Gene Level Regulation of Na,K-ATPase in the Renal Proximal Tubule Is Controlled by Two Independent but Interacting Regulatory Mechanisms Involving Salt Inducible Kinase 1 and CREB-Regulated Transcriptional Coactivators


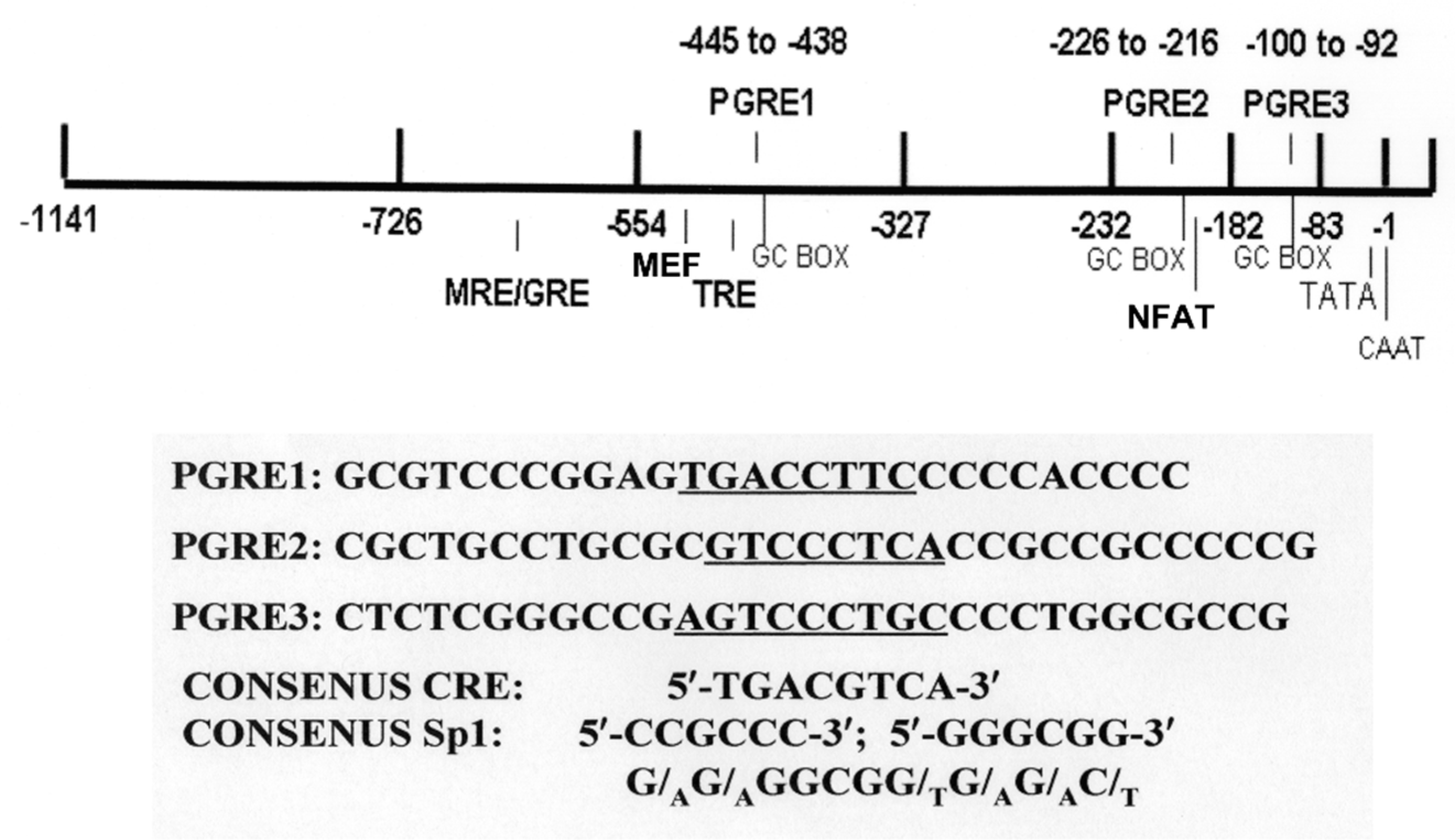{kind=link}
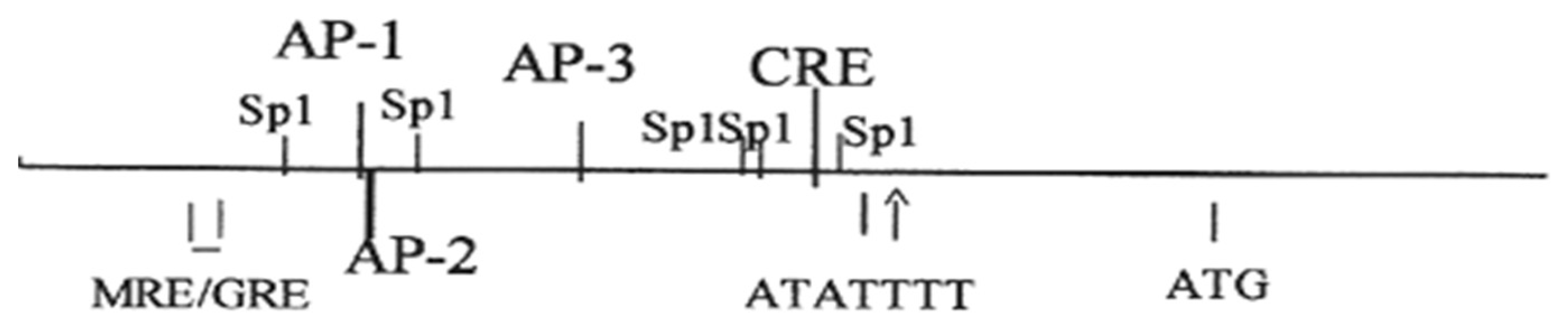{kind=link}
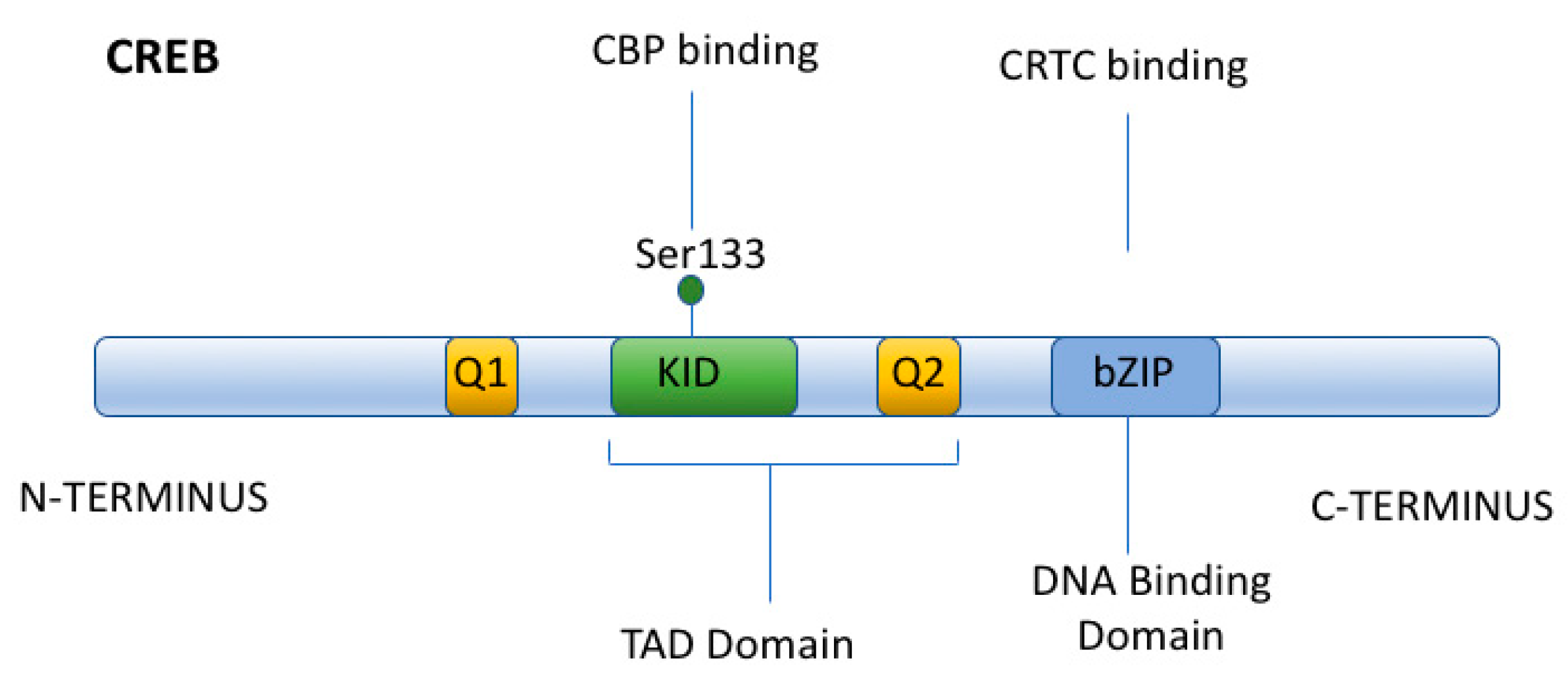{kind=link}
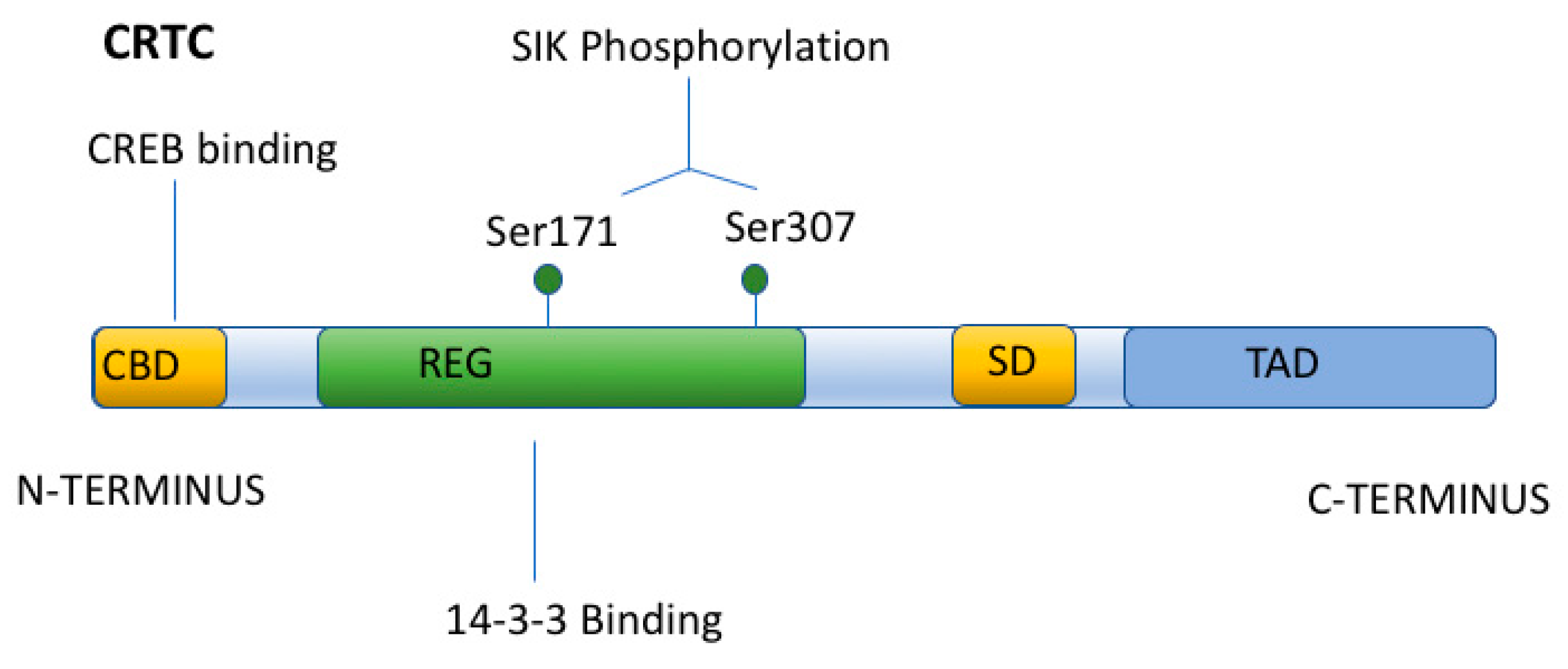{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
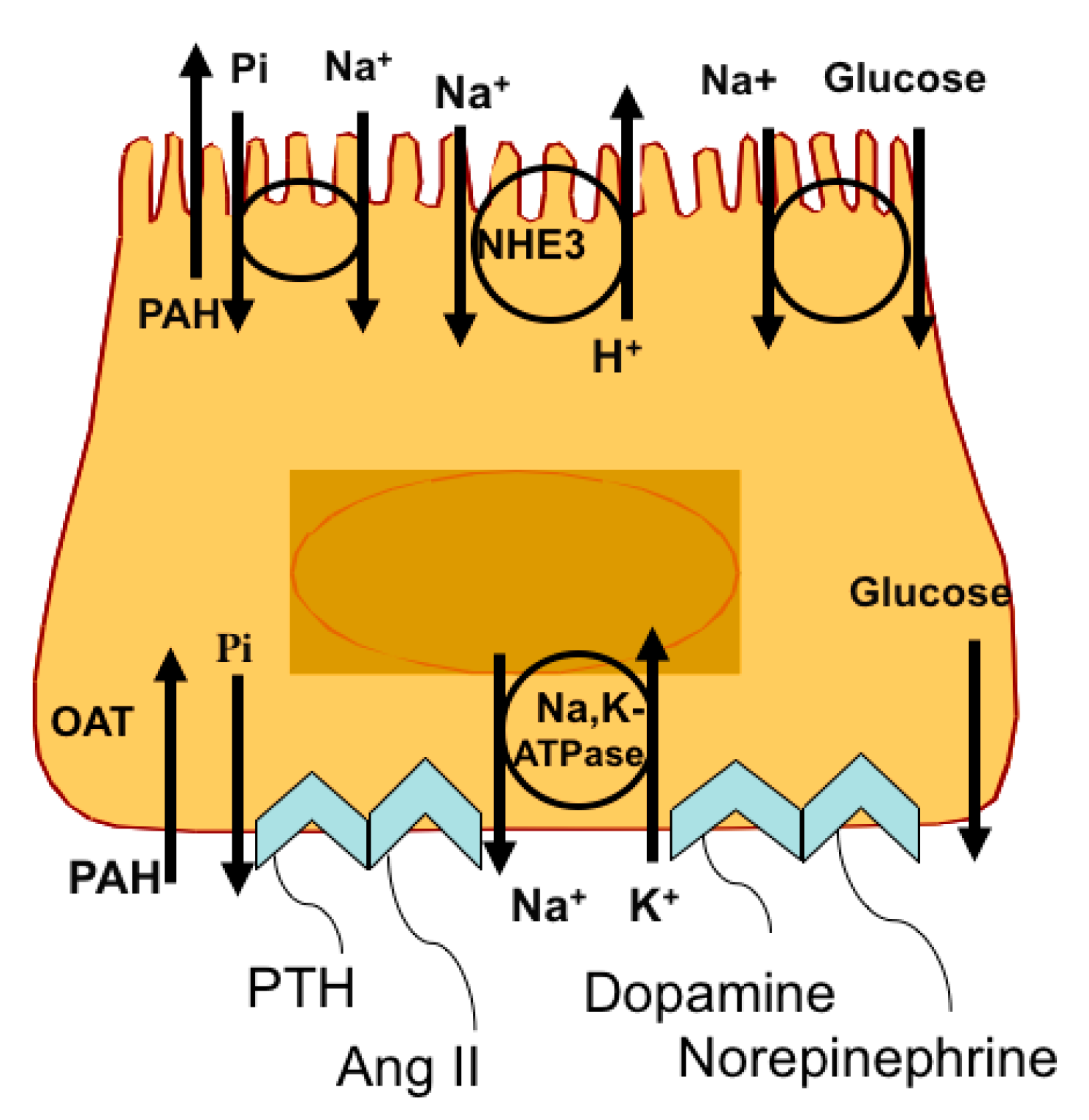
2. Response of the RPT to Changes in Na+ Intake
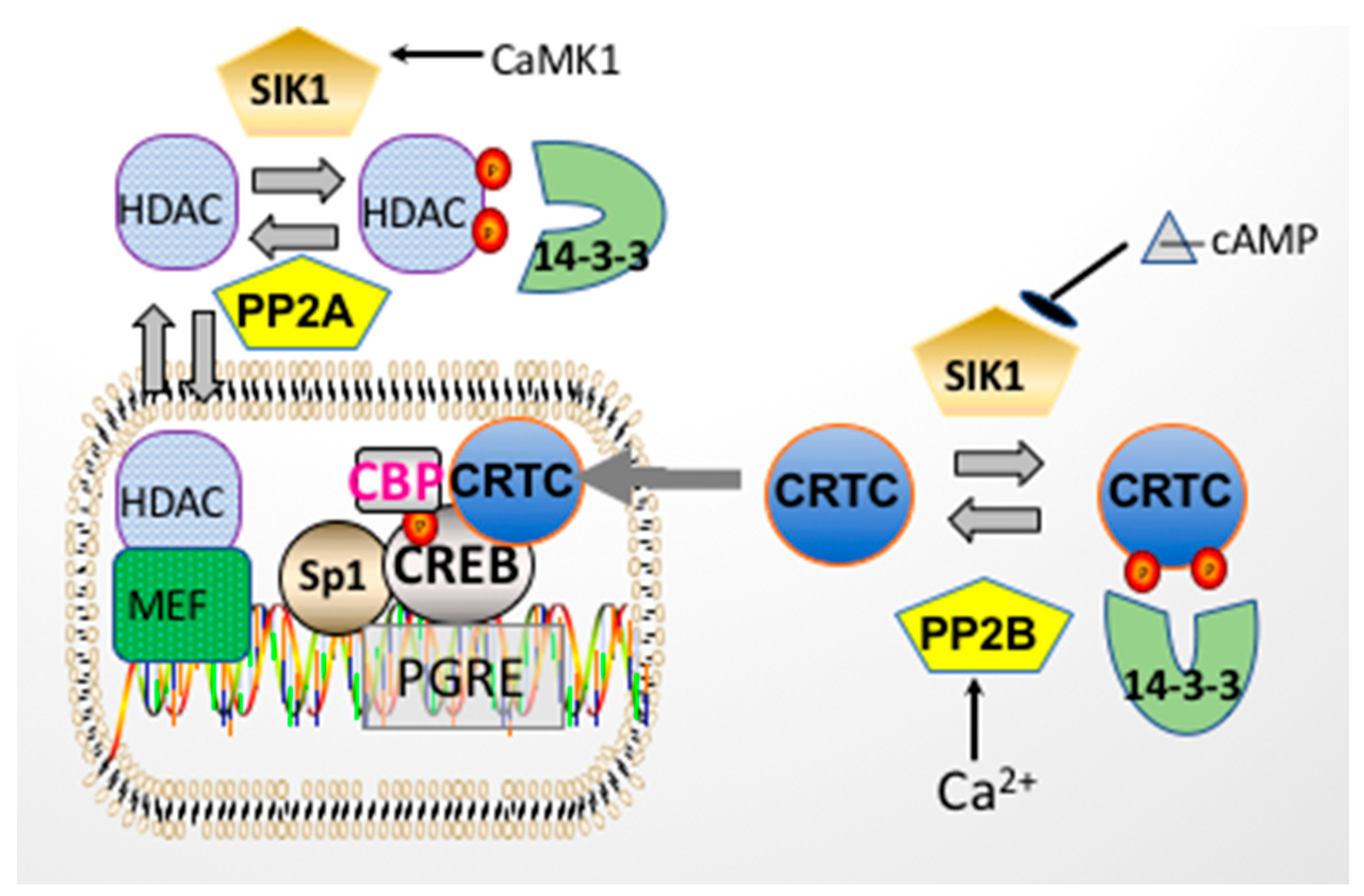
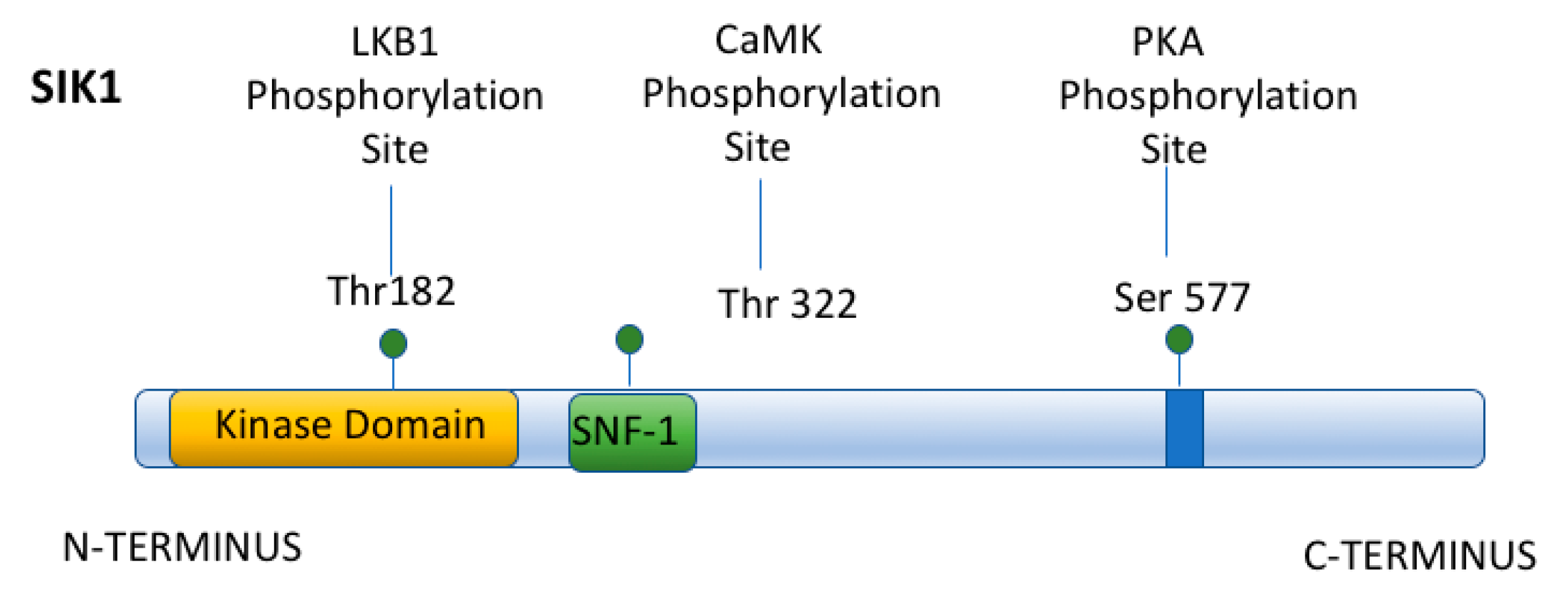
3. Acute Regulation of Na,K-ATPase by Salt Inducible Kinase 1 (SIK1)
4. Chronic Regulation of Na,K-ATPase by CREB and other Transcription Factors
5. Regulation of CREB by Transcriptional Coactivators
6. Involvement of CREB and CRTCs in Transcriptional Regulation of atp1b1
7. Regulation of the Na,K-ATPase β1 Subunit Gene atp1b1 in the Renal Proximal Tubule
8. Regulation of Na,K-ATPase Gene Expression by Adrenergic Agonists
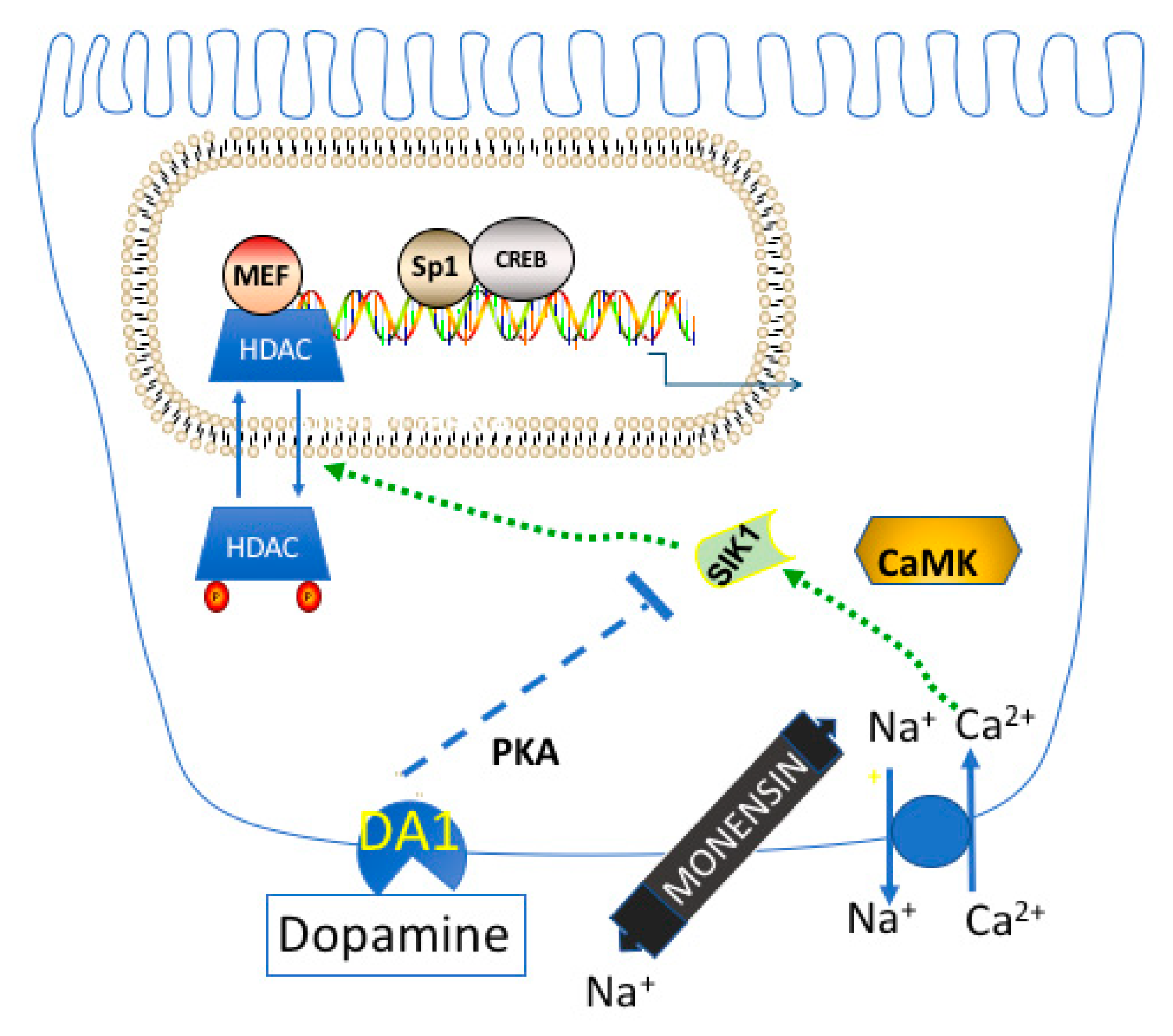
9. Regulation of Na,K-ATPase Gene Expression by Dopamine
10. Overall Role of CRTCs and SIKs in Regulating Sodium Homeostasis and Metabolism
11. Physiologic Consequences of the HDAC Kinase Activity of SIK1
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Subramanian, B.; Ko, W.C.; Yadav, V.; DesRochers, T.M.; Perrone, R.D.; Zhou, J.; Kaplan, D.L. The regulation of cystogenesis in a tissue engineered kidney disease system by abnormal matrix interactions. Biomaterials 2012, 33, 8383–8394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Askari, A. Na+/K+-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Garamella, S.; Kim, D.; Rajkhowa, T.; Cutuli, F. Renal Proximal Tubule Na,K-ATPase is Controlled by CREB Regulated Transcriptional CoActivators as well as Salt Inducible Kinase 1. Cell. Signal. 2015, 27, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.A. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R851–R861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrallah, R.; Hassouneh, R.; Hebert, R.L. Chronic kidney disease: Targeting prostaglandin E2 receptors. Am. J. Physiol. Ren. Physiol. 2014, 307, F243–F250. [Google Scholar] [CrossRef] [PubMed]
- Gurley, S.B.; Riquier-Brison, A.D.; Schnermann, J.; Sparks, M.A.; Allen, A.M.; Haase, V.H.; Snouwaert, J.N.; Le, T.H.; McDonough, A.A.; Koller, B.H.; et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011, 13, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Yao, B.; Wang, S.; Fan, X.; Wu, G.; Yang, H.; Yin, H.; Yang, S.; Harris, R.C. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J. Clin. Investig. 2011, 121, 2845–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Lokhandwala, M.F. Renal dopamine receptors and hypertension. Exp. Biol. Med. 2003, 228, 134–142. [Google Scholar] [CrossRef]
- Katholi, R.E. Renal nerves and hypertension: An update. Fed. Proc. 1985, 44, 2846–2850. [Google Scholar] [PubMed]
