Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration?


Abstract
:1. Introduction
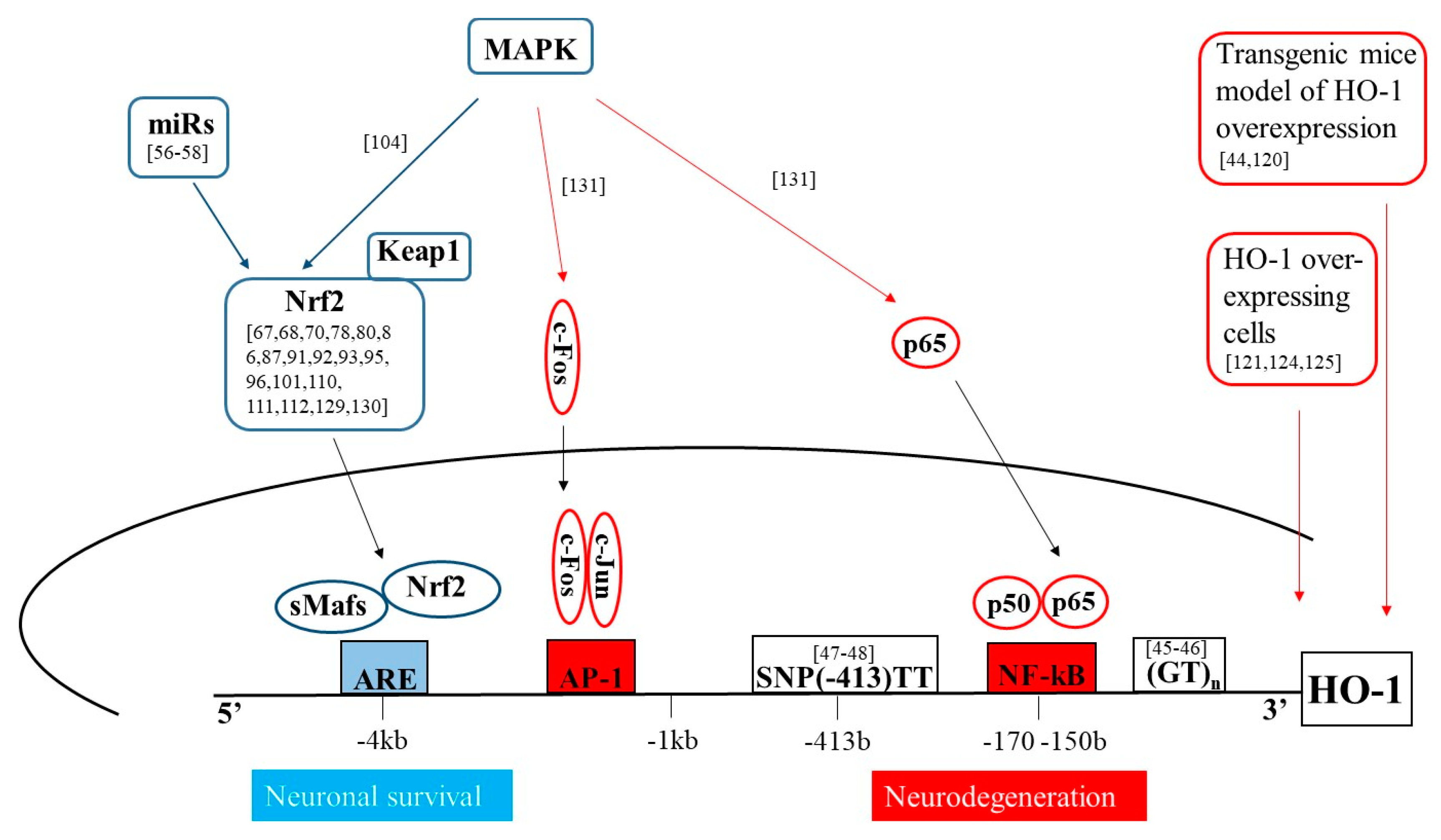
2. Molecular Mechanisms of HO-1 Induction
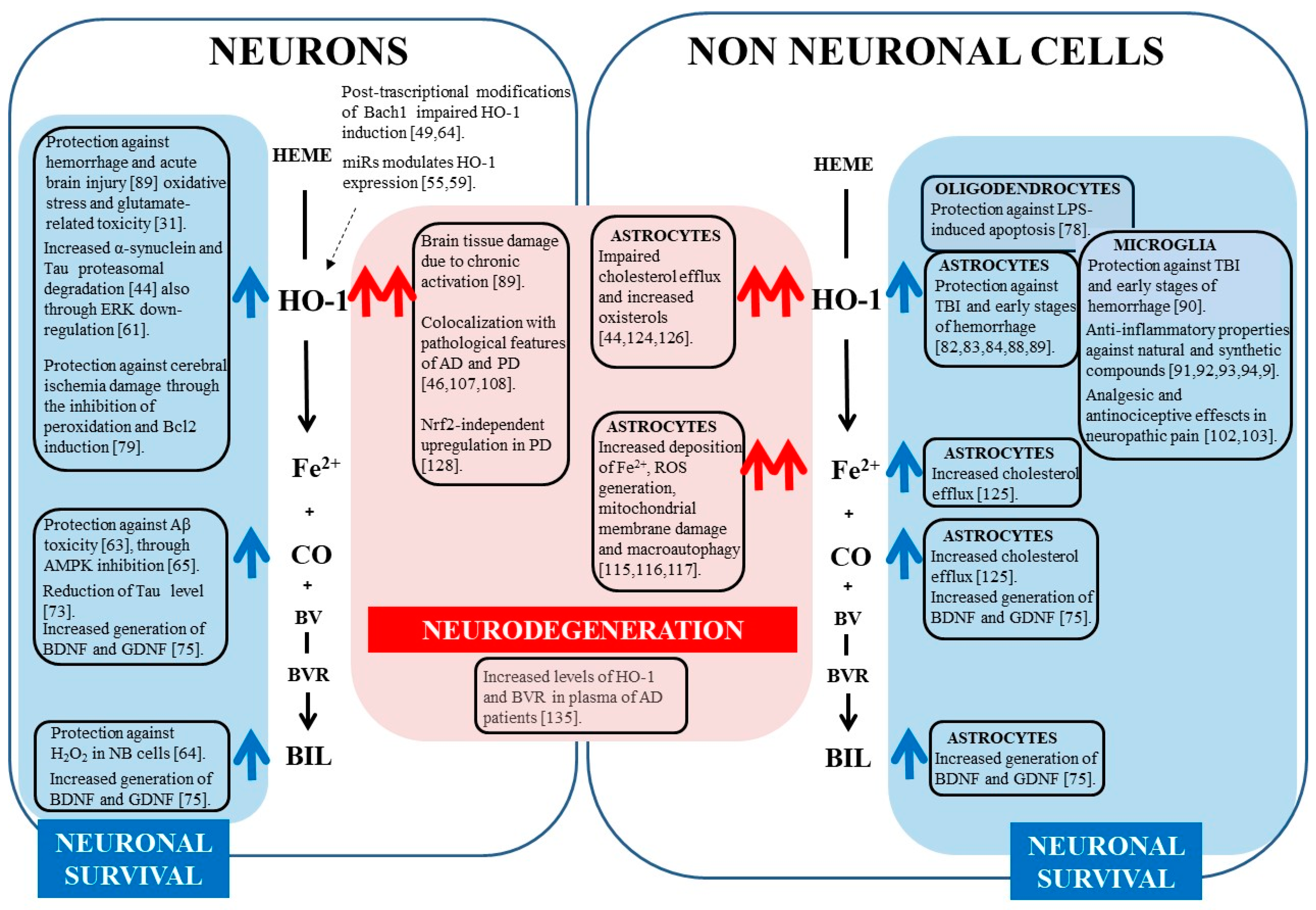
3. HO-1 Protective Effects in Nervous System
4. HO-1 Up-Regulation in Neurodegeneration
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| Akt | Serine/threonine kinase |
| AMPK | AMP-activated protein kinase |
| AP-1/2 | Activator protein 1/2 |
| ARE | Antioxidant Response Elements |
| ATR-I | Atractylenolide-I |
| Bach1 | BTB domain and CNC homolog 1 |
| BBB | Blood brain barrier |
| Bcl2 | Anti-apoptotic factor B-cell lymphoma 2 |
| BDNF | Brain-derived neurotrophic factor |
| BVR | Biliverdin reductase |
| CART | Cocaine- and amphetamine-regulated transcript |
| CNS | Central Nervous System |
| CO | Carbon monoxide |
| CoPPIX | Cobalt protoporphyrin IX |
| DDC | 2′,3′-dihydroxy-4′,6′-dimethoxychalcone |
| EAE | Experimental autoimmune encephalomyelitis |
| EPO | Erythropoietin |
| ERK | Extracellular signal-regulated kinase |
| Fe2+ | Free ferrous iron |
| GBS | Guillain-Barré syndrome |
| GCLM | Glutamate-Cysteine Ligase Modified subunit |
| GCLC | Glutamate-Cysteine Ligase Catalitic subunit |
| GDNF | Glial cell-derived neurotrophic factor |
| H2O2 | Hydrogen peroxide |
| HIF1 | Hypoxia-inducible factor 1 |
| HMOX1 | Heme oxygenase 1 gene |
| HO | Heme oxygenase |
| I/R | Ischemia/reperfusion |
| ICH | Intracerebral hemorrhage |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LPS | Lipopolysaccharide |
| LXR | Liver-x receptor |
| MAPK | Mitogen Activated Protein Kinases |
| MCI | Mild Cognitive Impairment |
| miRs | microRNAs |
| MPP+ | 1-methyl-4-phenylpyridine |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NB | Neuroblastoma |
| NF-κB | Nuclear factor kappa light- chain-enhancer of activated B cells |
| NOS | Nitric oxide synthase |
| NQO1 | NAD(P)H dehydrogenase [quinone] 1 |
| Nrf2 | Nuclear factor erythroid-derived 2-like 2 |
| PD | Parkinson’s Disease |
| PI3K | Phosphoinositide 3-kinase |
| PPARα | Peroxisome proliferator-activated receptors alpha |
| ROS | Reactive oxygen species |
| SAH | Subarachnoid hemorrhage |
| sGC | Soluble guanylyl cyclase |
| STZ | Streptozotocin |
| t-BHQ | Tert-butylhydroquinone |
| TBI | Traumatic brain injury |
| TrkB | Tropomyosinreceptorkinase B |
| 6-OHDA | 6-hydroxydopamine |
References
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [PubMed]
- Ewing, J.F.; Weber, C.M.; Maines, M.D. Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J. Neurochem. 1993, 61, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- McCoubrey, W.K.; Huang, T.J.; Maines, M.D. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur. J. Biochem. 1997, 247, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO system in tumor growth, angiogenesis and metabolism—Targeting HO-1 as an anti-tumor therapy. Vasc. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. Faseb J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, L.B.; Khrapova, N.G. Characterization of bilirubin inhibitory properties in free radical oxidation reactions. Membr. Cell Biol. 1998, 12, 233–240. [Google Scholar] [PubMed]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Nitti, M.; Piras, S.; Furfaro, A.L.; Traverso, N.; Pronzato, M.A.; Mann, G.E. Heme oxygenase-1-derived bilirubin protects endothelial cells against high glucose-induced damage. Free Radic. Biol. Med. 2015, 89, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [PubMed]
- Baker, H.M.; Anderson, B.F.; Baker, E.N. Dealing with iron: Common structural principles in proteins that transport iron and heme. Proc. Natl. Acad. Sci. USA 2003, 100, 3579–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Colombrita, C.; Calabrese, V.; Stella, A.M.G.; Mattei, F.; Alkon, D.L.; Scapagnini, G. Regional rat brain distribution of heme oxygenase-1 and manganese superoxide dismutase mRNA: Relevance of redox homeostasis in the aging processes. Exp. Biol. Med. 2003, 228, 517–524. [Google Scholar] [CrossRef]
- Takahashi, M.; Doré, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.H.; Thinakaran, G.; et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [PubMed]
