Inherited and Acquired Decrease in Complement Receptor 1 (CR1) Density on Red Blood Cells Associated with High Levels of Soluble CR1 in Alzheimer’s Disease

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Association between the Genetic CR1 Density Polymorphism and the CR1 Density Phenotypic Polymorphism
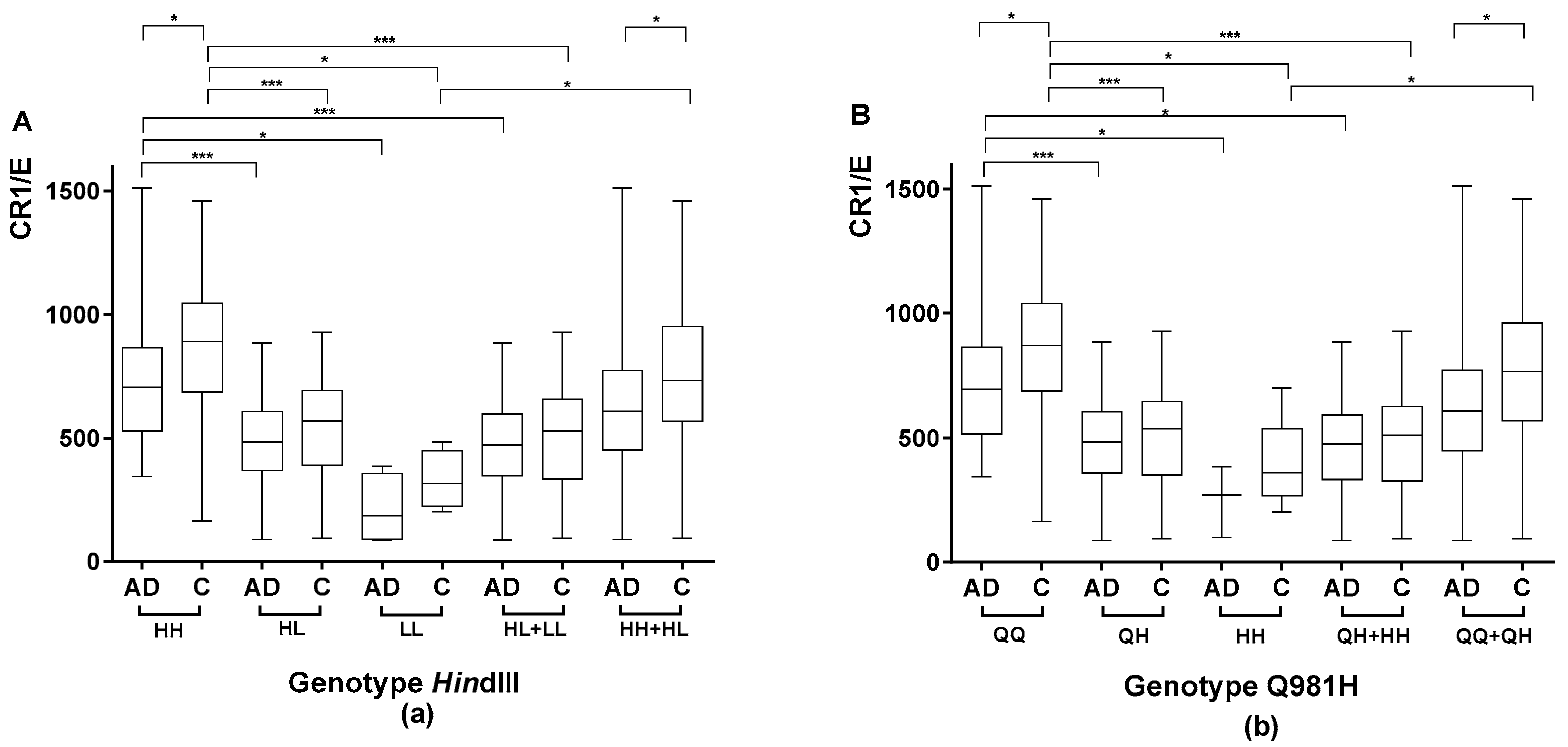
2.1.1. Association between the Genetic CR1 Density Polymorphism, HindIII, and the CR1 Density Phenotypic Polymorphism
2.1.2. Association between the CR1 Density Genetic Polymorphism Encoding Q981H and the CR1 Density Phenotypic Polymorphism
2.1.3. Study of the Agreement between the HindIII Genotype and Q981H
2.1.4. Comparison of CR1 Density Using HindIII and Q981H Genotype in AD Patients vs. Controls
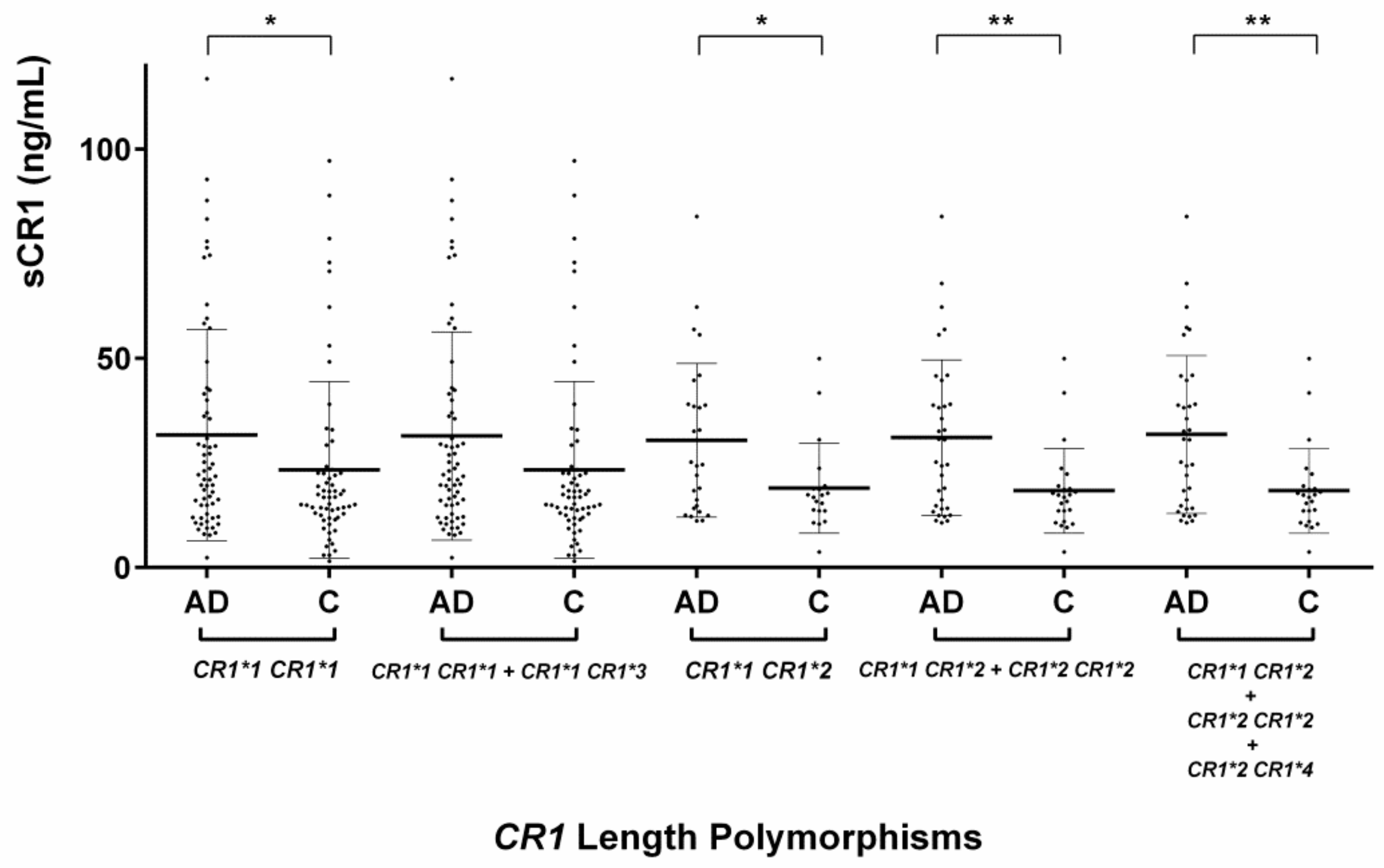
2.2. Evaluation of the CR1 Length Polymorphisms
2.3. Evaluation of the Serum Levels of sCR1
2.3.1. Serum sCR1 Levels According to CR1 Length Polymorphisms
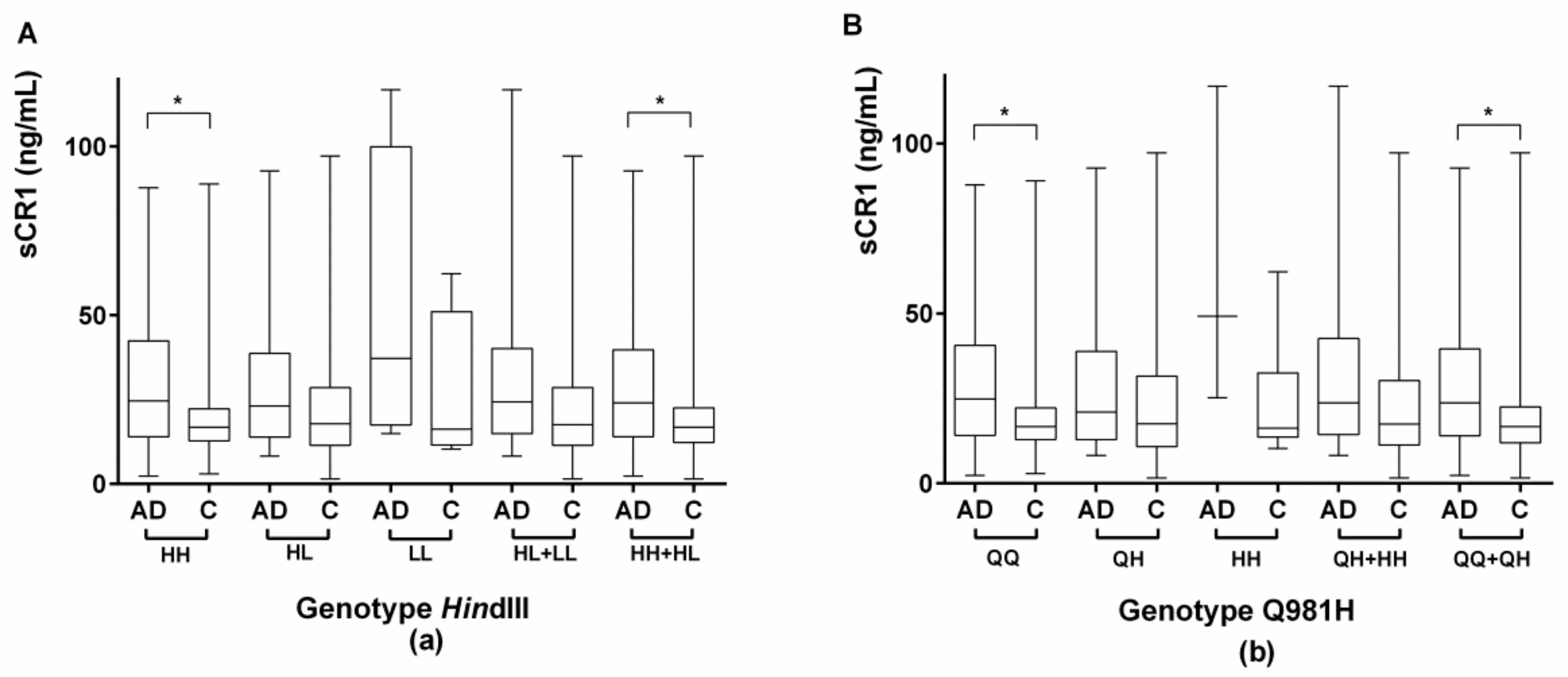
2.3.2. Evaluation of Serum sCR1 Levels According to the CR1 Density Polymorphisms HindIII and Q981H
2.4. Association between CR1/E and sCR1 and the Stage of AD
2.5. CR1 Methylation
2.6. Assessment of Factors Associated with AD
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Quantification of CR1 Density Using Flow Cytometry
4.3. DNA Extraction
4.3.1. APOE Genotyping using Amplification and High-Resolution Melting Analyses
4.3.2. Assessment of the CR1 Density Genetic Polymorphism Using HindIII RFLP
4.3.3. Determination of the CR1 Density Genetic Polymorphism by Pyrosequencing
Q981H (Exon 19) Amplification for Pyrosequencing
Pyrosequencing
4.3.4. Determination of the rate of CR1 methylation by Pyrosequencing
Amplification of the CR1 gene (LHR-B segment and LHR-C segment) for Pyrosequencing
Pyrosequencing
4.3.5. Determination of the CR1 Length Genetic Polymorphisms Using High-Resolution Melting PCR (HRM-PCR)
4.4. Quantification of sCR1 Using ELISA
4.5. Statistical Analyses
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AIDS | Acquired immunodeficiency syndrome |
| APOE | Apolipoprotein E |
