Characterization of the Complete Mitochondrial Genome Sequences of Three Croakers (Perciformes, Sciaenidae) and Novel Insights into the Phylogenetics

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
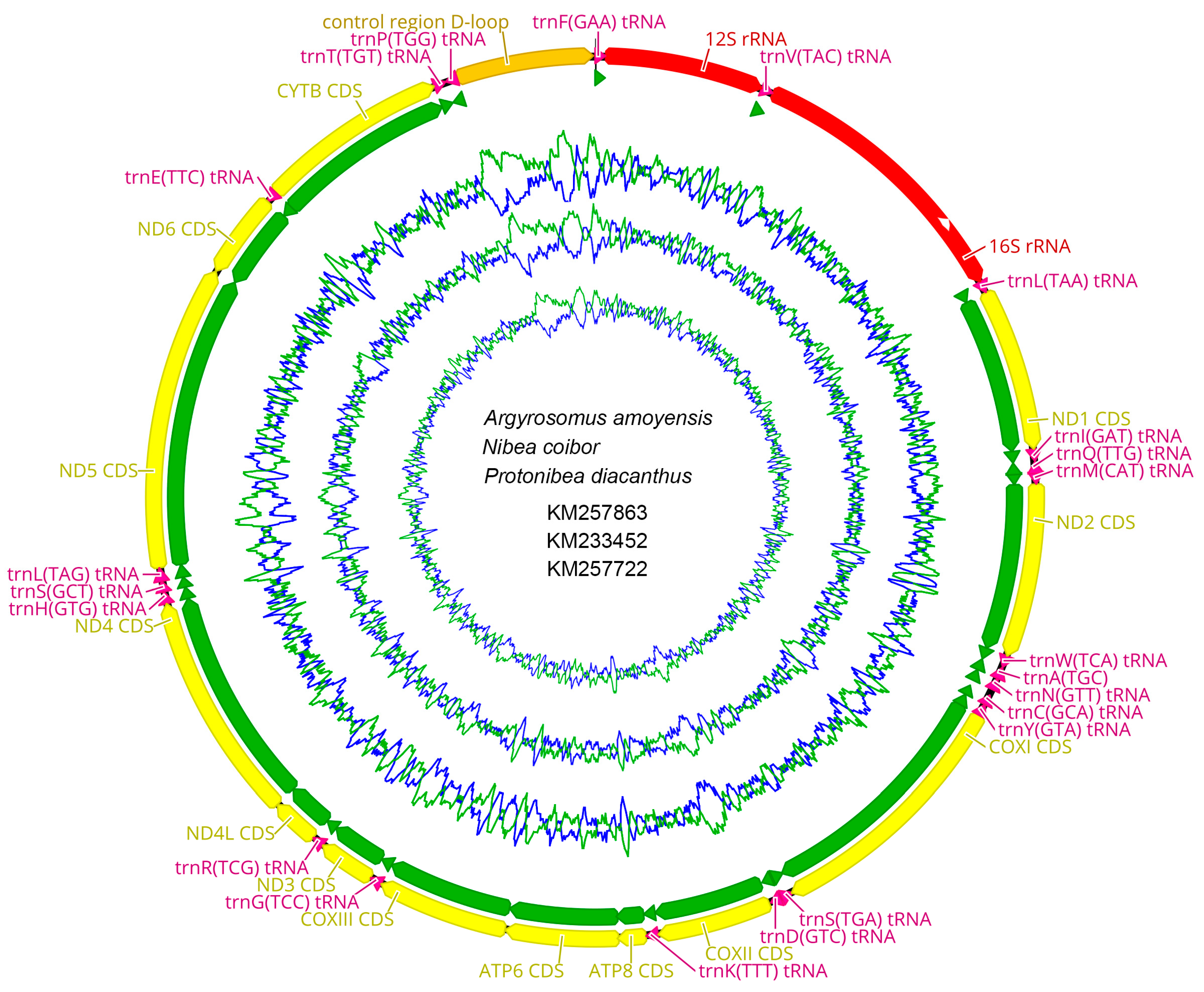
2.1. Genome Organization and Composition
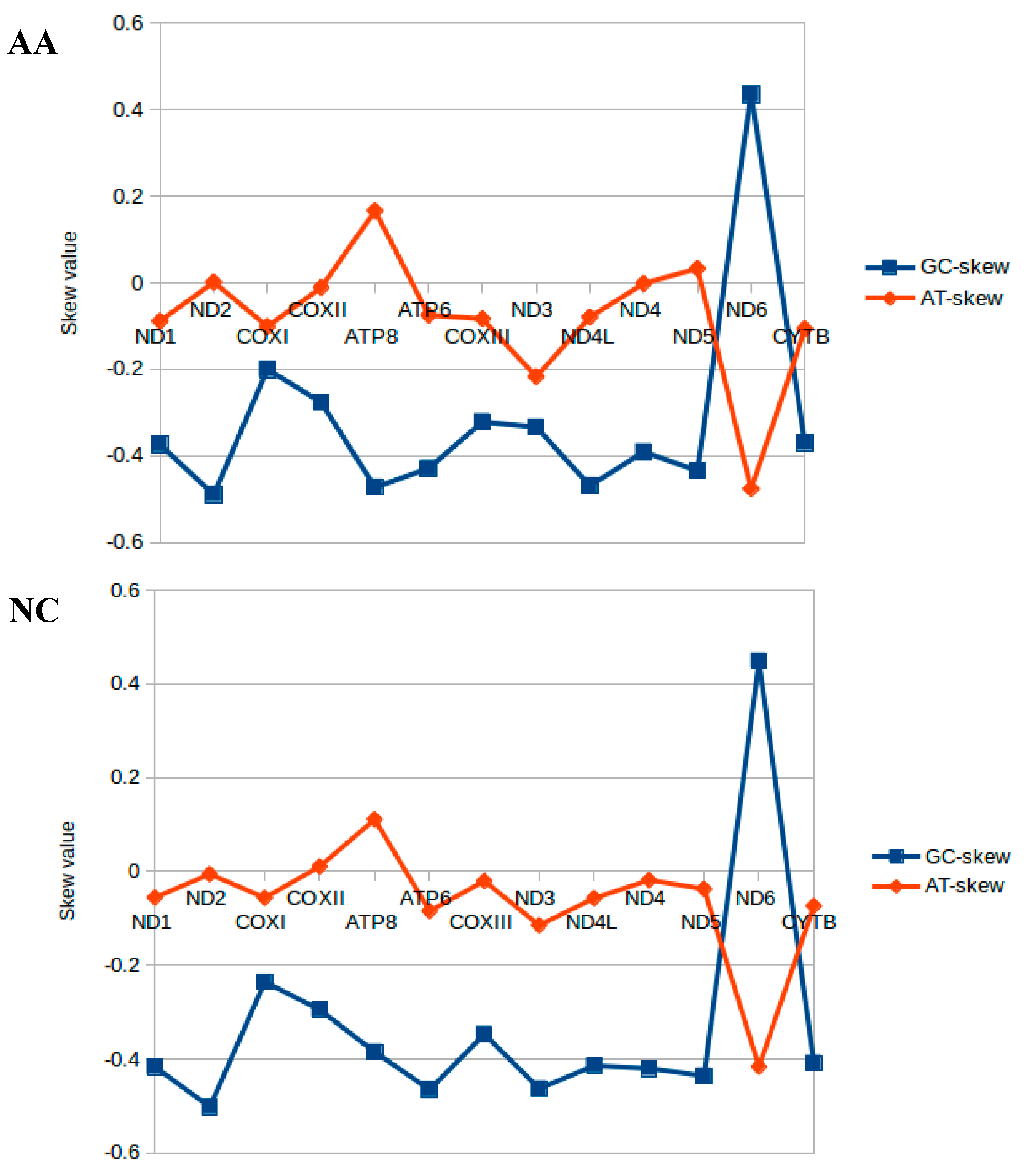
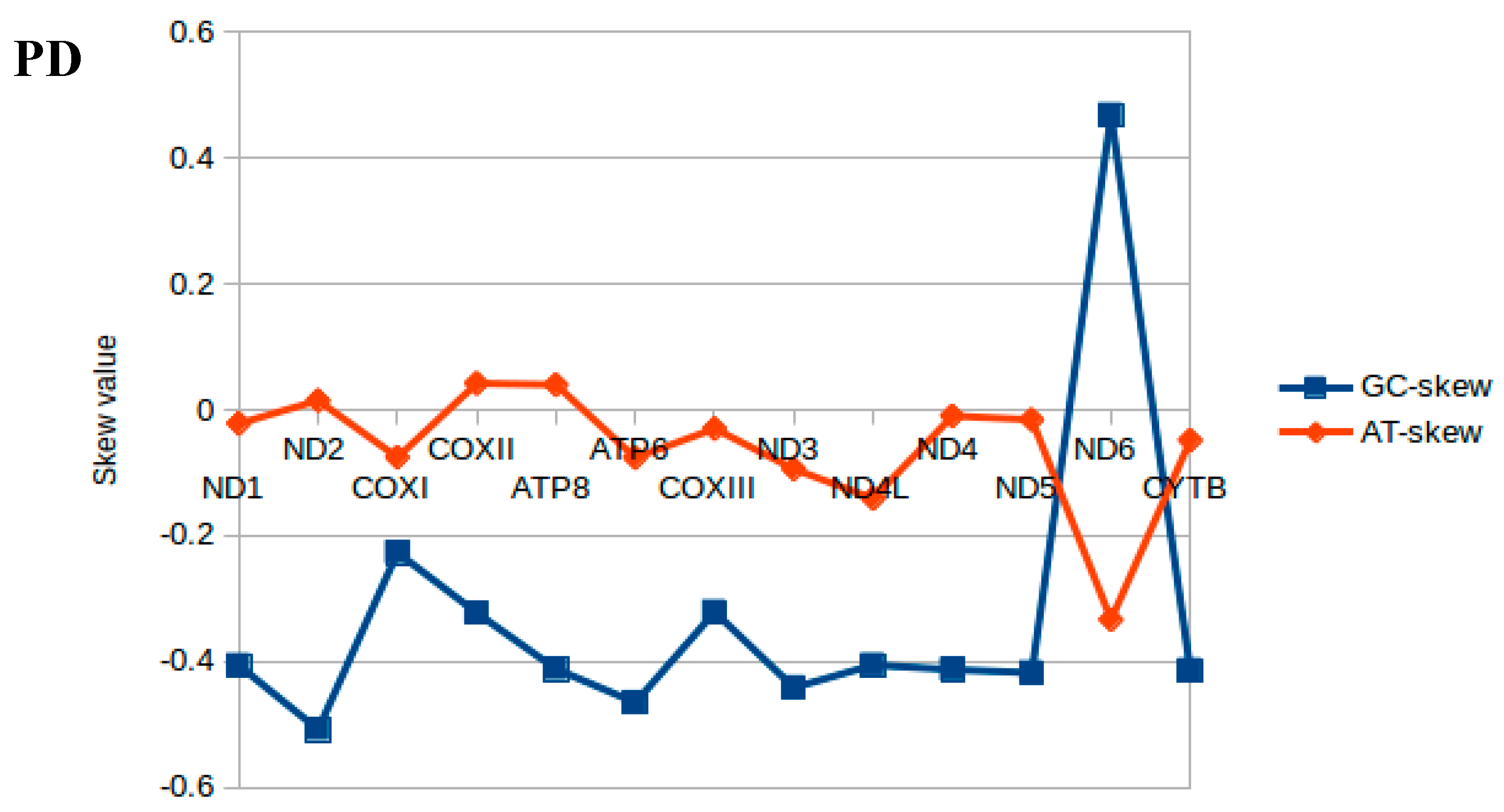
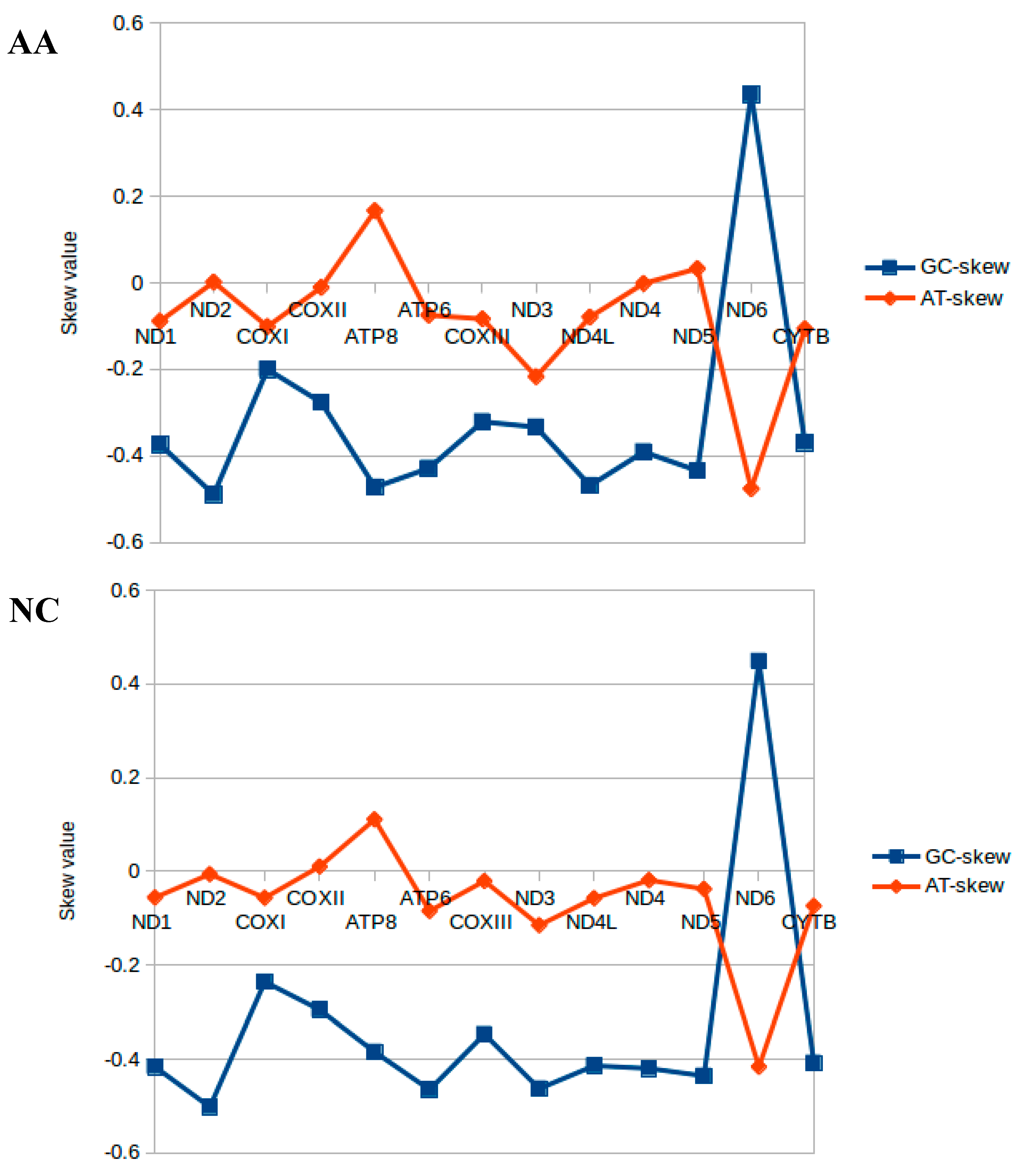
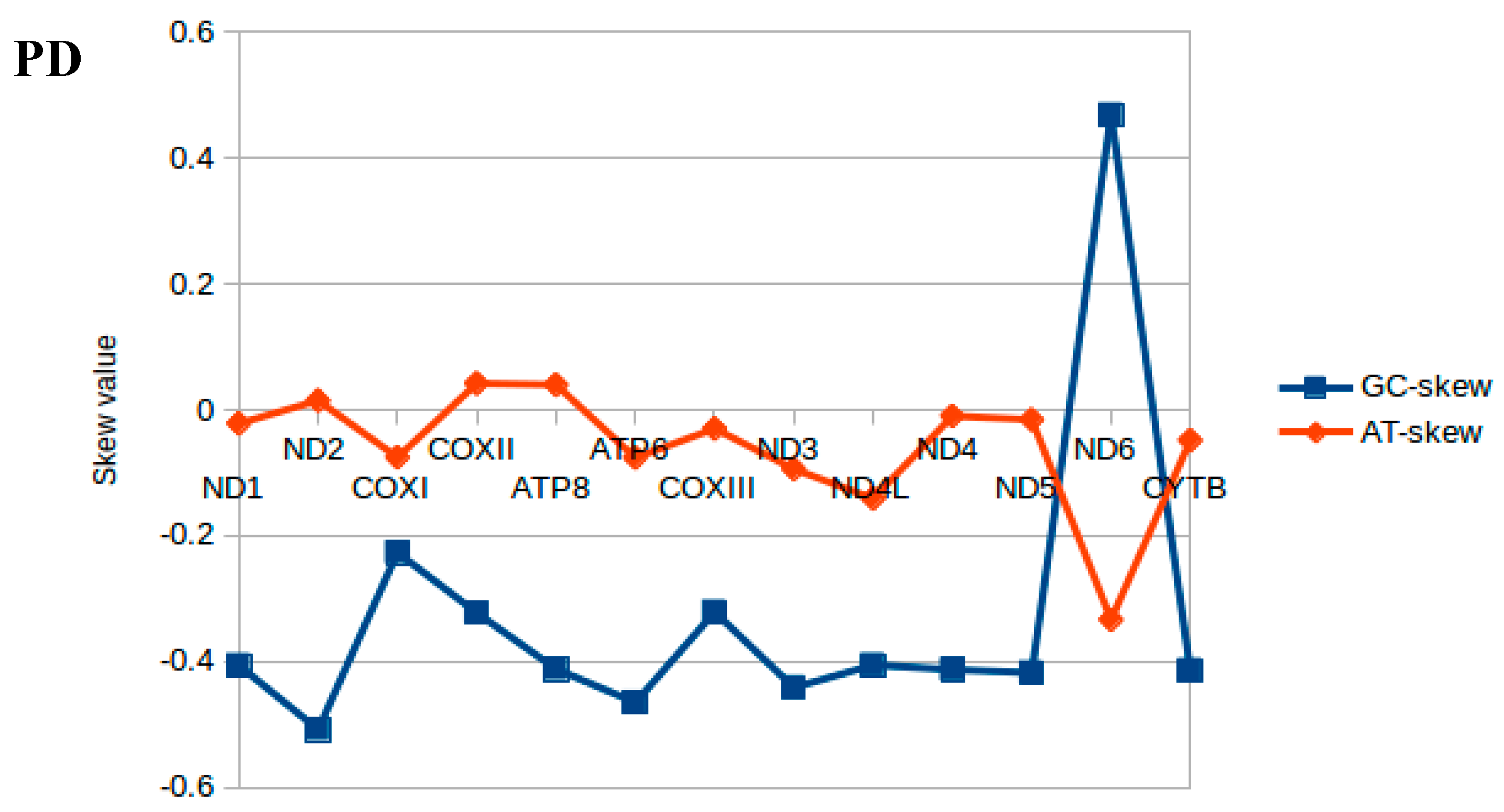
2.2. Nucleotide Composition
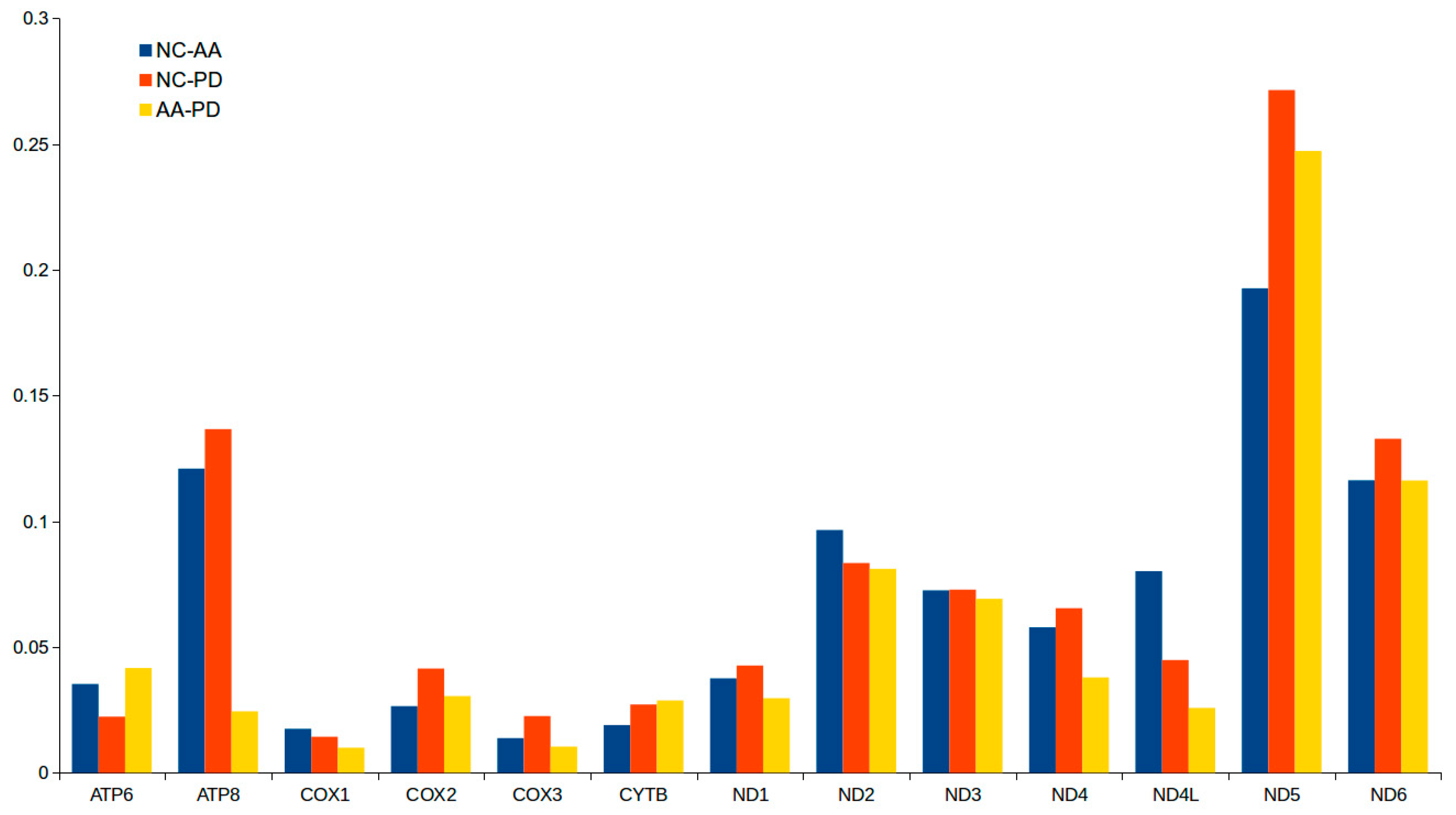
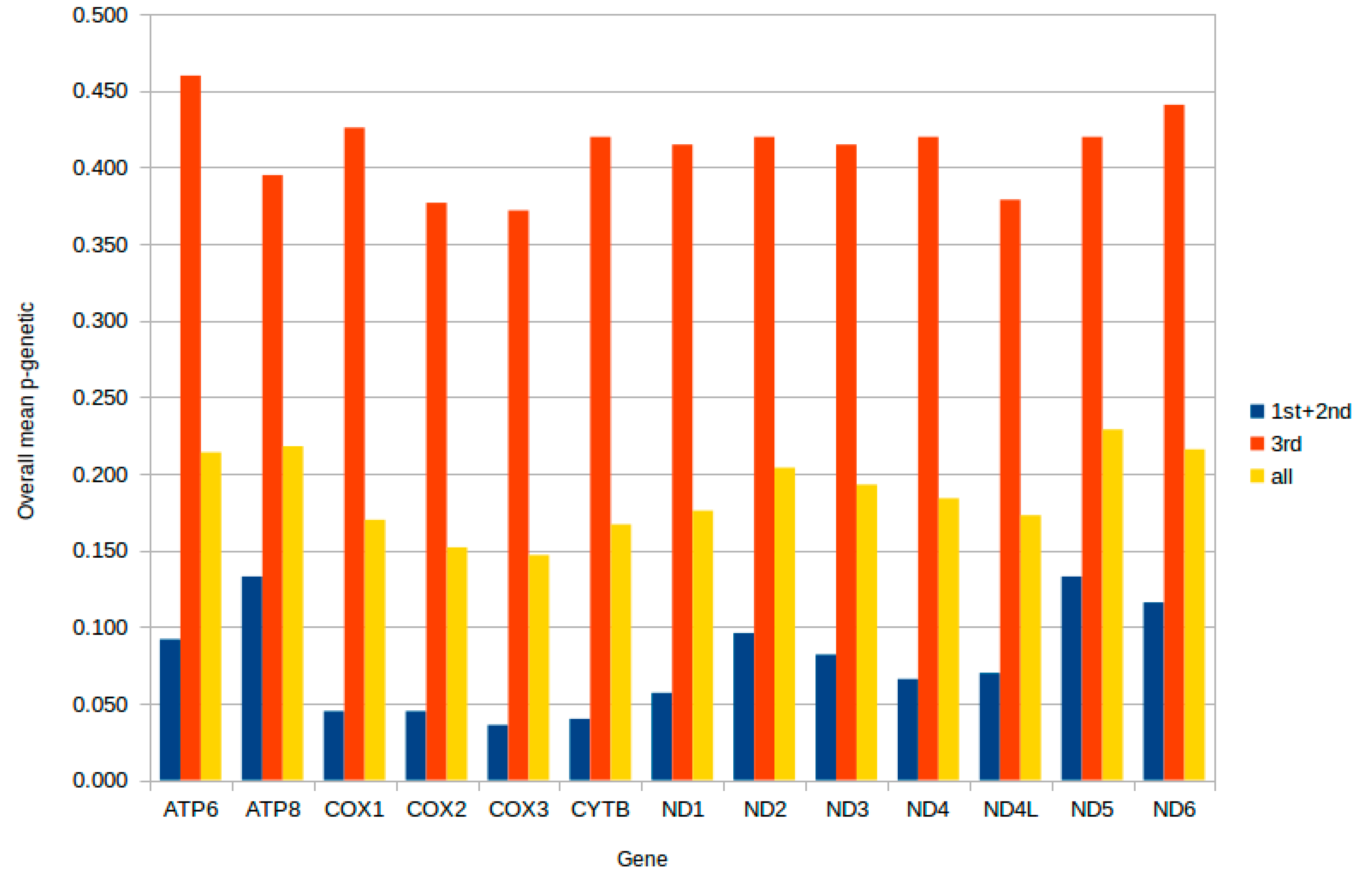
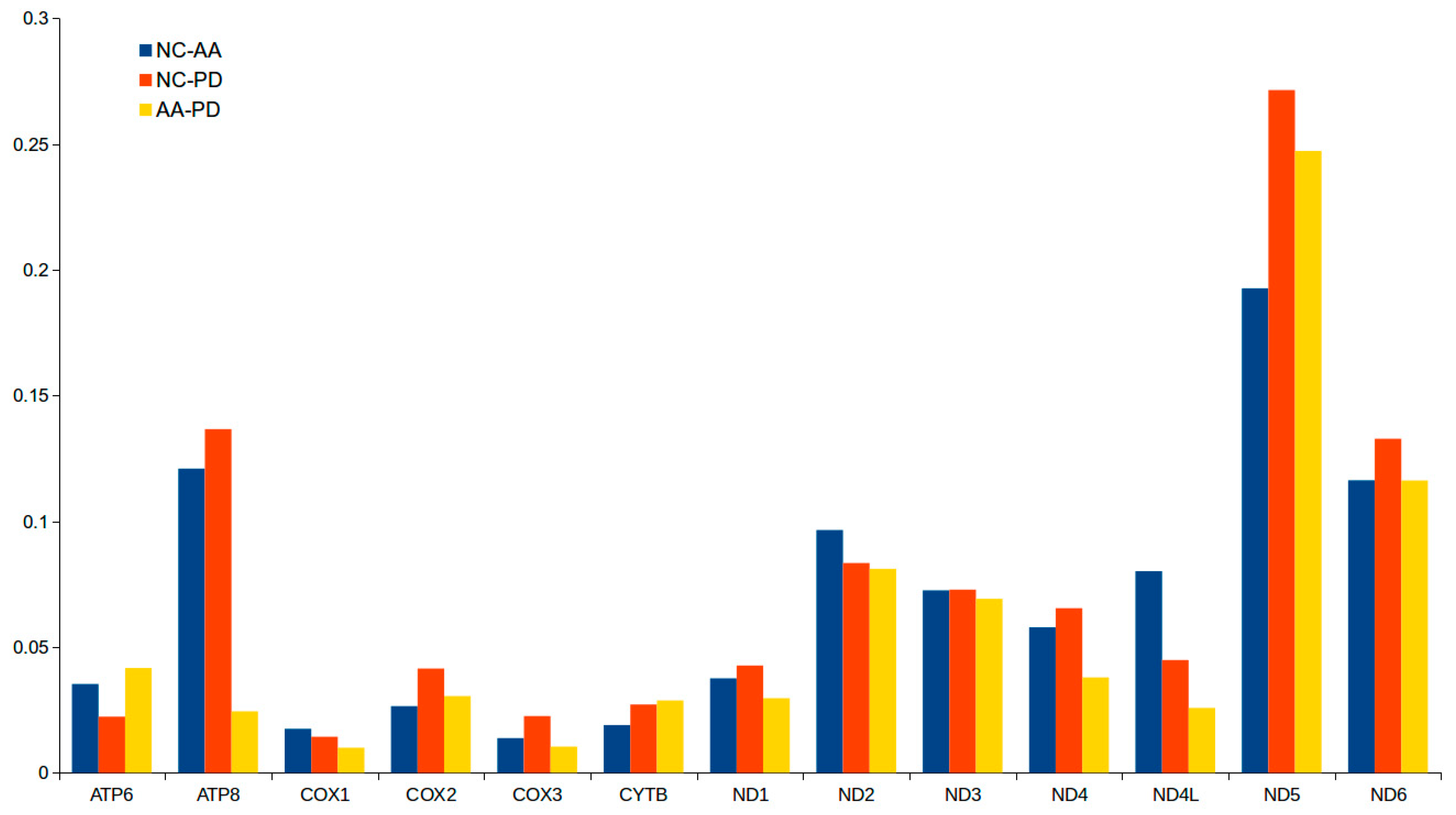
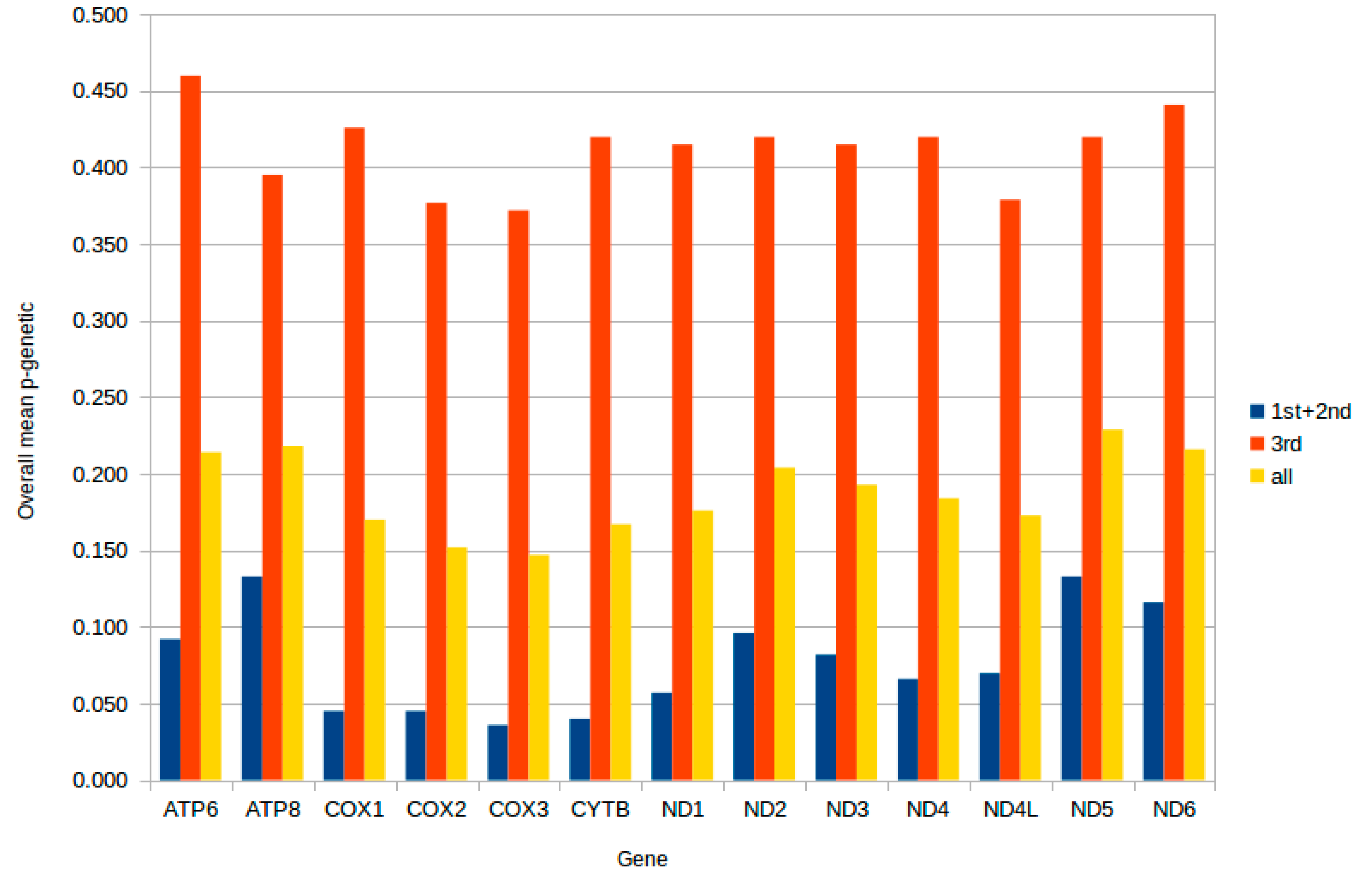
2.3. Protein-Coding Genes
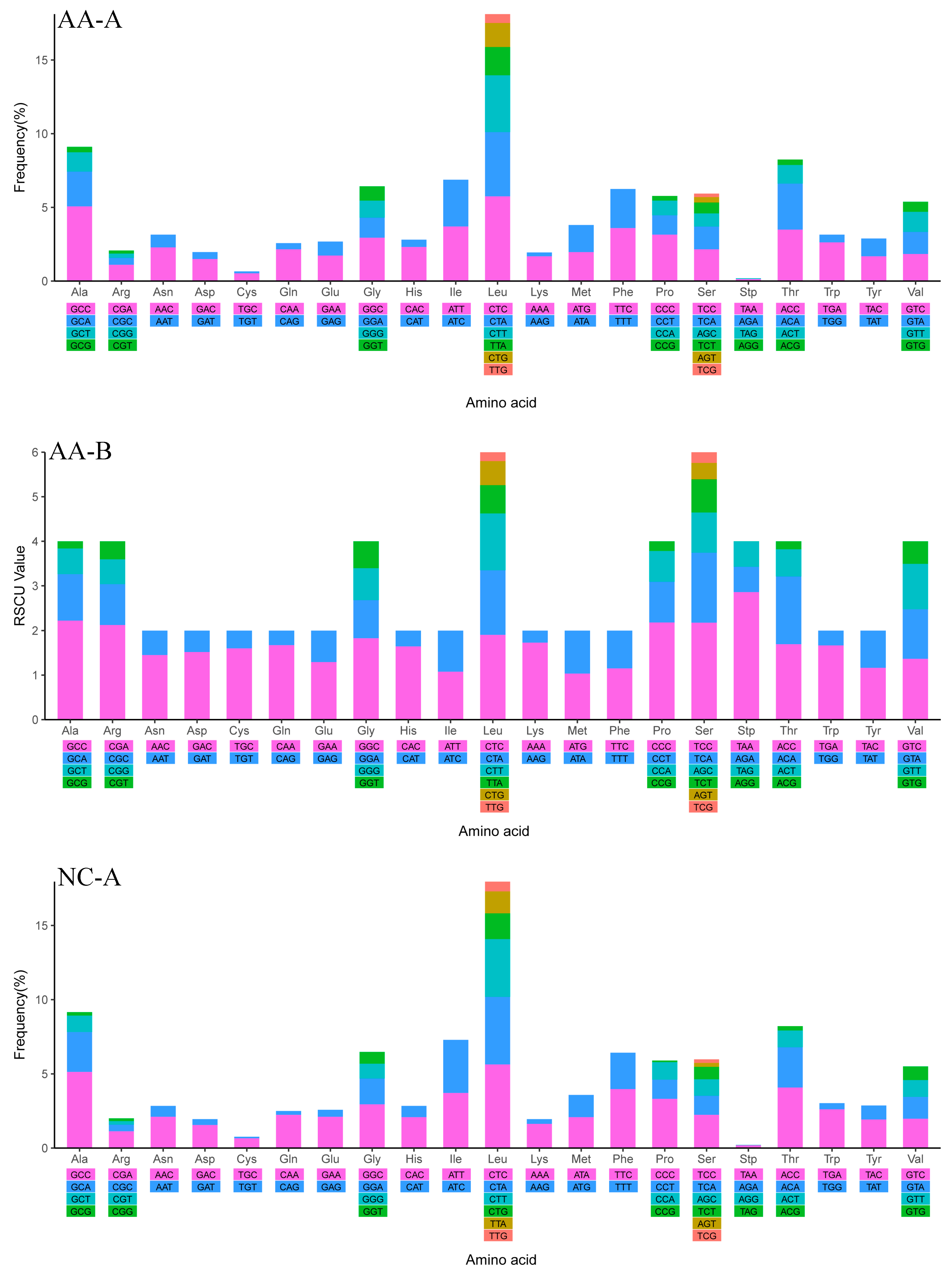
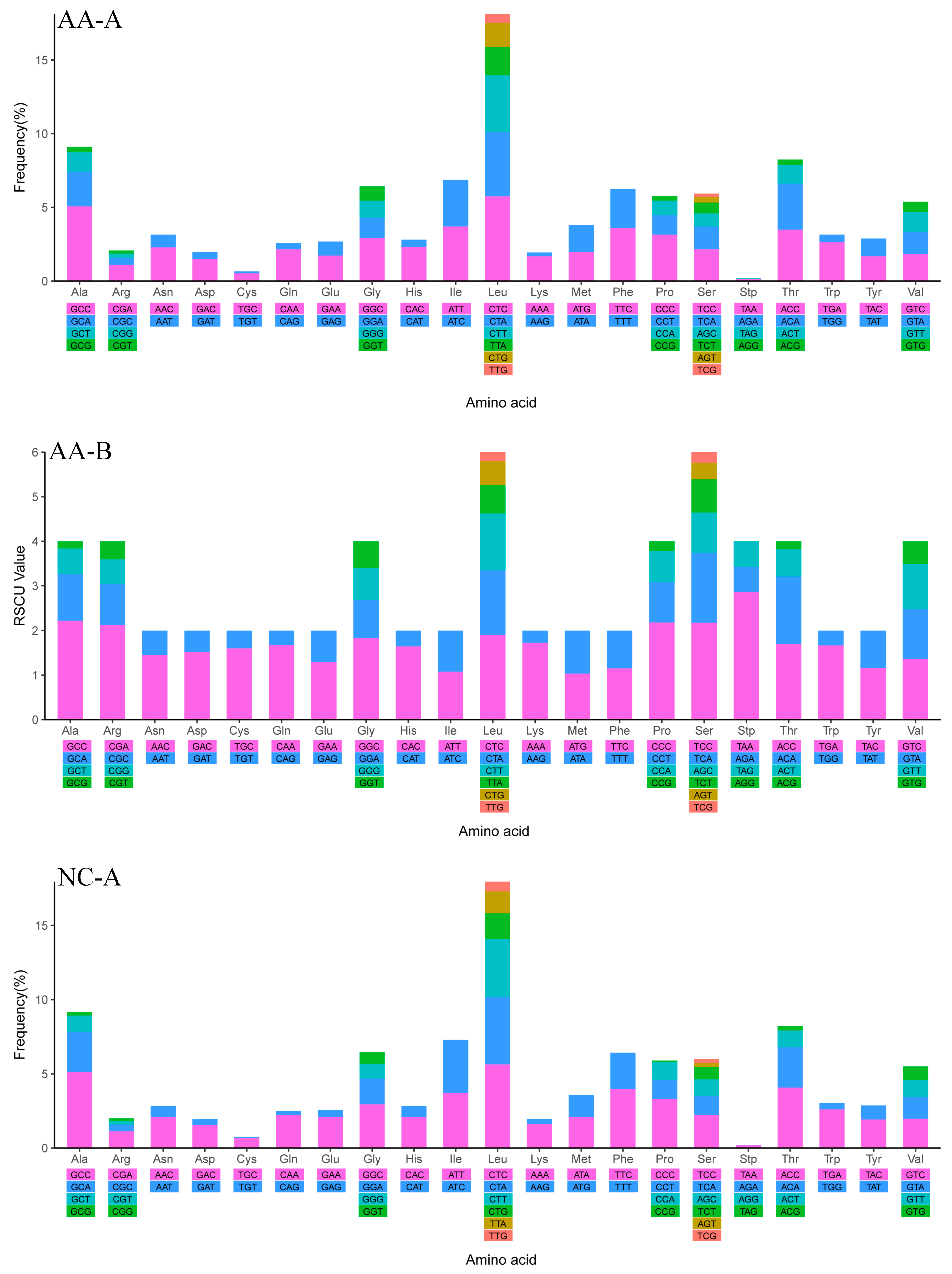
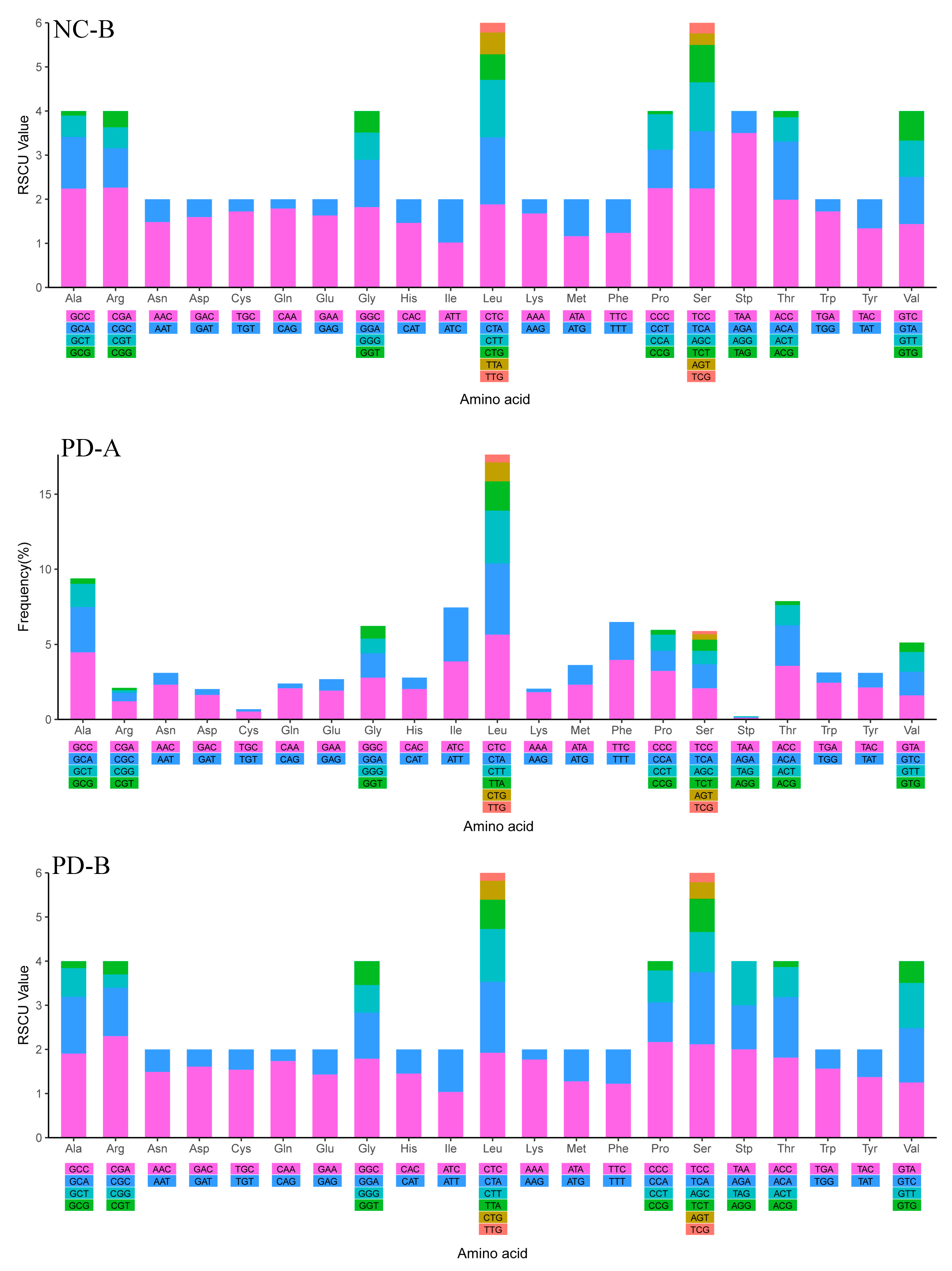
2.4. Mitochondrial Gene Codon Usage
2.5. Transfer and Ribosomal RNA Genes
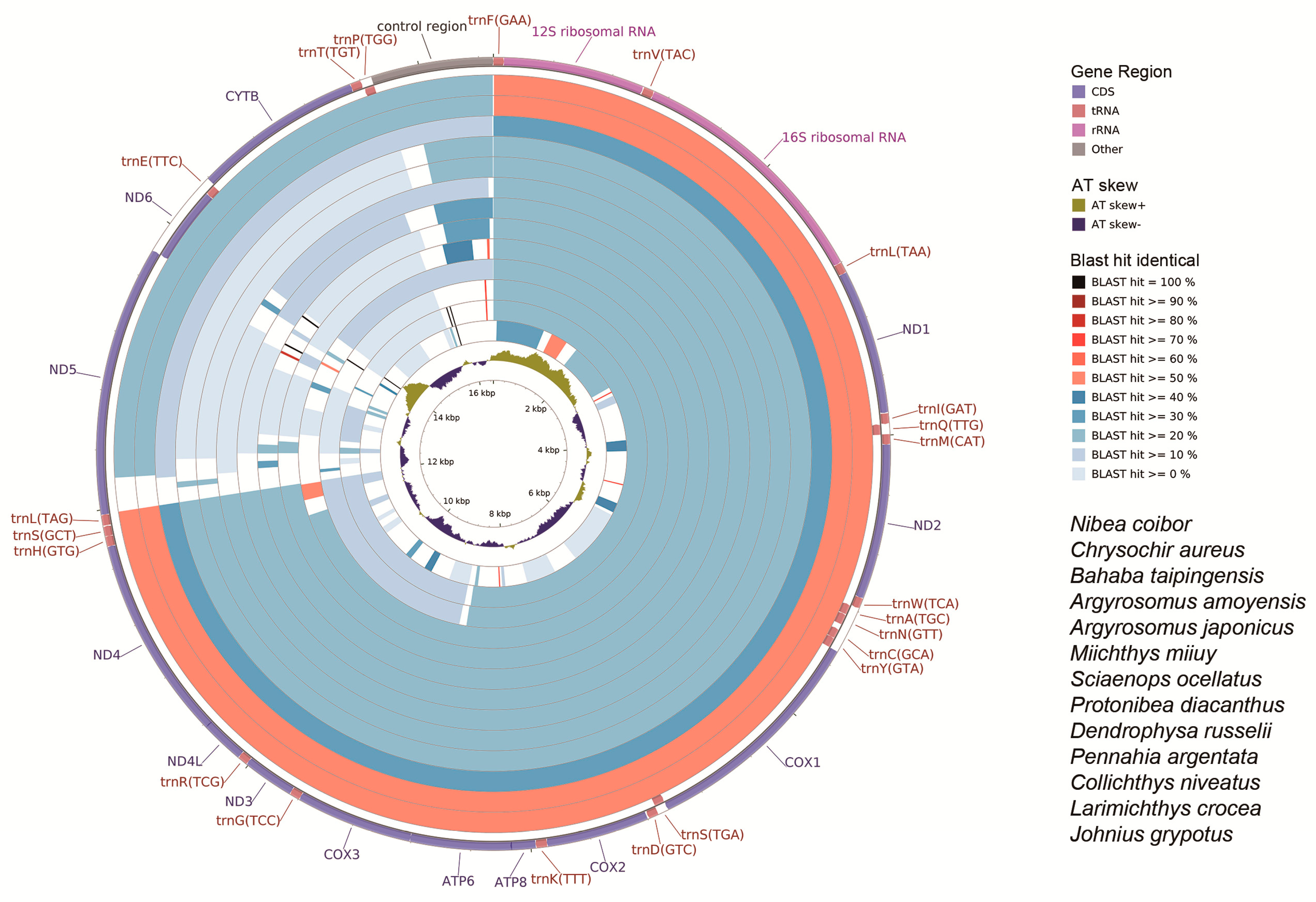
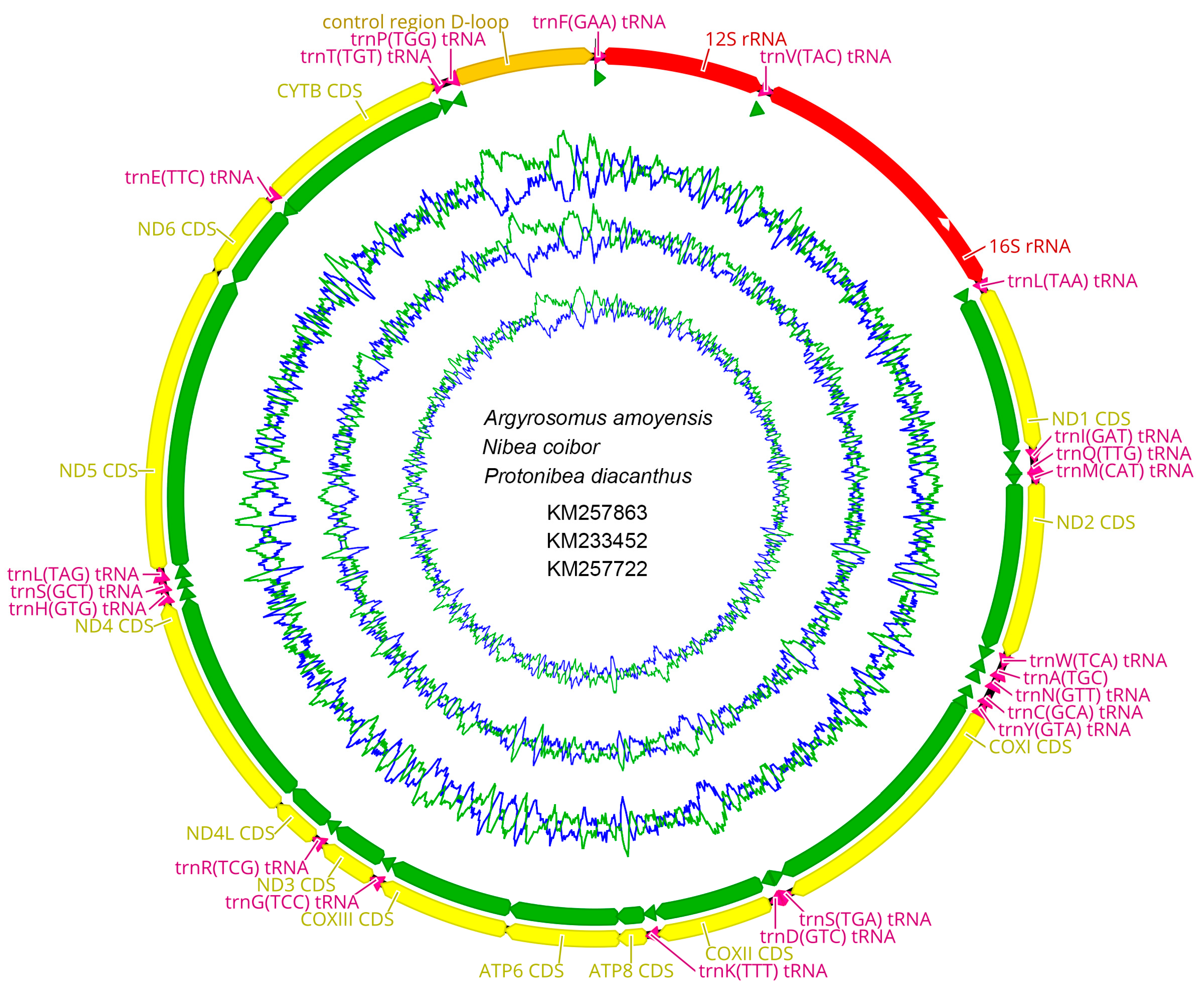
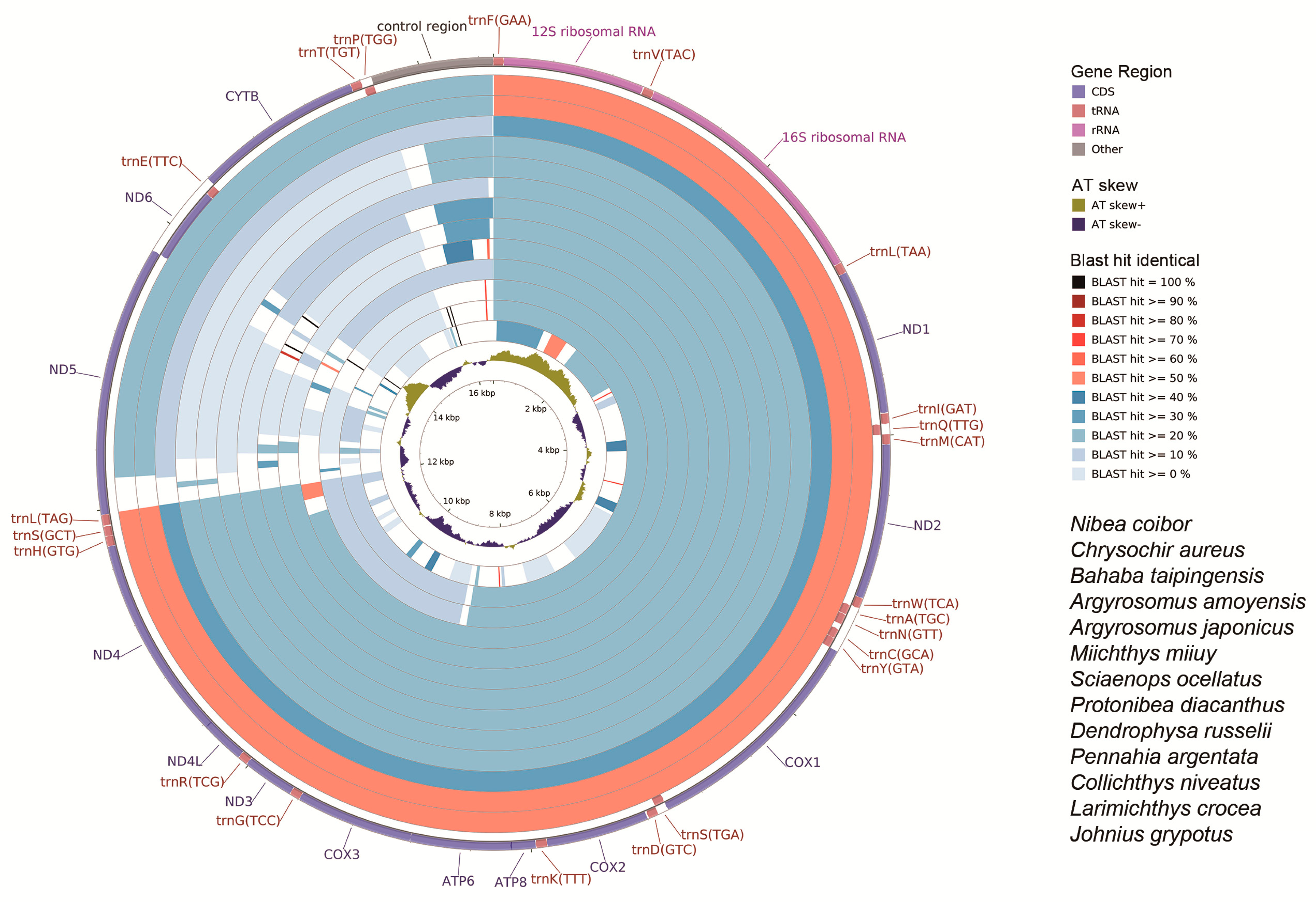
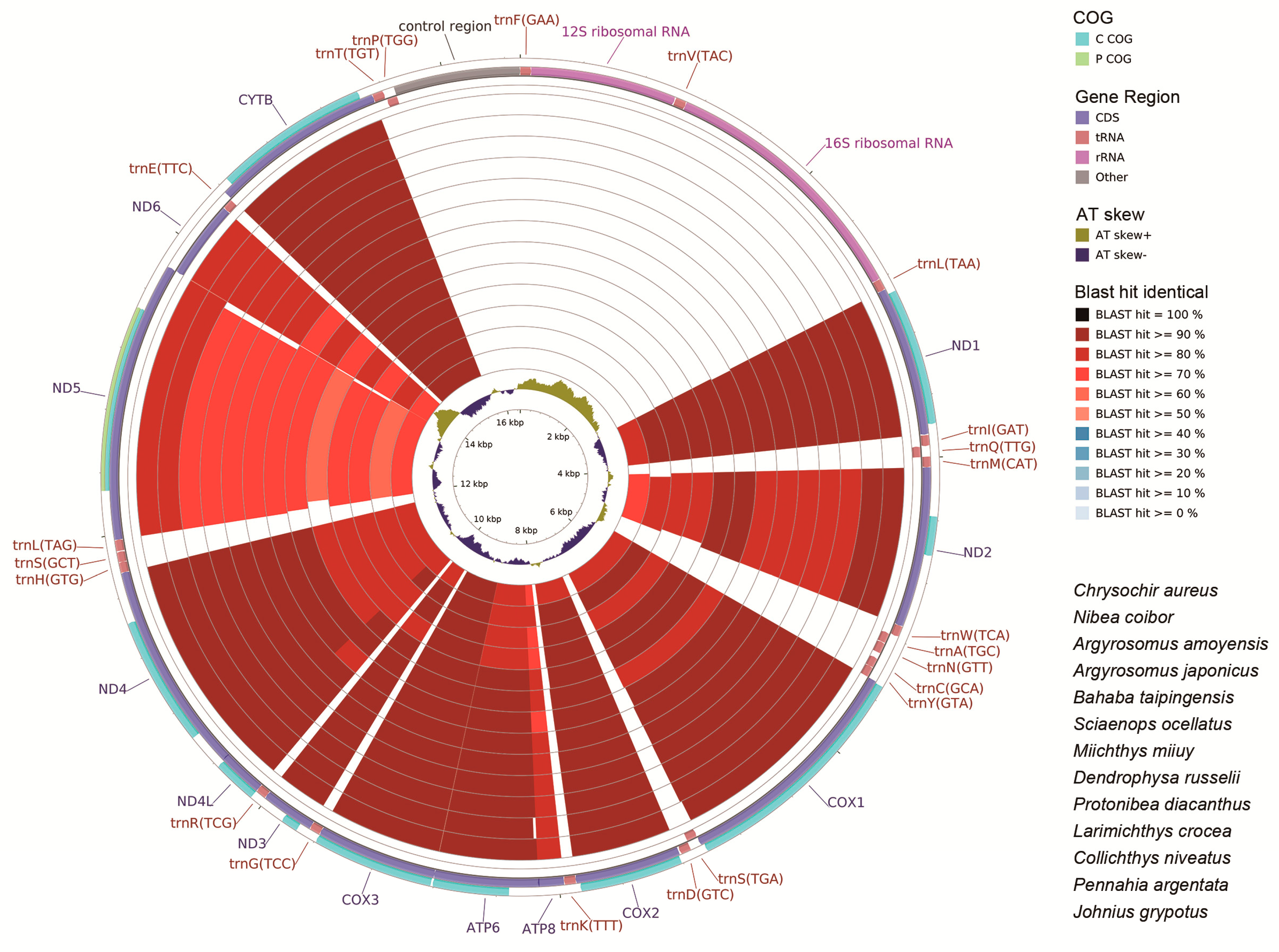
2.6. CG View Comparison Tool (CCT) Map
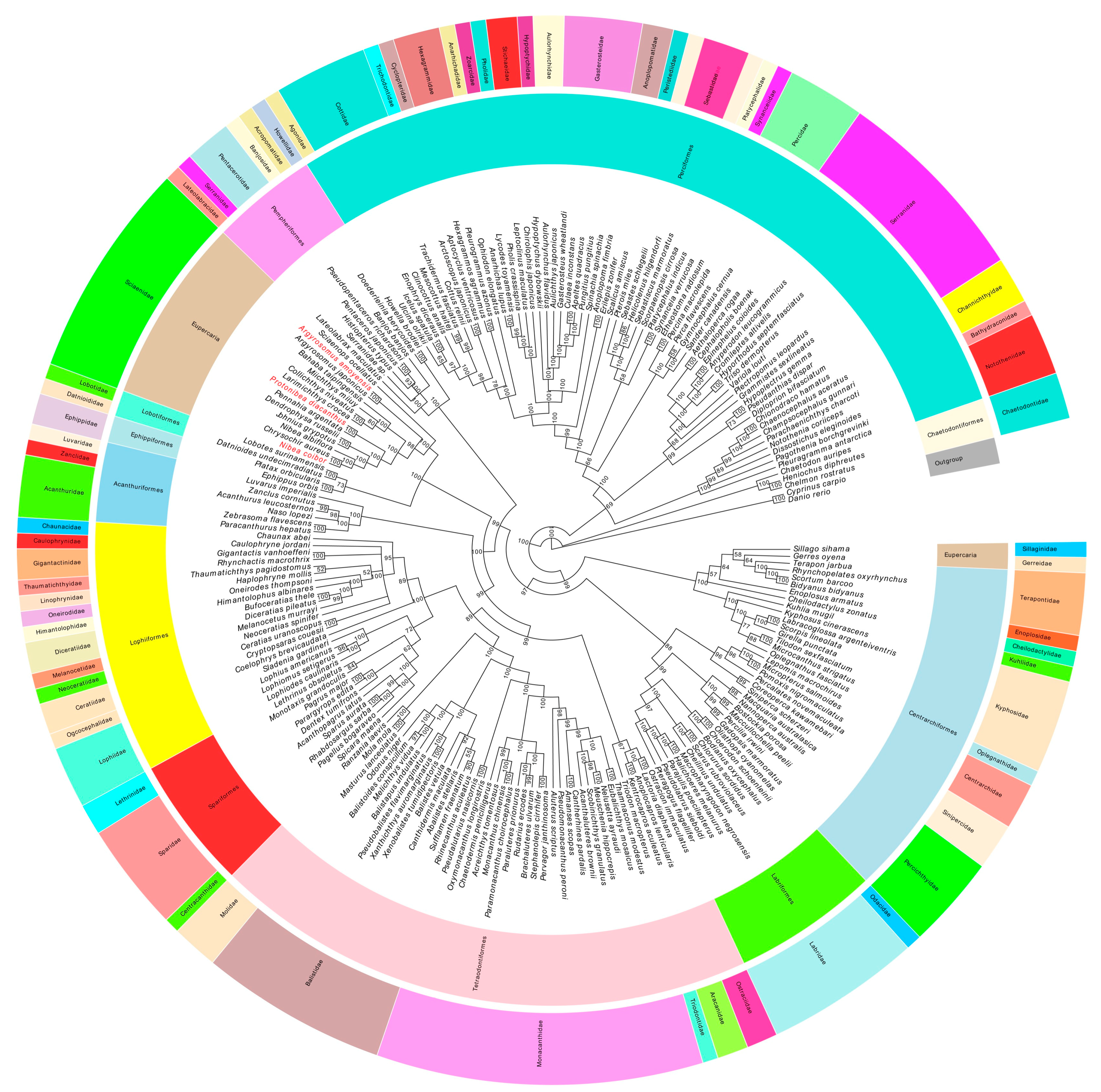
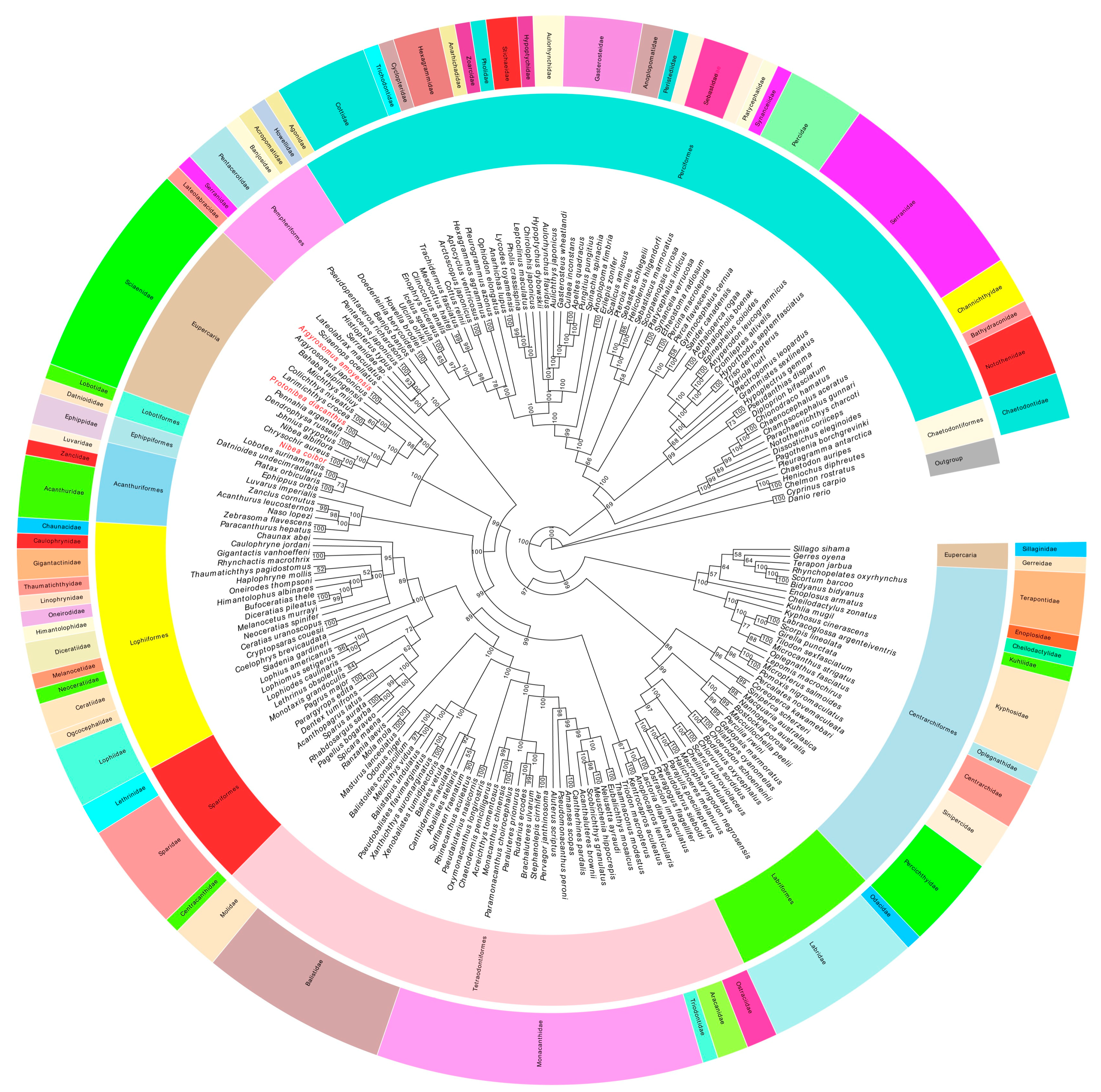
2.7. Phylogenetic Analyses
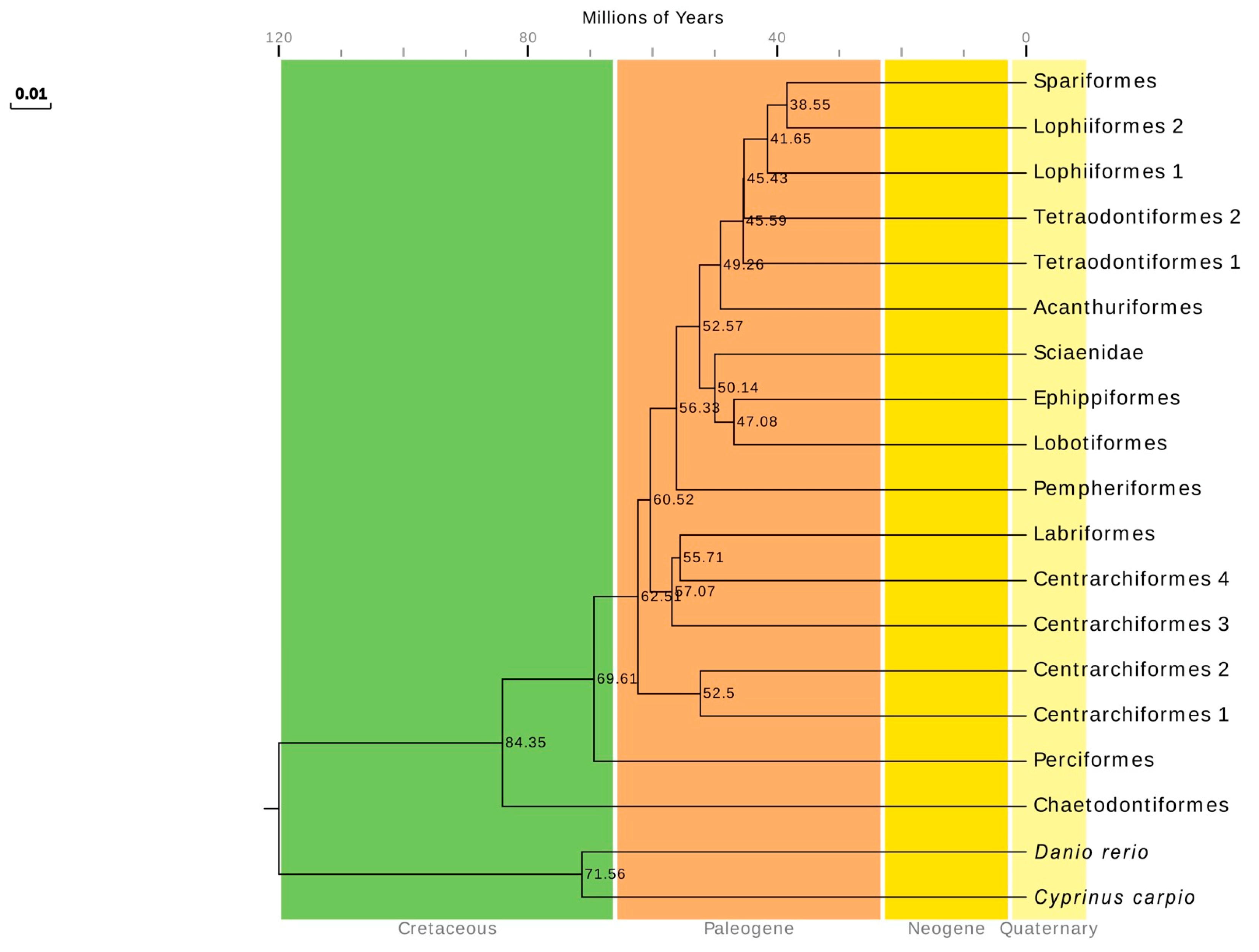
2.8. Divergence Times of the Series Eupercaria
3. Materials and Methods
3.1. Sampling and Materials
3.2. Library Preparation for Sequencing
3.3. Sequence Analysis and Annotation
3.4. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lal Mohan, R.S. Fao Species Identification Sheets for Fishery Purposes; FAO: Rome, Italy, 1984; Volume 4, pp. 2069–2790. [Google Scholar]
- Nguyen, H.P.; Trong, P.L.; Nguyen, N.T.; Nguyen, P.D.; Do, T.N.N.; Nguyen, V.L. Checklist of Marine Fishes in Vietnam; Science and Technics Publishing House: Ha Noi, Vietnam, 1995; Volume 3. [Google Scholar]
- Sasaki, K. Fao Species Identification Guide for Fishery Purposes; FAO: Rome, Italy, 2001; Volume 5, pp. 2791–3380. [Google Scholar]
- Shao, Y.Q.; Zhang, D.C.; Su, T.F.; Lv, J.L.; Jiang, S.G. The construction of pituitary gland cDNA library of nibea coibor. South China Fish. Sci. 2006, 2, 8–12. [Google Scholar]
- Jian, L.J.; Yang, Y.K.; Liu, X.D.; Chen, Q.K.; Wang, Z.Y. The cross breeding and genetic analysis of hybrids of Larimichthys crocea (♀) and Nibea miichthioides (♂). J. Fish. China 2013, 37, 801–808. [Google Scholar] [CrossRef]
- Meng, Q.W. Fish Taxonomy; China Agriculture Press: Beijing, China, 1996; Volume 2. [Google Scholar]
- Cheng, Q.T.; Zheng, B.S. Systematic Synopsis of Chinese Fishes; Science Press: Beijing, China, 1987; Volume 1. [Google Scholar]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Podsiadlowski, L.; Kohlhagen, H.; Koch, M. The complete mitochondrial genome of Scutigerella causeyae (myriapoda: Symphyla) and the phylogenetic position of symphyla. Mol. Phylogenet. Evol. 2007, 45, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kartavtsev, Y.P.; Jung, S.O.; Lee, Y.M.; Byeon, H.K.; Lee, J.S. Complete mitochondrial genome of the bullhead torrent catfish, Liobagrus obesus (siluriformes, amblycipididae): Genome description and phylogenetic considerations inferred from the CYT b and 16s rRNA genes. Gene 2007, 396, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Moritz, C.; Dowling, T.; Brown, W. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Stepien, C.A.; Kocher, T.D. Molecules and Morphology in Studies of Fish Evolution. In Molecular Systematics of Fishes; Elsevier: New York, NY, USA, 1997; pp. 1–11. [Google Scholar]
- Miya, M.; Nishida, M. Use of mitogenomic information in teleostean molecular phylogenetics: A tree-based exploration under the maximumparsimony optimality criterion. Mol. Phylogenet. Evol. 2000, 17, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, J.K.; Basu, S.; Sasidharan, S.; Chouksey, M.K.; Gudipati, V. Optimization of process parameters for gelatin extraction from the skin of blackspotted croaker using response surface methodology. J. Food Sci. Technol. 2014, 51, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Lakra, W.S.; Goswami, M.; Gopalakrishnan, A. Molecular identification and phylogenetic relationships of seven Indian Sciaenids (pisces: Perciformes, sciaenidae) based on 16s rRNA and cytochrome c oxidase subunit i mitochondrial genes. Mol. Biol. Rep. 2009, 36, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, H.; Yang, Z.; Zhao, H.; Sun, J.; Xiao, S.; Yang, X.; Li, G. The complete mitochondrial genome of the blackspotted croaker Protonibea diacanthus (perciformes, sciaenidae). Mitochondrial DNA. Part A 2016, 27, 1671–1673. [Google Scholar]
- Moravec, F.; Barton, D.P. Two gonad-infecting species of Philometra (nematoda: Philometridae) from marine fishes off the northern coast of Australia. Parasite 2015, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.; Zhao, L.; Gao, T.; Lu, H.; Yan, Y. The complete mitochondrial genome of Nibea coibor (perciformes: Sciaenidae). Mitochondrial DNA. Part A 2016, 27, 1681–1682. [Google Scholar]
- Taillebois, L.; Crook, D.; Saunders, T.; Ovenden, J. The complete mitochondrial genome of the black jewfish Protonibea diacanthus (perciformes: Sciaenidae). Mitochondrial DNA. Part A 2016, 27, 779–780. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, H.; Sun, J.; Yang, Z.; Xiao, S.; Li, G.; Liu, L. The complete mitochondrial genome of the amoy croaker Argyrosomus amoyensis (Perciformes, Sciaenidae). Mitochondrial DNA Part A 2016, 27, 1530–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shao, Y.; Jiang, S.; Li, J.; Xu, X. Nibea coibor growth hormone gene: Its phylogenetic significance, microsatellite variation and expression analysis. Gen. Comp. Endocrinol. 