- Chen, J.; Tu, Y.; Connolly, E.C.; Ronnett, G.V. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2005, 2, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014, 2014, 604981. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933. [Google Scholar] [PubMed]
- Foresti, R.; Hoque, M.; Bains, S.; Green, C.J.; Motterlini, R. Haem and nitric oxide: Synergism in the modulation of the endothelial haem oxygenase-1 pathway. Biochem. J. 2003, 372, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Barañano, D.E.; Snyder, S.H. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 10996–11002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Fariello, R.G.; Giuffrida Stella, A.M.; Abraham, N.G. Regional distribution of heme oxygenase, HSP70, and glutathione in brain: Relevance for endogenous oxidant/antioxidant balance and stress tolerance. J. Neurosci. Res. 2002, 68, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; D’Agata, V.; Calabrese, V.; Pascale, A.; Colombrita, C.; Alkon, D.; Cavallaro, S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002, 954, 51–59. [Google Scholar] [CrossRef]
- Le, W.D.; Xie, W.J.; Appel, S.H. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J. Neurosci. Res. 1999, 56, 652–658. [Google Scholar] [CrossRef]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Biomarker potential of heme oxygenase-1 in Alzheimer’s disease and mild cognitive impairment. Biomark. Med. 2007, 1, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Foresti, R.; Clark, J.E.; Green, C.J.; Motterlini, R. Thiol compounds interact with nitric oxide in regulating heme oxygenase-1 induction in endothelial cells. Involvement of superoxide and peroxynitrite anions. J. Biol. Chem. 1997, 272, 18411–18417. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Faraonio, R.; Vergara, P.; Marzo, D.D.; Napolitano, M.; Russo, T.; Cimino, F. Transcription regulation in NIH3T3 cell clones resistant to diethylmaleate-induced oxidative stress and apoptosis. Antioxid. Redox Signal. 2006, 8, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef] [PubMed]
- Davudian, S.; Mansoori, B.; Shajari, N.; Mohammadi, A.; Baradaran, B. BACH1, the master regulator gene: A novel candidate target for cancer therapy. Gene 2016, 588, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med. 2004, 37, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Kimpara, T.; Takeda, A.; Watanabe, K.; Itoyama, Y.; Ikawa, S.; Watanabe, M.; Arai, H.; Sasaki, H.; Higuchi, S.; Okita, N.; et al. Microsatellite polymorphism in the human heme oxygenase-1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum. Genet. 1997, 100, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Sánchez-Juan, P.; Rodríguez-Rodríguez, E.; Infante, J.; Vázquez-Higuera, J.L.; García-Gorostiaga, I.; Berciano, J.; Combarros, O. Synergistic effect of heme oxygenase-1 and tau genetic variants on Alzheimer’s disease risk. Dement. Geriatr. Cogn. Disord. 2008, 26, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.; Rodríguez-Rodríguez, E.; Mateo, I.; Llorca, J.; Vázquez-Higuera, J.L.; Berciano, J.; Combarros, O. Gene-gene interaction between heme oxygenase-1 and liver X receptor-beta and Alzheimer’s disease risk. Neurobiol. Aging 2010, 31, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Okita, Y.; Kamoshida, A.; Suzuki, H.; Itoh, K.; Motohashi, H.; Igarashi, K.; Yamamoto, M.; Ogami, T.; Koinuma, D.; Kato, M. Transforming growth factor-β induces transcription factors MafK and Bach1 to suppress expression of the heme oxygenase-1 gene. J. Biol. Chem. 2013, 288, 20658–20667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuoka, F.; Motohashi, H.; Tamagawa, Y.; Kure, S.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Small Maf compound mutants display central nervous system neuronal degeneration, aberrant transcription, and Bach protein mislocalization coincident with myoclonus and abnormal startle response. Mol. Cell. Biol. 2003, 23, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ku, C.H.; Siow, R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Le, T.; Wei, R.; Jiao, Y. Knockdown of JMJD1C, a target gene of hsa-miR-590-3p, inhibits mitochondrial dysfunction and oxidative stress in MPP+-treated MES23.5 and SH-SY5Y cells. Cell. Mol. Biol. 2016, 62, 39–45. [Google Scholar] [PubMed]