| CR1 | Complement component (C3b/C4b) receptor 1 |
| GWAS | Genome-wide association studies |
| HRM | High-resolution melting |
| LHR | Long homologous repeat |
| SCR | Short consensus repeat |
| sCR1 | Soluble form of CR1 |
| SNP | Single-nucleotide polymorphism |
References
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, S.L.; Kamboh, M.I. Late-onset Alzheimer’s disease genes and the potentially implicated pathways. Curr. Genet. Med. Rep. 2014, 2, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Li, R.; Mastroeni, D.; Grover, A.; Leonard, B.; Ahern, G.; Cao, P.; Kolody, H.; Vedders, L.; Kolb, W.P.; et al. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol. Aging 2006, 27, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Schifferli, J.A.; Ng, Y.C.; Estreicher, J.; Walport, M.J. The clearance of tetanus toxoid/anti-tetanus toxoid immune complexes from the circulation of humans. Complement- and erythrocyte complement receptor 1-dependent mechanisms. J. Immunol. 1988, 140, 899–904. [Google Scholar] [PubMed]
- Cosio, F.G.; Shen, X.P.; Birmingham, D.J.; Van Aman, M.; Hebert, L.A. Evaluation of the mechanisms responsible for the reduction in erythrocyte complement receptors when immune complexes form in vivo in primates. J. Immunol. 1990, 145, 4198–4206. [Google Scholar] [PubMed]
- Cornacoff, J.B.; Hebert, L.A.; Smead, W.L.; VanAman, M.E.; Birmingham, D.J.; Waxman, F.J. Primate erythrocyte-immune complex-clearing mechanism. J. Clin. Invest. 1983, 71, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, W.D.; Crane, A.; Johansson, J.U.; Yen, K.; Garfinkel, K.; Mastroeni, D.; Asok, P.; Bradt, B.; Sabbagh, M.; Wallace, T.L.; et al. Peripheral complement interactions with amyloid β peptides: Erythrocyte clearance mechanisms. Alzheimers Dement. 2017, 13, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.U.; Brubaker, W.D.; Javitz, H.; Bergen, A.W.; Nishita, D.; Trigunaite, A.; Crane, A.; Ceballos, J.; Mastroeni, D.; Tenner, A.J.; et al. Peripheral complement interactions with amyloid β peptide in Alzheimer’s disease: Polymorphisms, structure, and function of complement receptor 1. Alzheimers Dement. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Krych-Goldberg, M.; Atkinson, J.P. Structure-function relationships of complement receptor type 1. Immunol. Rev. 2001, 180, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, D.J.; Chen, W.; Liang, G.; Schmitt, H.C.; Gavit, K.; Nagaraja, H.N. A CR1 polymorphism associated with constitutive erythrocyte CR1 levels affects binding to C4b but not C3b. Immunology 2003, 108, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Niu, Z.X. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35). Immunopharm. Immunot. 2009, 31, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.H.; Lutz, H.U.; Pennaforte, J.L.; Bouchard, A.; Kazatchkine, M.D. Peripheral catabolism of CR1 (the C3b receptor, CD35) on erythrocytes from healthy individuals and patients with systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 1992, 87, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Cornillet, P.; Philbert, F.; Kazatchkine, M.D.; Cohen, J.H. Genomic determination of the CR1 (CD35) density polymorphism on erythrocytes using polymerase chain reaction amplification and HindIII restriction enzyme digestion. J. Immunol. Methods 1991, 136, 193–197. [Google Scholar] [CrossRef]
- He, J.R.; Xi, J.; Ren, Z.F.; Qin, H.; Zhang, Y.; Zeng, Y.X.; Mo, H.Y.; Jia, W.H. Complement receptor 1 expression in peripheral blood mononuclear cells and the association with clinicopathological features and prognosis of nasopharyngeal carcinoma. Asian Pac. J. Cancer Prev. 2012, 13, 6527–6531. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, R.; Kisserli, A.; Novella, J.L.; Donvito, B.; Drame, M.; Reveil, B.; Duret, V.; Jolly, D.; Pham, B.N.; Cohen, J.H. Alzheimer’s disease is associated with low density of the long CR1 isoform. Neurobiol. Aging 2015, 36, 1766.e5–1766.e12. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Murphy, E.E.; Wong, W.W.; Klickstein, L.B.; Weis, J.H.; Fearon, D.T. Identification of a restriction fragment length polymorphism by a CR1 cDNA that correlates with the number of CR1 on erythrocytes. J. Exp. Med. 1986, 164, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.W.; Cahill, J.M.; Rosen, M.D.; Kennedy, C.A.; Bonaccio, E.T.; Morris, M.J.; Wilson, J.G.; Klickstein, L.B.; Fearon, D.T. Structure of the human CR1 gene. Molecular basis of the structural and quantitative polymorphisms and identification of a new CR1-like allele. J. Exp. Med. 1989, 169, 847–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, L.; Rundles, J.R.; Hamilton, D.R.; Wilson, J.G. Quantitative alleles of CR1: Coding sequence analysis and comparison of haplotypes in two ethnic groups. J. Immunol. 1999, 163, 4939–4945. [Google Scholar] [PubMed]
- Herrera, A.H.; Xiang, L.; Martin, S.G.; Lewis, J.; Wilson, J.G. Analysis of complement receptor type 1 (CR1) expression on erythrocytes and of (CR1) allelic markers in Caucasian and African American populations. Clin. Immunol. Immunopathol. 1998, 87, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Pham, B.N.; Kisserli, A.; Donvito, B.; Duret, V.; Reveil, B.; Tabary, T.; Le Pennec, P.Y.; Peyrard, T.; Rouger, P.; Cohen, J.H. Analysis of complement receptor type 1 expression on red blood cells in negative phenotypes of the Knops blood group system, according to CR1 gene allotype polymorphisms. Transfusion 2010, 50, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Fearon, D.T. Characterization of a soluble form of the C3b/C4b receptor (CR1) in human plasma. J. Immunol. 1985, 134, 3332–3338. [Google Scholar] [PubMed]
- Ehlers, M.R.; Riordan, J.F. Membrane proteins with soluble counterparts: Role of proteolysis in the release of transmembrane proteins. Biochem. 1991, 30, 10065–10074. [Google Scholar] [CrossRef]
- Dervillez, X.; Oudin, S.; Libyh, M.T.; Tabary, T.; Reveil, B.; Philbert, F.; Bougy, F.; Pluot, M.; Cohen, J.H. Catabolism of the human erythrocyte C3b/C4b receptor (CR1, CD35): Vesiculation and/or proteolysis? Immunopharmacology 1997, 38, 129–140. [Google Scholar] [CrossRef]