- Burke, S.L.; Head, G.A.; Lambert, G.W.; Evans, R.G. Renal sympathetic neuroeffector function in renovascular and angiotensin II-dependent hypertension in rabbits. Hypertension 2007, 49, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Wu, J.; Qi, Z.; Yang, G.; Dou, D.; Gao, Y.; Chen, L.; Zhang, X.; Davis, L.S.; et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J. Clin. Investig. 2007, 117, 2496–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraja, A.T.; Hunt, S.C.; Rao, D.C.; Davila-Roman, V.G.; Arnett, D.K.; Province, M.A. Genetics of hypertension and cardiovascular disease and their interconnected pathways: Lessons from large studies. Curr. Hypertens. Rep. 2011, 13, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A.C. Intrarenal dopamine: A key signal in the interactive regulation of sodium metabolism. Annu. Rev. Physiol. 2000, 62, 621–647. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B.; Kuntzweiler, T. Na+,K+-ATPase. J. Biol. Chem. 1994, 269, 19659–19662. [Google Scholar] [PubMed]
- Derfoul, A.; Robertson, N.M.; Lingrel, J.B.; Hall, D.J.; Litwack, G. Regulation of the human Na/K-ATPase β1 gene promoter by mineralocorticoid and glucocorticoid receptors. J. Biol. Chem. 1998, 273, 20702–20711. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.A.; Poulsen, S.B.; de la Mona Chavez, S.; Soleimani, M.; Dominguez Rieg, J.A.; Rieg, T. Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 2017, 92, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Gildea, J.J.; Xu, P.; Kemp, B.A.; Carlson, J.M.; Tran, H.T.; Bigler Wang, D.; Langouet-Astrie, C.J.; McGrath, H.E.; Carey, R.M.; Jose, P.A.; et al. Sodium bicarbonate cotransporter NBCe2 gene variants increase sodium and bicarbonate transport in human renal proximal tubule cells. PLoS ONE 2018, 13, e0189464. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.G.; Flores-Maldonado, C.; Lazaro, A.; Shoshani, L.; Flores-Benitez, D.; Larre, I.; Cereijido, M. Ouabain binding to Na+,K+-ATPase relaxes cell attachment and sends a specific signal (NACos) to the nucleus. J. Membr. Biol. 2004, 198, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Blantz, R.C. Glomerulotubular balance, tubuloglomerular feedback, and salt homeostasis. J. Am. Soc. Nephrol. 2008, 19, 2272–2275. [Google Scholar] [CrossRef] [PubMed]
- Gullner, H.G. The role of the adrenergic nervous system in sodium and water excretion. Klin. Wochenschr. 1983, 61, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Snavely, M.D. Catecholamines and the kidney: Receptors and renal function. Annu. Rev. Physiol. 1981, 43, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A.; Ibarra, F.; Svensson, L.B.; Klee, C.; Greengard, P. Calcineurin mediates alpha-adrenergic stimulation of Na+,K+-ATPase activity in renal tubule cells. Proc. Natl. Acad. Sci. USA 1992, 89, 7394–7397. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A.; Holtbèack, U.; Syrâen, M.L.; Svensson, L.B.; Fryckstedt, J.; Greengard, P. Activation/deactivation of renal Na+,K+-ATPase: A final common pathway for regulation of natriuresis. FASEB J. 1994, 8, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Ogimoto, G.; Pedemonte, C.H.; Pressley, T.A.; Katz, A.I.; Fâeraille, E.; Berggren, P.O.; Bertorello, A.M. Dopamine-induced endocytosis of Na+,K+-ATPase is initiated by phosphorylation of Ser-18 in the rat alpha subunit and Is responsible for the decreased activity in epithelial cells. J. Biol. Chem. 1999, 274, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, A.R.; Efendiev, R.; Pedemonte, C.H. Trafficking of Na-K-ATPase and dopamine receptor molecules induced by changes in intracellular sodium concentration of renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F1117–F1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Li, H.; Xie, Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J. Mol. Cell. Cardiol. 