Protective Effects of 6-(Methylsulfinyl)hexyl Isothiocyanate on Aβ1-42-Induced Cognitive Deficit, Oxidative Stress, Inflammation, and Apoptosis in Mice

 , , , ,
, , , ,  , and
, and
Abstract
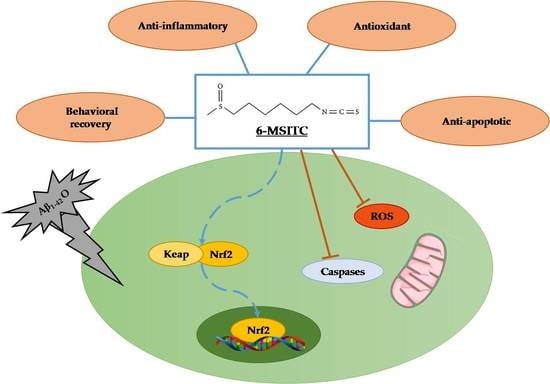
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
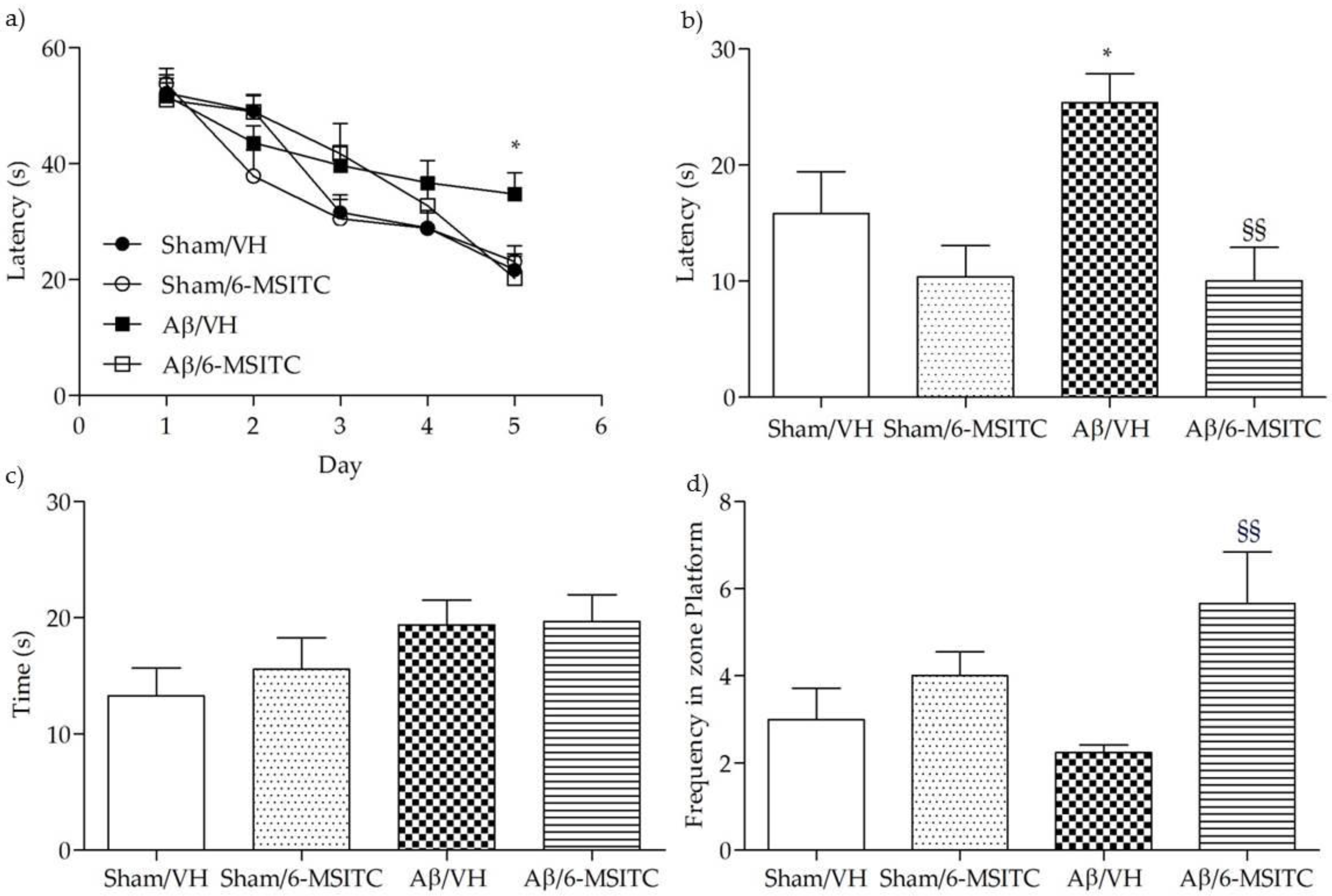
2.1. Effects of 6-MSITC on the Cognitive Functions After Aβ1-42O Injection
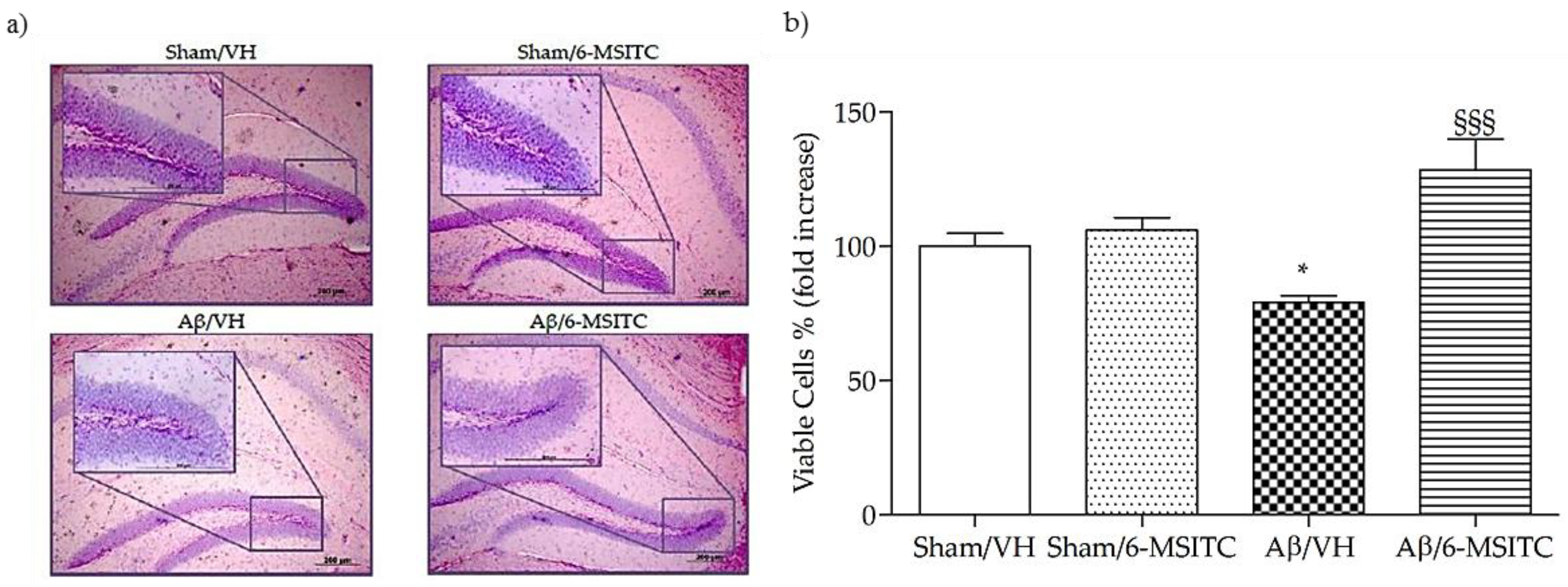
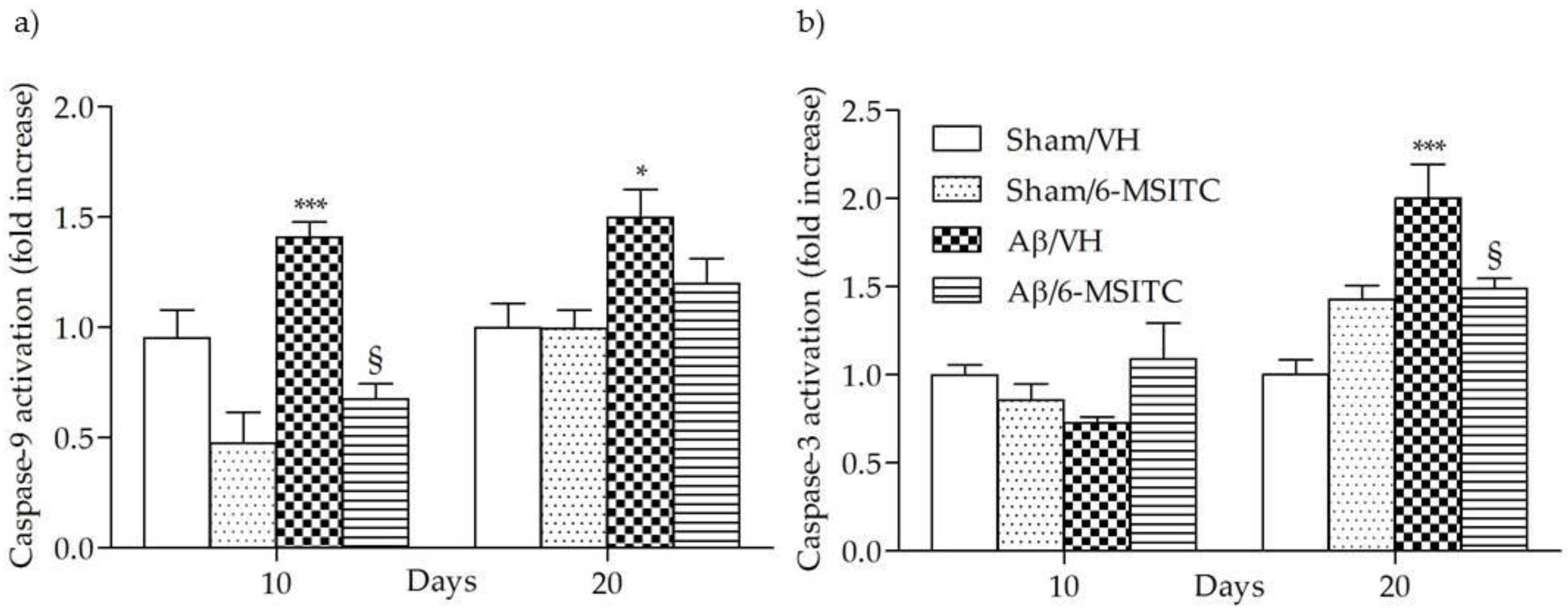
2.2. Effects of 6-MSITC on Hippocampal Cell Death After Aβ1-42O Injection
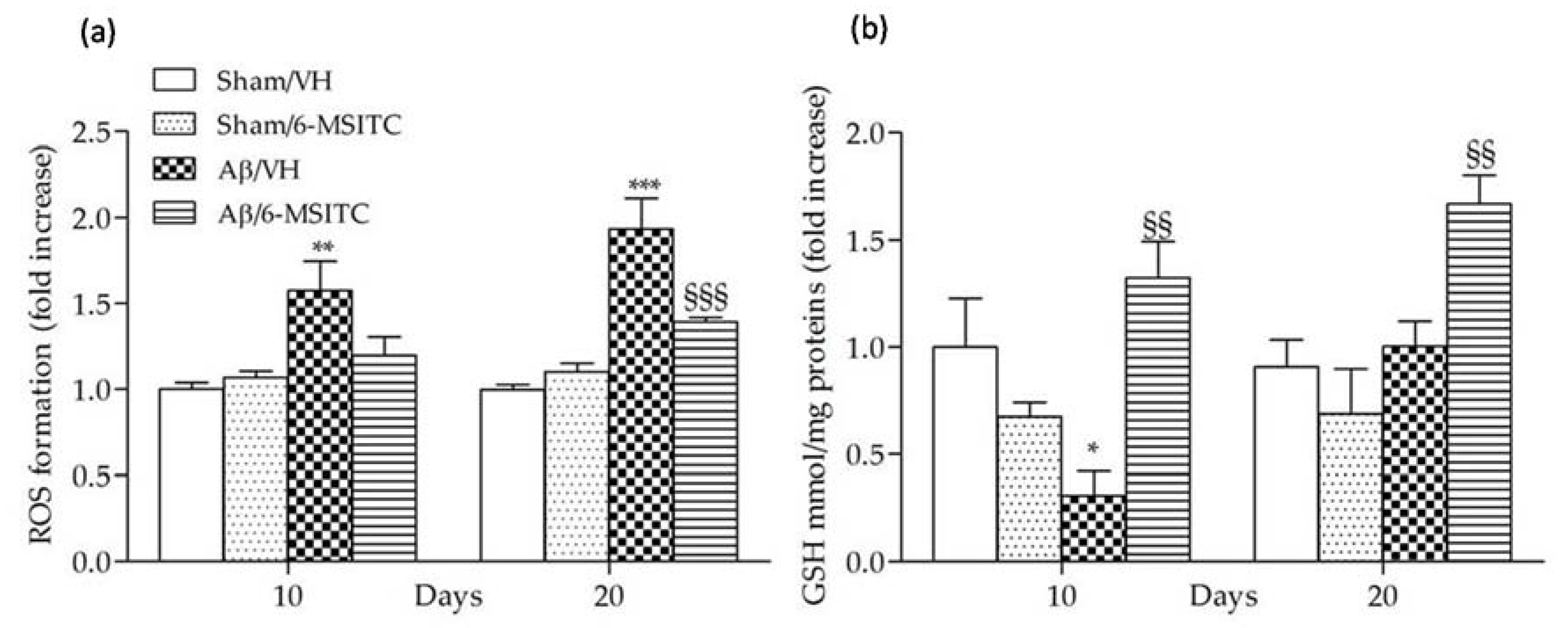
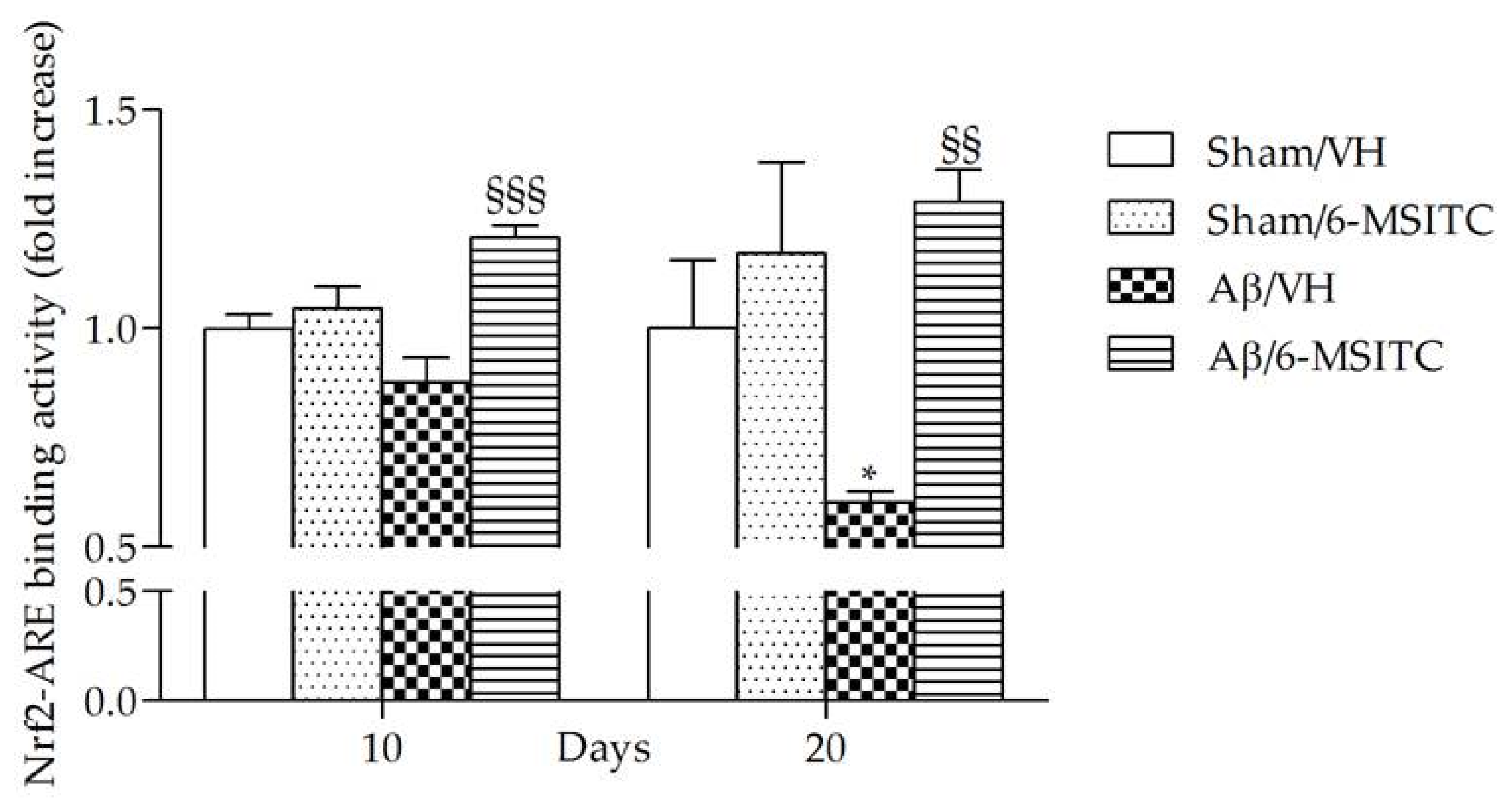
2.3. Effects of 6-MSITC on Oxidative Stress After Aβ1-42O Injection
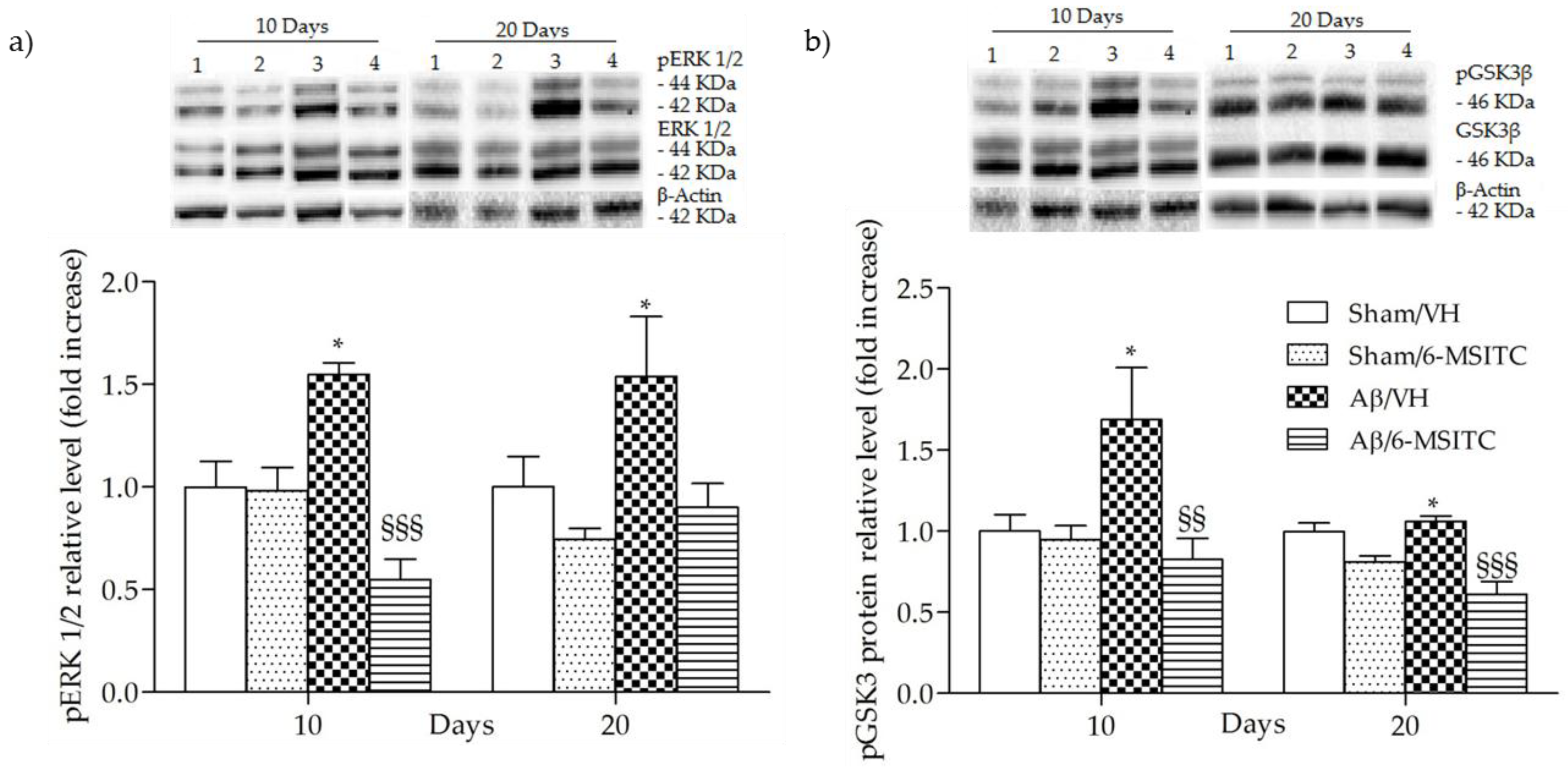
2.4. Effects of 6-MSITC on ERK1/2 and GSK3β Activities After Aβ1-42O Injection
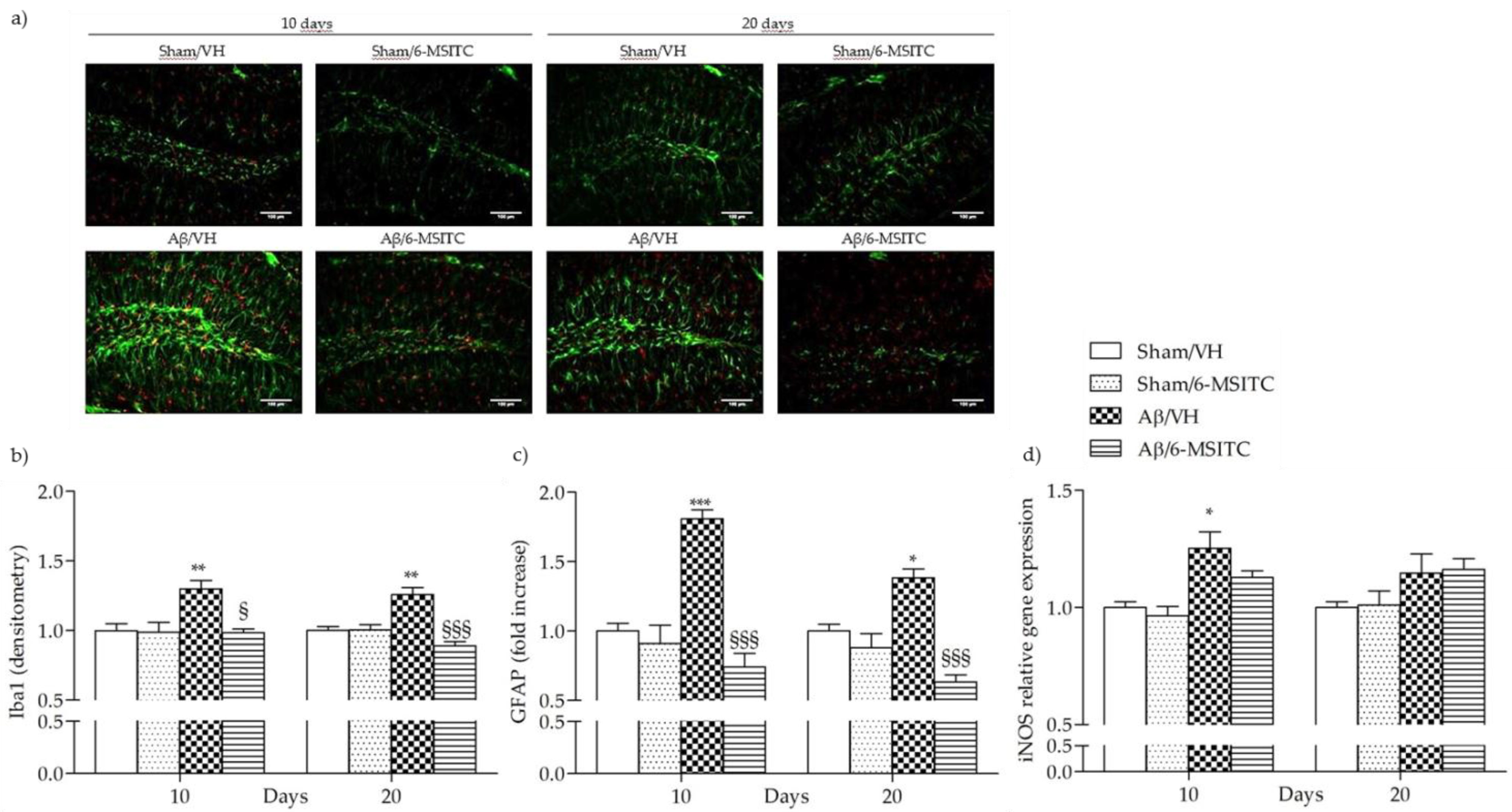
2.5. Effects of 6-MSITC on Neuroinflammation After Aβ1-42O Injection
3. Discussion
4. Materials and Methods
4.1. Animals
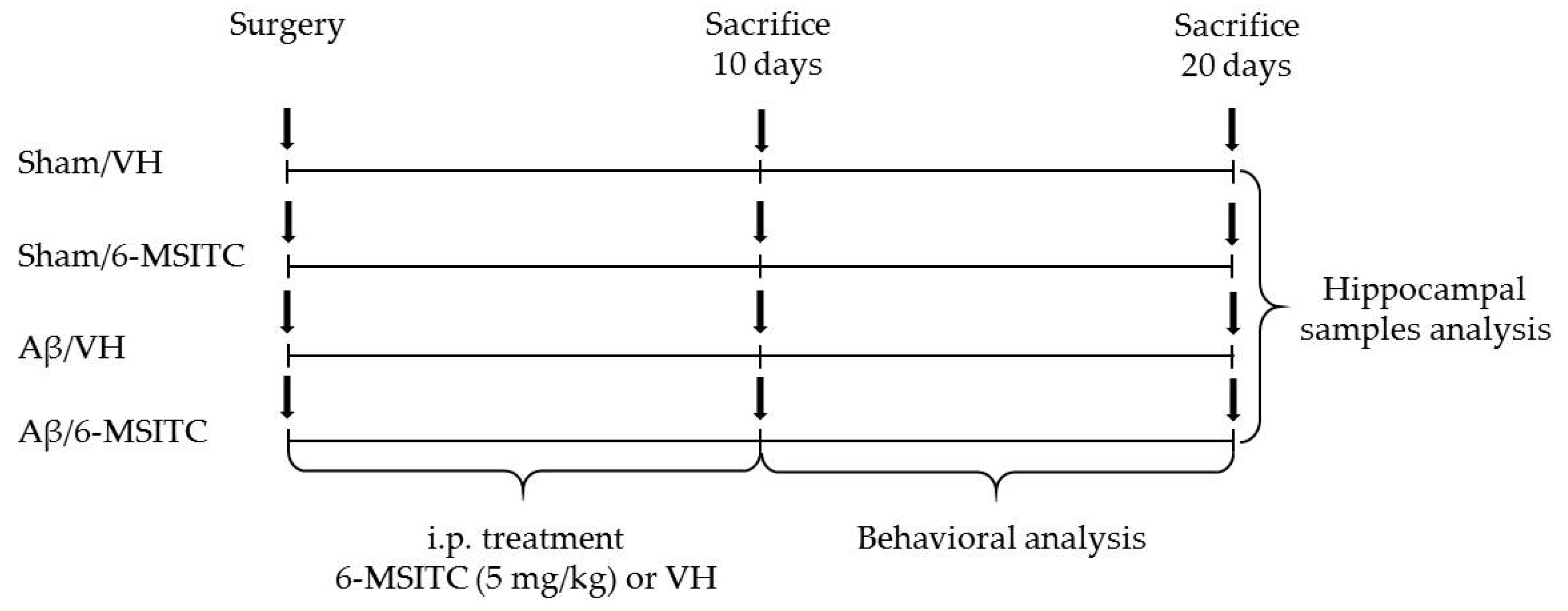
4.2. Experimental Design
4.3. Aβ1-42O Preparation and Injection
4.4. Behavioral Analysis
4.5. MWM Test
4.6. Passive Avoidance Test
4.7. Tissue Preparation for Immunohistochemistry and Neurochemical Analysis
4.8. H&E Staining
4.9. Determination of Caspases Activation
4.10. Determination of Redox Status
4.11. Determination of GSH Content
4.12. Determination of Nrf2 Activation
4.13. Western Blotting
4.14. GFAP and Iba1 Staining
4.15. RNA Extraction
RNA Reverse Transcription and Real Time PCR
4.16. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 6-MSITC | 6-(Methylsulfinyl)hexyl isothiocyanate |
| Aβ1-42O | β-Amyloid oligomers |
| AP-1 | Activator protein-1 |
| AD | Alzheimer’s disease |
| ARE | Antioxidant responsive element |
| JNK | c-Jun N-terminal kinases |
| COX-2 | Cyclooxygenase-2 |
| CNS | Central Nervous System |
| DCF | 2′7′-dichlorofluorescein |
| DCFH-DA | 2′7′-dichlorodihydrofluorescein diacetate |
| DNA-PK | DNA-Dependent Protein Kinase |
| ERK12 | Extracellular signal-regulated protein kinases 1 and 2 |
| GFAP | Glial Fibrillary Acidic Protein |
| GSH | Glutathione |