- Ji, Q.; Gao, J.; Zheng, Y.; Liu, X.; Zhou, Q.; Shi, C.; Yao, M.; Chen, X. Inhibition of microRNA-153 protects neurons against ischemia/reperfusion injury in an oxygen-glucose deprivation and reoxygenation cellular model by regulating Nrf2/HO-1 signaling. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, H.; Wang, R.; Wang, P.; Tao, Z.; Gao, L.; Yan, F.; Liu, X.; Yu, S.; Ji, X.; et al. MicroRNA-424 protects against focal cerebral ischemia and reperfusion injury in mice by suppressing oxidative stress. Stroke 2015, 46, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Piras, S.; Furfaro, A.L.; Caggiano, R.; Brondolo, L.; Garibaldi, S.; Ivaldo, C.; Marinari, U.M.; Pronzato, M.A.; Faraonio, R.; Nitti, M. microRNA-494 Favors HO-1 Expression in Neuroblastoma Cells Exposed to Oxidative Stress in a Bach1-Independent Way. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-H.; Song, W.; Cressatti, M.; Zukor, H.; Wang, E.; Schipper, H.M. Heme oxygenase-1 modulates microRNA expression in cultured astroglia: Implications for chronic brain disorders. Glia 2015, 63, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Perry, G.; Abraham, N.G.; Dwyer, B.E.; Kutty, R.K.; Laitinen, J.T.; Petersen, R.B.; Smith, M.A. Overexpression of heme oxygenase in neuronal cells, the possible interaction with Tau. J. Biol. Chem. 2000, 275, 5395–5399. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.; Gao, F.; Ou, L.; Yu, J.; Li, M.; Wei, P.; Miao, F. Tetrahydroxy stilbene glycoside (TSG) antagonizes Aβ-induced hippocampal neuron injury by suppressing mitochondrial dysfunction via Nrf2-dependent HO-1 pathway. Biomed. Pharmacother. 2017, 96, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Catino, S.; Paciello, F.; Miceli, F.; Rolesi, R.; Troiani, D.; Calabrese, V.; Santangelo, R.; Mancuso, C. Ferulic Acid Regulates the Nrf2/Heme Oxygenase-1 System and Counteracts Trimethyltin-Induced Neuronal Damage in the Human Neuroblastoma Cell Line SH-SY5Y. Front. Pharmacol. 2015, 6, 305. [Google Scholar] [CrossRef] [PubMed]
- Piras, S.; Furfaro, A.L.; Brondolo, L.; Passalacqua, M.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. Differentiation impairs Bach1 dependent HO-1 activation and increases sensitivity to oxidative stress in SH-SY5Y neuroblastoma cells. Sci. Rep. 2017, 7, 7568. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.; Wang, Y.; Kong, L.; Ou-Yang, T.; Meng, Q.; Fu, Q.; Hu, Z. CART peptide activates the Nrf2/HO-1 antioxidant pathway and protects hippocampal neurons in a rat model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 501, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Kim, B.R.; Oh, J.; Kim, J.-S. Soybean-Derived Phytoalexins Improve Cognitive Function through Activation of Nrf2/HO-1 Signaling Pathway. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Li, X.; Li, L.; Yuan, J.; Chen, J. t-BHQ Provides Protection against Lead Neurotoxicity via Nrf2/HO-1 Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 2075915. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin Inhibits Activation of NADPH Oxidase/p38 MAPK Pathway and Enhances Expression of Antioxidant Protein in Parkinson Disease Models. Front. Mol. Neurosci. 2018, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Masaki, Y.; Izumi, Y.; Matsumura, A.; Akaike, A.; Kume, T. Protective effect of Nrf2-ARE activator isolated from green perilla leaves on dopaminergic neuronal loss in a Parkinson’s disease model. Eur. J. Pharmacol. 2017, 798, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.; Wiggert, B.; Petersen, R.B.; Perry, G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1994, 145, 42–47. [Google Scholar] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Patel, A.; Qureshi, H.Y.; Han, D.; Schipper, H.M.; Paudel, H.K. The Parkinson disease-associated A30P mutation stabilizes alpha-synuclein against proteasomal degradation triggered by heme oxygenase-1 over-expression in human neuroblastoma cells. J. Neurochem. 2009, 110, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Liou, H.-C.; Fu, W.-M. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 2010, 58, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Capone, C.; Ranieri, S.C.; Fusco, S.; Calabrese, V.; Eboli, M.L.; Preziosi, P.; Galeotti, T.; Pani, G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 2008, 86, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Ouyang, C.; Wang, Y.; Zhang, S.; Ma, X.; Song, Y.; Yu, H.; Tang, J.; Fu, W.; Sheng, L.; et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 2014, 1577, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.M.; Martins, L.A.M.; Souza, D.O.; Quincozes-Santos, A. Glioprotective Effect of Resveratrol: An Emerging Therapeutic Role for Oligodendroglial Cells. Mol. Neurobiol. 