2009, 163, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, T.; Jin, X.; Shi, G.; Wang, R. The complete mitochondrial genome of silver croaker Argyrosomus argentatus (perciforems; sciaenidae): Genome characterization and phylogenetic consideration. Molekuliarnaia Biologiia 2012, 46, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Tang, D.; Jin, X. A surprising arrangement pattern and phylogenetic consideration: The complete mitochondrial genome of belanger’s croaker Johnius belangerii (percoidei: Sciaenidae). Mitochondrial DNA 2015, 26, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.J.; Sampaio, I.; Schneider, H.; Santos, S. Molecular phylogeny of weakfish species of the Stellifer group (sciaenidae, perciformes) of the western south Atlantic based on mitochondrial and nuclear data. PLoS ONE 2014, 9, e102250. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.; Gomes Mde, F.; Ferreira, A.R.; Sampaio, I.; Schneider, H. Molecular phylogeny of the western south Atlantic sciaenidae based on mitochondrial and nuclear data. Mol. Phylogenet. Evol. 2013, 66, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, R.; Sun, Y.; Xu, T. The complete mitochondrial genome of the small yellow croaker and partitioned bayesian analysis of sciaenidae fish phylogeny. Genet. Mol. Biol. 2012, 35, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ma, G.Q.; Song, N.; Gao, T.X. Complete mitochondrial genome sequence of bighead croaker Collichthys niveatus (perciformes, sciaenidae): A mitogenomic perspective on the phylogenetic relationships of pseudosciaeniae. Gene 2012, 491, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, R.; Xu, T. The mitochondrial genome of the spinyhead croaker collichthys lucida: Genome organization and phylogenetic consideration. Mar. Genom. 2011, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.S.; Grande, T.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; Volume 4. [Google Scholar]
- Betancur, R.R.; Wiley, E.O.; Arratia, G.; Acero, A.; Bailly, N.; Miya, M.; Lecointre, G.; Orti, G. Phylogenetic classification of bony fishes. BMC Evol. Biol. 2017, 17, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miya, M.; Takeshima, H.; Endo, H.; Ishiguro, N.B.; Inoue, J.G.; Mukai, T.; Satoh, T.P.; Yamaguchi, M.; Kawaguchi, A.; Mabuchi, K.; et al. Major patterns of higher teleostean phylogenies: A new perspective based on 100 complete mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2003, 26, 121–138. [Google Scholar] [CrossRef]
- Near, T.J.; Dornburg, A.; Eytan, R.I.; Keck, B.P.; Smith, W.L.; Kuhn, K.L.; Moore, J.A.; Price, S.A.; Burbrink, F.T.; Friedman, M.; et al. Phylogeny and tempo of diversification in the superradiation of spiny-rayed fishes. Proc. Natl. Acad. Sci. USA 2013, 110, 12738–12743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.L.; Wheeler, W.C. Venom evolution widespread in fishes: A phylogenetic road map for the bioprospecting of piscine venoms. J. Hered. 