| GSK3 | Glycogen synthase kinase 3 |
| H&E | Hematoxylin-Eosin |
| Iba1 | Ionized calcium-binding adaptor molecule 1 |
| i.c.v. | Intracerebroventricular |
| iNOS | Inducible Nitric Oxide Synthase |
| i.p. | Intraperitoneal |
| ITCs | Isothiocyanates |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MAPK | Mitogen-activated protein kinase |
| Morris Water Maze | MWM |
| NGS | Normal Goat Serum |
| NO | Nitric Oxide |
| Nrf2 | Nuclear factor E2-related factor 2 |
| PARP | Poly ADP Ribose Polymerase |
| PBS | Phosphate Buffer Saline |
| pNA | p-nitroaniline |
| ROS | Reactive Oxygen Species |
| TBS | Tris-buffered saline |
| VH | Vehicle |
References
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Giliberto, L.; Tamagno, E.; Tabaton, M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front. Aging Neurosci. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer’s disease models: The control of neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Tarozzi, A.; Rimondini, R.; Hrelia, P. Early effects of Aβ1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav. Brain Res. 2016, 314, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.M.; Massa, S.M. Neuroprotective strategies in Alzheimer’s disease. NeuroRx 2004, 1, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, L.; Venkatesan, R.; Kim, S.Y. Neuroprotective and anti-inflammatory activities of allyl isothiocyanate through attenuation of JNK/NF-κB/TNF-α signaling. Int. J. Mol. Sci. 2017, 18, 1423. [Google Scholar] [CrossRef] [PubMed]
- Sita, G.; Hrelia, P.; Tarozzi, A.; Morroni, F. Isothiocyanates are promising compounds against oxidative stress, neuroinflammation and cell death that may benefit neurodegeneration in Parkinson’s disease. Int. J. Mol. Sci. 2016, 17, E1454. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.; Seo, S.G.; Choi, B.R.