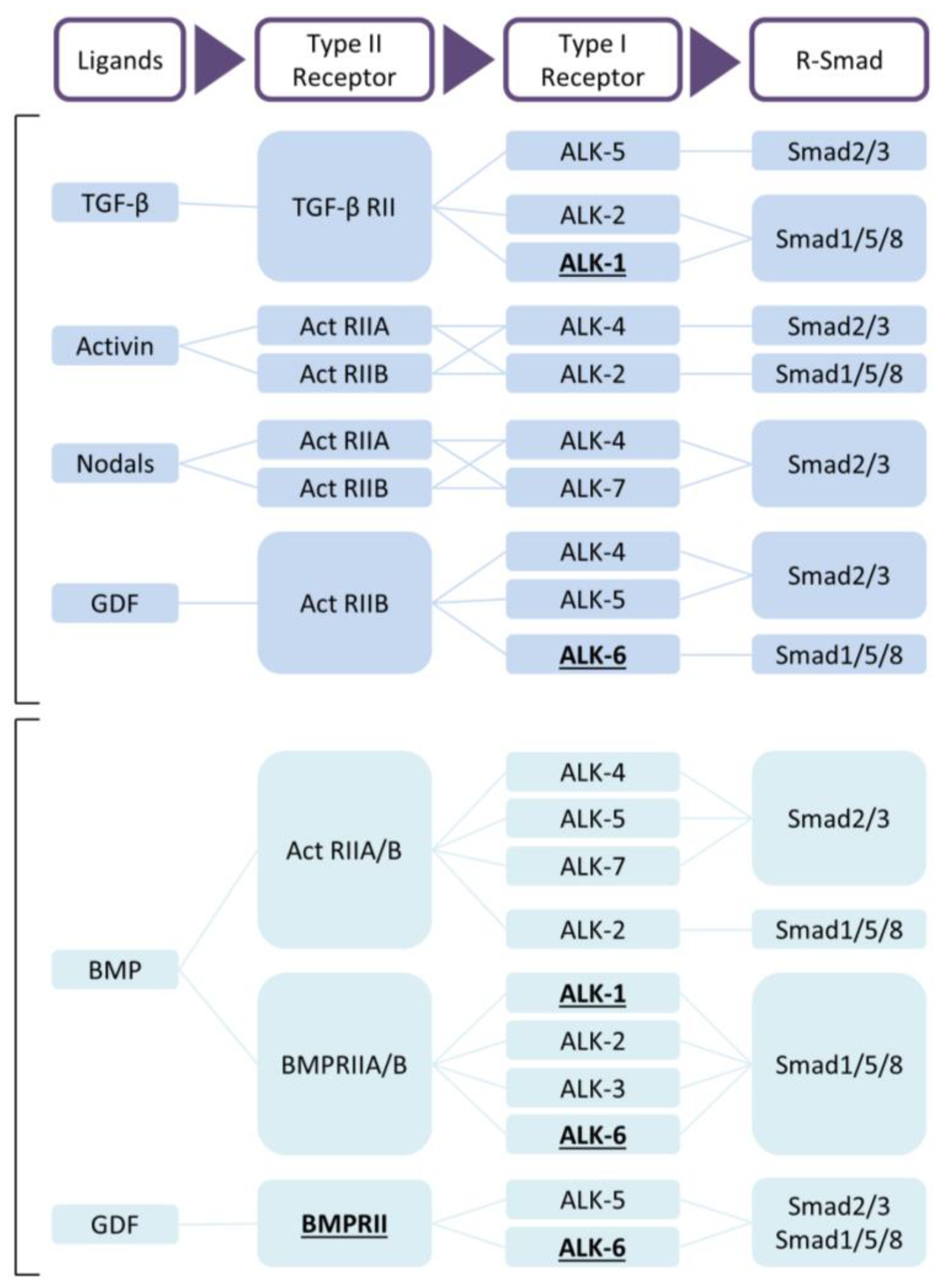
Figure 2.
Proposed mechanism of TGF-β signaling in the pathogenesis of pulmonary arterial hypertension. Abbreviations: ActRII, Activin receptor type II; AKT, protein kinase B; ALK1, activin receptor-like kinase 1; CTGF, connective tissue growth factor; ERK, extracellular signal-regulated kinases; GDF, growth/differentiation factor; JNK, c-Jun N-terminal kinases; MAPK, mitogen-activated protein kinase; PAI-1, plasminogen activator inhibitor-1; TGF-β, transforming growth factor β. TGFBRII, TGF-β receptor type II.
Figure 2.
Proposed mechanism of TGF-β signaling in the pathogenesis of pulmonary arterial hypertension. Abbreviations: ActRII, Activin receptor type II; AKT, protein kinase B; ALK1, activin receptor-like kinase 1; CTGF, connective tissue growth factor; ERK, extracellular signal-regulated kinases; GDF, growth/differentiation factor; JNK, c-Jun N-terminal kinases; MAPK, mitogen-activated protein kinase; PAI-1, plasminogen activator inhibitor-1; TGF-β, transforming growth factor β. TGFBRII, TGF-β receptor type II.