2006, 97, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Betancur, R.R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; Lopez, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Cureton Ii, J.C.; et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, H.; Zhao, H.; Sun, J.; Han, X.; Zhu, S.; Li, G. The complete mitochondrial genome of the Plectorhinchus cinctus (teleostei, haemulidae). Mitochondrial DNA Part A 2016, 27, 842–843. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sun, J.; Zhao, H.; Chen, Y.; Yang, Z.; Li, G.; Liu, L. The complete mitochondrial genome of the Clarias fuscus (Siluriformes, Clariidae). Mitochondrial DNA A 2016, 27, 1255–1256. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Z.; Li, S.; Wu, X.; Peng, C.; Zhang, Y.; Lin, H. The complete mitochondrial genome of the orange-spotted grouper Epinephelus coioides (perciformes, serranidae). Mitochondrial DNA Part A 2016, 27, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.E.; Luo, H.; Luo, M.; Guo, J.; Deng, Z.; Zhao, B. The complete mitochondrial genome of the mudsnail Cipangopaludina cathayensis (gastropoda: Viviparidae). Mitochondrial DNA Part A 2016, 27, 1892–1894. [Google Scholar]
- Yang, H.; Zhao, H.; Sun, J.; Chen, Y.; Liu, L.; Zhang, Y.; Liu, L. The complete mitochondrial genome of the Hemibarbus medius (cypriniformes, Cyprinidae). Mitochondrial DNA Part A 2016, 27, 1070–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, H.; Sun, J.; Xie, Z.; Yang, Z.; Liu, L. The complete mitochondrial genome of the Culter recurviceps (teleostei, cyprinidae). Mitochondrial DNA Part A 2016, 27, 762–763. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, H.; Sun, J.; Zhang, Y.; Yang, Z.; Liu, L. The complete mitochondrial genome of the Hemibagrus wyckioides (siluriformes, Bagridae). Mitochondrial DNA Part A 2016, 27, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, H.; Xie, Z.; Sun, J.; Yang, Z.; Liu, L. The complete mitochondrial genome of the Hemibagrus guttatus (teleostei, Bagridae). Mitochondrial DNA Part A 2016, 27, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, H.; Sun, J.; Chen, Y.; Liu, L.; Li, G.; Liu, L. The complete mitochondrial genome of the Anabas testudineus (perciformes, Anabantidae). Mitochondrial DNA Part A 2016, 27, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.E.; Guo, J.; Deng, Z.; Luo, H.; Luo, M.; Zhao, B. The complete mitochondrial genome of the giant African snail Achatina fulica (mollusca: Achatinidae). Mitochondrial DNA Part A 2016, 27, 1622–1624. [Google Scholar]
- Shi, X.; Tian, P.; Lin, R.; Huang, D.; Wang, J. Characterization of the complete mitochondrial genome sequence of the globose head whiptail Cetonurus globiceps (gadiformes: Macrouridae) and its phylogenetic analysis. PLoS ONE 2016, 11, e0153666. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Qu, M.; Zhang, X.; Ding, S. A comprehensive description and evolutionary analysis of 22 grouper (perciformes, Epinephelidae) mitochondrial genomes with emphasis on two novel genome organizations. PLoS ONE 2013, 8, e73561. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P.; Ochman, H. Strand asymmetries in DNA evolution. Trends Genet. TIG 1997, 13, 240–245. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.L.; Li, Q.; Gu, Y.; Shi, B.C.; van Achterberg, C.; Wei, S.J.; Chen, X.X. The complete mitochondrial genome of Taeniogonalos taihorina (bischoff) (hymenoptera: Trigonalyidae) reveals a novel gene rearrangement pattern in the hymenoptera. Gene 2014, 543, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Zeng, H.H.; Huang, Y.; Zheng, Z.M. The complete mitochondrial genomes of three grasshoppers, Asiotmethis zacharjini, Filchnerella helanshanensis and Pseudotmethis rubimarginis (orthoptera: Pamphagidae). Gene 2013, 517, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Chao, Q.J.; Li, Y.D.; Geng, X.X.; Zhang, L.; Dai, X.; Zhang, X.; Li, J.; Zhang, H.J. Complete mitochondrial genome sequence of Marmota himalayana (rodentia: Sciuridae) and phylogenetic analysis within rodentia. Genet. Mol. Res. GMR 2014, 13, 2739–2751. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.P.; Miya, M.; Endo, H.; Nishida, M. Round and pointed-head grenadier fishes (actinopterygii: Gadiformes) represent a single sister group: Evidence from the complete mitochondrial genome sequences. Mol. Phylogenet. Evol. 2006, 40, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Catanese, G.; Manchado, M.; Infante, C. Evolutionary relatedness of mackerels of the genus scomber based on complete mitochondrial genomes: Strong support to the recognition of Atlantic scomber colias and pacific scomber japonicus as distinct species. Gene 2010, 452, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Xia, K.; Jiang, S. Complete mitochondrial DNA sequence of the yellowfin seabream acanthopagrus latus and a genomic comparison among closely related sparid species. Mitochondrial DNA 2008, 19, 385–393. [Google Scholar] [PubMed]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial trnas of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Ji, R.; Ding, F.; Qi, D.; Gao, H.; Meng, H.; Yu, J.; Hu, S.; Zhang, H. A complete mitochondrial genome sequence of the wild two-humped camel (camelus bactrianus ferus): An evolutionary history of camelidae. BMC Genom. 2007, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, J.Y.; Zheng, R.Q.; Yu, B.G.; Yang, G. Complete nucleotide sequence and gene organization of the mitochondrial genome of Paa spinosa (anura: Ranoidae). Gene 2009, 447, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal trna: Unique structure of ascaris suum mitochondrial tRNA(SER)(UCU) having a short t arm and lacking the entire d arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef]
- Zardoya, R.; Meyer, A. The complete DNA sequence of the mitochondrial genome of a “living fossil,” the coelacanth (latimeria chalumnae). Genetics 1997, 146, 995–1010. [Google Scholar] [PubMed]
