Complement Activation in Liver Transplantation: Role of Donor Macrosteatosis and Implications in Delayed Graft Function


Abstract
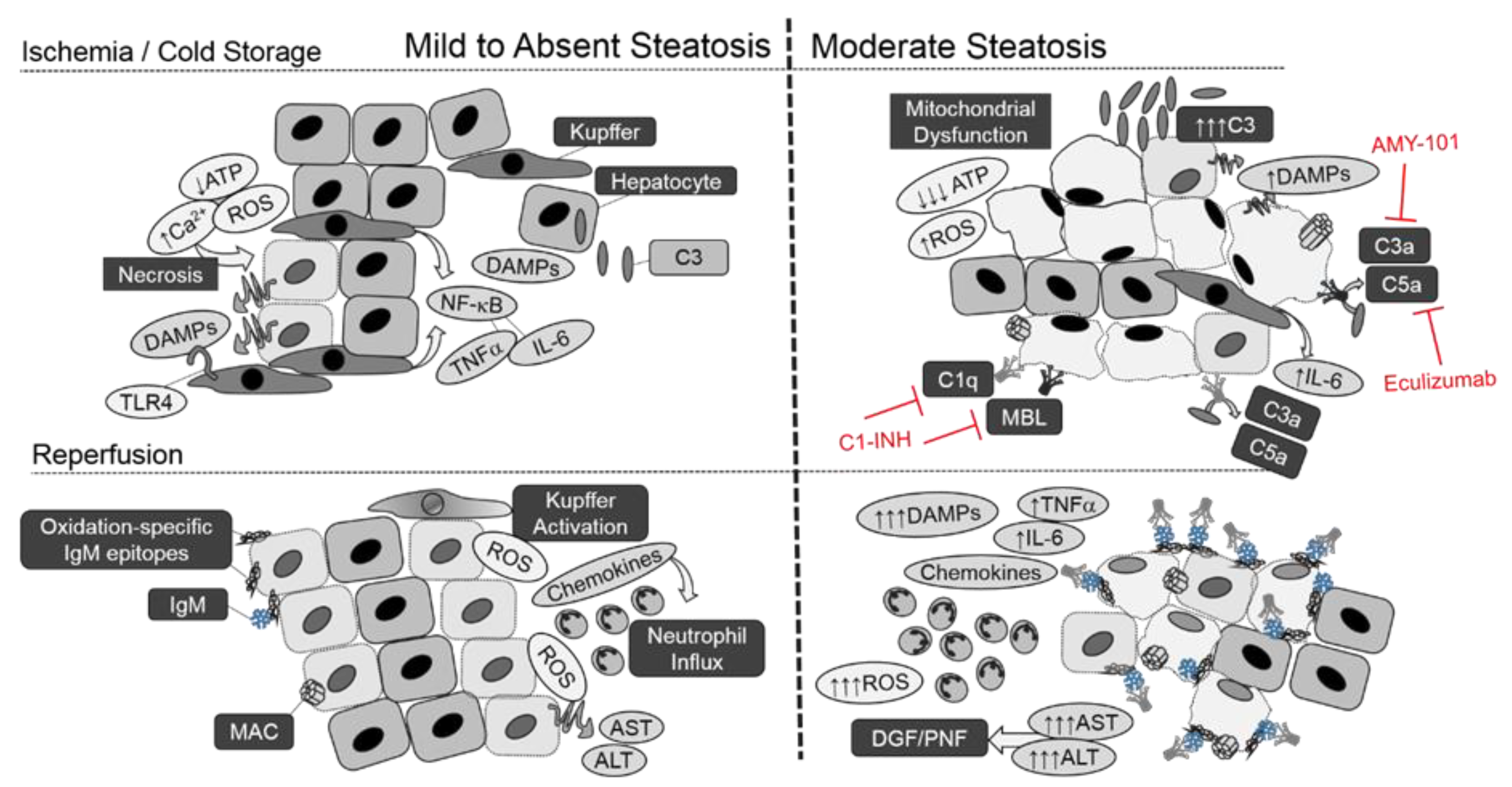
:1. Introduction
2. I/R Injury in Liver Procurement and Transplantation
2.1. Ischemic Phase—Organ Procurement and Cold Storage
2.2. Reperfusion Phase—Transplantation of the Graft
3. Mechanisms of Steatotic Graft Dysfunction after I/R Injury
3.1. Mechanisms of Increased Susceptibility to Injury
3.2. Postreperfusion Syndrome with Steatotic Grafts
4. The Complement System and Liver I/R Injury
4.1. Overview of the Complement System
4.2. Role of Complement Activation during I/R Injury
4.3. Strategies Targeting Complement Activation in I/R Injury
5. Role of Donor Liver Steatosis in Complement Activation
6. Role of Complement in Liver Donation after Brain Verses Cardiac Death
7. Steatosis in Living Donors
8. Targeting Complement Activation in Liver Transplantation
Complement-Directed Therapeutics in Liver Transplant I/R Injury
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| I/R | Ischemia/Reperfusion |
| ECD | Extended Criteria Donor |
| DGF | Delayed Graft Function |
| PNF | Primary Non-Function |
| MiS | Microsteatosis |
| MaS | Macrosteatosis |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| ROS | Reactive Oxygen Species |
| DAMP | Damage-Associated Molecular Pattern |
| HMGB1 | High-Mobility Group Protein 1 |
| TLR4 | Toll-Like Receptor 4 |
| TNFα | Tumor Necrosis Factor α |
| iNOS | Inducible Nitric Oxide Synthase |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| HO-1 | Heme-Oxygenase 1 |
| Nrf2 | Nuclear Factor Erythroid 2 |
| PPAR | Proliferator-Activated Receptor |
| Ig | Immunoglobulin |
| AST | Aspartate Aminotransferase |
| ALT | Alanine Aminotransferase |
| OLT | Orthotopic Liver Transplantation |
| MAC | Membrane Attack Complex |
| MBL | Mannose-Binding Lectin |
| DBD | Donation After Brain Death |
| DCD | Donation After Cardiac Death |
| NASH | Non-alcoholic Steatohepatitis |
References
- Kim, W.R.; Lake, J.R.; Smith, J.M.; Schladt, D.P.; Skeans, M.A.; Harper, A.M.; Wainright, J.L.; Snyder, J.J.; Israni, A.K.; Kasiske, B.L. OPTN/SRTR 2016 annual data report: Liver. Am. J. Transplant. 2018, 18, 172–253. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.W.; Tanaka, K. The utility of marginal donors in liver transplantation. Liver Transplant. 2003, 9, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, J.F.; Kin, C.; Kinkhabwala, M.; Jan, D.; Varadarajan, R.; Goldstein, M.; Brown, R., Jr.; Emond, J.C. Utilization of extended donor criteria liver allografts maximizes donor use and patient access to liver transplantation. Ann. Surg. 2005, 242, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.E.; Kreke, J.E.; Malek, F.A.; Schaefer, A.J.; Vacanti, J. Consequences of cold-ischemia time on primary nonfunction and patient and graft survival in liver transplantation: A meta-analysis. PLoS ONE 2008, 3, e2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, M.J.; Dare, A.J.; Phillips, A.R.; Bartlett, A.S. Donor hepatic steatosis and outcome after liver transplantation: A systematic review. J. Gastrointest. Surg. 2015, 19, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Dimou, F.M.; Mehta, H.B.; Adhikari, D.; Harland, R.C.; Riall, T.S.; Kuo, Y.F. The role of extended criteria donors in liver transplantation for nonalcoholic steatohepatitis. Surgery 2016, 160, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.C.; Feng, S.; Roberts, J.P. An examination of liver offers to candidates on the liver transplant wait-list. Gastroenterology 2012, 143, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.C.; Fung, J.Y.; Chok, K.S.; Cheung, T.T.; Chan, A.C.; Sharr, W.W.; Dai, W.C.; Chan, S.C.; Lo, C.M. Excellent outcomes of liver transplantation using severely steatotic grafts from brain-dead donors. Liver Transplant. 2016, 22, 226–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavin, K.D.; Taber, D.J.; Norcross, M.; Pilch, N.A.; Crego, H.; McGillicuddy, J.W.; Bratton, C.F.; Lin, A.; Baliga, P.K. Safe use of highly steatotic livers by utilizing a donor/recipient clinical algorithm. Clin. Transplant. 2013, 27, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Dutkowski, P.; Schlegel, A.; Slankamenac, K.; Oberkofler, C.E.; Adam, R.; Burroughs, A.K.; Schadde, E.; Mullhaupt, B.; Clavien, P.A. The use of fatty liver grafts in modern allocation systems: Risk assessment by the balance of risk (BAR) score. Ann. Surg. 2012, 256, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Escartin, A.; Castro, E.; Dopazo, C.; Bueno, J.; Bilbao, I.; Margarit, C. Analysis of discarded livers for transplantation. Transplant. Proc. 2005, 37, 3859–3860. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, N.; Shiffman, M.L. Impact of the donor liver with steatosis in patients with hepatitis c virus: Not so fast. Liver Transplant. 2009, 15, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Nativ, N.I.; Maguire, T.J.; Yarmush, G.; Brasaemle, D.L.; Henry, S.D.; Guarrera, J.V.; Berthiaume, F.; Yarmush, M.L. Liver defatting: An alternative approach to enable steatotic liver transplantation. Am. J. Transplant. 2012, 12, 3176–3183. [Google Scholar] [CrossRef] [PubMed]