2018, 55, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Song, T.; Yang, L.; Wang, X.; Yang, C.; Jiang, Y. Neuroprotective actions of pterostilbene on hypoxic-ischemic brain damage in neonatal rats through upregulation of heme oxygenase-1. Int. J. Dev. Neurosci. 2016, 54, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Shah, Z.A.; Li, R.-C.; Doré, S. Heme oxygenase 1 is associated with ischemic preconditioning-induced protection against brain ischemia. Neurobiol. Dis. 2009, 35, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Richmon, J.D.; Sato, M.; Sharp, F.R.; Panter, S.S.; Noble, L.J. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. 1996, 736, 68–75. [Google Scholar] [CrossRef]
- Chen-Roetling, J.; Kamalapathy, P.; Cao, Y.; Song, W.; Schipper, H.M.; Regan, R.F. Astrocyte heme oxygenase-1 reduces mortality and improves outcome after collagenase-induced intracerebral hemorrhage. Neurobiol. Dis. 2017, 102, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Chen-Roetling, J.; Song, W.; Schipper, H.M.; Regan, C.S.; Regan, R.F. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 2015, 46, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen-Roetling, J.; Regan, R.F. Systemic hemin therapy attenuates blood-brain barrier disruption after intracerebral hemorrhage. Neurobiol. Dis. 2014, 70, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ji, C.; Wu, L.; Qiu, J.; Li, Q.; Shao, Z.; Chen, G. Tert-butylhydroquinone alleviates early brain injury and cognitive dysfunction after experimental subarachnoid hemorrhage: Role of Keap1/Nrf2/ARE pathway. PLoS ONE 2014, 9, e97685. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-C.; Kong, Y.-Y.; Li, G.-Q.; Guan, Y.-F.; Wang, P.; Miao, C.-Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, M.; Ma, Y.; Wang, R.; Liu, W.; Xia, W.; Guan, A.; Xing, C.; Lu, F.; Ji, X. Fenofibrate Increases Heme Oxygenase 1 Expression and Astrocyte Proliferation While Limits Neuronal Injury during Intracerebral Hemorrhage. Curr. Neurovasc. Res. 2017, 14, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, Q.; Li, G.; Wang, L.; Hu, W.; Tang, Q.; Li, D.; Sun, Z. Time course of heme oxygenase-1 and oxidative stress after experimental intracerebral hemorrhage. Acta Neurochir. 2011, 153, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Yang, L.; Wan, C.-X.; Xia, Y.-Z.; Zhang, C.; Chen, M.-H.; Wang, Z.-D.; Li, Z.-R.; Li, X.-M.; Geng, Y.-D.; et al. Anti-neuroinflammatory effect of Sophoraflavanone G from Sophora alopecuroides in LPS-activated BV2 microglia by MAPK, JAK/STAT and Nrf2/HO-1 signaling pathways. Phytomedicine 2016, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Choi, D.-K. Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease. Nutrients 2017, 9. [Google Scholar] [CrossRef]
- Kwon, Y.-W.; Cheon, S.Y.; Park, S.Y.; Song, J.; Lee, J.-H. Tryptanthrin Suppresses the Activation of the LPS-Treated BV2 Microglial Cell Line via Nrf2/HO-1 Antioxidant Signaling. Front. Cell. Neurosci. 2017, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Lim, J.; Bang, Y.; Gal, J.; Lee, S.-U.; Cho, Y.-C.; Yoon, G.; Kang, B.Y.; Cheon, S.H.; Choi, H.J. Licochalcone E activates Nrf2/antioxidant response element signaling pathway in both neuronal and microglial cells: Therapeutic relevance to neurodegenerative disease. J. Nutr. Biochem. 2012, 23, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Kim, J.H.; Woo, S.Y.; Son, H.J.; Han, S.H.; Jang, B.K.; Choi, J.W.; Kim, D.J.; Park, K.D.; Hwang, O. A novel compound VSC2 has anti-inflammatory and antioxidant properties in microglia and in Parkinson’s disease animal model. Br. J. Pharmacol. 2015, 172, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Z.; Wu, S.; Li, X.; Sang, Y.; Li, J.; Niu, Y.; Ding, H. Myricetin alleviates cuprizone-induced behavioral dysfunction and demyelination in mice by Nrf2 pathway. Food Funct. 2016, 7, 4332–4342. [Google Scholar] [CrossRef] [PubMed]
