Transmembrane TNF and Partially TNFR1 Regulate TNFR2 Expression and Control Inflammation in Mycobacterial-Induced Pleurisy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
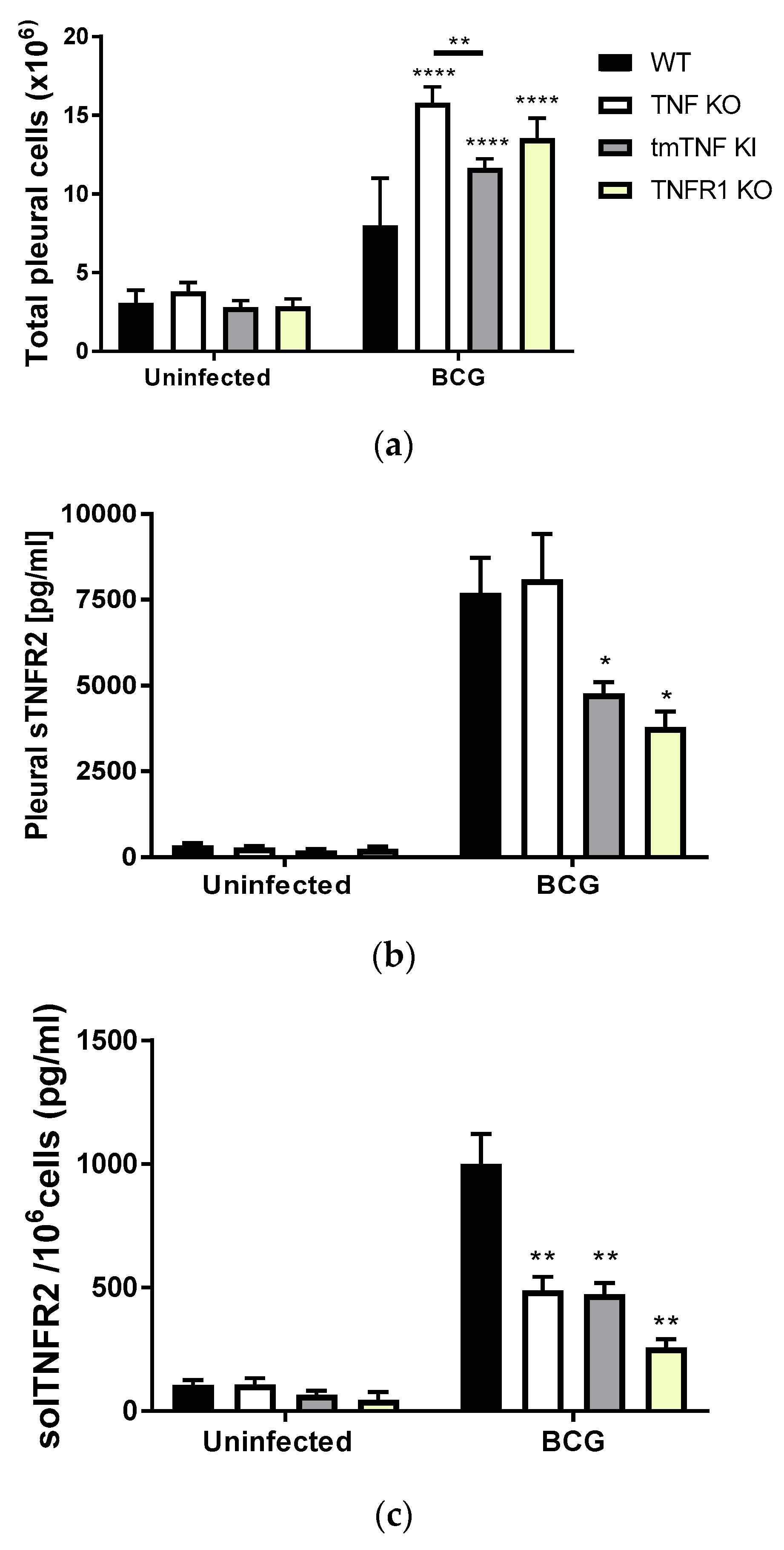
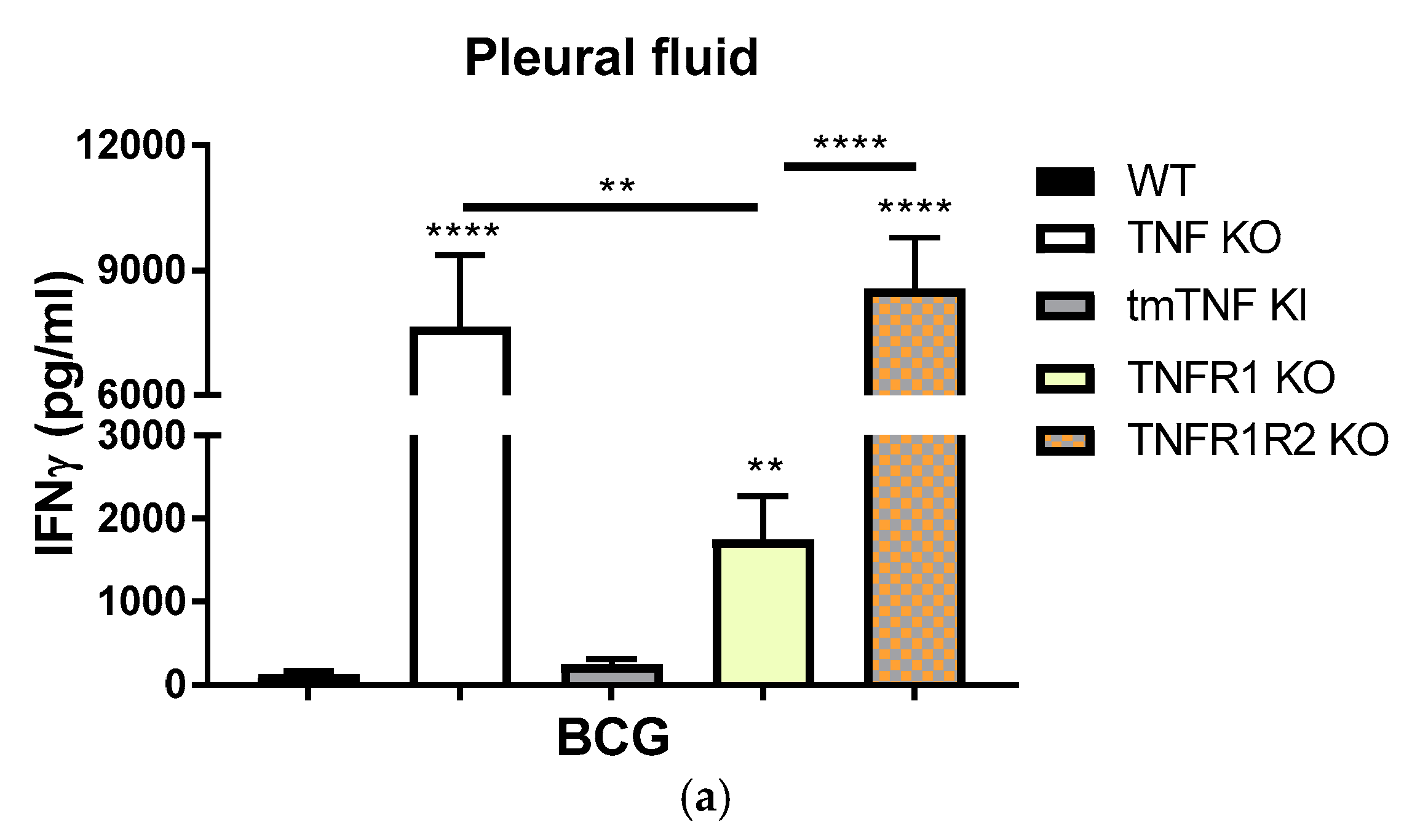
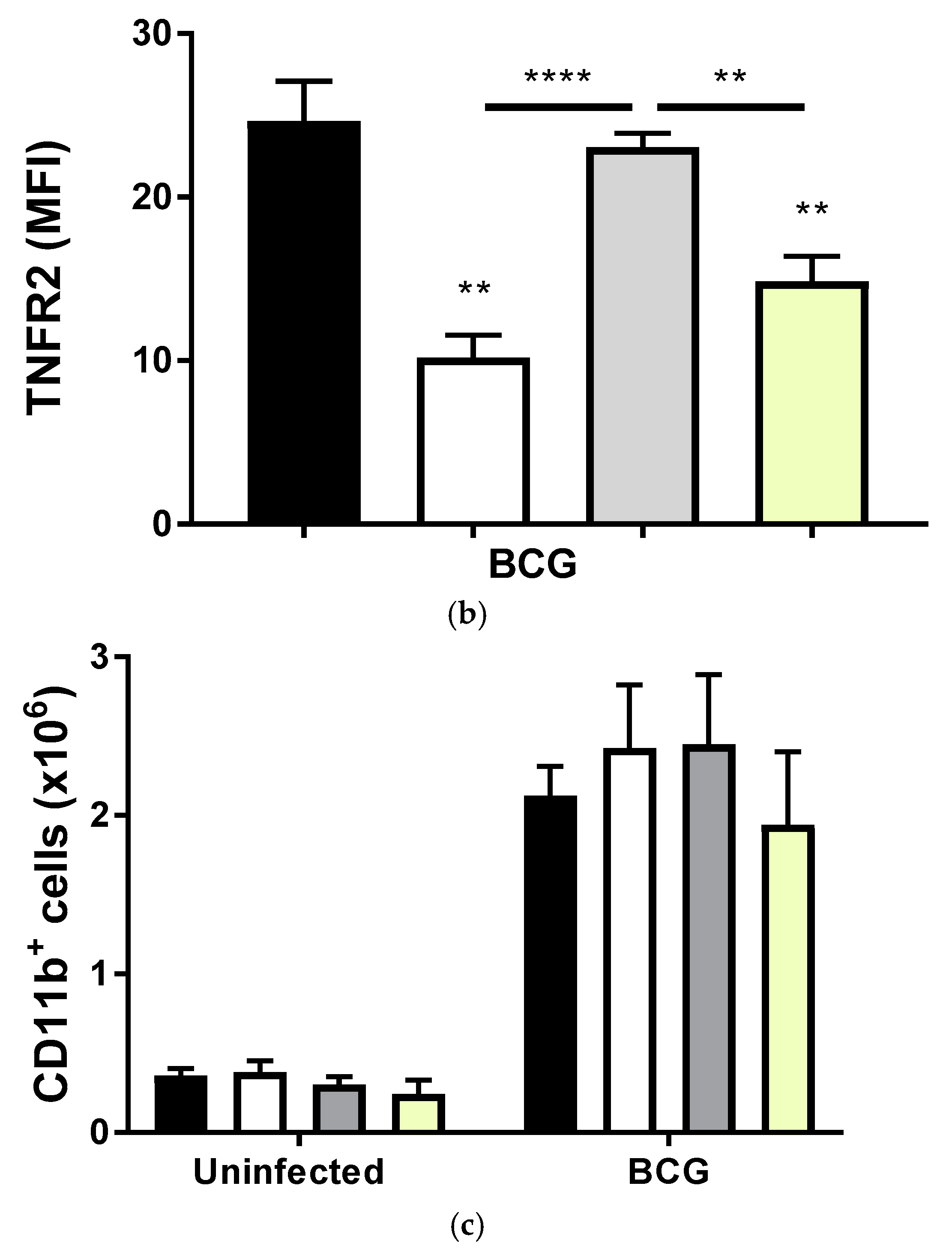
2.1. BCG-Induced Pleurisy Triggers the Accumulation of Inflammatory Cells Releasing Soluble TNFR2
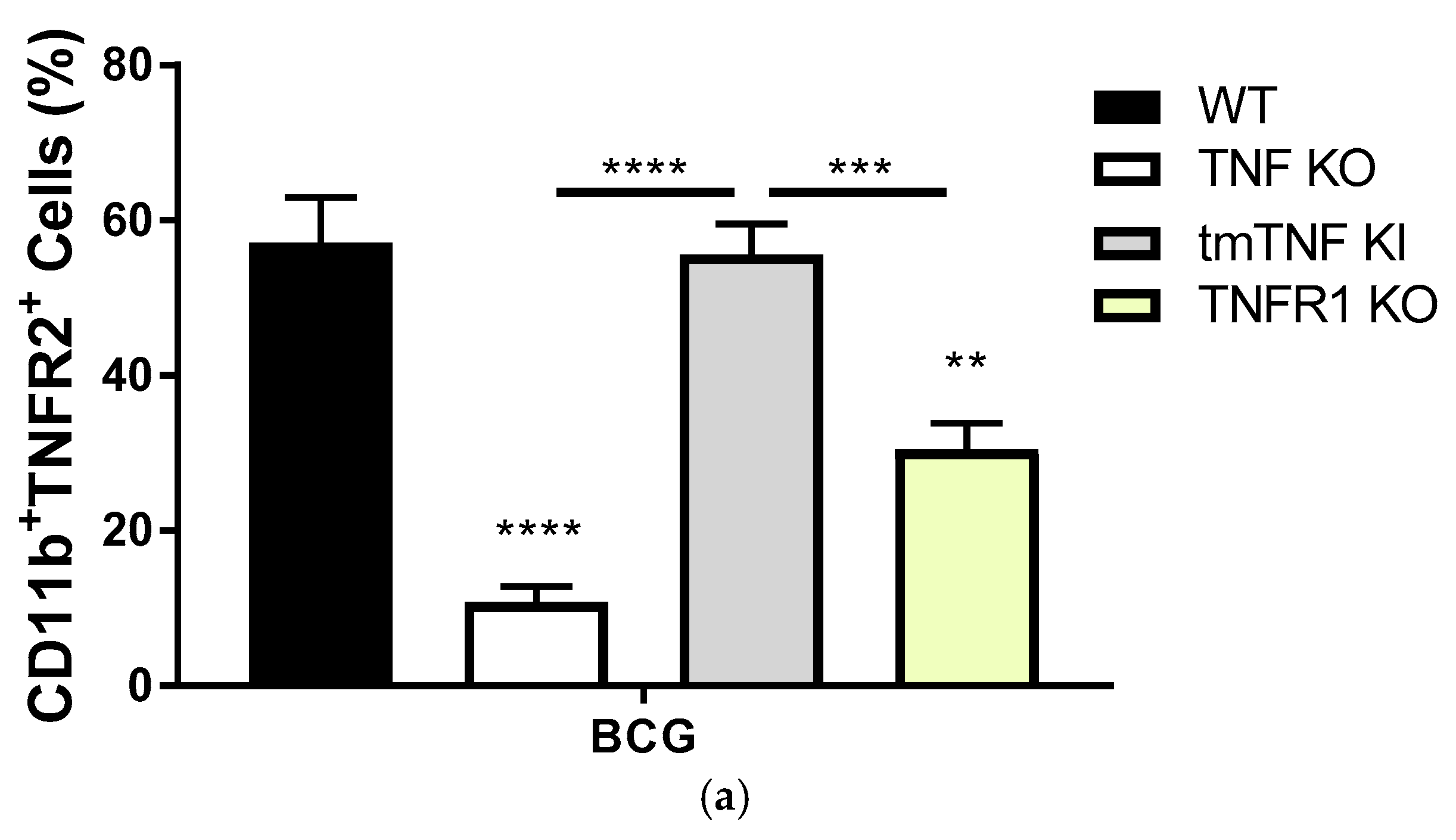
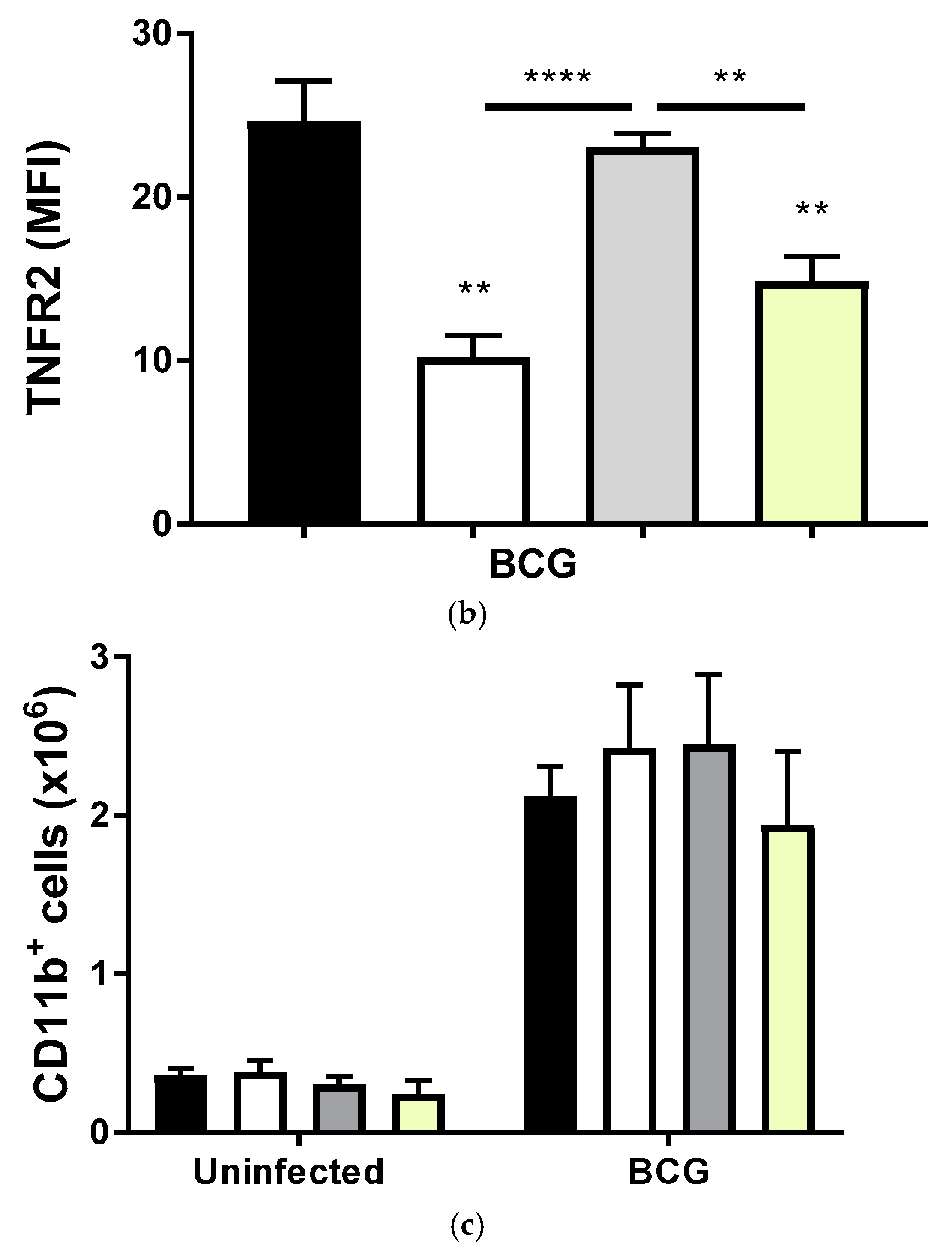
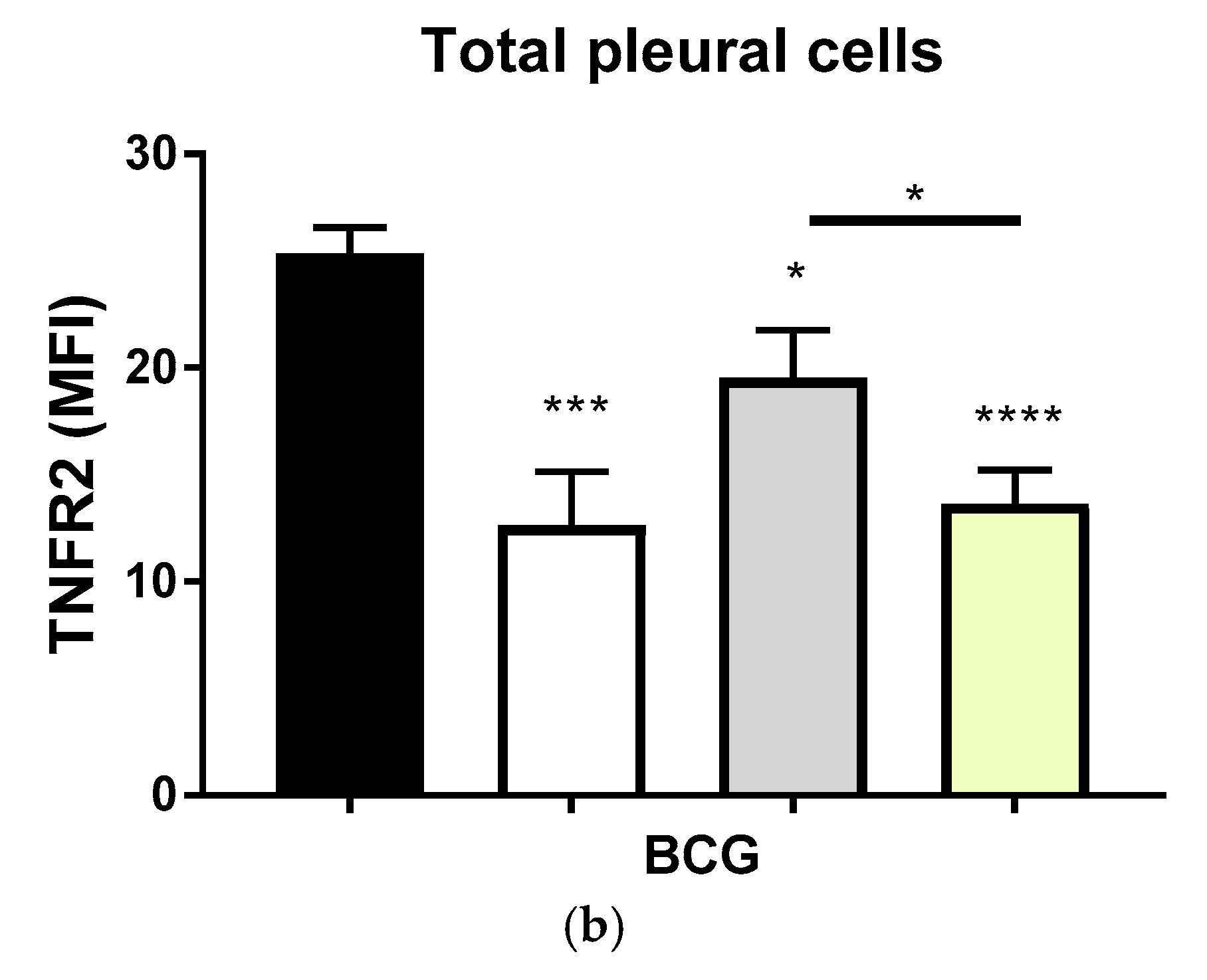
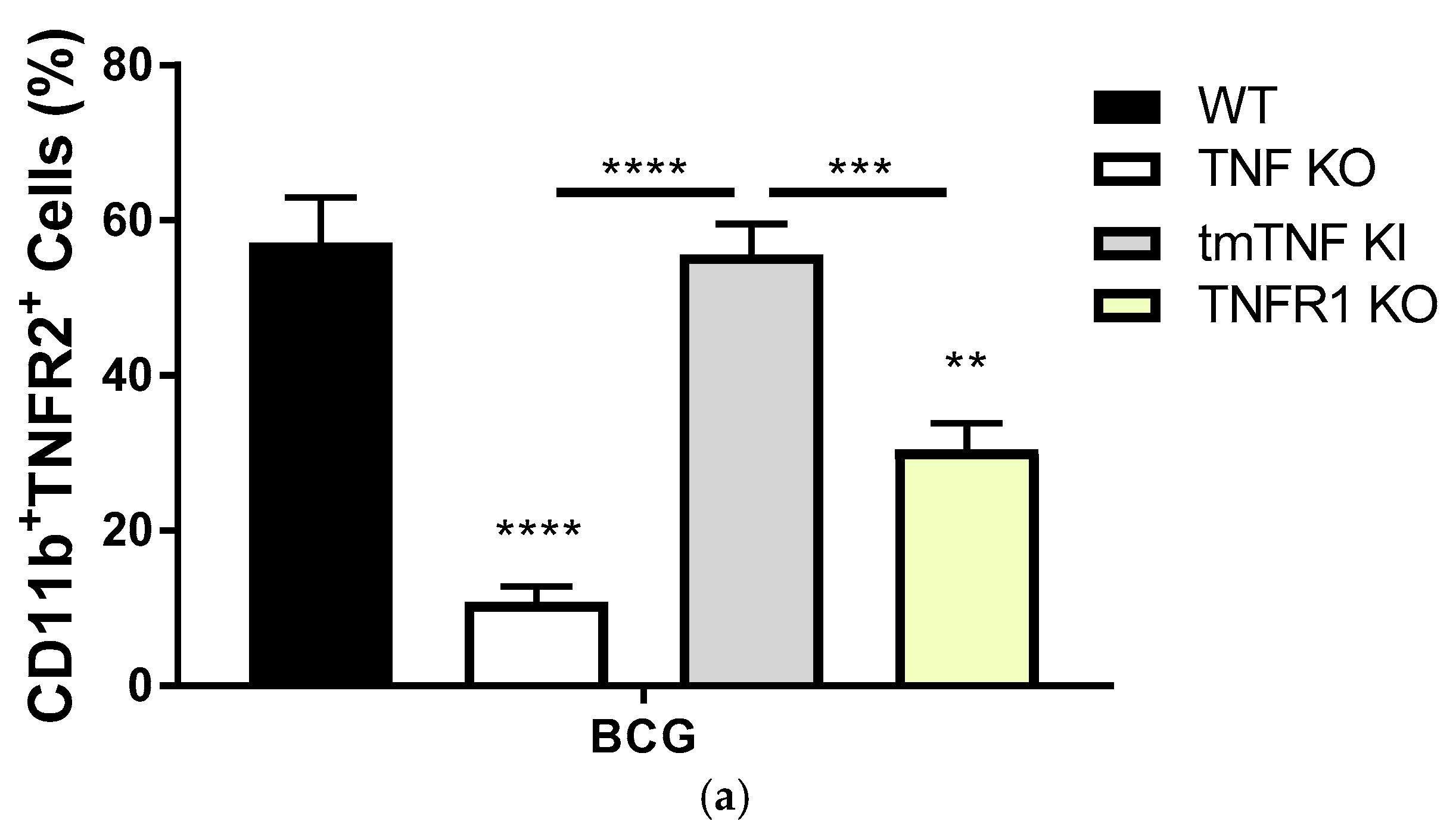
2.2. Transmembrane TNF Controls the Expression of Membrane Bound TNFR2 in Pleural Myeloid Cells
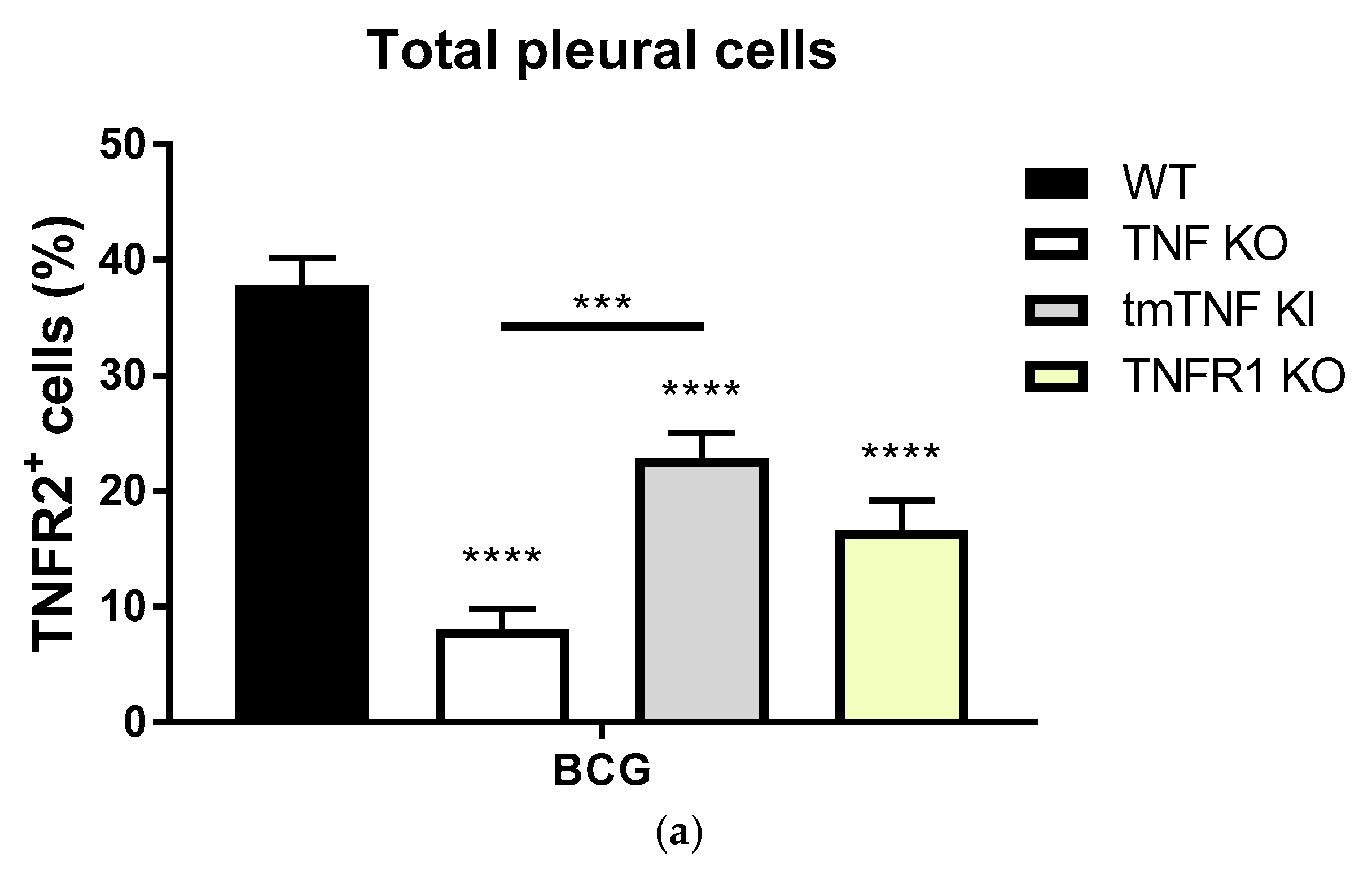
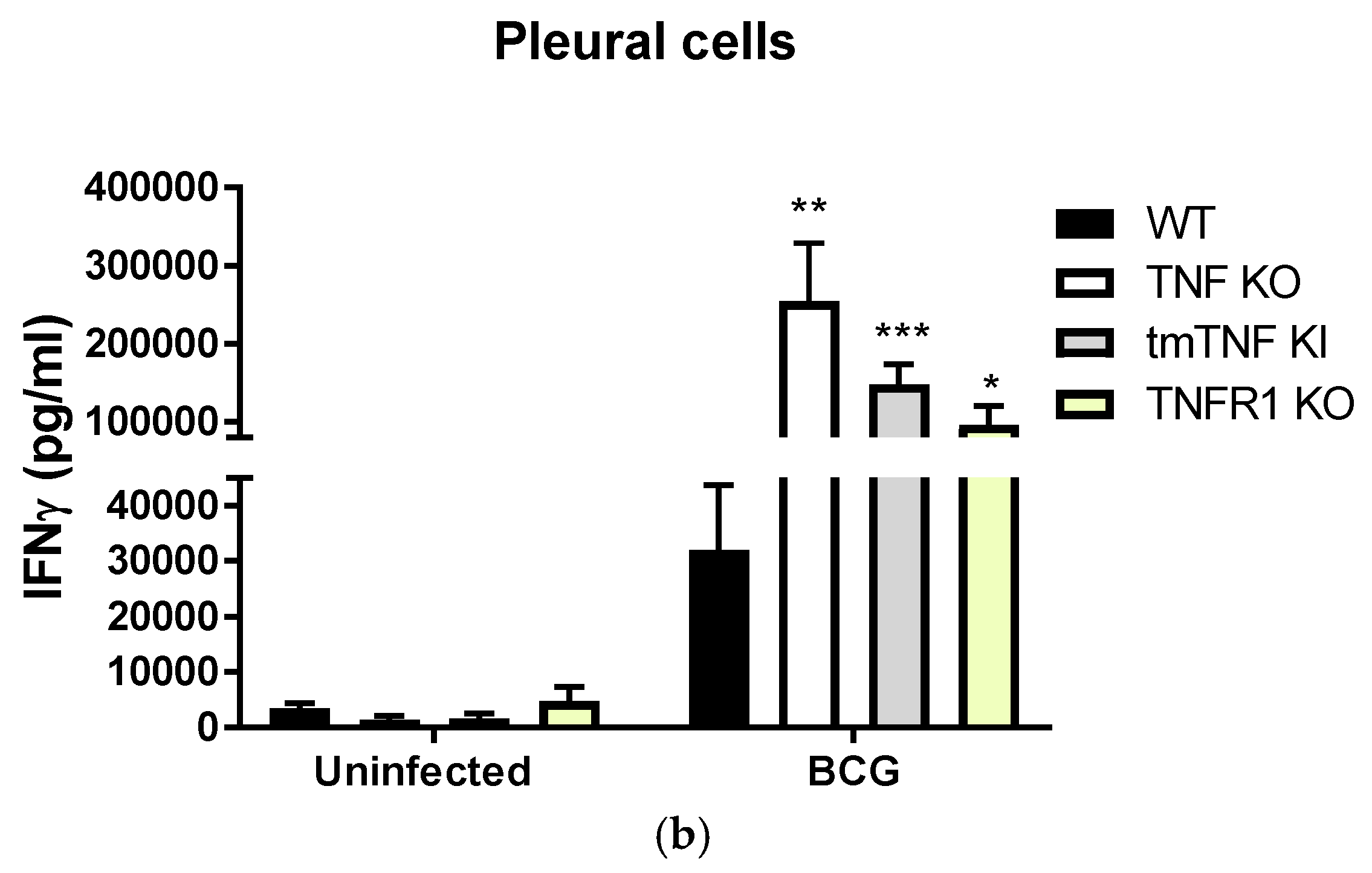
2.3. BCG-Induced Pleurisy Causes Inflammation That is Controlled by Transmembrane TNF, TNFR2 and Partially by TNFR1
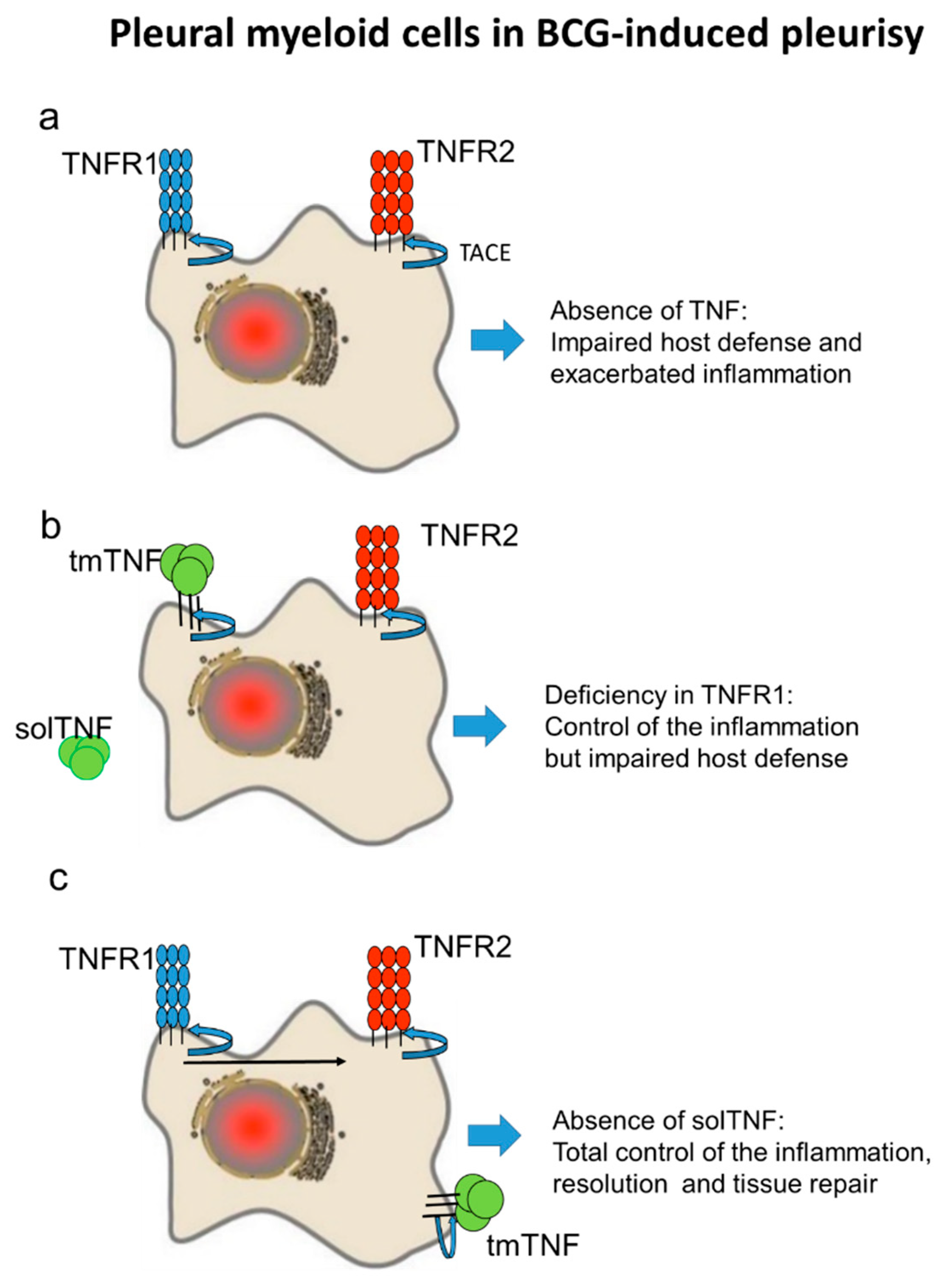
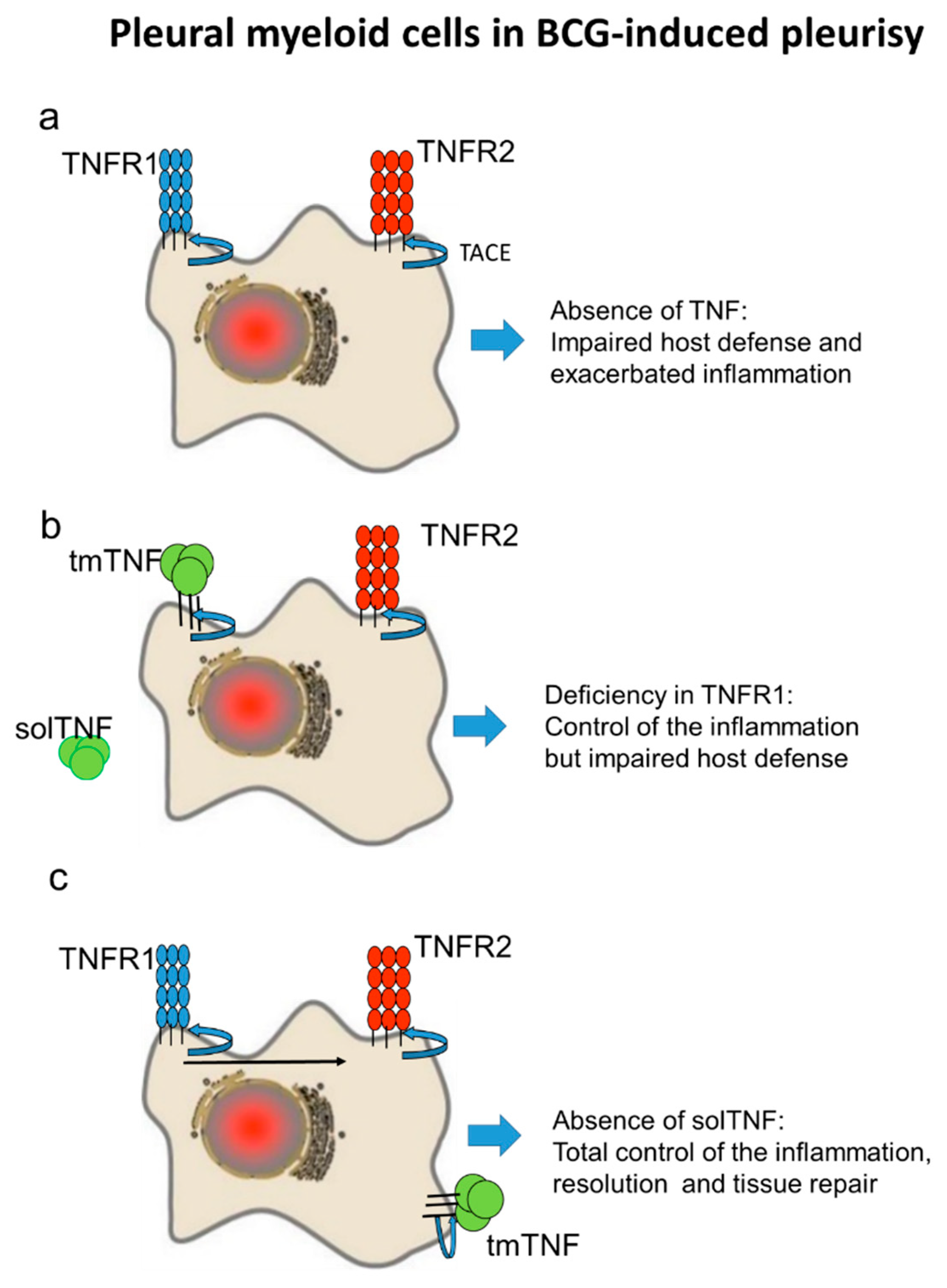
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. M. bovis BCG and Infection
4.3. Pleural Cell and Fluid Preparation
4.4. Multiparametric Flow Cytometry Analysis
4.5. Cytokine Evaluation Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. TNF Ex Vivo Stimulation of Pleural Cells
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TNF | Tumor Necrosis Factor |