{kind=link}
{kind=link}
{kind=link}
Table 1.
TGF-β signaling in pulmonary arterial hypertension in human tissue and animal models.
| Serum | Lung Tissue/Vessel | Heart Tissue | EC | SMC | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligands | ||||||||||||
| TGF-β1 | mRNA | = | = | ↑ a,b,d | ↑ b | = | ↑ | [12,18,19,20,21,22,23,24,25,26] | ||||
| Protein | ↑ | ↑ | ↑ a,b,d | ↑ b | ||||||||
| TGF-β2 | mRNA | ↓ b | [12,20] | |||||||||
| Protein | = | |||||||||||
| TGF-β3 | mRNA | ↓ | ↑ | [12,20,27,28,29,30] | ||||||||
| Protein | ↑ | = b,c ↑ a | ||||||||||
| Activin A | mRNA | [31] | ||||||||||
| Protein | ↑ | ↑ a | ||||||||||
| GDF 9/15 | mRNA | ↑ | [32,33,34] | |||||||||
| Protein | ↑ | ↑ | ||||||||||
| Type I receptors | ||||||||||||
| ALK1 | mRNA | ↑ | ↑ | = | [19,35,36,37] | |||||||
| Protein | ↑ | ↑↓ b | ↑ | = | ||||||||
| ALK5 | mRNA | =↑ | = | = | = | [19,21,38,39,40,41] | ||||||
| Protein | = | = | ||||||||||
| Type II receptors | ||||||||||||
| TGFBRII | mRNA | ↓ b↑ a,c | [25,42,43,44] | |||||||||
| Protein | ↓ b | = | = | |||||||||
| ActRII | mRNA | [31] | ||||||||||
| Protein | ↑ a | |||||||||||
| Co-receptors | ||||||||||||
| β-glycan | mRNA | = | ↓ | [28,38] | ||||||||
| Protein | ||||||||||||
| Endoglin | mRNA | = | ↓ b | ↑ | = | [19,43] | ||||||
| Protein | ↑ | ↑ | = | |||||||||
| Canonical signaling | ||||||||||||
| Smad2 | mRNA | ↓ | [12,27,31,37,39,40,41,43,44,45,46] | |||||||||
| Protein | ↑↓= a,b | ↓ | ↑ | |||||||||
| Smad3 | mRNA | ↓ | ↓ b | [12,13,27,31,41,43] | ||||||||
| Protein | ↑ a,b↓ b | |||||||||||
| Smad4 | mRNA | ↓ | ↓ b | [27,37,39,43,47,48] | ||||||||
| Protein | ↓ b | = | = | |||||||||
| Smad6/7 | mRNA | ↑ a↓ b | [37,44,48] | |||||||||
| Protein | ↑ a | |||||||||||
| PAI-1 | mRNA | ↑ | ↓ | ↑ b | [12,13,49,50] | |||||||
| Protein | ↓ | ↑ b | ||||||||||
| Non-canonical signaling | ||||||||||||
| MAPKs | mRNA | ↑ | [51] | |||||||||
| Protein | ||||||||||||
| Cav1 | mRNA | [36,37,52] | ||||||||||
| Protein | ↓ | = b | ||||||||||
| CTGF | mRNA | ↓ b | ↑ c,e | [43,53,54] | ||||||||
| Protein | ↓ b | ↑ c,e | ||||||||||
Grey boxes indicate findings in tissue of pulmonary arterial hypertension patients; white boxes indicate findings in experimental animal models. ↑, increase in comparison to controls; ↓, decreased in comparison to controls; =, no change in comparison to controls. (a) Hypoxia-induced PH in rat, (b) monocrotaline-induced PH in rat, (c) Sugen hypoxia-induced PH in rat, (d) Schistosoma-induced PH in mice, (e) pulmonary artery banding in rats. Increases in Smad protein regard phosphorylation. Abbreviations: ActRII, Activin receptor type II; ALK1, activin receptor-like kinase 1; ALK5, activin receptor-like kinase 5; Cav1, caveolin-1; CTGF, connective tissue growth factor; EC, endothelial cell; GDF, growth/differentiation factor; MAPKs, mitogen-activated protein kinase; PAI-1, plasminogen activator inhibitor-1; SMC, smooth muscle cell; TGF-β, transforming growth factor β; TGFBRII, TGF-β receptor type II.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.-J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. https://doi.org/10.3390/ijms19092585
AMA Style
Rol N, Kurakula KB, Happé C, Bogaard HJ, Goumans M-J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. International Journal of Molecular Sciences. 2018; 19(9):2585. https://doi.org/10.3390/ijms19092585
Chicago/Turabian StyleRol, Nina, Konda Babu Kurakula, Chris Happé, Harm Jan Bogaard, and Marie-José Goumans. 2018. "TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family" International Journal of Molecular Sciences 19, no. 9: 2585. https://doi.org/10.3390/ijms19092585
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.