- Li, T.; Yang, J.; Li, Y.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. A mitochondrial genome of Rhyparochromidae (hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.D.; Yang, R.H.; Hsiang, T.; Wang, K.; Liang, D.Q.; Liang, F.; Cao, D.M.; Zhou, F.; Wen, G.; et al. Complete mitochondrial genome of the medicinal fungus Ophiocordyceps sinensis. Sci. Rep. 2015, 5, 13892. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Nadler, S.A.; Liu, S.S.; Podolska, M.; D’Amelio, S.; Shao, R.; Gasser, R.B.; Zhu, X.Q. Mitochondrial phylogenomics yields strongly supported hypotheses for Ascaridomorph nematodes. Sci. Rep. 2016, 6, 39248. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.L.; Shao, G.Z.; Lai, C.F. Latin-Chinese International Fishes Manual; Aquatic Press: Taiwan, 1999. [Google Scholar]
- Betancur, R.R.; Orti, G.; Pyron, R.A. Fossil-based comparative analyses reveal ancient marine ancestry erased by extinction in ray-finned fishes. Ecol. Lett. 2015, 18, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012. [Google Scholar]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. Mitofish and mitoannotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. Trnascan-se on-line: Integrating search and context for analysis of transfer rna genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. Arwen: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, A.C.; Lessinger, A.C.; Torres, T.T.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Azeredo Espin, A.M. The mitochondrial genome of the blowfly Chrysomya chloropyga (diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Issac, B.; Raghava, G.P.; Ramaswamy, R. Spectral repeat finder (SRF): Identification of repetitive sequences using fourier transformation. Bioinformatics 2004, 20, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation inde—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 2012, 40, W569–W572. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. Timetree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession Number | Length (bp) | Entire Genome | Protein-Coding Gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (%) | T (%) | C (%) | G (%) | AT (%) | AT-Skew | GC-Skew | Length (aa) | AT (%) (all) | AT (%) (3rd) | AT-Skew | GC-Skew | |||
| Protonibea diacanthus | KM257722.1 | 16,535 | 27.54 | 25.34 | 31.09 | 16.03 | 52.88 | 0.0416 | −0.3195 | 3795 | 51.83 | 47.28 | −0.0123 | −0.3904 |
| Argyrosomus amoyensis | KM257863.1 | 16,490 | 26.96 | 25.31 | 31.02 | 16.71 | 52.27 | 0.0316 | −0.2998 | 3802 | 51.23 | 51.43 | −0.0205 | −0.371 |
| Nibea coibor | KM233452.1 | 16,502 | 26.88 | 25.19 | 31.62 | 16.31 | 52.07 | 0.0324 | −0.3193 | 3791 | 50.91 | 49.3 | −0.0191 | −0.3955 |
| Johnius belangerii | NC_022464.1 | 19,154 | 24.96 | 36.79 | 17.59 | 20.67 | 61.75 | −0.1916 | 0.0805 | 3802 | 60.89 | 62.19 | −0.3011 | 0.0273 |