1997, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, M.; Stenstrom, K.; Eneling, K.; Zwiller, J.; Katz, A.I.; Takemori, H.; Bertorello, A.M. SIK1 is part of a cell sodium-sensing network that regulates active sodium transport through a calcium-dependent process. Proc. Natl. Acad. Sci. USA 2007, 104, 16922–16927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freis, E.D. The role of salt in hypertension. Blood Press. 1992, 1, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr.; Roman, R.J. The role of the kidney in hypertension. JAMA 1996, 275, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B.; Orlowski, J.; Shull, M.M.; Price, E.M. Molecular genetics of Na,K-ATPase. Prog. Nucleic Acid Res. Mol. Biol. 1990, 38, 37–89. [Google Scholar] [PubMed]
- Geering, K. The functional role of the β-subunit in the maturation and intracellular transport of Na,K-ATPase. FEBS Lett. 1991, 285, 189–193. [Google Scholar] [CrossRef] [Green Version]
- McDonough, A.A.; Geering, K.; Farley, R.A. The sodium pump needs its beta subunit. FASEB J. 1990, 4, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G. Na,K-ATPase subunit heterogenity as a mechanism for tissue specific ion regulation. Semin. Nephrol. 2005, 25, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Kolla, V.; Robertson, N.M.; Litwack, G. Identification of a mineralocorticoid/glucocorticoid response element in the human Na/K ATPase α1 gene promoter. Biochem. Biophys. Res. Commun. 1999, 266, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Orlowski, J.; Lingrel, J.B. Identification of a functional thyroid hormone response element in the upstream flanking region of the human Na,K-ATPase β 1 gene. Nucleic Acids Res. 1993, 21, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Kawakami, K. ATF-1CREB heterodimer is involved in constitutive expression of the housekeeping Na,K-ATPase alpha 1 subunit gene. Nucleic Acids Res. 1995, 23, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Borsick, M.; Rajkhowa, T.; Taub, M. Identification of a Prostaglandin-responsive Element in the Na,K-ATPase β1 Promoter That Is Regulated by cAMP and Ca2+: Evidence for an interactive role of cAMP regulatory element-bindin protein and Sp1. J. Biol. Chem. 2005, 280, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Taub, M. Regulation of the Na-K-ATPase β(1)-subunit promoter by multiple prostaglandin-responsive elements. Am. J. Physiol. Ren. Physiol. 2006, 291, F635–F646. [Google Scholar] [CrossRef] [PubMed]
- Jancic, D.; Lopez de Armentia, M.; Valor, L.M.; Olivares, R.; Barco, A. Inhibition of cAMP Response Element-Binding Protein Reduces Neuronal Excitability and Plasticity, and Triggers Neurodegeneration. Cereb. Cortex 2009, 19, 2535–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, G.S.; Lepore, J.J.; Greytak, S.; Patten, R.; McNamee, R.; Aronovitz, M.; Wang, P.J.; Reed, G.L. The CREB leucine zipper regulates CREB phosphorylation, cardiomyopathy, and lethality in a transgenic model of heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1877–H1882. [Google Scholar] [CrossRef] [PubMed]
- Mantamadiotis, T.; Kretz, O.; Ridder, S.; Bleckmann, S.C.; Bock, D.; Grone, H.J.; Malaterre, J.; Dworkin, S.; Ramsay, R.G.; Schutz, G. Hypothalamic 3′,5′-cyclic adenosine monophosphate response element-binding protein loss causes anterior pituitary hypoplasia and dwarfism in mice. Mol. Endocrinol. 2006, 20, 204–211. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, D.; Sassone-Corsi, P. Transcriptional regulation by cyclic AMP-responsive factors. Prog. Nucleic Acid. Res. Mol. Biol. 2000, 64, 343–369. [Google Scholar] [PubMed]