| TNFR | Tumor necrosis Factor Receptor |
| tmTNF | Transmembrane TNF |
| solTNF | Soluble TNF |
| solTNFR | Soluble TNFR |
| BCG | Bacillus Calmette-Guérin |
| TACE | TNF-α-converting enzyme |
References
- Garcia, I.; Olleros, M.L.; Quesniaux, V.F.; Jacobs, M.; Allie, N.; Nedospasov, S.A.; Szymkowski, D.E.; Ryffel, B. Roles of soluble and membrane TNF and related ligands in mycobacterial infections: Effects of selective and non-selective TNF inhibitors during infection. Adv. Exp. Med. Biol. 2011, 691, 187–201. [Google Scholar] [PubMed]
- Ruddle, N.H. Lymphotoxin and TNF: How it all began-a tribute to the travelers. Cytokine Growth Factor Rev. 2014, 25, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Aderka, D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996, 7, 231–240. [Google Scholar] [CrossRef]
- Aderka, D.; Engelmann, H.; Shemer-Avni, Y.; Hornik, V.; Galil, A.; Sarov, B.; Wallach, D. Variation in serum levels of the soluble TNF receptors among healthy individuals. Lymphokine Cytokine Res. 1992, 11, 157–159. [Google Scholar] [PubMed]
- Dye, C.; Raviglione, M. Perspective: Weigh all TB risks. Nature 2013, 502, S13. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Garcia, I. The roles of tumor necrosis factor and other macrophage-derived cytokines in host defense mechanisms during the course of mycobacterium tuberculosis infection. In Current Topics on the Profiles of Host Immunological Response to Mycobacterial Infections; Singpost Special Immunol book Ed.: Tomioka, Japan, 2009. [Google Scholar]
- Roca, F.J.; Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; Syrbe, U.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeton, R.; Allie, N.; Dambuza, I.; Abel, B.; Hsu, N.-J.; Sebesho, B.; Randall, P.; Burger, P.; Fick, E.; Quesniaux, V.F.J.; et al. Soluble TNFRp75 regulates host protective immunity against Mycobacterium tuberculosis. J. Clin. Investig. 2014, 124, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segueni, N.; Benmerzoug, S.; Rose, S.; Gauthier, A.; Bourigault, M.-L.; Reverchon, F.; Philippeau, A.; Erard, F.; le Bert, M.; Bouscayrol, H.; et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci. Rep. 2016, 6, 22454. [Google Scholar] [CrossRef] [PubMed]
- Keane, J.; Gershon, S.; Wise, R.P.; Mirabile-Levens, E.; Kasznica, J.; Schwieterman, W.D.; Siegel, J.N.; Braun, M.M. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 2001, 345, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Kulchavenya, E. Extrapulmonary tuberculosis: Are statistical reports accurate? Ther. Adv. Infect. Dis. 2014, 2, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Antony, V.B. Immunological mechanisms in pleural disease. Eur. Respir. J. 2003, 21, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, A.; Gustafson, P.; Nhaga, A.; Djana, Q.; Poulsen, A.; Garly, M.-L.; Jensen, H.; Sodemann, M.; Rodriques, A.; Aaby, P. BCG vaccination scar associated with better childhood survival in Guinea-Bissau. Int. J. Epidemiol. 2005, 34, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandroff, A.B.; Jackson, A.M.; O’Donnell, M.A.; James, K. BCG immunotherapy of bladder cancer: 20 years on. Lancet 1999, 353, 1689–1694. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Vesin, D.; Segueni, N.; Prasad, P.; Buser-Llinares, R.; Blaser, G.; Pache, J.-C.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Tumor Necrosis Factor and Its Receptors Are Crucial to Control Mycobacterium Bovis Bacillus Calmette-Guerin Pleural Infection in a Murine Model. Am. J. Pathol. 2016, 186, 2364–2377. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Vesin, D.; Uysal, H.; Blaser, G.; Benkhoucha, M.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Transmembrane Tumor Necrosis Factor Controls Myeloid-Derived Suppressor Cell Activity via TNF Receptor 2 and Protects from Excessive Inflammation during BCG-Induced Pleurisy. Front. Immunol. 2017, 8, 999. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Vesin, D.; Bisig, R.; Santiago-Raber, M.L.; Schuepbach-Mallepell, S.; Kollias, G.; Gaide, O.; Garcia, I. Membrane-bound TNF induces protective immune responses to M. bovis BCG infection: Regulation of memTNF and TNF receptors comparing two memTNF molecules. PLoS ONE 2012, 7, e31469. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P.; Aderka, D.; Doherty, M.; Engelmann, H.; Gibbons, D.; Jones, A.C.; Brennan, F.M.; Maini, R.N.; Wallach, D.; Feldmann, M. Increased levels of soluble tumor necrosis factor receptors in the sera and synovial fluid of patients with rheumatic diseases. Arthritis Rheum. 1992, 35, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, I.; Guler, R.; Vesin, D.; Olleros, M.L.; Vassalli, P.; Chvatchko, Y.; Jacobs, M.; Ryffel, B. Lethal Mycobacterium bovis Bacillus Calmette Guerin infection in nitric oxide synthase 2-deficient mice: Cell-mediated immunity requires nitric oxide synthase 2. Lab. Investig. 2000, 80, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Chavez-Galan, L.; Segueni, N.; Bourigault, M.L.; Vesin, D.; Kruglov, A.A.; Drutskaya, M.S.; Bisig, R.; Ehlers, S.; Aly, S.; et al. Nedospasov, and I. Garcia, Control of Mycobacterial Infections in Mice Expressing Human Tumor Necrosis Factor (TNF) but Not Mouse TNF. Infect. Immun. 