- Chora, A.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-J.; Wang, Y.-L.; Lo, W.-T.; Wu, C.-C.; Hsieh, C.-W.; Huang, C.-F.; Lan, Y.-H.; Wang, C.-C.; Chang, D.-M.; Sytwu, H.-K. Erythropoietin enhances endogenous HAEM oxygenase-1 and represses immune responses to ameliorate experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cui, W.; Liu, J.; Li, R.; Liu, Q.; Xie, X.-H.; Ge, X.-L.; Zhang, J.; Song, X.-J.; Wang, Y.; et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp. Neurol. 2013, 250, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Fiebiger, S.; Bros, H.; Hertwig, L.; Romero-Suarez, S.; Hamann, I.; Chanvillard, C.; Bellmann-Strobl, J.; Paul, F.; Millward, J.M.; et al. Treatment of Chronic Experimental Autoimmune Encephalomyelitis with Epigallocatechin-3-Gallate and Glatiramer Acetate Alters Expression of Heme-Oxygenase-1. PLoS ONE 2015, 10, e0130251. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Xiao, J.; Zhai, H.; Hao, J. Dimethyl fumarate attenuates experimental autoimmune neuritis through the nuclear factor erythroid-derived 2-related factor 2/hemoxygenase-1 pathway by altering the balance of M1/M2 macrophages. J. Neuroinflamm. 2016, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Cheng, Z.; Zhang, J.; Xu, S.; Liu, H.; Jia, H.; Jin, Y. Spinal Heme Oxygenase-1 (HO-1) Exerts Antinociceptive Effects Against Neuropathic Pain in a Mouse Model of L5 Spinal Nerve Ligation. Pain Med. 2016, 17, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Castany, S.; Carcolé, M.; Leánez, S.; Pol, O. The Induction of Heme Oxygenase 1 Decreases Painful Diabetic Neuropathy and Enhances the Antinociceptive Effects of Morphine in Diabetic Mice. PLoS ONE 2016, 11, e0146427. [Google Scholar] [CrossRef] [PubMed]
- Riego, G.; Redondo, A.; Leánez, S.; Pol, O. Mechanism implicated in the anti-allodynic and anti-hyperalgesic effects induced by the activation of heme oxygenase 1/carbon monoxide signaling pathway in the central nervous system of mice with neuropathic pain. Biochem. Pharmacol. 2018, 148, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Syapin, P.J. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br. J. Pharmacol. 2008, 155, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Hirose, W.; Ikematsu, K.; Tsuda, R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg. Med. 2003, 5 (Suppl. S1), S360–S366. [Google Scholar] [CrossRef]
- Schipper, H.M.; Cissé, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, S.; Furfaro, A.L.; Cosso, L.; Maineri, E.P.; Balbis, E.; Domenicotti, C.; Nitti, M.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A.; et al. Heme oxygenase 1 expression in rat liver during ageing and ethanol intoxication. Biogerontology 2007, 8, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Wang, S.; Wang, Y.; Yang, X.; Jiang, J.; Wu, D.; Qu, X.; Fan, H.; Yao, R. Quercetin ameliorates learning and memory via the Nrf2-ARE signaling pathway in d-galactose-induced neurotoxicity in mice. Biochem. Biophys. Res. Commun. 2017, 491, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ren, B.; Guo, R.; Zhang, W.; Ma, S.; Yao, Y.; Yuan, T.; Liu, Z.; Liu, X. Supplementation of lycopene attenuates oxidative stress induced neuroinflammation and cognitive impairment via Nrf2/NF-κB transcriptional pathway. Food Chem. Toxicol. 2017, 109, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, A.; Bombicz, M.; Kiss, R.; Priksz, D.; Varga, B.; Hortobágyi, T.; Trencsényi, G.; Szabó, R.; Pósa, A.; Gesztelyi, R.; et al. Heme Oxygenase-1 Activity as a Correlate to Exercise-Mediated Amelioration of Cognitive Decline and Neuropathological Alterations in an Aging Rat Model of Dementia. Biomed. Res. Int. 2018, 2018, 7212861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Su, H.; Song, S.; Paudel, H.K.; Schipper, H.M. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J. Cell. Physiol. 2006, 206, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zukor, H.; Lin, S.-H.; Liberman, A.; Tavitian, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.-D.; et al. Unregulated brain iron deposition in transgenic mice over-expressing HMOX1 in the astrocytic compartment. J. Neurochem. 2012, 123, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zukor, H.; Liberman, A.; Kaduri, S.; Arvanitakis, Z.; Bennett, D.A.; Schipper, H.M. Astroglial heme oxygenase-1 and the origin of corpora amylacea in aging and degenerating neural tissues. Exp. Neurol. 