- Li, Z.; Langhans, S.A. Transcriptional regulators of Na,K-ATPase subunits. Front. Cell Dev. Biol. 2015, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kawakami, K. Synergism of the ATF/CRE site and GC box in the housekeeping Na,K-ATPase α1 subunit gene is essential for constitutive expression. Biochem. Biophys. Res. Commun. 1997, 241, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Borsick, M.; Geisel, J.; Matlhagela, K.; Rajkhowa, T.; Allen, C. Regulation of the Na,K-ATPase in MDCK cells by prostaglandin E1: A role for calcium as well as cAMP. Exp. Cell Res. 2004, 299, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Matlhagela, K.; Taub, M. Involvement of EP1 and EP2 receptors in the regulation of the Na,K-ATPase by prostaglandins in MDCK cells. Prostaglandins Other Lipid Mediat. 2006, 79, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlhagela, K.; Taub, M. Prostaglandins regulate transcription by means of prostaglandin response elements located in the promoters of mammalian Na,K-ATPase β 1 subunit genes. Ann. N. Y. Acad. Sci. 2006, 1091, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.K.; Gonzalez, G.A.; Biggs, W.H., 3rd; Montminy, M.R. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature 1988, 334, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.A. CREB-binding protein and p300: Molecular integrators of hematopoietic transcription. Blood 2000, 95, 745–755. [Google Scholar] [PubMed]
- Asahara, H.; Santoso, B.; Guzman, E.; Du, K.; Cole, P.A.; Davidson, I.; Montminy, M. Chromatin-dependent cooperativity between constitutive and inducible activation domains in CREB. Mol. Cell. Biol. 2001, 21, 7892–7900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Odom, D.T.; Koo, S.H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conkright, M.D.; Canettieri, G.; Screaton, R.; Guzman, E.; Miraglia, L.; Hogenesch, J.B.; Montminy, M. TORCs: Transducers of regulated CREB activity. Mol. Cell 2003, 12, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Altarejos, J.Y.; Goebel, N.; Conkright, M.D.; Inoue, H.; Xie, J.; Arias, C.M.; Sawchenko, P.E.; Montminy, M. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat. Med. 2008, 14, 1112–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Screaton, R.A.; Conkright, M.D.; Katoh, Y.; Best, J.L.; Canettieri, G.; Jeffries, S.; Guzman, E.; Niessen, S.; Yates, J.R., 3rd; Takemori, H.; et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004, 119, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, R.; Martinez, A.; Doskeland, S.O.; Haavik, J. The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 2011, 22, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, K.A.; Steullet, P.; Steinmann, M.; Do, K.Q.; Magistretti, P.J.; Halfon, O.; Cardinaux, J.R. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. USA 2007, 104, 4700–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Takemori, H.; Lin, X.Z.; Tamura, M.; Muraoka, M.; Satoh, T.; Tsuchiya, Y.; Min, L.; Doi, J.; Miyauchi, A.; et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006, 273, 2730–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.; Unterman, T.G. TORCs rev up gluconeogenesis. Cell Metab. 2005, 2, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Akhmedov, D.; Robb, C.; Leiter, C.; Berdeaux, R. Regulation of SIK1 abundance and stability is critical for myogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Cutulli, F. Experimental evidence for chronic effects of norepinephrine and dopamine on Na,K-ATPase in Primary Cultures of Renal Proximal Tubule Cells. Data Brief 2016. [Google Scholar] [CrossRef] [PubMed]