- Hamer, I.; Paccaud, J.P.; Belin, D.; Maeder, C.; Carpentier, J.L. Soluble form of complement C3b/C4b receptor (CR1) results from a proteolytic cleavage in the C-terminal region of CR1 transmembrane domain. Biochem. J. 1998, 329 Pt 1, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquet, M.; Lacroix, M.; Ancelet, S.; Gout, E.; Gaboriaud, C.; Thielens, N.M.; Rossi, V. Deciphering complement receptor type 1 interactions with recognition proteins of the lectin complement pathway. J. Immunol. 2013, 190, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Kubiak-Wlekly, A.; Perkowska-Ptasinska, A.; Olejniczak, P.; Rochowiak, A.; Kaczmarek, E.; Durlik, M.; Czekalski, S.; Niemir, Z.I. The comparison of the podocyte expression of synaptopodin, CR1 and neprilysin in human glomerulonephritis: Could the expression of CR1 be clinically relevant? Int. J. Biomed. Sci. 2009, 5, 28–36. [Google Scholar] [PubMed]
- Fonseca, M.I.; Chu, S.; Pierce, A.L.; Brubaker, W.D.; Hauhart, R.E.; Mastroeni, D.; Clarke, E.V.; Rogers, J.; Atkinson, J.P.; Tenner, A.J. Analysis of the putative role of CR1 in Alzheimer’s disease: Genetic association, expression and function. PLoS ONE 2016, 11, e0149792. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, N.; Van Cauwenberghe, C.; Engelborghs, S.; Lambert, J.C.; Bettens, K.; Le Bastard, N.; Pasquier, F.; Montoya, A.G.; Peeters, K.; Mattheijssens, M.; et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 2012, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Hazrati, L.N.; Van Cauwenberghe, C.; Brooks, P.L.; Brouwers, N.; Ghani, M.; Sato, C.; Cruts, M.; Sleegers, K.; St George-Hyslop, P.; Van Broeckhoven, C.; et al. Genetic association of CR1 with Alzheimer’s disease: A tentative disease mechanism. Neurobiol. Aging 2012, 33, 2949.e5–2949.e12. [Google Scholar] [CrossRef] [PubMed]
- Kucukkilic, E.; Brookes, K.; Barber, I.; Guetta-Baranes, T.; Morgan, K.; Hollox, E.J. Complement receptor 1 gene (CR1) intragenic duplication and risk of Alzheimer’s disease. Hum. Genet. 2018, 137, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, M.S.; Vala, M.; Pisetsky, D.S.; Greenberg, C.S.; Crawford, J.; Cohen, H.J. Correlation between erythrocyte CR1 reduction and other blood proteinase markers in patients with malignant and inflammatory disorders. Blood 1990, 75, 1699–1704. [Google Scholar] [PubMed]
