Differences in Functional Expression of Connexin43 and NaV1.5 by Pan- and Class-Selective Histone Deacetylase Inhibition in Heart


Abstract
:
1. Introduction
2. Results
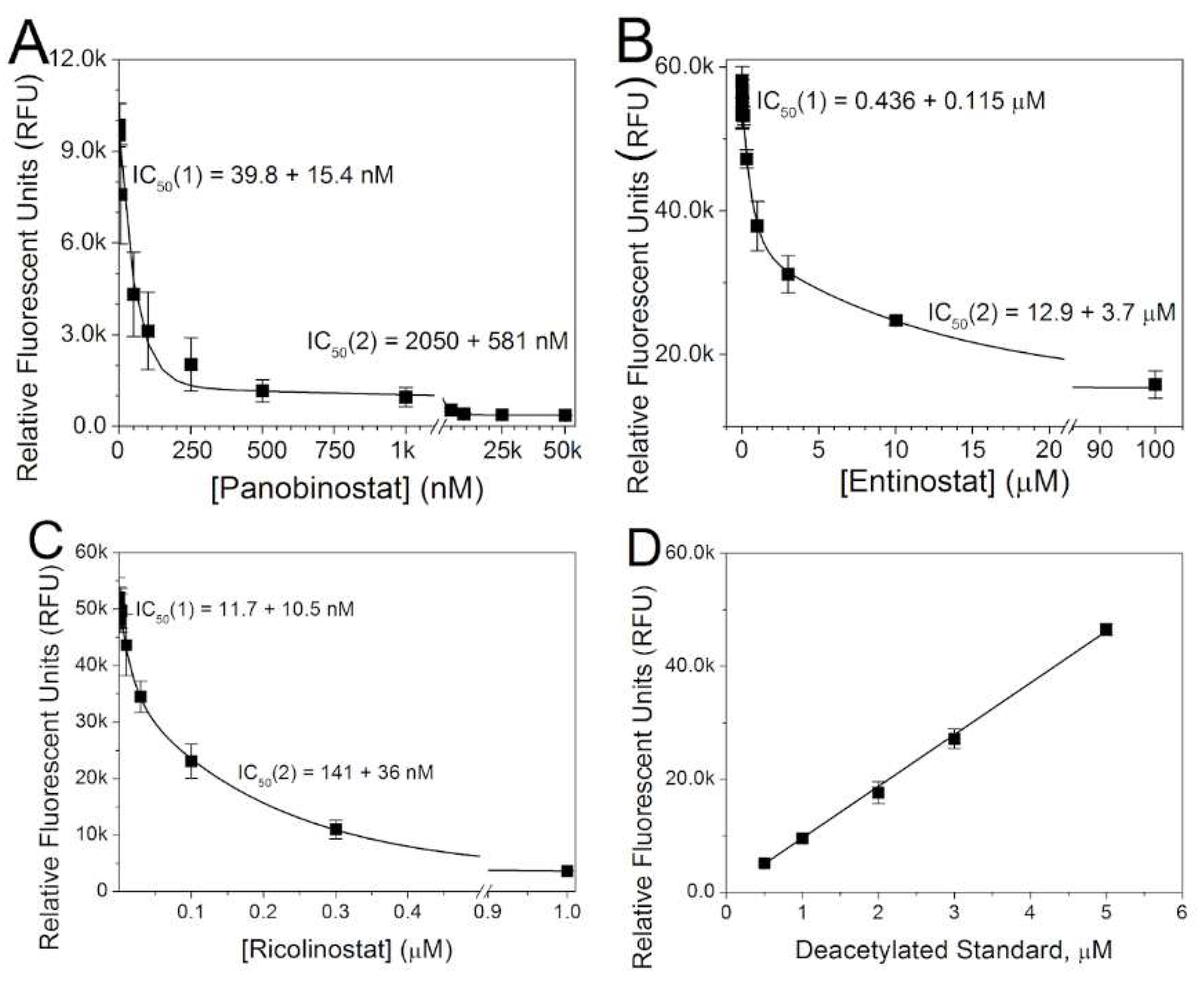
2.1. Inhibition of Cardiac HDAC Activity by Pan- and Class-Selective HDAC Inhibitors
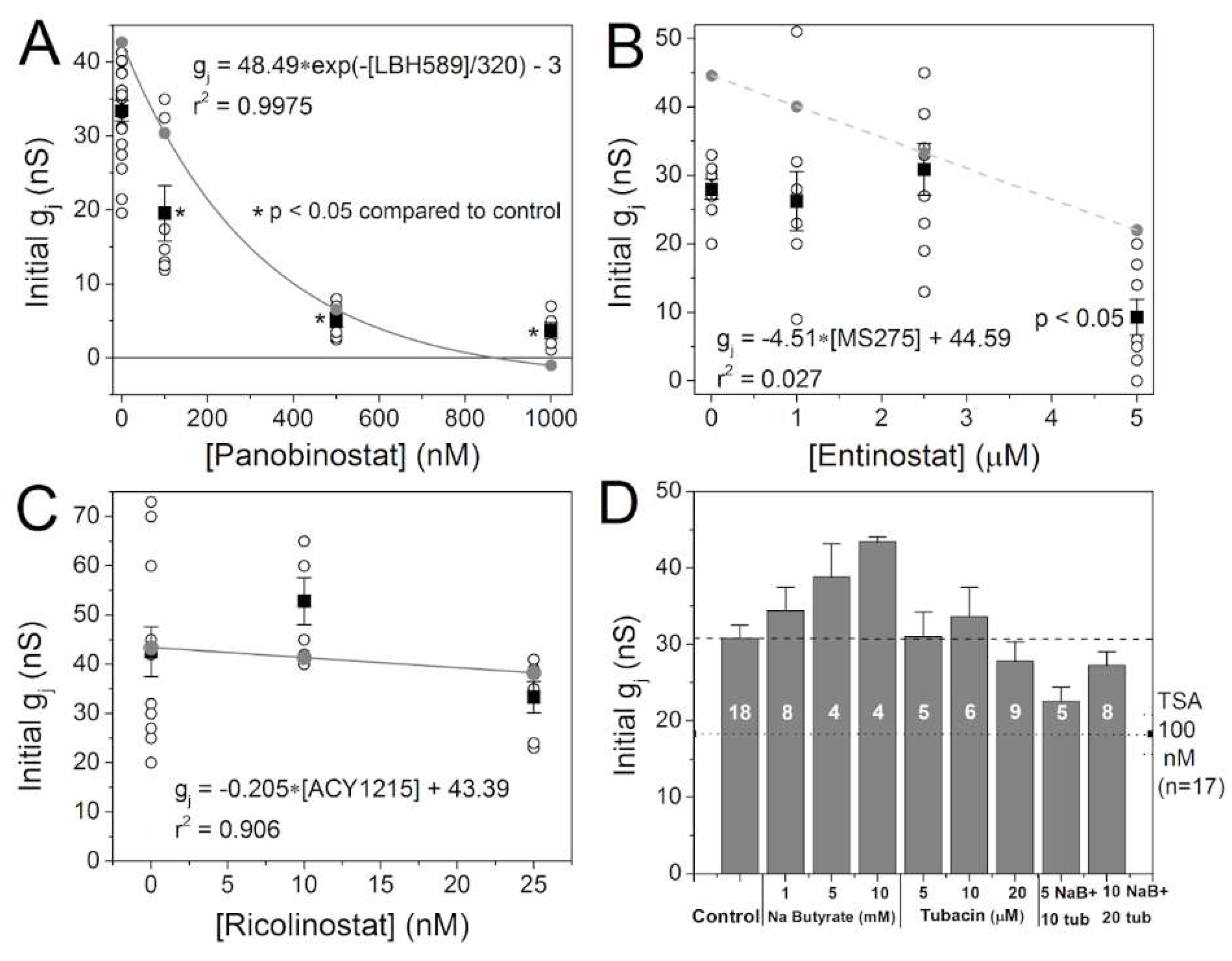
2.2. Effects of Class-Selective HDAC Inhibitors on Gap Junction Coupling
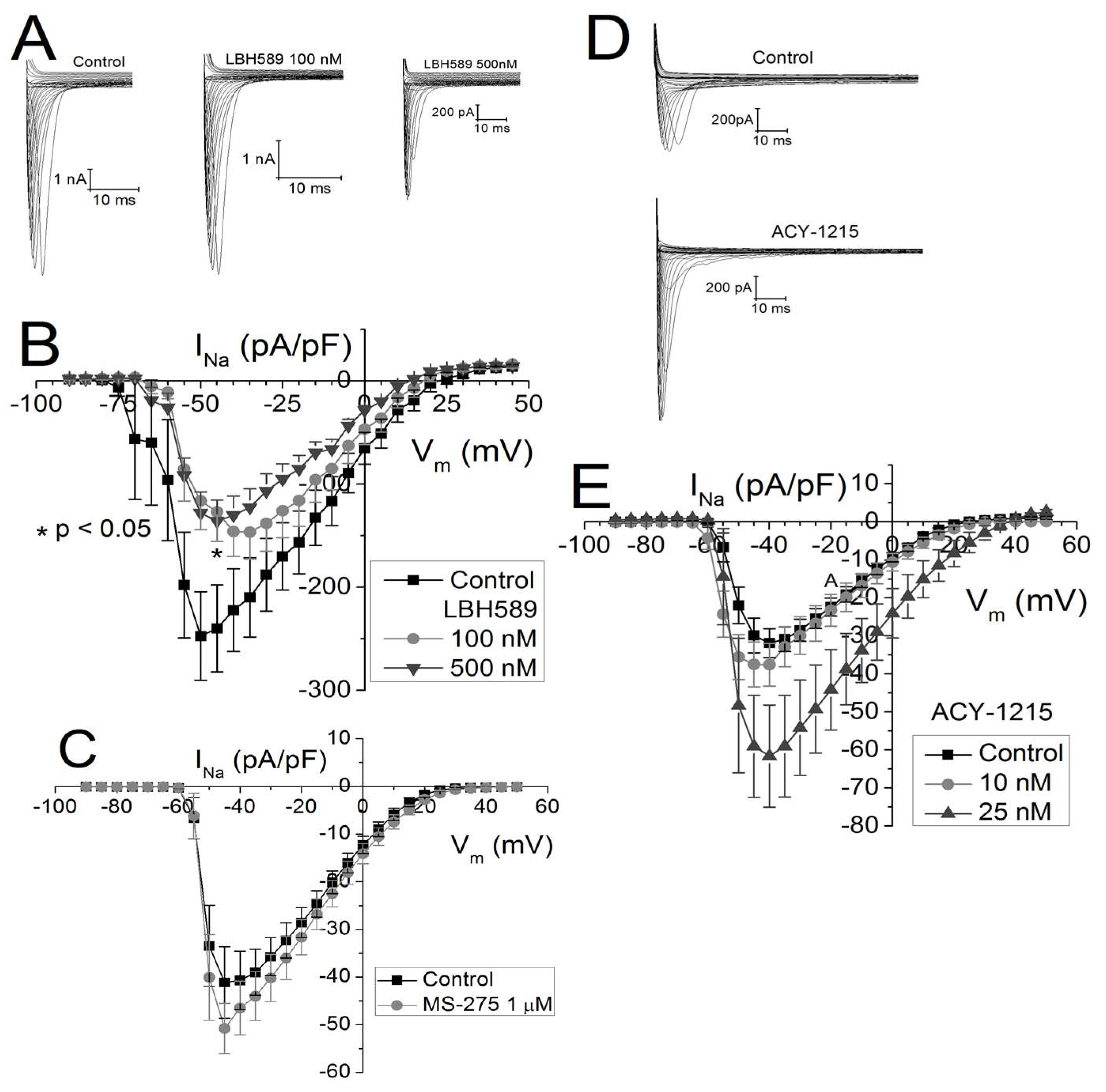
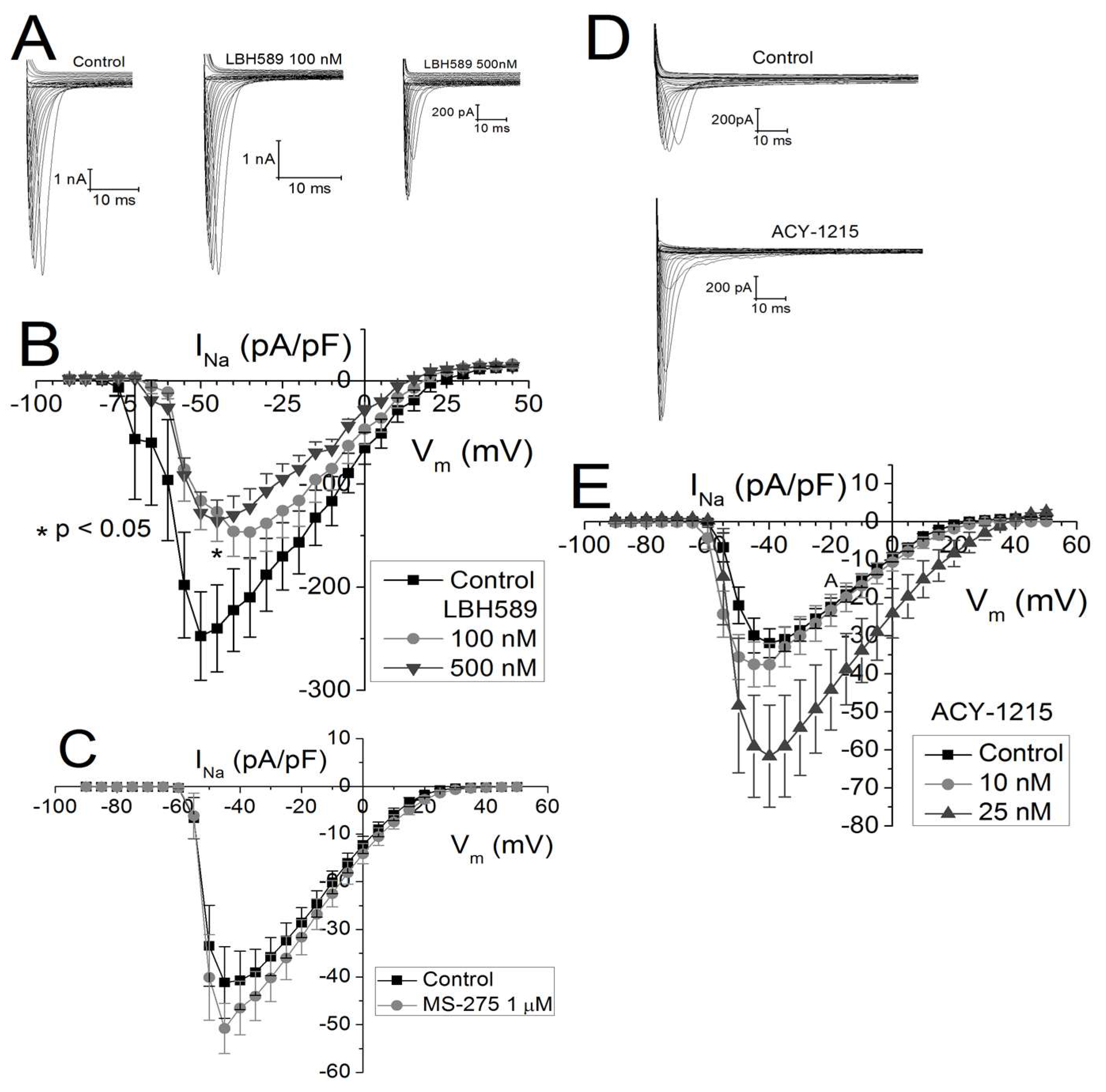
2.3. Effects of Class-Selective HDAC Inhibitors on Peak Cardiac Na+ Current Density
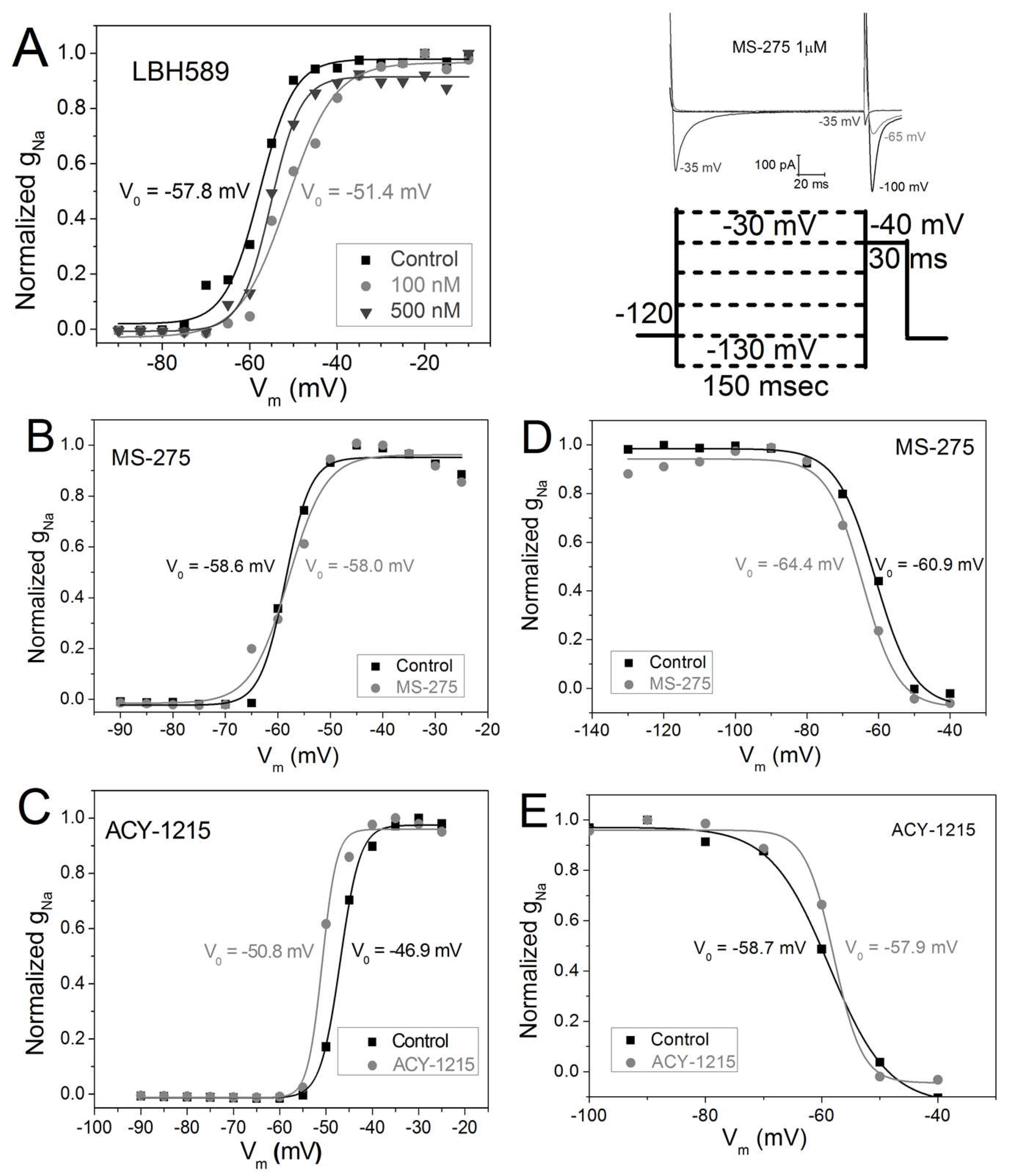
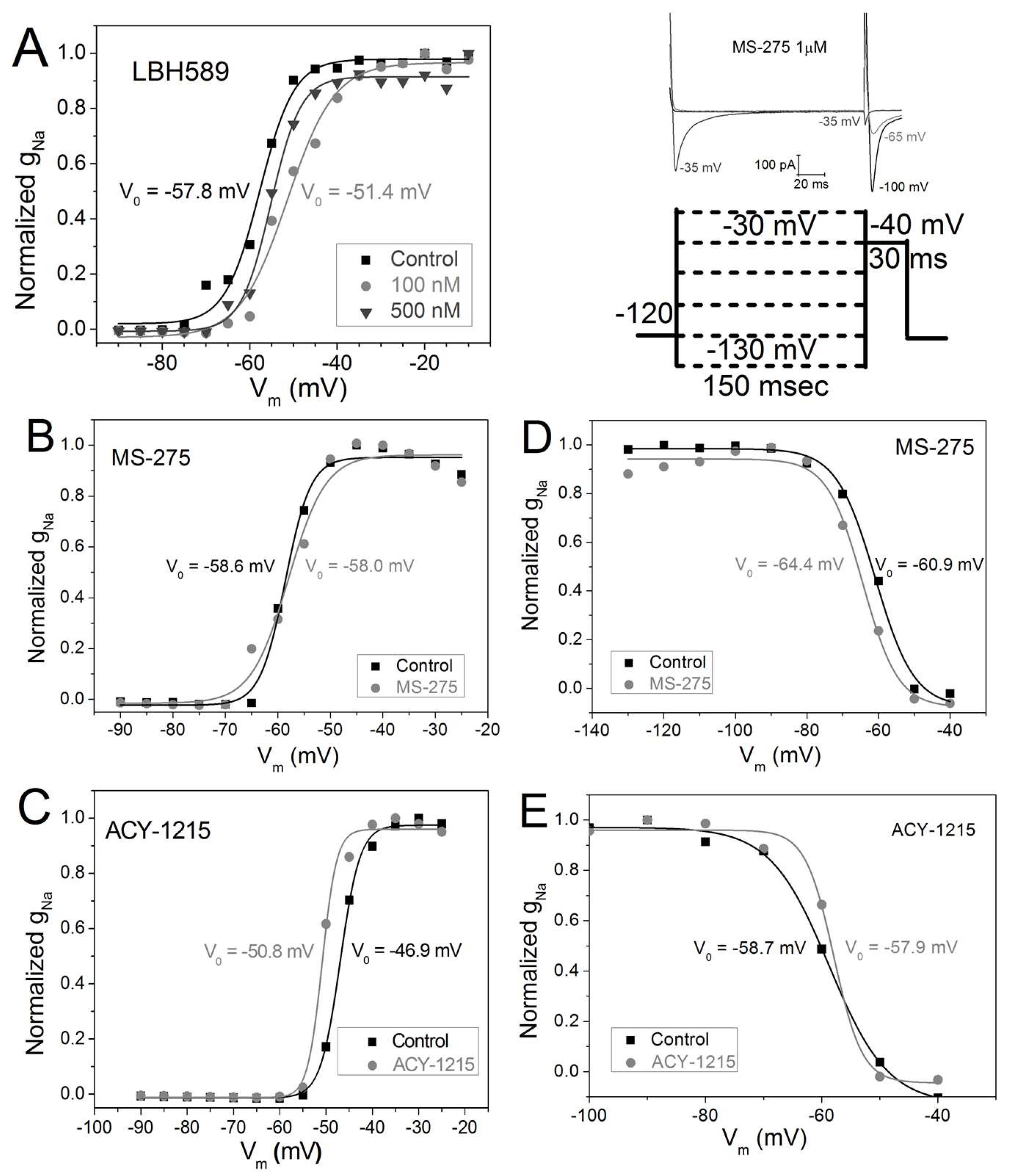
2.4. Vm-Dependence of gNa Activation and Inactivation
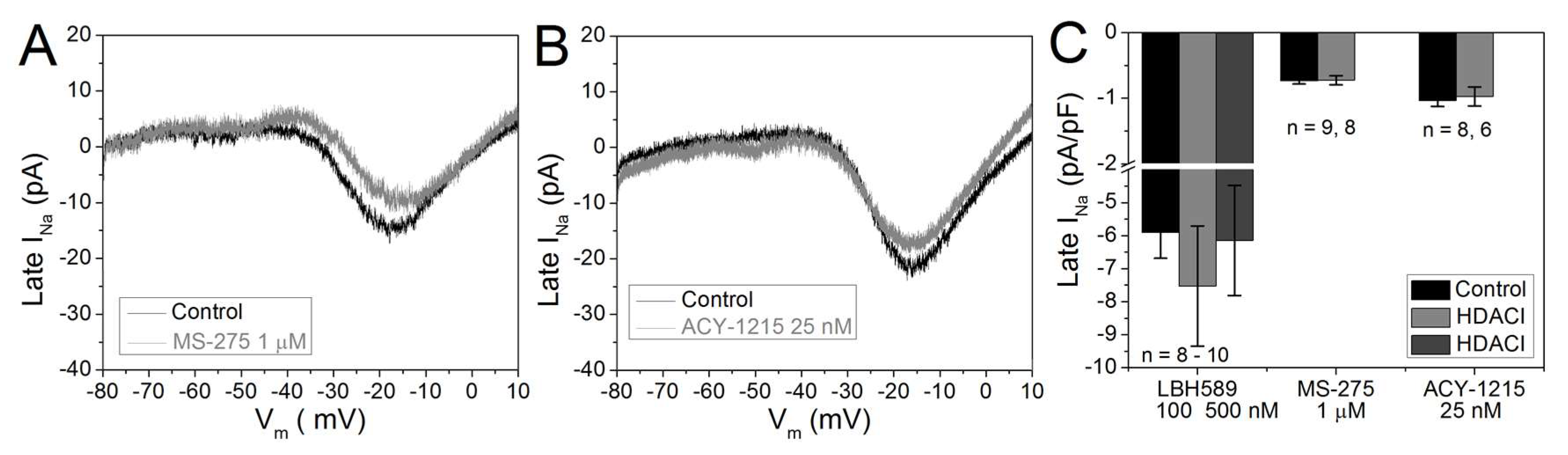
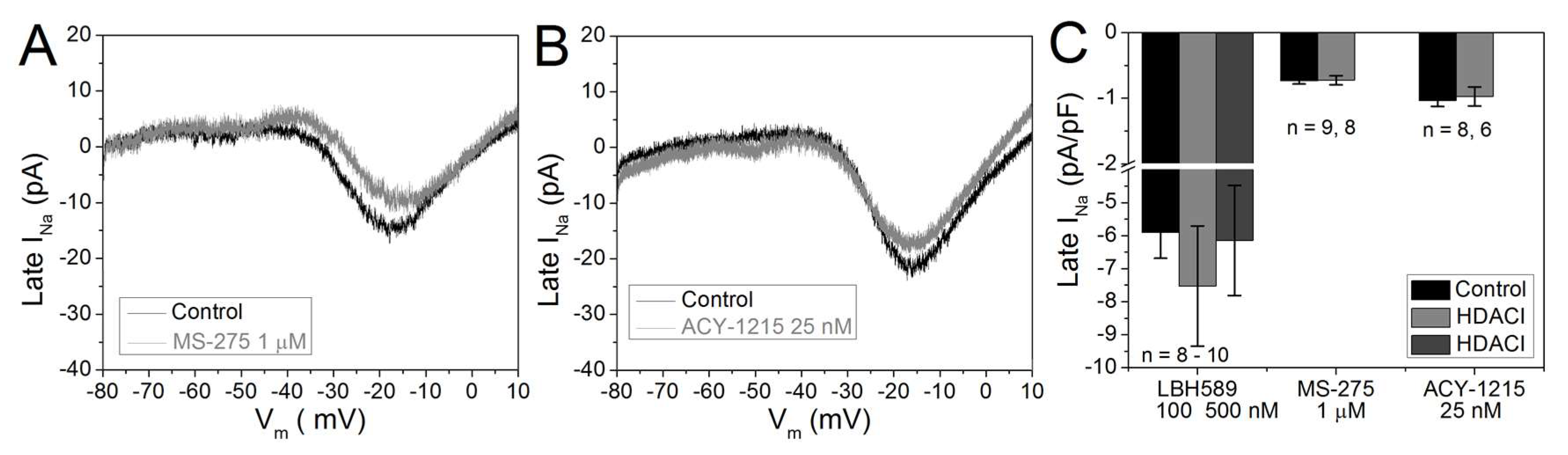
2.5. Cardiac Late INa
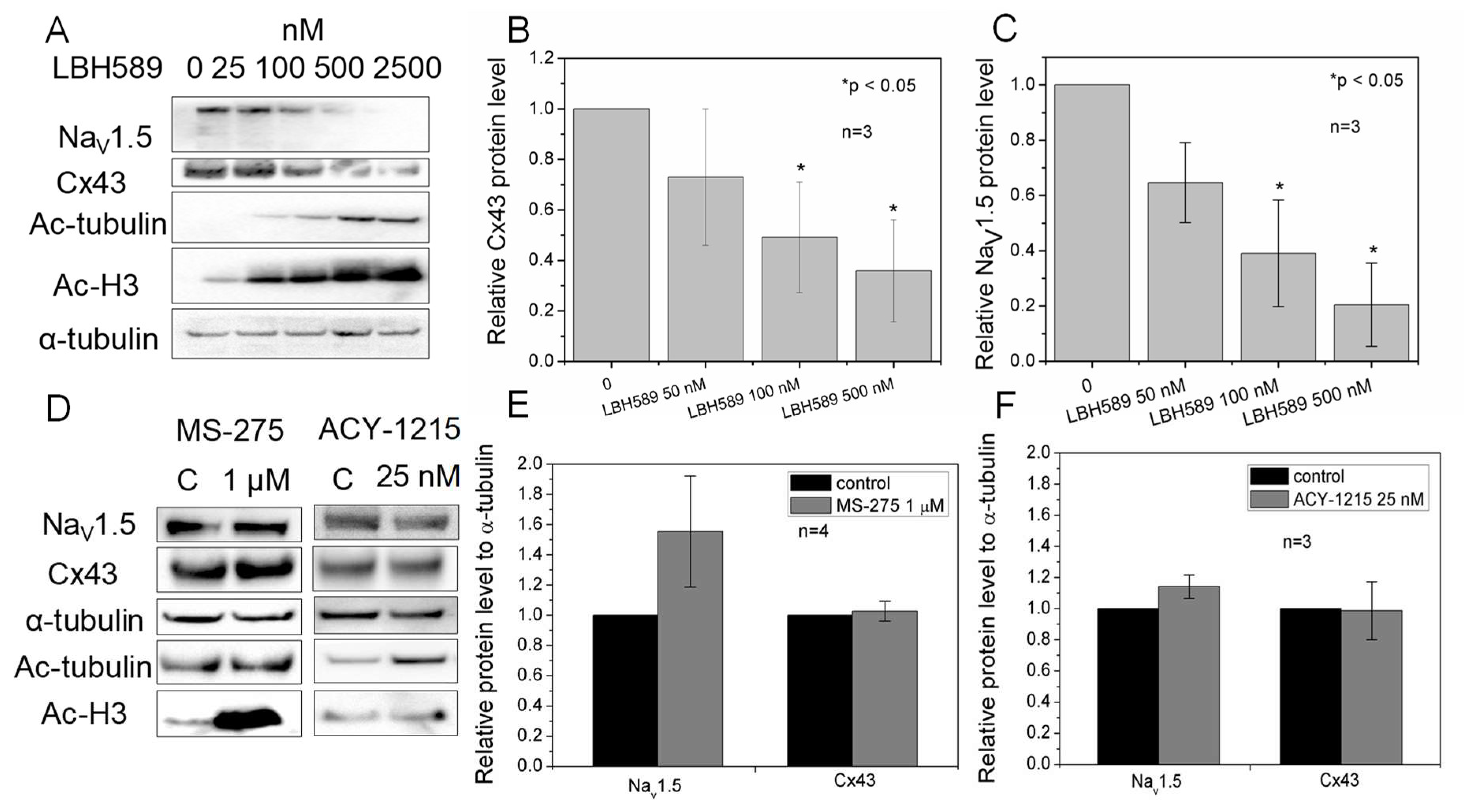
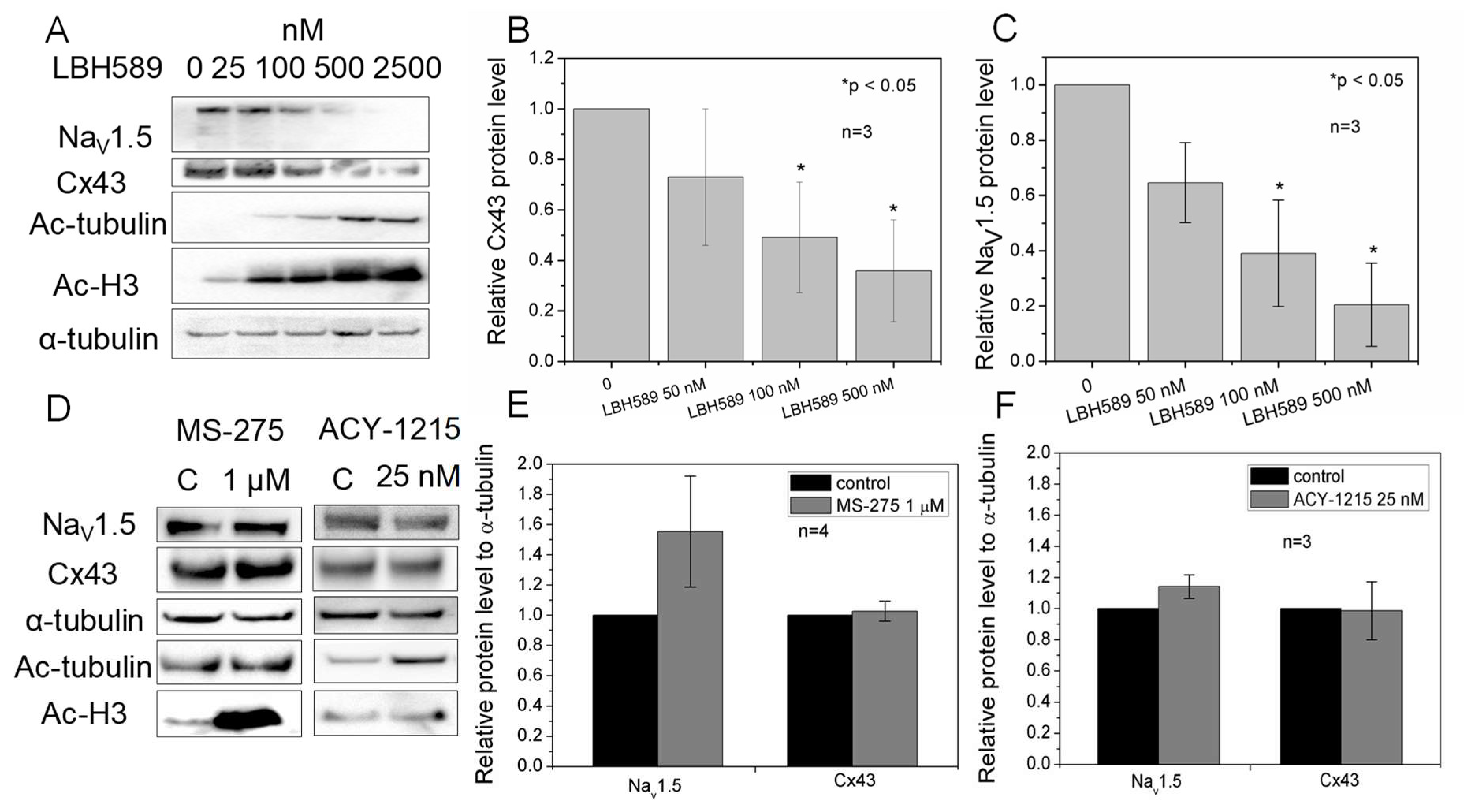
2.6. Cx43 and NaV1.5 Expression
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. HDAC Inhibitors
4.3. HDAC Activity Assay
4.4. Dual Whole Cell Patch Clamping
4.5. Whole Cell Patch Clamping
4.6. Western Blot
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Peng, L.; Seto, E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011, 206, 39–56. [Google Scholar] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R., Jr.; Washam, J.B.; Newby, L.K. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: A case report and review of the literature. Cardiol. J. 2012, 19, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar] [PubMed]