- Perez-Daga, J.A.; Santoyo, J.; Suarez, M.A.; Fernandez-Aguilar, J.A.; Ramirez, C.; Rodriguez-Canete, A.; Aranda, J.M.; Sanchez-Perez, B.; Montiel, C.; Palomo, D.; et al. Influence of degree of hepatic steatosis on graft function and postoperative complications of liver transplantation. Transplant. Proc. 2006, 38, 2468–2470. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, E.L.; Kench, J.; Dilworth, P.; Shackel, N.A.; Strasser, S.I.; Joseph, D.; Pleass, H.; Crawford, M.; McCaughan, G.W.; Verran, D.J. Grade of deceased donor liver macrovesicular steatosis impacts graft and recipient outcomes more than the donor risk index. J. Gastroenterol. Hepatol. 2012, 27, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, M.; Moisan, F.; Vidal, M.; Duarte, I.; Jimenez, M.; Izquierdo, G.; Dominguez, P.; Mendez, J.; Soza, A.; Benitez, C.; et al. Steatotic livers. Can we use them in OLTX? Outcome data from a prospective baseline liver biopsy study. Ann. Hepatol. 2012, 11, 891–898. [Google Scholar] [PubMed]
- Frongillo, F.; Avolio, A.W.; Nure, E.; Mule, A.; Pepe, G.; Magalini, S.C.; Agnes, S. Quantification of degree of steatosis in extended criteria donor grafts with standardized histologic techniques: Implications for graft survival. Transplant. Proc. 2009, 41, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, H.; Gruenberger, T.; Soliman, T.; Rockenschaub, S.; Langle, F.; Steininger, R. Organ survival after primary dysfunction of liver grafts in clinical orthotopic liver transplantation. Transpl. Int. 2000, 13, S154–S157. [Google Scholar] [CrossRef] [PubMed]
- Todo, S.; Demetris, A.J.; Makowka, L.; Teperman, L.; Podesta, L.; Shaver, T.; Tzakis, A.; Starzl, T.E. Primary nonfunction of hepatic allografts with preexisting fatty infiltration. Transplantation 1989, 47, 903–905. [Google Scholar] [CrossRef] [PubMed]
- McCormack, L.; Petrowsky, H.; Jochum, W.; Mullhaupt, B.; Weber, M.; Clavien, P.A. Use of severely steatotic grafts in liver transplantation: A matched case-control study. Ann. Surg. 2007, 246, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Xu, X.; Ling, Q.; Wu, J.; Zhou, L.; Xie, H.Y.; Wang, H.P.; Zheng, S.S. Efficacy and safety of moderately steatotic donor liver in transplantation. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 29–33. [Google Scholar] [PubMed]
- Avolio, A.W.; Frongillo, F.; Nicolotti, N.; Mule, A.; Vennarecci, G.; De Simone, P.; Agnes, S. Successful use of extended criteria donor grafts with low to moderate steatosis in patients with model for end-stage liver disease scores below 27. Transplant. Proc. 2009, 41, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.F.; Schwabauer, E.; Martus, P.; Videv, N.; Pratschke, J.; Malinowski, M.; Neuhaus, P.; Stockmann, M. Early diagnosis of primary nonfunction and indication for reoperation after liver transplantation. Liver Transplant. 2010, 16, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch-Puy, E.; Panisello, A.; Oliva, J.; Lopez, A.; Castro Benitez, C.; Adam, R.; Rosello-Catafau, J. Relevance of endoplasmic reticulum stress cell signaling in liver cold ischemia reperfusion injury. Int. J. Mol. Sci. 2016, 17, 807. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am. J. Transplant. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Zhang, Y.; Loughran, P.A.; Wang, Q.; Tsung, A.; Billiar, T.R. TIFA upregulation after hypoxia-reoxygenation is TLR4- and MyD88-dependent and associated with HMGB1 upregulation and release. Free Radic. Biol. Med. 2013, 63, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Shen, X.D.; O’Connell, R.; Gao, F.; Lassman, C.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J. Immunol. 2004, 173, 7115–7119. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.S.; Zhang, J.X.; Wang, L.; Tian, Y.; Wang, H.; Rotstein, O. Toll-like receptor 4 involvement in hepatic ischemia/reperfusion injury in mice. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 250–253. [Google Scholar] [PubMed]
- Nace, G.W.; Huang, H.; Klune, J.R.; Eid, R.E.; Rosborough, B.R.; Korff, S.; Li, S.; Shapiro, R.A.; Stolz, D.B.; Sodhi, C.P.; et al. Cellular-specific role of toll-like receptor 4 in hepatic ischemia-reperfusion injury in mice. Hepatology 2013, 58, 374–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. Hmgb1 release induced by liver ischemia involves toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Nace, G.W.; McDonald, K.A.; Tai, S.; Klune, J.R.; Rosborough, B.R.; Ding, Q.; Loughran, P.; Zhu, X.; Beer-Stolz, D.; et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014, 59, 1984–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Lou, C.; Wang, W. Stim1 deficiency protects the liver from ischemia/reperfusion injury in mice. Biochem. Biophys. Res. Commun. 2018, 496, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Pan, G.; Thobe, B.M.; Choudhry, M.A.; Matsutani, T.; Samy, T.S.; Kang, S.C.; Bland, K.I.; Chaudry, I.H. MyD88 and SRC are differentially regulated in Kupffer cells of males and proestrus females following hypoxia. Mol. Med. 2006, 12, 65–73. [Google Scholar] [PubMed]
- Jaeschke, H.; Lemasters, J.J. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 2003, 125, 1246–1257. [Google Scholar] [CrossRef]