- Ross, G.D.; Yount, W.J.; Walport, M.J.; Winfield, J.B.; Parker, C.J.; Fuller, C.R.; Taylor, R.P.; Myones, B.L.; Lachmann, P.J. Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J. Immunol. 1985, 135, 2005–2014. [Google Scholar] [PubMed]
- Pascual, M.; Lutz, H.U.; Steiger, G.; Stammler, P.; Schifferli, J.A. Release of vesicles enriched in complement receptor 1 from human erythrocytes. J. Immunol. 1993, 151, 397–404. [Google Scholar] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. CSH Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.N.; Donvito, B.; Cockburn, I.; Fandeur, T.; Rowe, J.A.; Cohen, J.H.; Moulds, J.M. A complement receptor-1 polymorphism with high frequency in malaria endemic regions of Asia but not Africa. Genes. Immun. 2005, 6, 31–36. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Press: Washington, DC, USA, 1994. [Google Scholar]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Fischer, E.; Boucheix, C.; Mirsrahi, M.; Jouvin, M.H.; Weiss, L.; Jack, R.M.; Kazatchkine, M.D. Mouse monoclonal antibodies to the human C3b receptor. Mol. Immunol. 1985, 22, 531–539. [Google Scholar] [CrossRef]
- Kiss, E.; Csipo, I.; Cohen, J.H.; Reveil, B.; Kavai, M.; Szegedi, G. CR1 density polymorphism and expression on erythrocytes of patients with systemic lupus erythematosus. Autoimmunity 1996, 25, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Kisserli, A.; Tabary, T.; Cohen, J.H.M.; Duret, V.; Mahmoudi, R. High-resolution melting PCR for complement receptor 1 length polymorphism genotyping: An innovative tool for Alzheimer’s disease gene susceptibility assessment. J. Vis. Exp. 2017, 125, 56012. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.; Brubaker, W.D.; Johansson, J.U.; Trigunaite, A.; Ceballos, J.; Bradt, B.; Glavis-Bloom, C.; Wallace, T.L.; Tenner, A.J.; Rogers, J. Peripheral complement interactions with amyloid β peptide in Alzheimer’s disease: 2. Relationship to amyloid β immunotherapy. Alzheimers Dement. 2018, 14, 243–252. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Variable | AD Patients (n = 100) | Controls (n = 87) | p |