2015, 83, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, H.; Novick, D.; Wallach, D. Two tumor necrosis factor-binding proteins purified from human urine. Evidence for immunological cross-reactivity with cell surface tumor necrosis factor receptors. J. Biol. Chem. 1990, 265, 1531–1536. [Google Scholar] [PubMed]
- Balcewicz-Sablinska, M.K.; Keane, J.; Kornfeld, H.; Remold, H.G. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J. Immunol. 1998, 161, 2636–2641. [Google Scholar] [PubMed]
- Higuchi, M.; Aggarwal, B.B. TNF induces internalization of the p60 receptor and shedding of the p80 receptor. J. Immunol. 1994, 152, 3550–3558. [Google Scholar] [PubMed]
- Polz, J.; Remke, A.; Weber, S.; Schmidt, D.; Weber-Steffens, D.; Pietryga-Krieger, A.; Muller, N.; Ritter, U.; Mostbock, S.; Mannel, D.N. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun. Inflamm. Dis. 2014, 2, 121–1230. [Google Scholar] [CrossRef] [PubMed]
- Hamano, R.; Huang, J.; Yoshimura, T.; Oppenheim, J.J.; Chen, X. TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4–1BB and OX40. Eur. J. Immunol. 2011, 41, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Zganiacz, A.; Santosuosso, M.; Wang, J.; Yang, T.; Chen, L.; Anzulovic, M.; Alexander, S.; Gicquel, B.; Wan, Y.; Bramson, J.; et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J. Clin. Investig. 