- Kim, J.S.; Qian, T.; Lemasters, J.J. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology 2003, 124, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Andrukhiv, A.; Costa, A.D.; West, I.C.; Garlid, K.D. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2067–H2074. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Farhood, A.; Bautista, A.P.; Spolarics, Z.; Spitzer, J.J. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am. J. Physiol. 1993, 264, G801–G809. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; McGuire, G.M.; Fisher, M.A.; Farhood, A.; Smith, C.W.; Jaeschke, H. Activation of Kupffer cells and neutrophils for reactive oxygen formation is responsible for endotoxin-enhanced liver injury after hepatic ischemia. Shock 1995, 3, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Farhood, A.; Smith, C.W. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J. 1990, 4, 3355–3359. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Bautista, A.P.; Spolarics, Z.; Spitzer, J.J. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic. Res. Commun. 1991, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Clemens, M.G. Kupffer cell exacerbation of hepatocyte hypoxia/reoxygenation injury. Circ. Shock 1992, 37, 245–252. [Google Scholar] [PubMed]
- Von Frankenberg, M.; Golling, M.; Mehrabi, A.; Nentwich, H.; Klar, E.; Kraus, T.W. Donor pretreatment with gadolinium chloride improves early graft function and survival after porcine liver transplantation. Transpl. Int. 2003, 16, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Prats, N.; Xaus, C.; Gelpi, E.; Rosello-Catafau, J. Protective effect of liver ischemic preconditioning on liver and lung injury induced by hepatic ischemia-reperfusion in the rat. Hepatology 1999, 30, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, N.Y.; Zhou, W.; Li, Q.; Zhang, Y.; Luo, M.; Yan, Z.; Lynch, T.J.; Abbott, D.; Banfi, B.; Engelhardt, J.F. Hepatocytes produce TNFα following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G84–G94. [Google Scholar] [CrossRef] [PubMed]
- Conzelmann, L.O.; Lehnert, M.; Kremer, M.; Zhong, Z.; Wheeler, M.D.; Lemasters, J.J. Graft tumor necrosis factor receptor-1 protects after mouse liver transplantation whereas host tumor necrosis factor receptor-1 promotes injury. Transplantation 2006, 82, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Frankenberg, M.V.; Weimann, J.; Fritz, S.; Fiedler, J.; Mehrabi, A.; Buchler, M.W.; Kraus, T.W. Gadolinium chloride-induced improvement of postischemic hepatic perfusion after warm ischemia is associated with reduced hepatic endothelin secretion. Transpl. Int. 2005, 18, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlmann, D.; Gaebel, G.; Armann, B.; Ludwig, S.; Hess, J.; Pietsch, U.C.; Fiedler, M.; Tannapfel, A.; Hauss, J.; Witzigmann, H. Attenuation of proinflammatory gene expression and microcirculatory disturbances by endothelin a receptor blockade after orthotopic liver transplantation in pigs. Surgery 2006, 139, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Liaw, J.M.; Jia, J.; Glasgow, S.C.; Liu, W.; Csontos, K.; Upadhya, G.A.; Mohanakumar, T.; Chapman, W.C. Ischemia-reperfusion injury in rat steatotic liver is dependent on NFκB P65 activation. Transpl. Immunol. 2012, 26, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Guo, Z.; Deng, W.; Fu, S.; Zhang, C.; Chen, M.; Ju, W.; Wang, D.; He, X. Calpain 2-mediated autophagy defect increases susceptibility of fatty livers to ischemia-reperfusion injury. Cell Death Dis. 2016, 7, e2186. [Google Scholar] [CrossRef] [PubMed]
- Gehrau, R.C.; Mas, V.R.; Dumur, C.I.; Suh, J.L.; Sharma, A.K.; Cathro, H.P.; Maluf, D.G. Donor hepatic steatosis induce exacerbated ischemia-reperfusion injury through activation of innate immune response molecular pathways. Transplantation 2015, 99, 2523–2533. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Caraceni, P.; Caraccio, G.; Domenicali, M.; Dall’Agata, M.; Trevisani, F.; Guerrieri, F.; Bernardi, M.; Altomare, E. Mitochondrial oxidative injury and energy metabolism alteration in rat fatty liver: Effect of the nutritional status. Hepatology 2001, 33, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraceni, P.; Domenicali, M.; Vendemiale, G.; Grattagliano, I.; Pertosa, A.; Nardo, B.; Morselli-Labate, A.M.; Trevisani, F.; Palasciano, G.; Altomare, E.; et al. The reduced tolerance of rat fatty liver to ischemia reperfusion is associated with mitochondrial oxidative injury. J. Surg. Res. 2005, 124, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Caraceni, P.; Bianchi, C.; Domenicali, M.; Maria Pertosa, A.; Maiolini, E.; Parenti Castelli, G.; Nardo, B.; Trevisani, F.; Lenaz, G.; Bernardi, M. Impairment of mitochondrial oxidative phosphorylation in rat fatty liver exposed to preservation-reperfusion injury. J. Hepatol. 2004, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, F.; Barbe, L.; Mokuno, Y.; MacDonald, A.D.; Jindal, R.; Yarmush, M.L. Steatosis reversibly increases hepatocyte sensitivity to hypoxia-reoxygenation injury. J. Surg. Res. 2009, 152, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, M.J.; Hickey, A.J.; Jiang, Y.; Petzer, A.; Bartlett, A.S.; Phillips, A.R. Mitochondrial dysfunction in steatotic rat livers occurs because a defect in complex I makes the liver susceptible to prolonged cold ischemia. Liver Transplant. 