- Parra-Damas, A.; Valero, J.; Chen, M.; Espana, J.; Martin, E.; Ferrer, I.; Rodriguez-Alvarez, J.; Saura, C.A. Crtc1 activates a transcriptional program deregulated at early Alzheimer’s disease-related stages. J. Neurosci. 2014, 34, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, C.E.; Fu, A.; Reeks, C.; Screaton, R.A. CRTC2 is required for β-cell function and proliferation. Endocrinology 2013, 154, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Wang, G.; Li, Q.; Ma, Z.; Dai, M.; Zou, F. CRTC3 polymorphisms were associated with the plasma level of total cholesterol and the risks of overweight and hypertriglyceridemia in a Chinese Han population. Mol. Biol. Rep. 2014, 41, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Alavi, N.; Livingston, D.; Hiller, S.; Taub, M. Characterization of primary rabbit kidney cultures that express proximal tubule functions in a hormonally defined medium. J. Cell Biol. 1982, 95, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taub, M.L.; Yang, I.S.; Wang, Y. Primary rabbit kidney proximal tubule cell cultures maintain differentiated functions when cultured in a hormonally defined serum-free medium. In Vitro Cell. Dev. Biol. 1989, 25, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Waqar, M.A.; Seto, J.; Chung, S.D.; Hiller-Grohol, S.; Taub, M. Phosphate uptake by primary renal proximal tubule cell cultures grown in hormonally defined medium. J. Cell. Physiol. 1985, 124, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.L.; Springate, J.E.; Cutuli, F. Reduced phosphate transport in the renal proximal tubule cells in cystinosis is due to decreased expression of transporters rather than an energy defect. Biochem. Biophys. Res. Commun. 2011, 407, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.S.; Goldinger, J.M.; Hong, S.K.; Taub, M. Preparation of basolateral membranes that transport p-aminohippurate from primary cultures of rabbit kidney proximal tubule cells. J. Cell. Physiol. 1988, 135, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.B.; Rajkhowa, T.; Cutuli, F.; Springate, J.E.; Taub, M. Regulation of renal proximal tubule Na-K-ATPase by prostaglandins. Am. J. Physiol. Ren. Physiol. 2010, 298, F1222–F1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Taub, M. Insulin and other regulatory factors modulate the growth and the phosphoenolpyruvate carboxykinase (PEPCK) activity of primary rabbit kidney proximal tubule cells in serum free medium. J. Cell. Physiol. 1991, 147, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.C.; Lee, S.M.; Kadakia, N.; Taub, M. Growth and function of primary rabbit kidney proximal tubule cells in glucose-free serum-free medium. J. Cell. Physiol. 1992, 150, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Park, S.H.; Koh, H.J.; Taub, M. Mechanism of regulation of Na+ transport by angiotensin II in primary renal cells. Kidney Int. 2000, 57, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Axelson, E.; Park, J.H. Colloidal silica-coated tissue culture dishes for primary cell cultures: Growth of rabbit renal proximal tubule cells. Biotechniques 1998, 25, 990–994, 996. [Google Scholar] [PubMed]