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane alleviates scopolamine-induced memory impairment in mice. Pharmacol. Res. 2014, 85, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Williamson, G. A critical review of the bioavailability of glucosinolates and related compounds. Nat. Prod. Rep. 2004, 21, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; De Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Liu, W.; Kang, Z.; Cai, J.; Wang, Q.; Cheng, N.; Wang, S.; Wang, S.; Zhang, J.H.; Sun, X. Sulforaphane protects brains against hypoxic-ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010, 1343, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Hou, D.X.; Morinaga, O.; Shoyama, Y. Molecular mechanisms underlying anti-inflammatory actions of 6-(methylsulfinyl)hexyl isothiocyanate derived from Wasabi (Wasabia japonica). Adv. Pharmacol. Sci. 2012, 2012, 614046. [Google Scholar] [CrossRef] [PubMed]
- Morimitsu, Y.; Hayashi, K.; Nakagawa, Y.; Fujii, H.; Horio, F.; Uchida, K.; Osawa, T. Antiplatelet and anticancer isothiocyanates in Japanese domestic horseradish, Wasabi. Mech. Ageing Dev. 2000, 116, 125–134. [Google Scholar] [CrossRef]
- Lu, Z.; Dockery, C.R.; Crosby, M.; Chavarria, K.; Patterson, B.; Giedd, M. Antibacterial activities of Wasabi against Escherichia coli O157:H7 and Staphylococcus aureus. Front. Microbiol. 2016, 7, 1403. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Fujii, M.; Hou, D.X. Effects of 6-(methylsulfinyl)hexyl isothiocyanate on cyclooxygenase-2 expression induced by lipopolysaccharide, interferon-γ and 12-O-tetradecanoylphorbol-13-acetate. Oncol. Rep. 2007, 17, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akita, N.; Nagai, M.; Hayashi, T.; Suzuki, K. 6-Methylsulfinylhexyl isothiocyanate modulates endothelial cell function and suppresses leukocyte adhesion. J. Nat. Med. 2014, 68, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Hsuan, S.W.; Chyau, C.C.; Hung, H.Y.; Chen, J.H.; Chou, F.P. The induction of apoptosis and autophagy by Wasabia japonica extract in colon cancer. Eur. J. Nutr. 2016, 55, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Shinoda, S.; Yamori, T.; Sawaki, S.; Nagata, I.; Ryoyama, K.; Fuke, Y. Selective sensitivity to wasabi-derived 6-(methylsulfinyl)hexyl isothiocyanate of human breast cancer and melanoma cell lines studied in vitro. Cancer Detect. Prev. 2005, 29, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.A.; Gerdes, R.G. Recent trends in glutathione biochemistry-glutathione-protein interactions: A molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 1998, 242, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Tarozzi, A.; Cantelli-Forti, G.; Hrelia, P. Neuroprotection by 6-(methylsulfinyl)hexyl isothiocyanate in a 6-hydroxydopamine mouse model of Parkinson’s disease. Brain Res. 2014, 1589, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T. The role of caspases in Alzheimer’s disease; potential novel therapeutic opportunities. Apoptosis 2010, 15, 1403–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubold, T.F.; Eschenburg, S. A molecular view on signal transduction by the apoptosome. Cell Signal. 2012, 24, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Rev. 2005, 49, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S. Du Amyloid-β-induced mitochondrial dysfunction. J. Alzheimers. Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis. 2018. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Kim, J.; Cho, C.H.; Hahn, H.G.; Choi, S.Y.; Cho, S.W. Neuroprotective effects of N-adamantyl-4-methylthiazol-2-amine against amyloid β-induced oxidative stress in mouse hippocampus. Brain Res. Bull. 2017, 128, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.M.; Hogg, E.L.; Collingridge, G.L.; Corrêa, S.A.L. Hippocampal metabotropic glutamate receptor long-term depression in health and disease: Focus on mitogen-activated protein kinase pathways. J. Neurochem. 2016, 139, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Wei, J.; Shang, Y.H.; Huang, H.C.; Lao, F.X. Modulation of AβPP and GSK3β by endoplasmic reticulum stress and involvement in Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Wang, Y.; Chen, C.; Hu, M.; Wang, L.; Fu, J.; Shi, G.; Zhang, D.; Zhang, T. Methyl salicylate lactoside protects neurons ameliorating cognitive disorder through inhibiting amyloid beta-induced neuroinflammatory response in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.T. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006, 83, 470S–474S. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Boucher, C.; Fontaine, B.; Delarasse, C. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer’s disease models: Effects of aging and amyloid pathology. Aging Cell 2017, 16, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Forloni, G. In vivo application of beta amyloid oligomers: A simple tool to evaluate mechanisms of action and new therapeutic approaches. Curr. Pharm. Des. 2014, 20, 2491–2505. [Google Scholar] [CrossRef] [PubMed]
- Waldmeier, P.C.; Tatton, W.G. Interrupting apoptosis in neurodegenerative disease: Potential for effective therapy? Drug Discov. Today 2004, 9, 210–218. [Google Scholar] [CrossRef]
- Zussy, C.; Brureau, A.; Delair, B.; Marchal, S.; Keller, E.; Ixart, G.; Naert, G.; Meunier, J.; Chevallier, N.; Maurice, T.; et al. Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid β fragment in rats. Am. J. Pathol. 2011, 179, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, Y.; Sun, J.; Xiang, Y.; Yang, J.; Dai, S.; Zhang, X. Neuroglobin attenuates beta amyloid-induced apoptosis through inhibiting caspases activity by activating PI3K/Akt signaling pathway. J. Mol. Neurosci. 2016, 58, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of amyloidosis by inflammation in the amyloid-beta Marmoset monkey model of Alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.-Z.; Ge, Q.H.; Li, Q.; Qu, R.; Ma, S.P. Peoniflorin attentuates Aβ1-42-mediated neurotoxicity by regulating calcium homeostasis and ameliorating oxidative stress in hippocampus of rats. J. Neurol. Sci. 2009, 280, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Costa, V.; Pomatto, L.C.D.; Davies, K.J.A. The proteasome and oxidative stress in Alzheimer’s disease. Antioxid. Redox Signal. 2016, 25, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Nicol, C.J.; Cheng, Y.C. Resveratrol activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced inflammation and oxidative stress. Neurochem. Int. 2018, 115, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates β-amyloid-induced oxidative stress via