2015, 21, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Nativ, N.I.; Yarmush, G.; So, A.; Barminko, J.; Maguire, T.J.; Schloss, R.; Berthiaume, F.; Yarmush, M.L. Elevated sensitivity of macrosteatotic hepatocytes to hypoxia/reoxygenation stress is reversed by a novel defatting protocol. Liver Transplant. 2014, 20, 1000–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliassotti, M.J. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2012, 32, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Bozaykut, P.; Sahin, A.; Karademir, B.; Ozer, N.K. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic steatohepatitis. Mech. Ageing Dev. 2016, 157, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Tiriveedhi, V.; Conzen, K.D.; Liaw-Conlin, J.; Upadhya, G.; Malone, J.; Townsend, R.R.; Kerns, R.; Jia, J.; Csontos, K.; Ramachandran, S.; et al. The role of molecular chaperonins in warm ischemia and reperfusion injury in the steatotic liver: A proteomic study. BMC Biochem. 2012, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Ben Mosbah, I.; Alfany-Fernandez, I.; Martel, C.; Zaouali, M.A.; Bintanel-Morcillo, M.; Rimola, A.; Rodes, J.; Brenner, C.; Rosello-Catafau, J.; Peralta, C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010, 1, e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.D.; Upadhya, G.; Conzen, K.D.; Jia, J.; Brunt, E.M.; Tiriveedhi, V.; Xie, Y.; Ramachandran, S.; Mohanakumar, T.; Davidson, N.O.; et al. Endoplasmic reticulum stress is a mediator of posttransplant injury in severely steatotic liver allografts. Liver Transplant. 2011, 17, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias-Miro, M.; Jimenez-Castro, M.B.; Mendes-Braz, M.; Casillas-Ramirez, A.; Peralta, C. The current knowledge of the role of PPAR in hepatic ischemia-reperfusion injury. PPAR Res. 2012, 2012, 802384. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Atkinson, C.; Evans, Z.; Ellett, J.D.; Southwood, M.; Elvington, A.; Chavin, K.D.; Tomlinson, S. A role for complement in the enhanced susceptibility of steatotic livers to ischemia and reperfusion injury. J. Immunol. 2009, 183, 4764–4772. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Shuai, X.R.; Yan, M.L.; Zhang, M.M.; Yan, L.N. Nuclear factor-κB decoy oligodeoxynucleotides attenuates ischemia/reperfusion injury in rat liver graft. World J. Gastroenterol. 2005, 11, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Ellett, J.D.; Evans, Z.P.; Atkinson, C.; Schmidt, M.G.; Schnellmann, R.G.; Chavin, K.D. Toll-like receptor 4 is a key mediator of murine steatotic liver warm ischemia/reperfusion injury. Liver Transplant. 2009, 15, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowicka, B.; Akar, R.A.; Olszewska, A.; Smoter, P.; Krawczyk, M. The occurrence of postreperfusion syndrome in orthotopic liver transplantation and its significance in terms of complications and short-term survival. Ann. Transplant. 2011, 16, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Siniscalchi, A.; Gamberini, L.; Laici, C.; Bardi, T.; Ercolani, G.; Lorenzini, L.; Faenza, S. Post reperfusion syndrome during liver transplantation: From pathophysiology to therapy and preventive strategies. World J. Gastroenterol. 2016, 22, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M. Postreperfusion syndrome during liver transplantation. Korean J. Anesthesiol. 2015, 68, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.S.; Kim, H.Y.; Shin, Y.H.; Ko, J.S.; Gwak, M.S.; Sim, W.S.; Kim, G.S.; Lee, S.K. Incidence and predictors of post-reperfusion syndrome in living donor liver transplantation. Clin. Transplant. 2012, 26, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.; Oppermann, M.; Czech, W.; Dobos, G.; Schopf, E.; Norgauer, J.; Kapp, A. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur. J. Immunol. 1994, 24, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ehrengruber, M.U.; Geiser, T.; Deranleau, D.A. Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett. 1994, 346, 181–184. [Google Scholar] [PubMed]
- Elsner, J.; Oppermann, M.; Czech, W.; Kapp, A. C3a activates the respiratory burst in human polymorphonuclear neutrophilic leukocytes via pertussis toxin-sensitive G-proteins. Blood 1994, 83, 3324–3331. [Google Scholar] [PubMed]
- Chenoweth, D.E.; Goodman, M.G. The C5a receptor of neutrophils and macrophages. Agents Actions Suppl. 1983, 12, 252–273. [Google Scholar] [PubMed]