- Shah, M.H.; Binkley, P.; Chan, K.; Xiao, J.; Arbogast, D.; Collamore, M.; Farra, Y.; Young, D.; Grever, M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 2006, 12, 3997–4003. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Cattaneo, F.; Trimarco, B.; Esposito, G.; Perrino, C. Cardiovascular effects of histone deacetylase inhibitors epigenetic therapies: Systematic review of 62 studies and new hypotheses for future research. Int. J. Cardiol. 2016, 219, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Disc. Today 2015, 20, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Ryan, Q.C.; Headlee, D.; Acharya, M.; Sparreboom, A.; Trepel, J.B.; Ye, J.; Figg, W.D.; Hwang, K.; Chung, E.J.; Murgo, A.; et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Klein, P.M. PD01–04: Entinostat, a novel histone deacetylase inhibitor, added to exemestane improves PFS in advanced breast cancer in a randomized, phase II, double-blind study. Cancer Res. 2011, 71, PD01–PD04. [Google Scholar] [CrossRef]
- Ruiz, R.; Raez, L.E.; Rolfo, C. Entinostat (SNDX-275) for the treatment of non-small cell lung cancer. Expert. Opin. Investig. Drugs 2015, 24, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Patel, D.; Zhang, X.; Veenstra, R.D. Changes in cardiac Nav 1.5 expression, function, and acetylation by pan-histone deacetylase inhibitors. Am. J. Physiol. Hear. Circ. Physiol. 