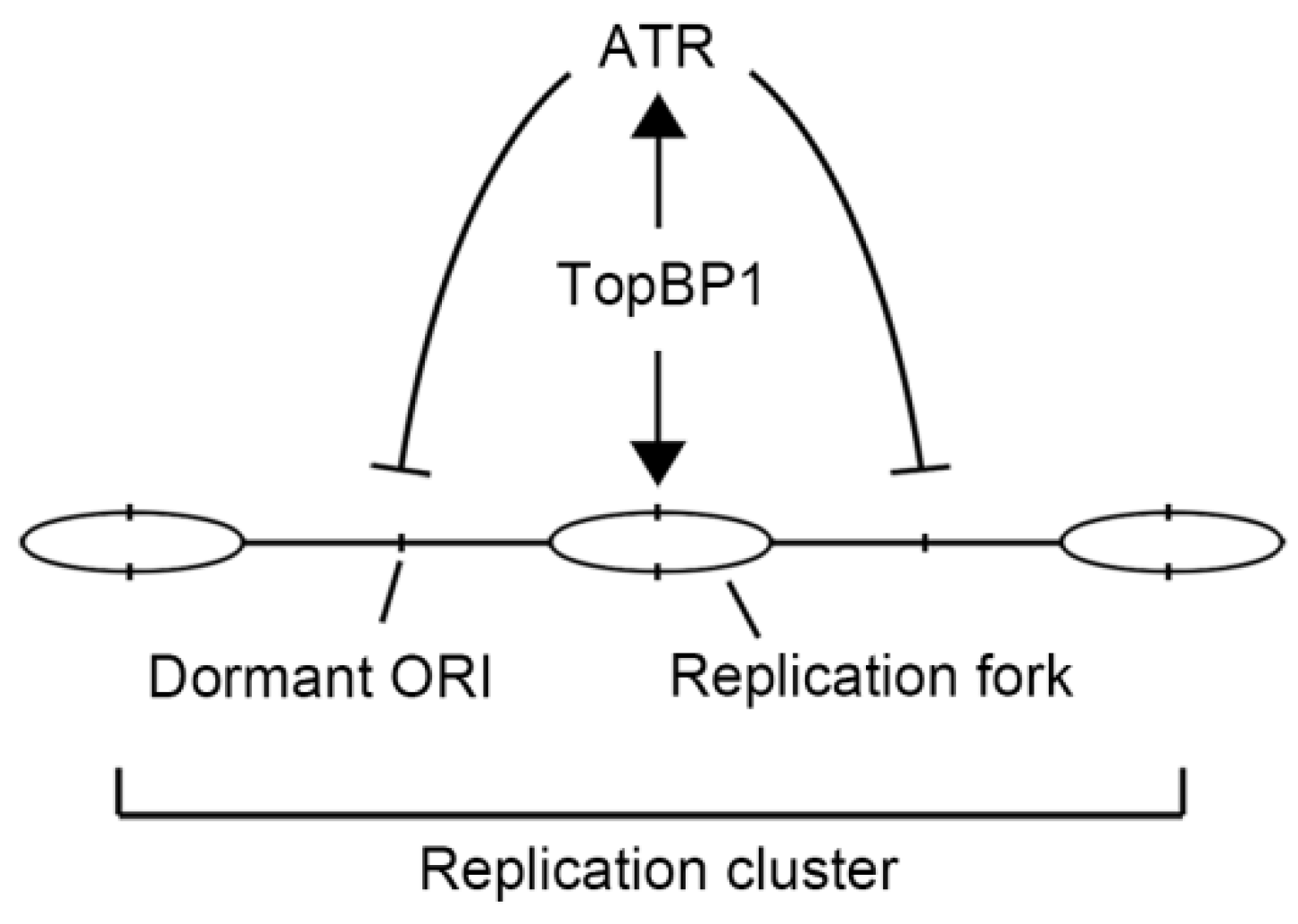
The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
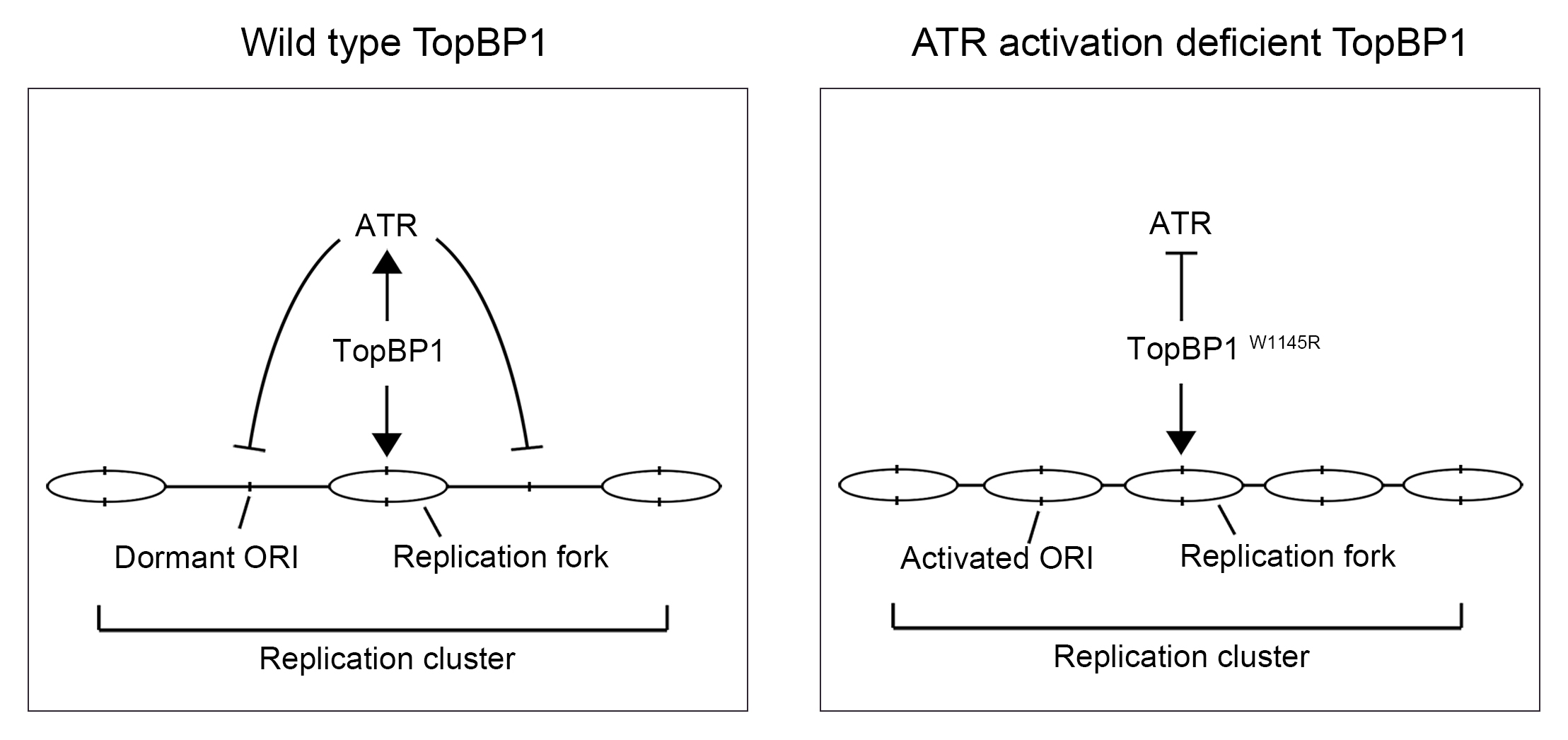
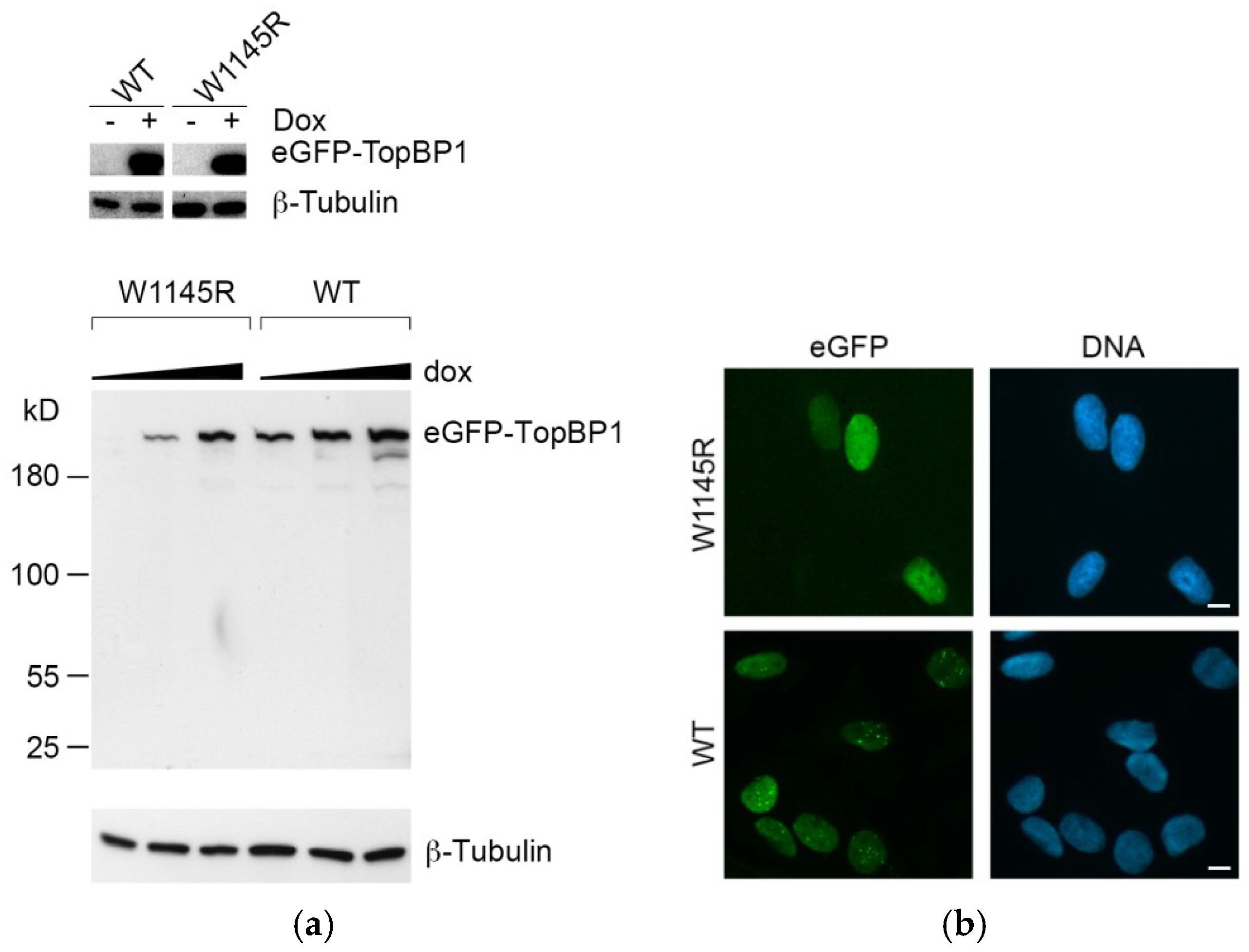
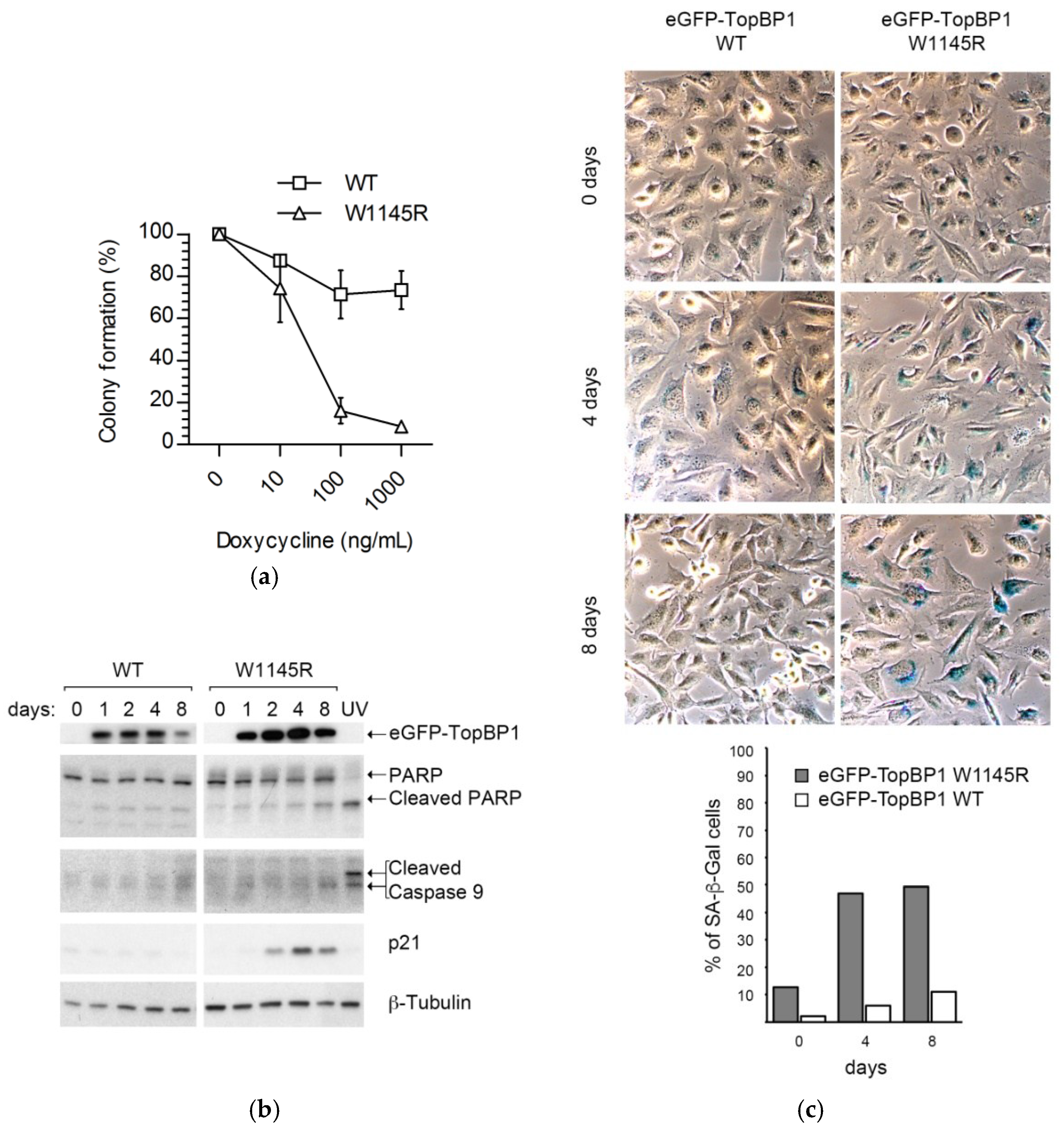
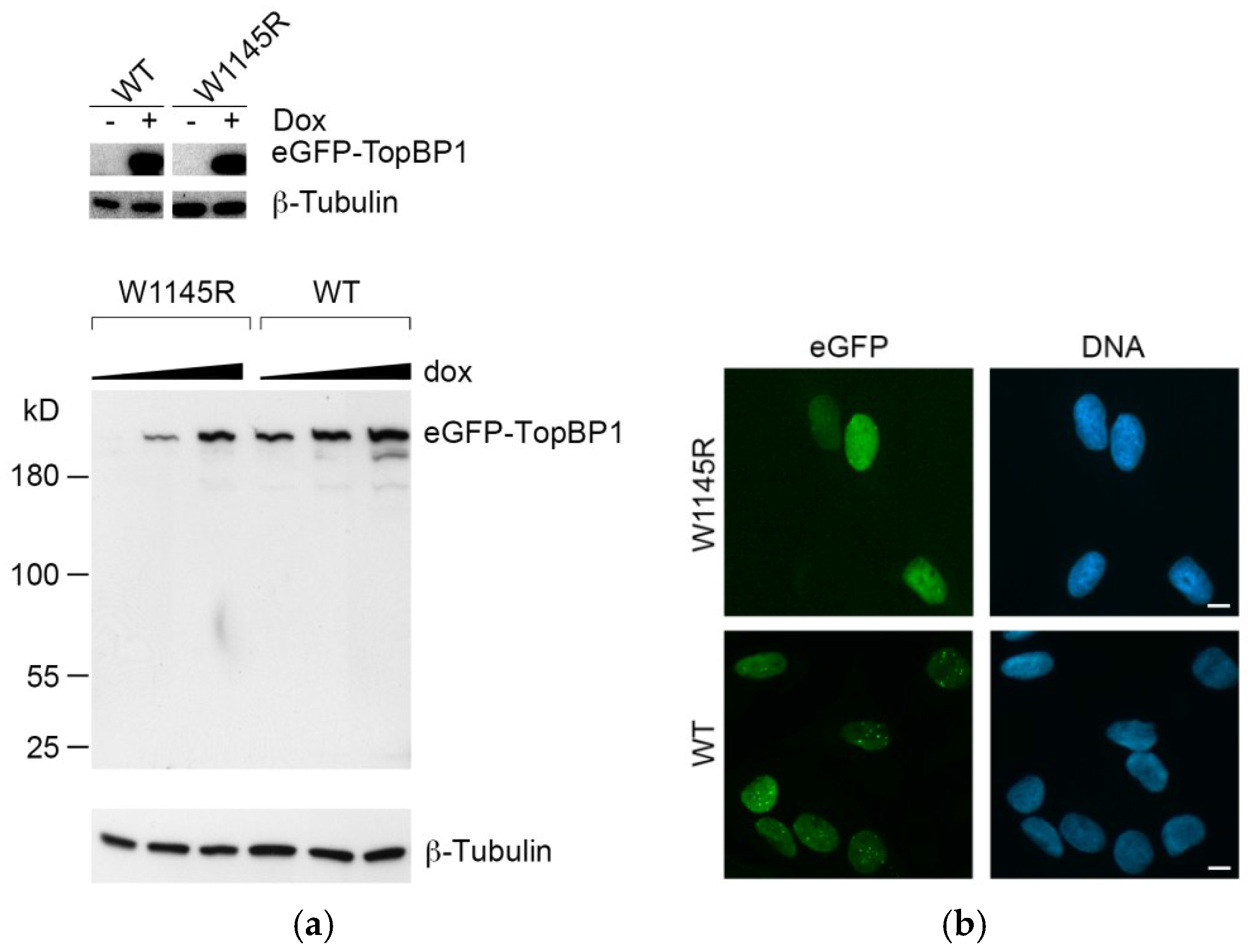
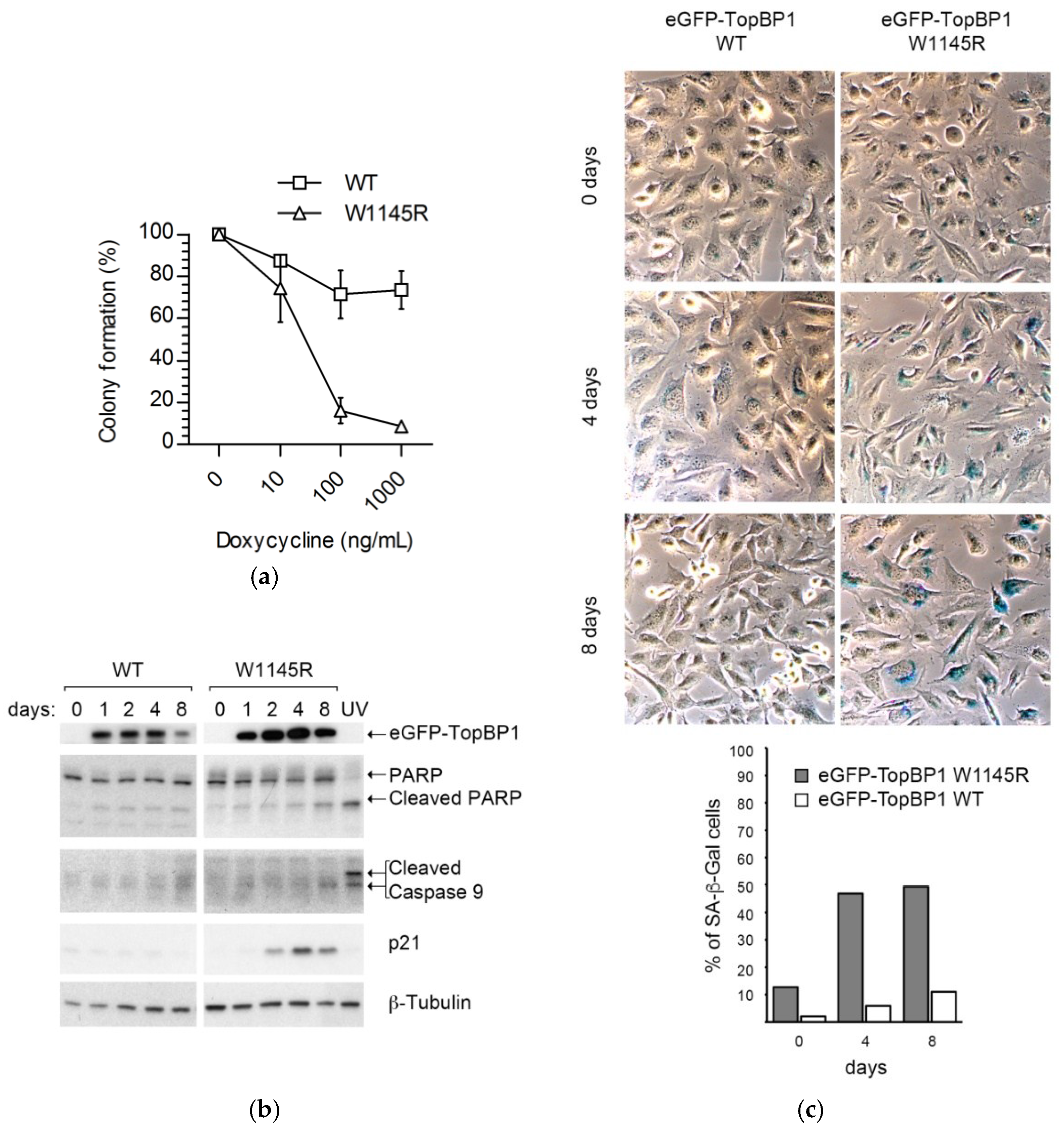
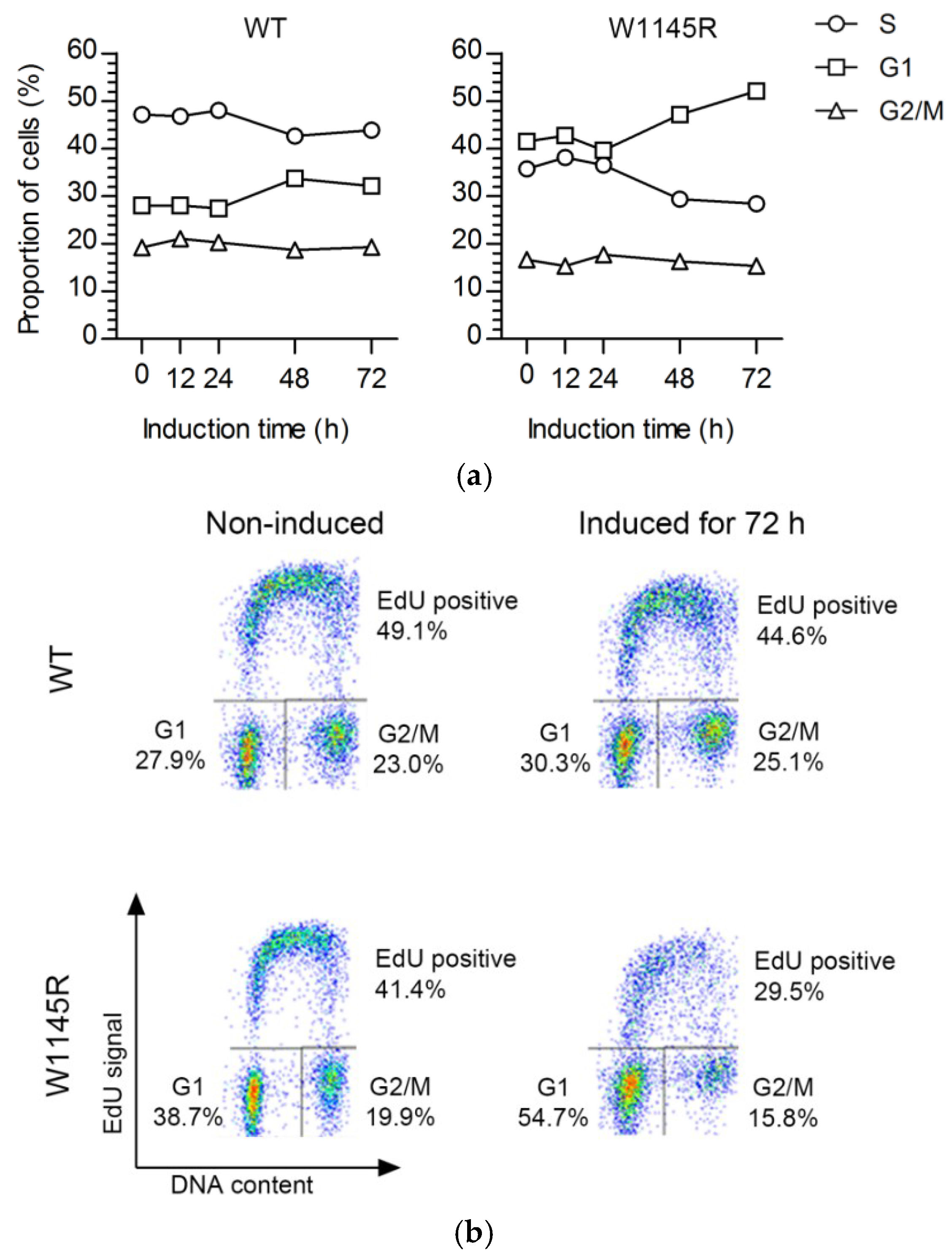
2.1. Cells Expressing the TopBP1 AAD Mutant Arrest at G1 and Enter Senescence
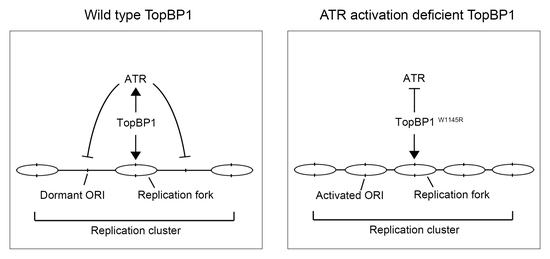
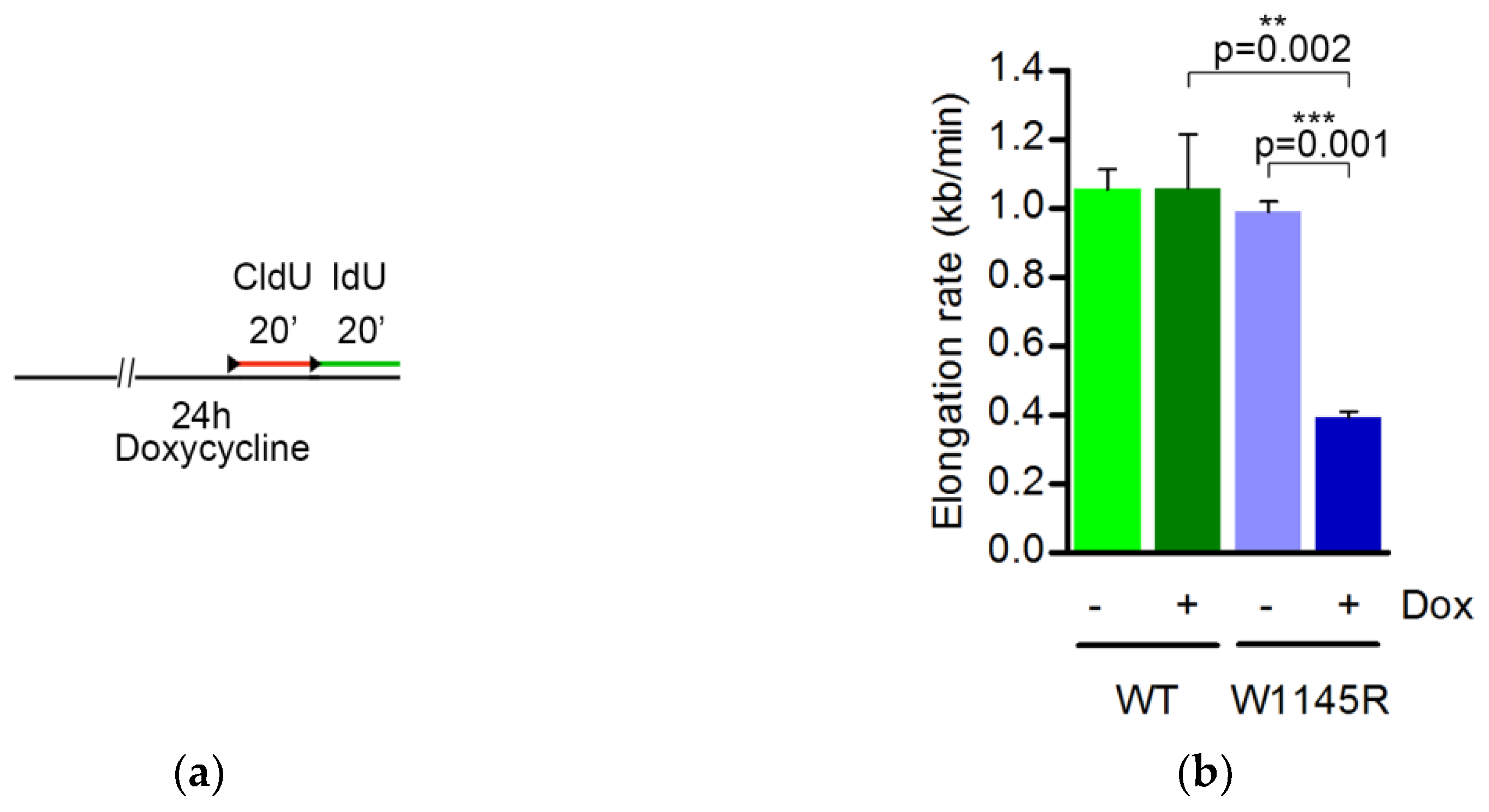
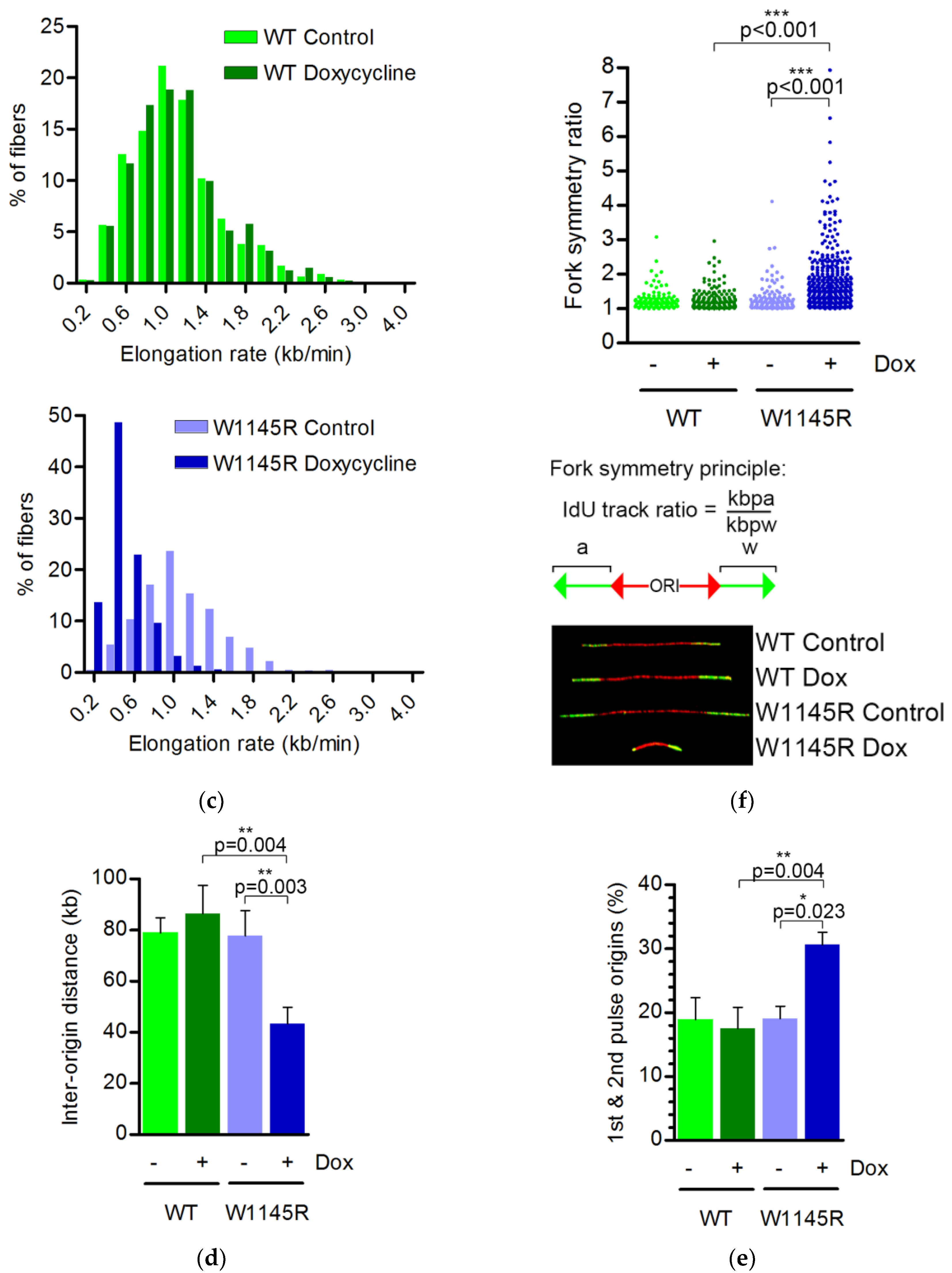
2.2. The TopBP1 AAD Mutant Slows Down the DNA Replication Elongation Rate and Increases the Number of Fired Origins
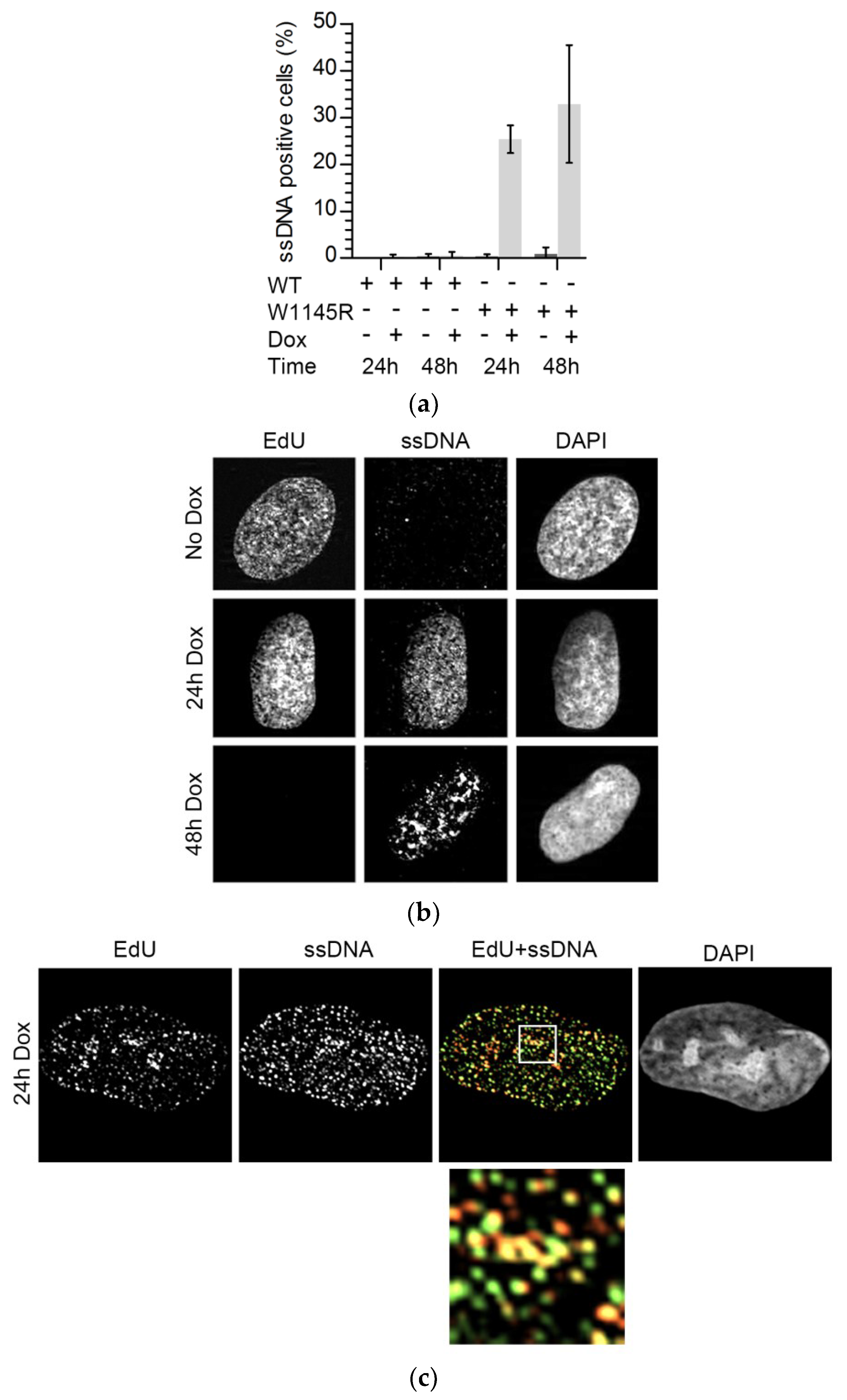
2.3. The TopBP1 AAD Mutant Induces Accumulation of Single-Stranded DNA
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Colony Formation Assay
4.3. Flow Cytometry
4.4. DNA Fiber Assays
4.5. Single-Stranded DNA (ssDNA) Analysis and Immunofluorescence Microscopy