- Chenoweth, D.E.; Hugli, T.E. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA 1978, 75, 3943–3947. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.; Larregina, A.; Chuluyan, I.; Kolkowski, E.; Fainboim, L. Expression and modulation of C5a receptor (CD88) on skin dendritic cells. Chemotactic effect of C5a on skin migratory dendritic cells. Immunology 1996, 89, 126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glovsky, M.M.; Hugli, T.E.; Ishizaka, T.; Lichtenstein, L.M.; Erickson, B.W. Anaphylatoxin-induced histamine release with human leukocytes: Studies of C3a leukocyte binding and histamine release. J. Clin. Investig. 1979, 64, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Lisewski, M.; Zwirner, J.; Mommert, S.; Diesel, C.; Wittmann, M.; Kapp, A.; Werfel, T. Human monocyte-derived dendritic cells are chemoattracted to C3a after up-regulation of the C3a receptor with interferons. Immunology 2004, 111, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayyazi, A.; Scheel, O.; Werfel, T.; Schweyer, S.; Oppermann, M.; Gotze, O.; Radzun, H.J.; Zwirner, J. The C5a receptor is expressed in normal renal proximal tubular but not in normal pulmonary or hepatic epithelial cells. Immunology 2000, 99, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasque, P.; Singhrao, S.K.; Neal, J.W.; Gotze, O.; Morgan, B.P. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am. J. Pathol. 1997, 150, 31–41. [Google Scholar] [PubMed]
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003, 17, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Langkabel, P.; Zwirner, J.; Oppermann, M. Ligand-induced phosphorylation of anaphylatoxin receptors C3aR and C5aR is mediated by “G protein-coupled receptor kinases. Eur. J. Immunol. 1999, 29, 3035–3046. [Google Scholar] [CrossRef]
- Takabayashi, T.; Vannier, E.; Burke, J.F.; Tompkins, R.G.; Gelfand, J.A.; Clark, B.D. Both C3a and C3a(desArg) regulate interleukin-6 synthesis in human peripheral blood mononuclear cells. J. Infect. Dis. 1998, 177, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Vannier, E.; Clark, B.D.; Margolis, N.H.; Dinarello, C.A.; Burke, J.F.; Gelfand, J.A. A new biologic role for C3a and C3a desArg: Regulation of TNF-α and IL-1β synthesis. J. Immunol. 1996, 156, 3455–3460. [Google Scholar] [PubMed]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Kohl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Bamberg, C.E.; Mackay, C.R.; Lee, H.; Zahra, D.; Jackson, J.; Lim, Y.S.; Whitfeld, P.L.; Craig, S.; Corsini, E.; Lu, B.; et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J. Biol. Chem. 2010, 285, 7633–7644. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.E., 3rd; Harrison, R.A. Structural characterization of factor I mediated cleavage of the third component of complement. Biochemistry 1982, 21, 5745–5749. [Google Scholar] [CrossRef] [PubMed]
- Camous, L.; Roumenina, L.; Bigot, S.; Brachemi, S.; Fremeaux-Bacchi, V.; Lesavre, P.; Halbwachs-Mecarelli, L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood 2011, 117, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, M.; Jozsi, M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J. Biol. Chem. 2012, 287, 19528–19536. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Cruz, M.A.; Zhang, H.; Lopez, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klickstein, L.B.; Bartow, T.J.; Miletic, V.; Rabson, L.D.; Smith, J.A.; Fearon, D.T. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J. Exp. Med. 1988, 168, 1699–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangburn, M.K.; Rawal, N. Structure and function of complement C5 convertase enzymes. Biochem. Soc. Trans. 2002, 30, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Rawal, N.; Pangburn, M.K. Formation of high affinity C5 convertase of the classical pathway of complement. J. Biol. Chem. 2003, 278, 38476–38483. [Google Scholar] [CrossRef] [PubMed]
- Scoazec, J.Y.; Borghi-Scoazec, G.; Durand, F.; Bernuau, J.; Pham, B.N.; Belghiti, J.; Feldmann, G.; Degott, C. Complement activation after ischemia-reperfusion in human liver allografts: Incidence and pathophysiological relevance. Gastroenterology 1997, 112, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Fondevila, C.; Shen, X.D.; Tsuchihashi, S.; Uchida, Y.; Freitas, M.C.; Ke, B.; Busuttil, R.W.; Kupiec-Weglinski, J.W. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury. Liver Transplant. 2008, 14, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straatsburg, I.H.; Boermeester, M.A.; Wolbink, G.J.; van Gulik, T.M.; Gouma, D.J.; Frederiks, W.M.; Hack, C.E. Complement activation induced by ischemia-reperfusion in humans: A study in patients undergoing partial hepatectomy. J. Hepatol. 2000, 32, 783–791. [Google Scholar] [CrossRef]
- Heijnen, B.H.; Straatsburg, I.H.; Padilla, N.D.; Van Mierlo, G.J.; Hack, C.E.; Van Gulik, T.M. Inhibition of classical complement activation attenuates liver ischaemia and reperfusion injury in a rat model. Clin. Exp. Immunol. 2006, 143, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diepenhorst, G.M.; de Graaf, W.; Niessen, H.W.; van Vliet, A.K.; Hack, C.E.; van Gulik, T.M. Immunoglobulin M, C-reactive protein and complement activation in rat hepatic ischemia-reperfusion injury. Eur. Surg. Res. 2014, 52, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Llacuna, L.; Mari, M.; Lluis, J.M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Morales, A. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-κB inactivation in prolonged ischemia/reperfusion. Am. J. Pathol. 2009, 174, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.; Walter, S.J.; Peutz-Kootstra, C.J.; Wolfs, T.G.; van Heurn, L.W.; Buurman, W.A. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am. J. Pathol. 2004, 165, 1677–1688. [Google Scholar] [CrossRef]