2004, 113, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Olleros, M.L.; Vesin, D.; Lambou, A.F.; Janssens, J.P.; Ryffel, B.; Rose, S.; Fremond, C.; Quesniaux, V.F.; Szymkowski, D.E.; Garcia, I. Dominant-negative tumor necrosis factor protects from Mycobacterium bovis Bacillus Calmette Guerin (BCG) and endotoxin-induced liver injury without compromising host immunity to BCG and Mycobacterium tuberculosis. J. Infect. Dis. 2009, 199, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Bourigault, M.-L.; Vacher, R.; Rose, S.; Olleros, M.L.; Janssens, J.P.; Quesniaux, V.F.; Garcia, I. Tumor necrosis factor neutralization combined with chemotherapy enhances Mycobacterium tuberculosis clearance and reduces lungpathology. Am. J. Clin. Exp. Immunol. 2013, 2, 124–134. [Google Scholar] [PubMed]
- Skerry, C.; Harper, J.; Klunk, M.; Bishai, W.R.; Jain, S.K. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS ONE 2012, 7, e39680. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.W.; Dunn, A.; Grail, D.; Inglese, M.; Noguchi, Y.; Richards, E.; Jungbluth, A.; Wada, H.; Moore, M.; Williamson, B.; et al. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8093–8098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruuls, S.R.; Hoek, R.M.; Ngo, V.N.; McNeil, T.; Lucian, L.A.; Janatpour, M.J.; Korner, H.; Scheerens, H.; Hessel, E.M.; Cyster, J.G.; et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity 2001, 15, 533–543. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uysal, H.; Chavez-Galan, L.; Vesin, D.; Blaser, G.; Benkhoucha, M.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Transmembrane TNF and Partially TNFR1 Regulate TNFR2 Expression and Control Inflammation in Mycobacterial-Induced Pleurisy. Int. J. Mol. Sci. 2018, 19, 1959. https://doi.org/10.3390/ijms19071959
Uysal H, Chavez-Galan L, Vesin D, Blaser G, Benkhoucha M, Ryffel B, Quesniaux VFJ, Garcia I. Transmembrane TNF and Partially TNFR1 Regulate TNFR2 Expression and Control Inflammation in Mycobacterial-Induced Pleurisy. International Journal of Molecular Sciences. 2018; 19(7):1959. https://doi.org/10.3390/ijms19071959
Chicago/Turabian StyleUysal, Husnu, Leslie Chavez-Galan, Dominique Vesin, Guillaume Blaser, Mahdia Benkhoucha, Bernhard Ryffel, Valérie F. J. Quesniaux, and Irene Garcia. 2018. "Transmembrane TNF and Partially TNFR1 Regulate TNFR2 Expression and Control Inflammation in Mycobacterial-Induced Pleurisy" International Journal of Molecular Sciences 19, no. 7: 1959. https://doi.org/10.3390/ijms19071959