2016, 311, H1139–H1149. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Lin, X.; Andrews, L.; Patel, D.; Lampe, P.D.; Veenstra, R.D. Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front. Pharmacol. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.S.C.; van Veen, T.A.B.; de Bakker, J.M.T.; van Rijen, H.V.M. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion channel macromolecular complexes in cardiomyocytes: Roles in sudden cardiac death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bonnin, B.; Rinné, S.; Moss, R.; Streit, A.K.; Scharf, M.; Richter, K.; Stöber, A.; Pfeufer, A.; Seemann, G.; Kääb, S.; et al. Electrophysiological characterization of a large set of novel variants in the SCN5A-gene: Identification of novel LQTS3 and BrS mutations. Pflügers. Arch Eur. J. Physiol. 2016, 468, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Kopljar, I.; Gallacher, D.J.; de Bondt, A.; Cougnaud, L.; Vlaminckx, E.; van den Wyngaert, I.; Lu, H.R. Functional and Transcriptional Characterization of Histone Deacetylase Inhibitor-Mediated Cardiac Adverse Effects in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Transl. Med. 2016, 5, 602–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert. Opin. Investig. Drugs 2007, 16, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Appelskog, I.B.; Ammerpohl, O.; Ekström, T.J.; Almqvist, P.M. Histone deacetylase inhibitor 