| Pennahia argentata | NC_015202.1 | 16,485 | 27.46 | 26.33 | 30.18 | 16.04 | 53.79 | 0.021 | −0.3059 | 3802 | 52.98 | 54.92 | −0.0357 | −0.3747 |
| Argyrosomus japonicus | NC_017610.1 | 16,496 | 26.99 | 25.29 | 31.02 | 16.69 | 52.28 | 0.0326 | −0.3002 | 3802 | 51.24 | 51.43 | −0.0196 | −0.372 |
| Dendrophysa russelii | NC_017606.1 | 16,626 | 27.28 | 26.12 | 30.4 | 16.2 | 53.4 | 0.0217 | −0.3048 | 3801 | 52.48 | 54.02 | −0.0302 | −0.3797 |
| Pennahia macrocephalus | NC_031409.1 | 16,508 | 27.51 | 25.28 | 31.22 | 15.99 | 52.79 | 0.0423 | −0.3227 | 3793 | 51.7 | 47.92 | −0.009 | −0.3975 |
| Bahaba taipingensis | NC_018347.1 | 16,500 | 27.55 | 25.15 | 31.41 | 15.9 | 52.7 | 0.0457 | −0.3279 | 3802 | 51.67 | 51.06 | 0 | −0.4051 |
| Nibea albiflora | NC_015205.1 | 16,499 | 26.4 | 25.88 | 30.81 | 16.91 | 52.28 | 0.0099 | −0.2913 | 3797 | 51.26 | 49.61 | −0.0477 | −0.3604 |
| Larimichthys crocea | NC_011710.1 | 16,466 | 27.55 | 25.46 | 30.69 | 16.3 | 53.01 | 0.0393 | −0.3062 | 3800 | 52.2 | 52.26 | −0.0169 | −0.3745 |
| Collichthys lucidus | NC_014350.1 | 16,442 | 27.95 | 25.63 | 30.69 | 15.73 | 53.58 | 0.0434 | −0.3223 | 3801 | 52.98 | 53.49 | −0.0081 | −0.3909 |
| Sciaenops ocellatus | NC_016867.1 | 16,500 | 27.47 | 25.47 | 30.72 | 16.35 | 52.94 | 0.0378 | −0.3054 | 3803 | 52 | 51.42 | −0.0118 | −0.3803 |
| Nibea miichthioides | NC_029875.1 | 16,490 | 26.96 | 25.32 | 31.04 | 16.69 | 52.28 | 0.0313 | −0.3006 | 3802 | 51.25 | 51.54 | −0.0189 | −0.3728 |
| Johnius grypotus | NC_021130.1 | 18,523 | 24.99 | 36.18 | 18.57 | 20.27 | 61.17 | −0.183 | 0.0439 | 3803 | 60.61 | 62.51 | −0.2738 | 0.0007 |
| Collichthys niveatus | NC_014263.1 | 16,469 | 27.59 | 24.96 | 31.3 | 16.16 | 52.55 | 0.0499 | −0.319 | 3801 | 51.61 | 51.76 | 0.0003 | −0.3876 |
| Larimichthys polyactis | NC_013754.1 | 16,470 | 27.55 | 25 | 31.27 | 16.18 | 52.55 | 0.0486 | −0.318 | 3801 | 51.65 | 51.84 | −0.0012 | −0.3866 |
| Chrysochir aureus | NC_016987.1 | 16,505 | 26.93 | 25.17 | 31.64 | 16.25 | 52.1 | 0.0337 | −0.3214 | 3802 | 50.9 | 49.3 | −0.0199 | −0.3947 |
| Miichthys miiuy | NC_014351.1 | 16,493 | 27.49 | 24.43 | 32.19 | 15.89 | 51.92 | 0.059 | −0.339 | 3802 | 50.63 | 49.93 | 0.0124 | −0.4165 |
| Gene/Element | Strand | Size (bp) | GC_Percent (%) | Amino Acids (aa) | Inferred Initiation Codon | Inferred Termination Codon | One Letter Code | Anti-Codon | Intergenic Nucleotide * (bp) |
|---|---|---|---|---|---|---|---|---|---|
| tRNA-Phe | H | 68–70 | 40.58–45.50 | F | GAA | 0–45 | |||
| 12SrRNA | H | 907–953 | 48.05–48.58 | 0 | |||||
| tRNA-Val | H | 71–72 | 46.48–50.00 | V | TAC | 0 | |||
| 16SrRNA | H | 1696–1720 | 46.74–47.59 | 0 | |||||
| tRNA-Leu | H | 74 | 44.59–48.65 | L | TAA | 0 | |||
| ND1 | H | 975 | 48.00–49.13 | 324 | ATG, GTG | TAG, TAA | 4 | ||
| tRNA-Ile | H | 70 | 42.86–51.43 | I | GAT | −1 | |||
| tRNA-Gln | L | 71 | 43.66–46.48 | Q | TTG | −1 | |||