4.6. Immunoblotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAD | ATR activating domain (in TopBP1) |
| ATM | Serine-protein kinase ATM |
| ATR | Serine/threonine-protein kinase ATR |
| BSA | Bovine serum albumin |
| Cdc45 | Cell division control protein 45 homolog |
| Cdc6 | Cell division control protein 6 homolog |
| Cdt1 | DNA replication factor Cdt1 |
| Chk1 | Serine/threonine-protein kinase Chk1 |
| CldU | 5-Chloro-2′-deoxyuridine |
| CMG | Cdc45-MCM-GINS complex |
| DDK | Dbf4-dependent kinase |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| ETAA1 | Ewing’s tumor-associated antigen 1 |
| eGFP | Enhanced green fluorescent protein |
| GEMC1 | Geminin coiled-coil domain-containing protein 1 |
| GINS | Sld5-Psf1-Psf2-Psf3 complex (go-ichi-ni-san) |
| H2A.X/γH2AX | Phosphorylated histone H2AX |
| IdU | 5-iodo-2′-deoxyuridine |
| kb | kilobase |
| MCM2-7 | DNA replication licensing factor MCM2-7 |
| ORI | Origin of DNA replication |
| p21 | Cyclin-dependent kinase inhibitor 1 |
| p27 | Cyclin-dependent kinase inhibitor 1B |