2014, 254, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Monte, D.A.; Schipper, H.M.; Hetts, S.; Langston, J.W. Iron-mediated bioactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures. Glia 1995, 15, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Pistel, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, N.T.; Boyle, J.P.; Dallas, M.L.; Al-Owais, M.M.; Scragg, J.L.; Peers, C. Heme oxygenase-1 derived carbon monoxide suppresses Aβ1-42 toxicity in astrocytes. Cell Death Dis. 2017, 8, e2884. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, N.; Wang, R.; Jiang, H.; Xie, J. Preferential Heme Oxygenase-1 Activation in Striatal Astrocytes Antagonizes Dopaminergic Neuron Degeneration in MPTP-Intoxicated Mice. Mol. Neurobiol. 2016, 53, 5056–5065. [Google Scholar] [CrossRef] [PubMed]
- Vaya, J.; Song, W.; Khatib, S.; Geng, G.; Schipper, H.M. Effects of heme oxygenase-1 expression on sterol homeostasis in rat astroglia. Free Radic. Biol. Med. 2007, 42, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Hascalovici, J.R.; Song, W.; Vaya, J.; Khatib, S.; Fuhrman, B.; Aviram, M.; Schipper, H.M. Impact of heme oxygenase-1 on cholesterol synthesis, cholesterol efflux and oxysterol formation in cultured astroglia. J. Neurochem. 2009, 108, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaya, J.; Schipper, H.M. Oxysterols, cholesterol homeostasis, and Alzheimer disease. J. Neurochem. 2007, 102, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Suttner, D.M.; Dennery, P.A. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999, 13, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.-H.; Shih, C.-T.; Ching, C.-H.; Huang, J.-Y.; Jen, C.J.; Yu, L.; Kuo, Y.-M.; Wu, F.-S.; Chuang, J.-I. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp. Neurol. 2015, 263, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin Mitigates MPTP-Induced Loss of Nigrostriatal Dopaminergic Neurons and Accumulation of α-Synuclein via the Nrf2/HO-1 Pathway. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hu, Y.; Liu, L.; Cai, J.; Peng, C.; Li, Q. Gastrodin protects against MPP(+)-induced oxidative stress by up regulates heme oxygenase-1 expression through p38 MAPK/Nrf2 pathway in human dopaminergic cells. Neurochem. Int. 2014, 75, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-M.; Lin, C.-C.; Hsieh, H.-L. High-Glucose-Derived Oxidative Stress-Dependent Heme Oxygenase-1 Expression from Astrocytes Contributes to the Neuronal Apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Chertkow, H.; Mehindate, K.; Frankel, D.; Melmed, C.; Bergman, H. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology 2000, 54, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Maes, O.C.; Kravitz, S.; Mawal, Y.; Su, H.; Liberman, A.; Mehindate, K.; Berlin, D.; Sahlas, D.J.; Chertkow, H.M.; Bergman, H.; et al. Characterization of α1-antitrypsin as a heme oxygenase-1 suppressor in Alzheimer plasma. Neurobiol. Dis. 2006, 24, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Infante, J.; Sánchez-Juan, P.; García-Gorostiaga, I.; Rodríguez-Rodríguez, E.; Vázquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Mancuso, C.; Perluigi, M.; Cocciolo, A.; Mecocci, P.; Butterfield, D.A.; Coccia, R. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: A potential biochemical marker for the prediction of the disease. J. Alzheimers Dis. 2012, 32, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease | Effect | Summary | Experimental Model | Refs |
|---|---|---|---|---|
| AD | Neuroprotection | HO-1 overexpression reduces tau expression and inactivates MAPK cascade. | NB cells | [61] |
| HO-1 reduces β-amyloid toxicity through CO generation and AMPK inhibition. | SH-SY5Y NB cells and rat primary neurons | [65] | ||
| HO-1 exerts cytoprotection promoting tau proteasomal degradation. | M17 NB cells | [44,71] | ||
| Tetrahydroxystilbene glucoside up-regulates HO-1 and increases neuronal survival. | HT-22 cells exposed to Aβ | [62] | ||
| CART neuropeptides up-regulate Nrf2/HO-1 axis favoring neuroprotection. | AD rat model (Aβ injection in brain) | [66] | ||
| Neurodegeneration | HO-1 is overexpressed in AD brains and co-localizes to neurons, astrocytes, choroid plexus epithelial cells, ependyma, corpora amylacea, neurofibrillary tangles and senile plaques. | [44,107,114] | ||