|---|---|---|---|
| Age (years) | 81.5 ± 7.2 | 74.3 ± 6.3 | <10−4 |
| Female sex | 66 (66.0%) | 50 (57.4%) | 0.23 |
| APOE-ε4+ (n = 73) | 48 (48.0%) | 25 (28.74%) | 0.0071 |
| Living at home | 88 (88.0%) | 83 (95.4%) | 0.73 |
| Comorbidities (Charlson) | 1.31 ± 1.26 | 1.12 ± 1.02 | 0.27 |
| Level of dependence | |||
| IADL | 4.81 ± 2.57 | 7.83 ± 0.86 | <10−4 |
| ADL | 5.38 ± 1.0 | 5.95 ± 0.25 | <10−4 |
| Cognitive status | |||
| MMSE | 19.2 ± 5.3 | 28.8 ± 1.3 | <10−4 |
| AD stage | |||
| Mild (MMSE ≥ 21) | 42 (42.00%) | – | – |
| Moderate (MMSE 10–20) | 55 (55.00%) | – | – |
| Severe (MMSE < 10) | 3 (3.00%) | – | – |
| CR1 Polymorphisms | Subjects | ||||
|---|---|---|---|---|---|
| All (n = 187), % | AD Patients (n = 100), % | Controls (n = 87), % | |||
| Density polymorphisms | HindIII | HH | 114 (61.0) | 59 (59.0) | 55 (63.2) |
| HL | 65 (34.8) | 37 (37.0) | 28 (32.2) | ||
| LL | 8 (4.3) | 4 (4.0) | 4 (4.6) | ||
| Q981H | 118 (63.1) | 62 (62.0) | 56 (64.4) | ||
| QH | 60 (32.1) | 35 (35.0) | 25 (28.7) | ||
| HH | 9 (4.8) | 3 (3.0%) | 6 (6.9) | ||
| Length polymorphisms | CR1*1 CR1*1 | 126 (67.4) | 63 (63.0) | 63 (72.4) | |
| CR1*1 CR1*1 + CR1*1 CR1*3 | 128 (68.5) | 65 (65.0) | 63 (72.4) | ||
| CR1*1 CR1*2 | 48 (25.7) | 28 (28.0) | 20 (23.0) | ||
| CR1*2 CR1*2 | 10 (5.4) | 6 (6.0 | 4 (4.6) | ||
| CR1*1 CR1*3 | 2 (1.1) | 2 (2.0) | 0 (0) | ||
| CR1*1 CR1*2 + CR1*2 CR1*2 | 58 (31.0) | 34 (34.0) | 24 (27.6) | ||
| CR1*2 CR1*4 | 1 (0.5) | 1 (1.0) | 0 (0) | ||
| CR1*1 CR1*2 + CR1*2 CR1*2 + CR1*2 CR1*4 | 59 (31.6) | 35 (35.0) | 24 (27.6) | ||
| CR1*2 CR1*2 + CR1*2 CR1*4 | 11 (5.9) | 7 (7.0) | 4 (4.6) | ||
| Variable | Unit | Estimate | 95% CIs | p |
|---|---|---|---|---|
| Alzheimer’s disease | – | −63.14 | −144.89, 18.6 | 0.129 |
| Age | 1 | −6.51 | −11.79, −1.23 | 0.016 |
| Density polymorphism HindIII (reference: HH) | <0.001 | |||
| HL | – | −311.67 | −389.16, −234.17 | |
| LL | – | −569.51 | −740.02, −398.99 | |