| p53 | Cellular tumor antigen p53 |
| PARP | Poly (ADP-ribose) polymerase |
| PBS | Phosphate-buffered saline |
| RecQL4 | ATP-dependent DNA helicase Q4 |
| RPA | Replication protein A complex |
| S-CDK | S-phase-specific cyclin dependent kinase |
| SA-β-Gal | Senescence-associated beta-galactosidase |
| ssDNA | Single-stranded DNA |
| TopBP1 | DNA topoisomerase 2-binding protein 1 |
| U2OS | Human osteosarcoma cell line |
| UV-C | Ultraviolet C (254 nm) |
| WT | Wild type |
References
- Aladjem, M.I.; Redon, C.E. Order from clutter: Selective interactions at mammalian replication origins. Nat. Rev. Genet. 2017, 18, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.A.; Shevchenko, A.A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, A.; Cosentino, C.; Errico, A.; Garner, E.; Costanzo, V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 2010, 12, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuno, K.; Kumano, M.; Kubota, Y.; Hashimoto, Y.; Takisawa, H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol. Cell. Biol. 2006, 26, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Sangrithi, M.N.; Bernal, J.A.; Madine, M.; Philpott, A.; Lee, J.; Dunphy, W.G.; Venkitaraman, A.R. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 2005, 121, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.D.; Muijtjens, M.; Os, R.V.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H.J. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef]
- Jeon, Y.; Ko, E.; Lee, K.Y.; Ko, M.J.; Park, S.Y.; Kang, J.; Jeon, C.H.; Lee, H.; Hwang, D.S. TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. J. Biol. Chem. 2011, 286, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-W.; Liu, C.; Li, T.-L.; Bruhn, C.; Krueger, A.; Min, W.; Wang, Z.-Q.; Carr, A.M. An Essential Function for the ATR-Activation-Domain (AAD) of TopBP1 in Mouse Development and Cellular Senescence. PLoS Genet. 2013, 9, e1003702. [Google Scholar] [CrossRef] [PubMed]