- Berger, S.P.; Daha, M.R. Emerging role of the mannose-binding lectin-dependent pathway of complement activation in clinical organ transplantation. Curr. Opin. Organ. Transplant. 2011, 16, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Raaschou-Jensen, N.; Roos, A.; Daha, M.R.; Madsen, H.O.; Borrias-Essers, M.C.; Ryder, L.P.; Koch, C.; Garred, P. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur. J. Immunol. 2003, 33, 2853–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hu, W.; Xing, W.; You, T.; Xu, J.; Qin, X.; Peng, Z. The protective role of CD59 and pathogenic role of complement in hepatic ischemia and reperfusion injury. Am. J. Pathol. 2011, 179, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschini, L.; Gobbo, G.; Gatti, S.; Caccamo, L.; Prato, P.; Maggioni, M.; Braidotti, P.; Di Stefano, R.; Fassati, L.R. Endothelial targeting with C1-inhibitor reduces complement activation in vitro and during ex vivo reperfusion of pig liver. Clin. Exp. Immunol. 2001, 126, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, T.G.; Heger, M.; Munch, S.; Kirschfink, M.; Klar, E. In vivo microscopy reveals that complement inhibition by C1-esterase inhibitor reduces ischemia/reperfusion injury in the liver. Transpl. Int. 2000, 13 (Suppl. 1), S547–S550. [Google Scholar] [CrossRef] [PubMed]
- Saidi, R.F.; Rajeshkumar, B.; Shariftabrizi, A.; Dresser, K.; Walter, O. Human C1 inhibitor attenuates liver ischemia-reperfusion injury and promotes liver regeneration. J. Surg. Res. 2014, 187, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Weisman, H.F.; Bartow, T.; Leppo, M.K.; Marsh, H.C., Jr.; Carson, G.R.; Concino, M.F.; Boyle, M.P.; Roux, K.H.; Weisfeldt, M.L.; Fearon, D.T. Soluble human complement receptor type 1: In vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990, 249, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Lindsay, T.F.; Ortiz, F.; Yeh, C.G.; Hechtman, H.B.; Moore, F.D., Jr. Soluble complement receptor type 1 ameliorates the local and remote organ injury after intestinal ischemia-reperfusion in the rat. J. Immunol. 1992, 149, 1723–1728. [Google Scholar] [PubMed]
- Lehmann, T.G.; Koeppel, T.A.; Munch, S.; Heger, M.; Kirschfink, M.; Klar, E.; Post, S. Impact of inhibition of complement by sCR1 on hepatic microcirculation after warm ischemia. Microvasc. Res. 2001, 62, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, T.G.; Koeppel, T.A.; Kirschfink, M.; Gebhard, M.M.; Herfarth, C.; Otto, G.; Post, S. Complement inhibition by soluble complement receptor type 1 improves microcirculation after rat liver transplantation. Transplantation 1998, 66, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Cartaya, R.E.; DeSola, G.P.; Wright, L.; Jamieson, N.V.; White, D.J. Regulation of the complement cascade by soluble complement receptor type 1. Protective effect in experimental liver ischemia and reperfusion. Transplantation 1995, 59, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.V.; Shiels, I.A.; Strachan, A.J.; Abbenante, G.; Fairlie, D.P.; Taylor, S.M. A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int. 2003, 63, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, T.V.; Woodruff, T.M.; Stocks, S.Z.; Proctor, L.M.; Pollitt, S.; Shiels, I.A.; Reid, R.C.; Fairlie, D.P.; Taylor, S.M. Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J. Hepatol. 2004, 40, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.D.; Shea-Donohue, T.; Guthridge, J.M.; Kulik, L.; Waldschmidt, T.J.; Gipson, M.G.; Tsokos, G.C.; Holers, V.M. Mice deficient in complement receptors 1 and 2 lack a tissue injury-inducing subset of the natural antibody repertoire. J. Immunol. 2002, 169, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Song, H.; Lu, B.; Qiao, F.; Burns, T.A.; Holers, V.M.; Tsokos, G.C.; Tomlinson, S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J. Clin. Investig. 2005, 115, 2444–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, K.; Jin, J.; Atkinson, C.; Alawieh, A.; Qiao, F.; Lei, B.; Chavin, K.D.; He, S.; Tomlinson, S. Natural immunoglobulin M initiates an inflammatory response important for both hepatic ischemia reperfusion injury and regeneration in mice. Hepatology 2017, 67, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.; Slaats, Y.; Driessen, A.; Peutz-Kootstra, C.J.; Nijhuis, J.; Steffensen, R.; Greve, J.W.; Buurman, W.A. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 2009, 50, 1809–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, T.V.; Marini, M.A.; Succurro, E.; Andreozzi, F.; Sciacqua, A.; Hribal, M.L.; Perticone, F.; Sesti, G. Association between hemoglobin glycation index and hepatic steatosis in non-diabetic individuals. Diabetes Res. Clin. Pract. 2017, 134, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Chen, Y.; Xu, L.; Miao, M.; Li, Y.; Yu, C. Serum complement C3 levels are associated with nonalcoholic fatty liver disease independently of metabolic features in Chinese population. Sci. Rep. 2016, 6, 23279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Li, C.; Xia, Y.; Zhang, Q.; Wu, H.; Du, H.; Liu, L.; Wang, C.; Shi, H.; Guo, X.; et al. Association between complement C3 and prevalence of fatty liver disease in an adult population: A cross-sectional study from the Tianjin Chronic Low-Grade Systemic Inflammation and Health (TCLSIHealth) cohort study. PLoS ONE 2015, 10, e0122026. [Google Scholar] [CrossRef] [PubMed]