4-phenylbutyrate modulates glial fibrillary acidic protein and connexin 43 expression, and enhances gap-junction communication, in human glioblastoma cells. Eur. J. Cancer 2004, 40, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; Gordon, C.; Bergamaschi, R.; Wang, H.Z.; Cohen, I.S.; Valiunas, V.; Brink, P.R. The effects of the histone deacetylase inhibitor 4-phenylbutyrate on gap junction conductance and permeability. Front. Pharmacol. 2013, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makielski, J.C. Late sodium current: A mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc. Med. 2016, 26, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, R.G.; Zheng, D.W.; Xing, J.L.; Zhang, X.J.; Song, Y.; Xie, Y.B.; Kuang, F.; Dong, H.; You, S.W.; Xu, H.; et al. Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS ONE 2011, 6, e18681. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- FDA Reference ID: 3699607. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/ 205353s000lbl.pdf (accessed on 3 August 2018).
- Hojayev, B.; Hill, J.A. HDACs and hypertrophy, kinases and cancer. Circulation 2011, 123, 2341–2343. [Google Scholar] [CrossRef] [PubMed]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Investig. 2006, 116, 1853–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, C.R.; Li, D.L.; Pedrozo, Z.; May, H.I.; Jiang, N.; Kyrychenko, V.; Cho, G.W.; Kim, S.Y.; Wang, Z.V.; Rotter, D.; et al. Inhibition of class I histone deacetylases blunts cardiac hypertrophy through TSC2-dependent mTOR repression. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieliauskas, A.V.; Pflum, M.K.H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Bowlby, M.R.; Peri, R.; Zhang, H.; Dunlop, J. hERG (KCNH2 or Kv11.1) K+ channels: Screening for cardiac arrhythmia risk. Curr. Drug Med. 2008, 9, 965–970. [Google Scholar] [CrossRef]
- Kerr, J.; Galloway, S.; Lagrutta, A.; Armstrong, M.; Miller, T.; Richon, V.; Andrews, P. Nonclinical Safety Assessment of the Histone Deacetylase Inhibitor Vorinostat. Int. J. Toxicol. 2010, 29, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, A.; Hatake, K.; Matsui, K.; Sakajiri, S.; Hirashima, T.; Tanii, H.; Kobayashi, K.; Yamamoto, N. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Investig. New Drugs 2011, 30, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; Seto, E.; Bhalla, K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.-Q.; Tu, R.-H.; Zeng, Z.-Y.; Li, Q.-J.; He, Y.; Li, S.; Xiao, F. Novel functional role of heat shock protein 90 in protein kinase C-mediated ischemic postconditioning. J. Surg. Res. 2014, 189, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Carystinos, G.D.; Kandouz, M.; Alaoui-Jamali, M.A.; Batist, G. Unexpected induction of the human connexin 43 promoter by the ras signaling pathway is mediated by a novel putative promoter sequence. Mol. Pharmacol. 