| Targeted suppression of glial HO-1 by using HO-1-inhibitors exerts neuroprotection. | APPswe/PS1ΔE9 transgenic mice, rat astrocytes overexpressing HO-1 | [121] | ||
| Transient transfection of rat astroglia with human HO-1 cDNA, significantly decreases intracellular cholesterol content and increases oxysterols levels. | Primary neonatal rat astrocytes | [124] | ||
| HO-1 overexpression and its byproducts stimulate cholesterol efflux via activation of liver-x-receptor. | Primary neonatal rat astrocytes | [125] | ||
| HO-1 overexpression in astroglia from AD brains promotes the oxidation of cholesterol to oxysterols. | Primary neonatal rat astrocytes | [44,126] | ||
| Plasma HO-1 protein levels are significantly decreased in patients with probable sporadic AD. | Sporadic AD and MCI Patients | [32,132] | ||
| The activity of the HO-1 suppressor α1-antitrypsin reduces HO-1 expression in AD plasma. | Sporadic AD patients | [133] | ||
| The HO-1/BVR status in plasma is a potential biomarker for the earliest stages of AD. | Plasma from probable AD patients | [135] | ||
| PD | Neuroprotection | HO-1 overexpression favors α-synuclein proteasomal degradation through the generation of iron and CO. | M17 NB cells | [44,73] |
| HO-1 induction exerts a neuroprotective effect in dopaminergic neurons and glia through the enhancement of neurotrophic factor generation. | Parkinsonian rat model | [73,74,75] | ||
| DDC prevents 6-OHDA-induced dopaminergic neuronal death through the up-regulation of glial expression of HO-1. | C57BL/6N mice and primary mesencephalic cultures from rat embryos | [70] | ||
| ATR-I reduces the inflammatory response exerting by inducing HO-1. | Male C57BL6/J mice, BV-2 mouse microglial cells | [92] | ||
| Vinyl sulfone activates Nrf2/HO-1 axis preventing neuroinflammation in microglia and in an animal model of PD. | Male C57Bl/6 mice, BV-2 mouse microglial cells | [95] | ||
| Simvastatin up-regulates HO-1 and increases antioxidant responses in PD. | Mice treated with 6-OHDA and SH-SY5Y cells exposed to 6-HODA | [69] | ||
| Neurodegeneration | HO-1 is overexpressed in nigral astroglia and in dopaminergic neuronal Lewy bodies. | [107,114] | ||
| HO-1 up-regulation is associated with the loss of nigral dopaminergic neurons. | Wistar rats exposed to 1-methyl-4-phenylpyridine (MPP+) | [119] | ||
| Neuronal injury/Neurotoxicity | Neuroprotection | HO-1 protects neurons against oxidative stress-induced injury. | SN56 NB cells | [30] |
| HO-1 overexpression protects from glutamate toxicity and H2O2-induced cell death. | HO-1(−/−) Tg mice | [31] | ||
| Glyceollin increases MAPK/Nrf2/HO-1 pathway and protects against glutamate-induced toxicity. | HT22 cells | [67] | ||
| Ferulic acid mediates neuroprotection through HO-1 up-regulation. | SH-SY5Y NB cells | [63] | ||
| t-BHQ-mediated induction of Nrf2/HO-1 pathway protects against lead neurotoxicity. | SH-SY5Y NB cells, cortex and hippocampus from rat | [68] | ||
| HO-1-derived bilirubin protects neuronal cells from oxidative stress but neuronal differentiation decreases HO-1 induction. | SH-SY5Y NB cells | [64] | ||
| Quercetin increases Nrf2/HO-1 favoring neuroprotection against galactose-induced damage. | D-galactose treated mice | [110] | ||
| Neurodegeneration | Lycopene reduces neuroinflammation and improves cognitive functions in a model of ageing-like neurodegeneration. | D-galactose treated mice | [111] | |
| Glial HO-1 up-regulation promotes abnormal patterns of iron deposition and mitochondrial insufficiency in different human neurodegenerative disorders. | Rat primary astrocytes and Tg mice over-expressing HO-1 | [114,116,117] | ||
| Inflammation | Neuroprotection | Resveratrol restores GDNF, BDNF and TGF-β production from oligodendrocytes by up-regulating Nrf2/HO-1 axis. | Oligodendrocyte progenitor cells from Wistar rats | [78] |
| LPS-induced neuroinflammation is inhibited by Sophoraflavanone G through nuclear translocation of Nrf2 and HO-1 upregulation. | BV2 mouse microglial cells | [91] | ||
| Tryptanthrin protects against LPS-induced inflammation via Nrf2/HO-1 antioxidant signaling. | C57BL/6 mice, BV2 mouse microglial cells | [93] | ||
| Licochalcone E exerts anti-inflammatory and cytoprotective effects activating the Nrf2/HO-1 pathway. | BV2, HEK293T and SH-SY5Y cells. | [94] | ||