- Segers, F.M.; Verdam, F.J.; de Jonge, C.; Boonen, B.; Driessen, A.; Shiri-Sverdlov, R.; Bouvy, N.D.; Greve, J.W.; Buurman, W.A.; Rensen, S.S. Complement alternative pathway activation in human nonalcoholic steatohepatitis. PLoS ONE 2014, 9, e110053. [Google Scholar] [CrossRef] [PubMed]
- Wlazlo, N.; van Greevenbroek, M.M.; Ferreira, I.; Jansen, E.H.; Feskens, E.J.; van der Kallen, C.J.; Schalkwijk, C.G.; Bravenboer, B.; Stehouwer, C.D. Activated complement factor 3 is associated with liver fat and liver enzymes: The CODAM study. Eur. J. Clin. Investig. 2013, 43, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.; Morrison, L.; MacSween, R.N.; Whaley, K. Biosynthesis of complement components by cultured rat hepatocytes. Biochem. J. 1985, 232, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadori, G.; Rasokat, H.; Burger, R.; Meyer Zum Buschenfelde, K.H.; Bitter-Suermann, D. Quantitative determination of complement components produced by purified hepatocytes. Clin. Exp. Immunol. 1984, 55, 189–196. [Google Scholar] [PubMed]
- Darlington, G.J.; Wilson, D.R.; Lachman, L.B. Monocyte-conditioned medium, interleukin-1, and tumor necrosis factor stimulate the acute phase response in human hepatoma cells in vitro. J. Cell Biol. 1986, 103, 787–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falus, A.; Rokita, H.; Walcz, E.; Brozik, M.; Hidvegi, T.; Meretey, K. Hormonal regulation of complement biosynthesis in human cell lines—II. Upregulation of the biosynthesis of complement components C3, factor B and C1 inhibitor by interleukin-6 and interleukin-1 in human hepatoma cell line. Mol. Immunol. 1990, 27, 197–201. [Google Scholar] [CrossRef]
- Paglialunga, S.; Fisette, A.; Yan, Y.; Deshaies, Y.; Brouillette, J.F.; Pekna, M.; Cianflone, K. Acylation-stimulating protein deficiency and altered adipose tissue in alternative complement pathway knockout mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E521–E529. [Google Scholar] [CrossRef] [PubMed]
- Bavia, L.; Cogliati, B.; Dettoni, J.B.; Ferreira Alves, V.A.; Isaac, L. The complement component C5 promotes liver steatosis and inflammation in murine non-alcoholic liver disease model. Immunol. Lett. 2016, 177, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.Y.; Fogelstrand, L.; Hartvigsen, K.; Hansen, L.F.; Woelkers, D.; Shaw, P.X.; Choi, J.; Perkmann, T.; Backhed, F.; Miller, Y.I.; et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Investig. 2009, 119, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Perkmann, T.; Afonyushkin, T.; Mangold, A.; Prohaska, T.A.; Papac-Milicevic, N.; Millischer, V.; Bartel, C.; Horkko, S.; Boulanger, C.M.; et al. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies. J. Lipid Res. 2015, 56, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Binder, C.J.; Miller, Y.I.; Subbanagounder, G.; Silverman, G.J.; Berliner, J.A.; Witztum, J.L. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J. Exp. Med. 2004, 200, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Bergmark, C.; Laurila, A.; Horkko, S.; Han, K.H.; Friedman, P.; Dennis, E.A.; Witztum, J.L. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA 1999, 96, 6353–6358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellusova, J.; Duber, S.; Guckel, E.; Binder, C.J.; Weiss, S.; Voll, R.; Nitschke, L. Siglec-G regulates B1 cell survival and selection. J. Immunol. 2010, 185, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Austen, W.G., Jr.; Zhang, M.; Chan, R.; Friend, D.; Hechtman, H.B.; Carroll, M.C.; Moore, F.D., Jr. Murine hindlimb reperfusion injury can be initiated by a self-reactive monoclonal IgM. Surgery 2004, 136, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Austen, W.G., Jr.; Chiu, I.; Alicot, E.M.; Hung, R.; Ma, M.; Verna, N.; Xu, M.; Hechtman, H.B.; Moore, F.D., Jr.; et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 3886–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.P.; Pechet, T.T.; Weiser, M.R.; Reid, R.; Kobzik, L.; Moore, F.D., Jr.; Carroll, M.C.; Hechtman, H.B. Intestinal reperfusion injury is mediated by IgM and complement. J. Appl. Physiol. 1999, 86, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.D.; Egan, R.P.; Chai, C.; Girardi, G.; Holers, V.M.; Salmon, J.; Monestier, M.; Tsokos, G.C. Anti-phospholipid antibodies restore mesenteric ischemia/reperfusion-induced injury in complement receptor 2/complement receptor 1-deficient mice. J. Immunol. 2004, 173, 7055–7061. [Google Scholar] [CrossRef] [PubMed]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Watzenbock, M.L.; Walenbergh, S.M.; Amir, S.; Gruber, S.; Kozma, M.O.; Grabsch, H.I.; Koek, G.H.; Pierik, M.J.; Staufer, K.; et al. Low levels of IgM antibodies recognizing oxidation-specific epitopes are associated with human non-alcoholic fatty liver disease. BMC Med. 2016, 14, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartier, P.; Potter, P.K.; Ehrenstein, M.R.; Walport, M.J.; Botto, M. Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur. J. Immunol. 2005, 35, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.; Bloks, V.W.; Daha, M.R.; van der Most, P.J.; Sanjabi, B.; van der Vlies, P.; Snieder, H.; Ploeg, R.J.; Krikke, C.; Leuvenink, H.G.; et al. Hypoxia and complement-and-coagulation pathways in the deceased organ donor as the major target for intervention to improve renal allograft outcome. Transplantation 2015, 99, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Naesens, M.; Li, L.; Ying, L.; Sansanwal, P.; Sigdel, T.K.; Hsieh, S.C.; Kambham, N.; Lerut, E.; Salvatierra, O.; Butte, A.J.; et al. Expression of complement components differs between kidney allografts from living and deceased donors. J. Am. Soc. Nephrol. 2009, 20, 1839–1851. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.; Seelen, M.A.; Moers, C.; Daha, M.R.; Rahmel, A.; Leuvenink, H.G.; Paul, A.; Pirenne, J.; Ploeg, R.J. Systemic complement activation in deceased donors is associated with acute rejection after renal transplantation in the recipient. Transplantation 2011, 92, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.; Nijboer, W.N.; Schuurs, T.A.; Leuvenink, H.G.; Morariu, A.M.; Tullius, S.G.; van Goor, H.; Ploeg, R.J.; Seelen, M.A. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol. Dial. Transplant. 2011, 26, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, R.A.; Liu, B.; Akhtar, M.Z.; Ottens, P.J.; Zhang, J.N.; Ploeg, R.J.; Leuvenink, H.G. Steroid anti-inflammatory effects did not improve organ quality in brain-dead rats. Biomed. Res. Int. 2015, 2015, 207534. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, R.; Liu, B.; Akhtar, M.Z.; Ottens, P.J.; Zhang, J.N.; Ploeg, R.J.; Leuvenink, H.G. Prednisolone has a positive effect on the kidney but not on the liver of brain dead rats: A potencial role in complement activation. J. Transl. Med. 2014, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Poppelaars, F.; Seelen, M.A. Complement-mediated inflammation and injury in brain dead organ donors. Mol. Immunol. 2017, 84, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.K.; Johnson, L.A.; Germin, B.I.; Marcos, A. One hundred consecutive hepatic biopsies in the workup of living donors for right lobe liver transplantation. Liver Transplant. 2002, 8, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Ko, J.S.; Kwon, G.; Park, C.; Lee, S.; Kim, J.; Kim, G.; Kwon, C.D.; Gwak, M.; Ha, S. Effect of pure microsteatosis on transplant outcomes after living donor liver transplantation: A matched case-control study. Liver Transplant. 2014, 20, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoazec, J.Y.; Delautier, D.; Moreau, A.; Durand, F.; Degott, C.; Benhamou, J.P.; Belghiti, J.; Feldmann, G. Expression of complement-regulatory proteins in normal and UW-preserved human liver. Gastroenterology 1994, 107, 505–516. [Google Scholar] [CrossRef]
- Kaabak, M.; Babenko, N.; Shapiro, R.; Zokoyev, A.; Dymova, O.; Kim, E. A prospective randomized, controlled trial of eculizumab to prevent ischemia-reperfusion injury in pediatric kidney transplantation. Pediatr. Transplant. 2018, 22, e13129. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.A.; Zeevi, A.; Choi, J.; Cisneros, K.; Toyoda, M.; Kahwaji, J.; Peng, A.; Villicana, R.; Puliyanda, D.; Reinsmoen, N.; et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation 2015, 99, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am. J. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Larsson, R.; Hong, J.; Elgue, G.; Ekdahl, K.N.; Sahu, A.; Lambris, J.D. Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulation. Blood 1998, 92, 1661–1667. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Hajishengallis, E.; Kajikawa, T.; Wang, B.; Yancopoulou, D.; Ricklin, D.; Lambris, J.D. Complement inhibition in pre-clinical models of periodontitis and prospects for clinical application. Semin. Immunol. 2016, 28, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Molecule | Target | Effect | References (Clinical Trials) | |
|---|---|---|---|---|
| Eculizumab | Humanized monoclonal antibody | C5 | Prevents C5 cleavage | [155] (NCT1756508) |
| C1-INH | Plasma purified protein | C1s | Prevents C2 and C4 formation | [156,157] (01134510) |
| AMY-101 | Synthetic peptide | C3 | Prevents cleavage of C3 | (NCT03316521) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, K.; Thevenot, P.; Alfadhli, A.; Cohen, A. Complement Activation in Liver Transplantation: Role of Donor Macrosteatosis and Implications in Delayed Graft Function. Int. J. Mol. Sci. 2018, 19, 1750. https://doi.org/10.3390/ijms19061750
Núñez K, Thevenot P, Alfadhli A, Cohen A. Complement Activation in Liver Transplantation: Role of Donor Macrosteatosis and Implications in Delayed Graft Function. International Journal of Molecular Sciences. 2018; 19(6):1750. https://doi.org/10.3390/ijms19061750
Chicago/Turabian StyleNúñez, Kelley, Paul Thevenot, Abeer Alfadhli, and Ari Cohen. 2018. "Complement Activation in Liver Transplantation: Role of Donor Macrosteatosis and Implications in Delayed Graft Function" International Journal of Molecular Sciences 19, no. 6: 1750. https://doi.org/10.3390/ijms19061750