- Han, H.J.; Sigurdson, W.J.; Nickerson, P.A.; Taub, M. Both mitogen activated protein kinase and the mammalian target of rapamycin modulate the development of functional renal proximal tubules in matrigel. J. Cell Sci. 2004, 117, 1821–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Han, H.J. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3β, Snail1, and β-catenin in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2010, 298, F1263–F1275. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Heo, J.S.; Lee, Y.J.; Lee, J.H.; Taub, M.; Han, H.J. Dopamine stimulates 45Ca2+ uptake through cAMP, PLC/PKC, and MAPKs in renal proximal tubule cells. J. Cell. Physiol. 2007, 211, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Suh, H.N.; Han, H.J. Effect of BSA-induced ER stress on SGLT protein expression levels and alpha-MG uptake in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2009, 296, F1405–F1416. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.F.; Hopfer, U.; Madhun, Z.T.; Zhou, W.; Douglas, J.G. Angiotensin II actions in the rabbit proximal tubule. Angiotensin II mediated signaling mechanisms and electrolyte transport in the rabbit proximal tubule. Ren. Physiol. Biochem. 1991, 14, 199–207. [Google Scholar] [PubMed]
- Andreatta-van Leyen, S.; Romero, M.F.; Khosla, M.C.; Douglas, J.G. Modulation of phospholipase A2 activity and sodium transport by angiotensin-(1-7). Kidney Int. 1993, 44, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Lee, Y.J.; Park, S.H.; Lee, J.H.; Taub, M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2005, 288, F988–F996. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Odejinmi, S.; Schnellmann, R.G. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J. Pharmacol. Exp. Ther. 2010, 333, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Tay, L.K.; Bregman, C.L.; Masters, B.A.; Williams, P.D. Effects of cis-diamminedichloroplatinum(II) on rabbit kidney in vivo and on rabbit renal proximal tubule cells in culture. Cancer Res. 1988, 48, 2538–2543. [Google Scholar] [PubMed]
- Morin, J.P.; Monteil, C.; Glomot, R.; Fillastre, J.P. Rabbit kidney proximal tubule cells in primary culture: Evaluation of the impact of expressed phenotype on cellular toxic response. Toxicol. In Vitro 1991, 5, 383–388. [Google Scholar] [CrossRef]
- Handler, J.S. Studies of kidney cells in culture. Kidney Int. 1986, 30, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.F.; Douglas, J.G.; Eckert, R.L.; Hopfer, U.; Jacobberger, J.W. Development and characterization of rabbit proximal tubular epithelial cell lines. Kidney Int. 1992, 42, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Han, H.J.; Rajkhowa, T.; Allen, C.; Park, J.H. Clonal analysis of immortalized renal proximal tubule cells: Na+/glucose cotransport system levels are maintained despite a decline in transport function. Exp. Cell Res. 2002, 281, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Sata, Y.; Head, G.A.; Denton, K.; May, C.N.; Schlaich, M.P. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front. Med. 2018, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Sonalker, P.A.; Tofovic, S.P.; Bastacky, S.I.; Jackson, E.K. Chronic noradrenaline increases renal expression of NHE-3, NBC-1, BSC-1 and aquaporin-2. Clin. Exp. Pharmacol. Physiol. 2008, 35, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lim, K.; Denton, K.M. Editorial: Function of Renal Sympathetic Nerves. Front. Physiol. 2017, 8, 642. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Stull, R.; Eisenhofer, G.; Gill, J.R., Jr. Urinary excretion of dihydroxyphenylalanine and dopamine during alterations of dietary salt intake in humans. Clin. Sci. 1989, 76, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Siragy, H.M.; Felder, R.A.; Carey, R.M. Intrarenal dopamine production and distribution in the rat. Physiological control of sodium excretion. Hypertension 1997, 29, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Di Sole, F.; Zhang, J.; McLeroy, P.; Moe, O.W. Chronic regulation of the renal Na+/H+ exchanger NHE3 by dopamine: Translational and posttranslational mechanisms. Am. J. Physiol. Ren. Physiol. 2013, 304, F1169–F1180. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Armando, I.; Luo, Y.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Dysregulation of dopamine-dependent mechanisms as a determinant of hypertension: Studies in dopamine receptor knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H551–H569. [Google Scholar] [CrossRef] [PubMed]
- Bruno, N.E.; Kelly, K.A.; Hawkins, R.; Bramah-Lawani, M.; Amelio, A.L.; Nwachukwu, J.C.; Nettles, K.W.; Conkright, M.D. Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J. 2014, 33, 1027–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanyo, R.; Price, D.M.; Chik, C.L.; Ho, A.K. Salt-inducible kinase 1 in the rat pinealocyte: Adrenergic regulation and role in arylalkylamine N-acetyltransferase gene transcription. Endocrinology 2009, 150, 4221–4230. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Bertorello, A.M.; Zandomeni, R.; Cinelli, A.R.; Pedemonte, C.H. Agonist-dependent regulation of renal Na+,K+-ATPase activity is modulated by intracellular sodium concentration. J. Biol. Chem. 2002, 277, 11489–11496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Takemori, H.; Halder, S.K.; Nonaka, Y.; Okamoto, M. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. 1999, 453, 135–139. [Google Scholar] [CrossRef]
- Okamoto, M.; Takemori, H.; Katoh, Y. Salt-inducible kinase in steroidogenesis and adipogenesis. Trends Endocrinol. Metab. 2004, 15, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Takemori, H.; Katoh, Y.; Doi, J.; Horike, N.; Makino, A.; Nonaka, Y.; Okamoto, M. Salt-inducible kinase is involved in the ACTH/cAMP-dependent protein kinase signaling in Y1 mouse adrenocortical tumor cells. Mol. Endocrinol. 2001, 15, 1264–1276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Poon, V.; Sanchez-Watts, G.; Watts, A.G.; Takemori, H.; Aguilera, G. Salt-inducible kinase is involved in the regulation of corticotropin-releasing hormone transcription in hypothalamic neurons in rats. Endocrinology 2012, 153, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.A.; Cardinaux, J.R. Emerging Roles of CREB-Regulated Transcription Coactivators in Brain Physiology and Pathology. Trends Neurosci. 2017, 40, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef] [PubMed]
- Berdeaux, R.; Goebel, N.; Banaszynski, L.; Takemori, H.; Wandless, T.; Shelton, G.D.; Montminy, M. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat. Med. 2007, 13, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.; Venetsanou, K.; Chedrese, P.J.; Pinto, V.; Takemori, H.; Franco-Cereceda, A.; Eriksson, P.; Mochizuki, N.; Soares-da-Silva, P.; Bertorello, A.M. Increases in intracellular sodium activate transcription and gene expression via the salt-inducible kinase 1 network in an atrial myocyte cell line. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H57–H65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, S.; Takemori, H.; Tokudome, T.; Mao, Y.; Otani, K.; Mochizuki, N.; Pires, N.; Pinho, M.J.; Franco-Cereceda, A.; Torielli, L.; et al. Lack of salt-inducible kinase 2 (SIK2) prevents the development of cardiac hypertrophy in response to chronic high-salt intake. PLoS ONE 2014, 9, e95771. [Google Scholar] [CrossRef] [PubMed]