2003, 63, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Saidi Brikci-Nigassa, A.; Clement, M.J.; Ha-Duong, T.; Adjadj, E.; Ziani, L.; Pastre, D.; Curmi, P.A.; Savarin, P. Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry 2012, 51, 4331–4342. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.-S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, S.C.; Alushin, G.M.; Shida, T.; Nachury, M.V.; Nogales, E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 2014, 25, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupkovitz, G.; Tischler, J.; Posch, M.; Sadzak, I.; Ramsauer, K.; Egger, G.; Grausenburger, R.; Schweifer, N.; Chiocca, S.; Decker, T.; et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell. Biol. 2006, 26, 7913–7928. [Google Scholar] [CrossRef] [PubMed]
- Greim, H.; Albertini, R.J. Cellular response to the genotoxic insult: The question of threshold for genotoxic carcinogens. Toxicol. Res. 2015, 4, 36–45. [Google Scholar] [CrossRef]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A.; et al. Nε-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Lewarchik, C.M.; Yoon, J.-Y.; Naqvi, A.; Kumar, S.; Morgan, G.M.; Jacobs, J.S.; Li, Q.; Kim, Y.-R.; Kassan, M.; et al. Sirtuin 1 regulates cardiac electrical activity by deacetylating the cardiac sodium channel. Nat. Med. 2017, 23, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, R.D. Voltage clamp limitations of dual whole-cell gap junction current and voltage recordings. I. Conductance measurements. Biophys. J. 2001, 80, 2231–2247. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDACI | A1 | C1 (nM) | IC50(1) (nM) | A2 | C2 (nM) | IC50(2) (nM) | B | R2 |