Akt/GSK-3β/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Trio, P.Z.; Fujisaki, S.; Tanigawa, S.; Hisanaga, A.; Sakao, K.; Hou, D.X. DNA microarray highlights Nrf2-mediated neuron protection targeted by Wasabi-derived isothiocyanates in IMR-32 cells. Gene Regul. Syst. Biol. 2016, 10, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.X.; Korenori, Y.; Tanigawa, S.; Yamada-Kato, T.; Nagai, M.; He, X.; He, J. Dynamics of Nrf2 and Keap1 in ARE-mediated NQO1 expression by wasabi 6-(methylsulfinyl)hexyl isothiocyanate. J. Agric. Food Chem. 2011, 59, 11975–11982. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Holzer, M.; Grossmann, A.; Zedlick, D.; Brückner, M.K. Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer’s disease. Neuroscience 1995, 68, 5–18. [Google Scholar] [CrossRef]
- Gärtner, U.; Holzer, M.; Heumann, R.; Arendt, T. Induction of p21ras in Alzheimer pathology. Neuroreport 1995, 6, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Lim, C.M.; Kim, J.B.; Shin, J.H.; Lee, S.; Lee, M.; Lee, J.K. Extracellular HMGB1 released by NMDA treatment confers neuronal apoptosis via RAGE-p38 MAPK/ERK signaling pathway. Neurotox. Res. 2011, 20, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, R.E.J.; King, A.E.; Dickson, T.C. α-Synuclein protects neurons from apoptosis downstream of free-radical production through modulation of the MAPK signalling pathway. Neurotox. Res. 2013, 23, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Oh-Hashi, K.; Kiuchi, K.; Hirata, Y. p44/42 MAP kinase and c-Jun N-terminal kinase contribute to the up-regulation of caspase-3 in manganese-induced apoptosis in PC12 cells. Brain Res. 2006, 1099, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.; Heizer, J.W.; Li, Y.; Chapman, K.; Ogden, C.A.; Andreasen, K.; Shapland, E.; Kucera, G.; Mogan, J.; Humann, J.; et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2011, 108, 11578–11583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, K.P.; Mizuno, K. The roles of protein kinases in learning and memory. Learn. Mem. 2013, 20, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Zhang, Y.; Zhang, J.; Wang, H.; Ren, B. ERK in learning and memory: A review of recent research. Int. J. Mol. Sci. 2010, 11, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Hernández, F.; Gómez-Ramos, P.; Morán, M.A.; Hen, R.; Avila, J. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, D.E.; Molina-Porcel, L.; Carroll, J.C.; Macdonald, C.; Aboagye, A.K.; Trojanowski, J.Q.; Lee, V.M.Y. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J. Neurosci. 2012, 32, 7392–7402. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Swatton, J.E.; Sellers, L.A.; Faull, R.L.M.; Holland, A.; Iritani, S.; Bahn, S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 2004, 19, 2711–2719. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venè, R.; Cardinali, B.; Arena, G.; Ferrari, N.; Benelli, R.; Minghelli, S.; Poggi, A.; Noonan, D.M.; Albini, A.; Tosetti, F. Glycogen synthase kinase 3 regulates cell death and survival signaling in tumor cells under redox stress. Neoplasia 2014, 16, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.F.; Ghirnikar, R.S. GFAP and astrogliosis. Brain Pathol. 1994, 4, 229–237. [Google Scholar] [CrossRef] [PubMed]
- González-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Uto, T.; Fujii, M.; Hou, D.X. 6-(Methylsulfinyl)hexyl isothiocyanate suppresses inducible nitric oxide synthase expression through the inhibition of Janus kinase 2-mediated JNK pathway in lipopolysaccharide-activated murine macrophages. Biochem. Pharmacol. 2005, 70, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Merlicco, A.; Morroni, F.; Franco, F.; Cantelli-Forti, G.; Teti, G.; Falconi, M.; Hrelia, P. Cyanidin 3-O-glucopyranoside protects and rescues SH-SY5Y cells against amyloid-beta peptide-induced toxicity. Neuroreport 2008, 19, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Maezawa, I.; Yao, N.; Xu, B.; Diaz-Avalos, R.; Rana, S.; Hua, D.H.; Cheng, R.H.; Lam, K.S.; Jin, L.W.; et al. Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Aβ oligomer-induced cytotoxicity. Brain Res. 2007, 1130, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maezawa, I.; Hong, H.S.; Liu, R.; Wu, C.Y.; Cheng, R.H.; Kung, M.P.; Kung, H.F.; Lam, K.S.; Oddo, S.; LaFerla, F.M.; et al. Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 2008, 104, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Santo-Yamada, Y.; Wada, K. Stress-induced impairment of inhibitory avoidance learning in female neuromedin B receptor-deficient mice. Physiol. Behav. 2003, 78, 303–309. [Google Scholar] [CrossRef]
- Akar, F.; Mutlu, O.; Komsuoglu Celikyurt, I.; Bektas, E.; Tanyeri, P.; Ulak, G.; Erden, F. Effects of 7-NI and ODQ on memory in the passive avoidance, novel object recognition, and social transmission of food preference tests in mice. Med. Sci. Monit. Basic Res. 2014, 20, 27–35. [Google Scholar] [PubMed] [Green Version]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Fischer, A.; Jacobson, K.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissueand cell sections. Cold Spring Harb. Protoc. 2008, 3, 3–5. [Google Scholar]
- Movsesyan, V.A.; Yakovlev, A.G.; Dabaghyan, E.A.; Stoica, B.A.; Faden, A.I. Ceramide induces neuronal apoptosis through the caspase-9/caspase-3 pathway. Biochem. Biophys. Res. Commun. 2002, 299, 201–207. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time Quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Protective Effects of 6-(Methylsulfinyl)hexyl Isothiocyanate on Aβ1-42-Induced Cognitive Deficit, Oxidative Stress, Inflammation, and Apoptosis in Mice. Int. J. Mol. Sci. 2018, 19, 2083. https://doi.org/10.3390/ijms19072083
Morroni F, Sita G, Graziosi A, Turrini E, Fimognari C, Tarozzi A, Hrelia P. Protective Effects of 6-(Methylsulfinyl)hexyl Isothiocyanate on Aβ1-42-Induced Cognitive Deficit, Oxidative Stress, Inflammation, and Apoptosis in Mice. International Journal of Molecular Sciences. 2018; 19(7):2083. https://doi.org/10.3390/ijms19072083
Chicago/Turabian StyleMorroni, Fabiana, Giulia Sita, Agnese Graziosi, Eleonora Turrini, Carmela Fimognari, Andrea Tarozzi, and Patrizia Hrelia. 2018. "Protective Effects of 6-(Methylsulfinyl)hexyl Isothiocyanate on Aβ1-42-Induced Cognitive Deficit, Oxidative Stress, Inflammation, and Apoptosis in Mice" International Journal of Molecular Sciences 19, no. 7: 2083. https://doi.org/10.3390/ijms19072083