| tRNA-Met | H | 69 | 39.13–44.93 | M | CAT | 0 | |||
| ND2 | H | 1045–1046 | 47.90–50.72 | 348 | ATG | TA, T | 0 | ||
| tRNA-Trp | H | 71 | 49.30–50.70 | W | TCA | 0–1 | |||
| tRNA-Ala | L | 69 | 37.68–42.03 | A | TGC | 2 | |||
| tRNA-Asn | L | 72–73 | 45.21–50.68 | N | GTT | 34–36 | |||
| tRNA-Cys | L | 66 | 43.94–53.03 | C | GCA | 0 | |||
| tRNA-Tyr | L | 70 | 45.71–50.00 | Y | GTA | 1 | |||
| COXI | H | 1557 | 49.13–49.90 | 518 | ATG | AGA | −5 | ||
| tRNA-Ser | L | 71 | 46.48–50.70 | S | TGA | 3 | |||
| tRNA-Asp | H | 69 | 40.58–46.38 | D | GTC | 8 | |||
| COXII | H | 687–691 | 44.69–45.59 | 228–230 | ATG | T, AGA | 0–5 | ||
| tRNA-Lys | H | 74–75 | 46.67–50.00 | K | TTT | 1 | |||
| ATP8 | H | 168 | 40.48–46.43 | 55 | ATG | TAA | (−10)–17 | ||
| ATP6 | H | 657–684 | 47.79–49.12 | 218–227 | ATG, ATA, GTG | TA, TAA | 0–1 | ||
| COXIII | H | 785–786 | 48.03–50.25 | 261 | ATG | TA, TAA | (−1)–0 | ||
| tRNA-Gly | H | 71 | 35.21–36.62 | G | TCC | 0 | |||
| ND3 | H | 349 | 50.14–52.44 | 116 | ATG | T | 0 | ||
| tRNA-Arg | H | 69 | 36.23–37.68 | R | TCG | 0 | |||
| ND4L | H | 297 | 49.83–53.20 | 98 | ATG | TAA | −7 | ||
| ND4 | H | 1381 | 48.08–49.67 | 460 | ATG | T | 0 | ||
| tRNA-His | H | 69–72 | 34.78–36.11 | H | GTG | (−1)–0 | |||
| tRNA-Ser | H | 68–71 | 54.41–57.35 | S | GCT | 2–5 | |||
| tRNA-Leu | H | 73 | 43.84–49.32 | L | TAG | 0–15 | |||
| ND5 | H | 1824–1839 | 47.47–47.80 | 607–612 | ATG, ATC | TAA, TAG | −4 | ||
| ND6 | L | 522 | 45.40–50.77 | 173 | ATG | TAA, TAG | 0–1 | ||
| tRNA-Glu | L | 69 | 39.13–46.38 | E | TTC | 3–5 | |||
| Cytb | H | 1137–1141 | 48.72–50.57 | 378–380 | ATG | T, TAA | 0 | ||
| tRNA-Thr | H | 72 | 52.78–56.94 | T | TGT | 3–5 | |||
| tRNA-Pro | L | 70 | 34.29–38.57 | P | TGG | 0 | |||
| D-loop | H | 821–838 | 35.92–37.50 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Xia, J.; Zhang, J.-e.; Yang, J.; Zhao, H.; Wang, Q.; Sun, J.; Xue, H.; Wu, Y.; Chen, J.; et al. Characterization of the Complete Mitochondrial Genome Sequences of Three Croakers (Perciformes, Sciaenidae) and Novel Insights into the Phylogenetics. Int. J. Mol. Sci. 2018, 19, 1741. https://doi.org/10.3390/ijms19061741
Yang H, Xia J, Zhang J-e, Yang J, Zhao H, Wang Q, Sun J, Xue H, Wu Y, Chen J, et al. Characterization of the Complete Mitochondrial Genome Sequences of Three Croakers (Perciformes, Sciaenidae) and Novel Insights into the Phylogenetics. International Journal of Molecular Sciences. 2018; 19(6):1741. https://doi.org/10.3390/ijms19061741
Chicago/Turabian StyleYang, Huirong, Jun Xia, Jia-en Zhang, Jinzeng Yang, Huihong Zhao, Qing Wang, Jijia Sun, Huayi Xue, Yuanyuan Wu, Jiehu Chen, and et al. 2018. "Characterization of the Complete Mitochondrial Genome Sequences of Three Croakers (Perciformes, Sciaenidae) and Novel Insights into the Phylogenetics" International Journal of Molecular Sciences 19, no. 6: 1741. https://doi.org/10.3390/ijms19061741