|---|---|---|---|---|---|---|---|---|
| TSA * (RFU) | 2829 ± 219 | 10.6 ± 1.3 | 7.3 ± 0.9 | 298 ± 71 | 258 ± 76 | 179 ± 53 | 84 ± 7 | 0.98 |
| VOR * (% RFU) | 48.8 ± 7.7 | 140 ± 30 | 97 ± 21 | 44.1 ± 7.1 | 1086 ± 312 | 753 ± 216 | 6.1 ± 2.4 | 0.99 |
| FK228 † (% RFU) | 63.5 ± 5.3 | 7.9 ± 0.9 | 5.5 ± 0.6 | 27.7 ± 2.8 | 2524 ± 678 | 1749 ± 470 | 8.8 ± 1.4 | 0.99 |
| LBH589 (RFU) | 8322 ± 711 | 57.4 ± 22.2 | 39.8 ± 15.4 | 927 ± 357 | 2959 ± 838 | 2050 ± 581 | 374 ± 3 | 0.96 |
| MS-275 (RFU) | 20,111 ± 3080 | 629 ± 166 | 436 ± 115 | 20,106 ± 2150 | 12,926 ± 3655 | 8958 ± 2533 | 15,408 ± 1138 | 0.97 |
| ACY-1215 (RFU) | 14,914 ± 6840 | 16.9 ± 15.1 | 11.7 ± 10.5 | 32,744 ± 6835 | 204 ± 52 | 141 ± 36 | 3369 ± 263 | 0.99 |
| HDACI | LBH589 | LBH589 | MS-275 | MS-275 | MS-275 | ACY-1215 | Tubacin | VPA |
|---|---|---|---|---|---|---|---|---|
| (nM) [23] | (nM) [26] | (nM) [19] | (nM) [23] | (nM) [26] | (nM) [20] | (nM) [26] | (nM) [23] | |
| HDAC | IC50 | KI | IC50 | IC50 | KI | IC50 | KI | IC50 |
| HDAC1 | 3 ± 0 | 1.00 ± 0.1 | 180 ± 70 | 181 ± 62 | 22 ± 2 | 58 | 28 ± 4 | 1.584 × 10+6 ± 0.3 |
| HDAC2 | 3 ± 0 | 0.65 ± 0.1 | --- | 1155 ± 134 | 65 ± 5 | 48 | 42 ± 3.5 | 3.07 × 10+6 ± 0 |