| HO-1 and its end-product CO have a protective effect against autoimmune neuroinflammation in experimental autoimmune encephalomyelitis. | C57BL/6 and SJL/J mice, BV2 mouse microglial cells | [97] | ||
| EPO up-regulates endogenous HO-1 and represses immune and inflammatory responses in experimental autoimmune encephalomyelitis. | C57BL/6 mice | [98] | ||
| Sulforaphane, by enhancing Nrf2/HO-1 activity, antagonizes autoimmune inflammation and inhibits EAE development and severity. | C57BL/6 mice | [99] | ||
| Myricetin improves motor functions and reduces demyelination in vivo. | Cuprizone-treated mice | [96] | ||
| Neuronal damage | Up-regulation of HO-1 contributes to diminish the neuroprotective effects of epigallocatechin-3-gallate in experimental autoimmune encephalomyelitis model. | C57BL/6 mice | [100] | |
| Ischemia | Neuroprotection | HO-1 overexpression in hippocampus protects against cerebral I/R activating the BDNF–TrkB–PI3K/Akt signaling pathway. | Sprague-Dawley rats | [77] |
| HO-1-mediated neuroprotection is related to enhanced expression of bcl-2, inhibition of nuclear localization of p53 and decreased levels of lipid peroxidation end-products. | HO-1 overexpressing mice | [79] | ||
| HO-1 is required for ischemic preconditioning-induced neuroprotection against brain ischemia. | HO-1 (−/−) Tg mice | [81] | ||
| Pterostilbene administration prevents ischemic brain injury in newborns. | Rat model of neonatal ischemic damage | [80] | ||
| Traumatic brain injury (TBI) | Neuroprotection | A prolonged glial induction of HO-1 exerts neuroprotection in animal models of traumatic brain injury. | Sprague-Dawley rats | [82] |
| Hemorrhage | Neuroprotection | Selective HO-1 overexpression in astrocytes exerts neuroprotection after intracerebral hemorrhage. | HO-1 overexpressing mice | [83,84] |
| Hemin administration reduces BBB damage and improves neurological outcome in experimental models of traumatic and ischemic CNS injury. | Swiss-Webster mice | [85] | ||
| Nrf2/HO-1 up-regulation mediated by t-BHQ administration decreases the development of early brain injury in a subarachnoid hemorrhage model. | Sprague-Dawley rats | [86] | ||
| Nrf2/HO-1 activation mediated by nicotinamide mononucleotide treatment induces neuroprotection after intracerebral hemorrhage. | CD1 mice | [87] | ||
| Fenofibrate reduces neuronal damage of ICH rat brain by increasing HO-1 expression level and decreasing NF-κB expression in PPARα-dependent manner. | ICH rat model, LN-18 glioblastoma cells, rat brain astrocytes | [88] | ||
| HO-1 overexpression and CO generation are necessary to reduce neuronal injury and cognitive dysfunction in a mouse model of subarachnoid hemorrhage. | SAH stroke murine model. | [90] | ||
| Neuroprotection/Neuronal damage | The early up-regulation of HO-1 has a protective role against oxidative stress, whereas late stage overexpression may result in dysfunctions and toxicity in intracerebral hemorrhage. | Sprague–Dawley rats | [89] | |
| Neuropatic pain | Neuroprotection | HO-1 up-regulation exerts analgesic effects against neuropathic pain inhibiting spinal microglia activation. | Mouse model of L5 spinal nerve ligation | [102] |
| HO-1 induction enhances the antinociceptive effects of morphine via inhibition of microglia activation in painful STZ-induced diabetic neuropathy. | STZ-treated C57BL/6J mice | [103] | ||
| Guillain-Barré syndrome (GBS) | Neuroprotection | In a mouse model of GBS, treatment with dimethyl fumarate favors macrophages M2 polarization through the up-regulation of Nrf2 and HO-1, preventing inflammation. | Lewis rats | [101] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. https://doi.org/10.3390/ijms19082260
Nitti M, Piras S, Brondolo L, Marinari UM, Pronzato MA, Furfaro AL. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? International Journal of Molecular Sciences. 2018; 19(8):2260. https://doi.org/10.3390/ijms19082260
Chicago/Turabian StyleNitti, Mariapaola, Sabrina Piras, Lorenzo Brondolo, Umberto Maria Marinari, Maria Adelaide Pronzato, and Anna Lisa Furfaro. 2018. "Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration?" International Journal of Molecular Sciences 19, no. 8: 2260. https://doi.org/10.3390/ijms19082260