- Sokka, M.; Rilla, K.; Miinalainen, I.; Pospiech, H.; Syväoja, J.E. High levels of TopBP1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation. Nucleic Acids Res. 2015, 43, 4975–4989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Maya-Mendoza, A.; Zachos, G.; Gillespie, D.A.F.; Jackson, D.A.; Caldecott, K.W. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol. Cell. Biol. 2006, 26, 3319–3326. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. PNAS 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moiseeva, T.; Hood, B.; Schamus, S.; O’Connor, M.J.; Conrads, T.P.; Bakkenist, C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017, 8, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, C.; Koalick, D.; Fabricius, A.; Parplys, A.C.; Borgmann, K.; Pospiech, H.; Grosse, F. Cdc45 is limiting for replication initiation in humans. Cell Cycle 2016, 15, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marheineke, K.; Hyrien, O.; Krude, T. Visualization of bidirectional initiation of chromosomal DNA replication in a human cell free system. Nucleic Acids Res. 2005, 33, 6931–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of Human Chk1 Causes Increased Initiation of DNA Replication, Phosphorylation of ATR Targets, and DNA Breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 2002, 22, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.F.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007, 26, 2719–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, J.A.; Conti, C.; Syed, A.; Aladjem, M.I.; Pommier, Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: Single-cell and -DNA fiber analyses. Mol. Cell. Biol. 2007, 27, 5806–5818. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Costanzo, V.; Gautier, J. ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 2004, 6, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase I inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014, 74, 6968–6978. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Zhao, Y.; Xu, Y.; Ning, S.; Huo, W.; Hou, M.; Gao, G.; Ji, J.; Guo, R.; Xu, D. Ewing tumor-associated antigen 1 interacts with replication protein A to promote restart of stalled replication forks. J. Biol. Chem. 2016, 291, 21956–21962. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.J.; Fangman, W.L. Initiation at closely spaced replication origins in a yeast chromosome. Science 1993, 262, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Marahrens, Y.; Stillman, B. Replicator dominance in a eukaryotic chromosome. EMBO J. 1994, 13, 3395–3400. [Google Scholar] [PubMed]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A. DNA replication origin interference increases the spacing between initiation events in human cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Takisawa, H. Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J. 2003, 22, 2526–2535. [Google Scholar] [CrossRef] [PubMed]
- Mäkiniemi, M.; Hillukkala, T.; Tuusa, J.; Reini, K.; Vaara, M.; Huang, D.; Pospiech, H.; Majuri, I.; Westerling, T.; Mäkelä, T.P.P.; et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001, 276, 30399–30406. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mcavoy, S.A.; Smith, D.I.; Chen, J. Human TopBP1 Ensures Genome Integrity during Normal S Phase. Mol. Cell. Biol. 2005, 25, 10907–10915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokka, M.; Koalick, D.; Hemmerich, P.; Syväoja, J.E.; Pospiech, H. The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. Int. J. Mol. Sci. 2018, 19, 2376. https://doi.org/10.3390/ijms19082376
Sokka M, Koalick D, Hemmerich P, Syväoja JE, Pospiech H. The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. International Journal of Molecular Sciences. 2018; 19(8):2376. https://doi.org/10.3390/ijms19082376
Chicago/Turabian StyleSokka, Miiko, Dennis Koalick, Peter Hemmerich, Juhani E. Syväoja, and Helmut Pospiech. 2018. "The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase" International Journal of Molecular Sciences 19, no. 8: 2376. https://doi.org/10.3390/ijms19082376