| HDAC3 | 4 ± 1 | 1.1 ± 0.15 | 740 ± 250 | 2311 ± 803 | 360 ± 15 | 51 | 275 ± 320 | 3.07× 10+6 ± 0 |
| HDAC8 | 248 ± 11 | 105 ± 20 | 44,900 ± 18,100 | >100,000 | --- | 100 | 170 ± 10 | 7.442 × 10+6 ± 2.74 |
| HDAC6 | 61 ± 1 | 1.50 ± 0.5 | >100,000 | >100,000 | --- | 4.7 | 16 ± 2 | >100,000 |
| HDAC4 | 12 ± 5 | 550 ± 50 | --- | >100,000 | --- | 7000 | 17,000 ± 2500 | --- |
| HDAC5 | --- | 80 ± 10 | --- | --- | --- | 5000 | 1500 ± 250 | --- |
| HDAC7 | 14 ± 7 | 4550 ± 315 | --- | >100,000 | --- | 1400 | 8500 ± 1500 | >100,000 |
| HDAC9 | 3 ± 2 | 3200 ± 200 | --- | 505 ± 37 | --- | >10,000 | --- | >100,000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Patel, D.; Xu, Q.; Veenstra, R. Differences in Functional Expression of Connexin43 and NaV1.5 by Pan- and Class-Selective Histone Deacetylase Inhibition in Heart. Int. J. Mol. Sci. 2018, 19, 2288. https://doi.org/10.3390/ijms19082288
Zhang X, Patel D, Xu Q, Veenstra R. Differences in Functional Expression of Connexin43 and NaV1.5 by Pan- and Class-Selective Histone Deacetylase Inhibition in Heart. International Journal of Molecular Sciences. 2018; 19(8):2288. https://doi.org/10.3390/ijms19082288
Chicago/Turabian StyleZhang, Xian, Dakshesh Patel, Qin Xu, and Richard Veenstra. 2018. "Differences in Functional Expression of Connexin43 and NaV1.5 by Pan- and Class-Selective Histone Deacetylase Inhibition in Heart" International Journal of Molecular Sciences 19, no. 8: 2288. https://doi.org/10.3390/ijms19082288