- Moe, O.W. Acute regulation of proximal tubule apical membrane Na/H exchanger NHE-3: Role of phosphorylation, protein trafficking, and regulatory factors. J. Am. Soc. Nephrol. 1999, 10, 2412–2425. [Google Scholar] [PubMed]
- Baum, M.; Quigley, R. Inhibition of proximal convoluted tubule transport by dopamine. Kidney Int. 1998, 54, 1593–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaine, J.; Weinman, E.J.; Cunningham, R. The regulation of renal phosphate transport. Adv. Chronic Kidney Dis. 2011, 18, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Murer, H.; Hernando, N.; Forster, I.; Biber, J. Regulation of Na/Pi transporter in the proximal tubule. Annu. Rev. Physiol. 2003, 65, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Biber, J.; Hernando, N.; Forster, I.; Murer, H. Regulation of phosphate transport in proximal tubules. Pflug. Arch. Eur. J. Physiol. 2009, 458, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Neri, E.A.; Bezerra, C.N.; Reboucas, N.A. Essential regulatory elements for NHE3 gene transcription in renal proximal tubule cells. Braz. J. Med. Biol. Res. 2011, 44, 514–523. [Google Scholar] [PubMed]
- Hilfiker, H.; Kvietikova, I.; Hartmann, C.M.; Stange, G.; Murer, H. Characterization of the human type II Na/Pi-cotransporter promoter. Pflug. Arch. 1998, 436, 591–598. [Google Scholar] [CrossRef]
- Bertorello, A.M.; Pires, N.; Igreja, B.; Pinho, M.J.; Vorkapic, E.; Wagsater, D.; Wikstrom, J.; Behrendt, M.; Hamsten, A.; Eriksson, P.; et al. Increased arterial blood pressure and vascular remodeling in mice lacking salt-inducible kinase 1 (SIK1). Circ. Res. 2015, 116, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Stow, L.R.; Gumz, M.L. The circadian clock in the kidney. J. Am. Soc. Nephrol. 2011, 22, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Stow, L.R.; Richards, J.; Cheng, K.Y.; Lynch, I.J.; Jeffers, L.A.; Greenlee, M.M.; Cain, B.D.; Wingo, C.S.; Gumz, M.L. The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension 2012, 59, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, T.; Yang, C.; Turner, D.A.; Giovannucci, D.R.; Xie, Z. Regulation of inositol 1,4,5-trisphosphate receptor-mediated calcium release by the Na/K-ATPase in cultured renal epithelial cells. J. Biol. Chem. 2008, 283, 1128–1136. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taub, M. Gene Level Regulation of Na,K-ATPase in the Renal Proximal Tubule Is Controlled by Two Independent but Interacting Regulatory Mechanisms Involving Salt Inducible Kinase 1 and CREB-Regulated Transcriptional Coactivators. Int. J. Mol. Sci. 2018, 19, 2086. https://doi.org/10.3390/ijms19072086
Taub M. Gene Level Regulation of Na,K-ATPase in the Renal Proximal Tubule Is Controlled by Two Independent but Interacting Regulatory Mechanisms Involving Salt Inducible Kinase 1 and CREB-Regulated Transcriptional Coactivators. International Journal of Molecular Sciences. 2018; 19(7):2086. https://doi.org/10.3390/ijms19072086
Chicago/Turabian StyleTaub, Mary. 2018. "Gene Level Regulation of Na,K-ATPase in the Renal Proximal Tubule Is Controlled by Two Independent but Interacting Regulatory Mechanisms Involving Salt Inducible Kinase 1 and CREB-Regulated Transcriptional Coactivators" International Journal of Molecular Sciences 19, no. 7: 2086. https://doi.org/10.3390/ijms19072086