| 2nd methylation site | 1% | −6.68 | −12.37, −0.99 | 0.022 |
| Variable | Unit | OR | 95% CIs | p |
|---|---|---|---|---|
| Age (years) | 1 | 1.182 | 1.118, 1.260 | <0.0001 |
| Sex (female) | – | 2.605 | 1.172, 6.050 | 0.0215 |
| APOE-ε4+ | – | 4.745 | 2.152, 11.199 | 0.0002 |
| Density (number of CR1 antigenic sites per erythrocyte) | 30 | 0.936 | 0.894, 0.975 | 0.0025 |
| 100 | 0.801 | 0.689, 0.920 | 0.0025 | |
| 200 | 0.641 | 0.475, 0.847 | 0.0025 | |
| 400 | 0.411 | 0.225, 0.718 | 0.0025 | |
| 500 | 0.329 | 0.155, 0.661 | 0.0025 | |
| Density polymorphism Q981H (Q vs. HH) | – | 12.416 | 1.603, 112.155 | 0.0193 |
| Serum level of soluble CR1 (ng/mL) | 1 | 1.032 | 1.013, 1.054 | 0.0015 |
| 10 | 1.369 | 1.139, 1.685 | 0.0015 | |
| 20 | 1.874 | 1.298, 2.840 | 0.0015 | |
| 30 | 2.565 | 1.479, 4.787 | 0.0015 | |
| 40 | 3.512 | 1.684, 8.068 | 0.0015 | |
| 50 | 4.807 | 1.919, 13.597 | 0.0015 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoudi, R.; Feldman, S.; Kisserli, A.; Duret, V.; Tabary, T.; Bertholon, L.-A.; Badr, S.; Nonnonhou, V.; Cesar, A.; Neuraz, A.; et al. Inherited and Acquired Decrease in Complement Receptor 1 (CR1) Density on Red Blood Cells Associated with High Levels of Soluble CR1 in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 2175. https://doi.org/10.3390/ijms19082175
Mahmoudi R, Feldman S, Kisserli A, Duret V, Tabary T, Bertholon L-A, Badr S, Nonnonhou V, Cesar A, Neuraz A, et al. Inherited and Acquired Decrease in Complement Receptor 1 (CR1) Density on Red Blood Cells Associated with High Levels of Soluble CR1 in Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(8):2175. https://doi.org/10.3390/ijms19082175
Chicago/Turabian StyleMahmoudi, Rachid, Sarah Feldman, Aymric Kisserli, Valérie Duret, Thierry Tabary, Laurie-Anne Bertholon, Sarah Badr, Vignon Nonnonhou, Aude Cesar, Antoine Neuraz, and et al. 2018. "Inherited and Acquired Decrease in Complement Receptor 1 (CR1) Density on Red Blood Cells Associated with High Levels of Soluble CR1 in Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 8: 2175. https://doi.org/10.3390/ijms19082175