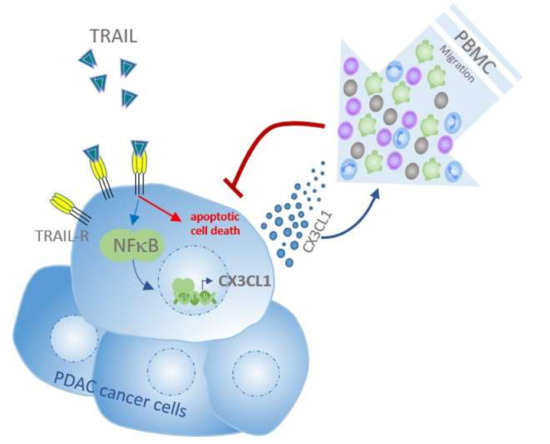
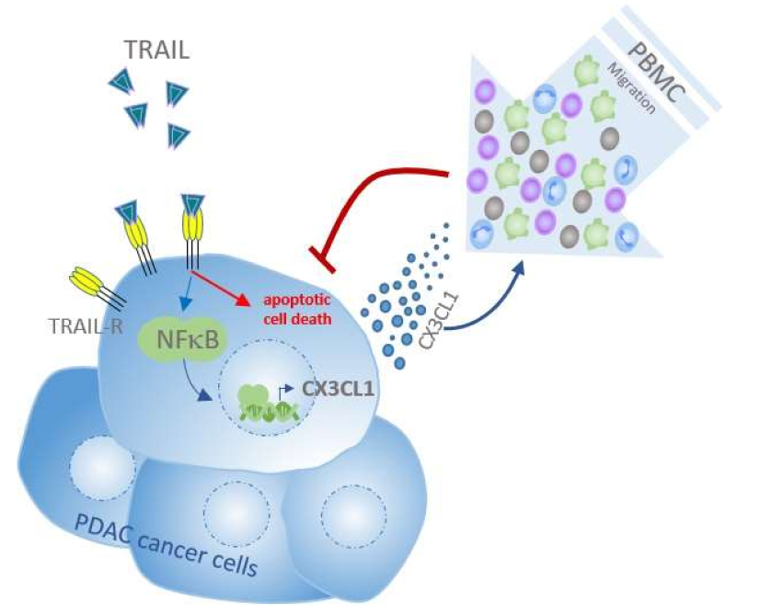
TRAIL/NF-κB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines

 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
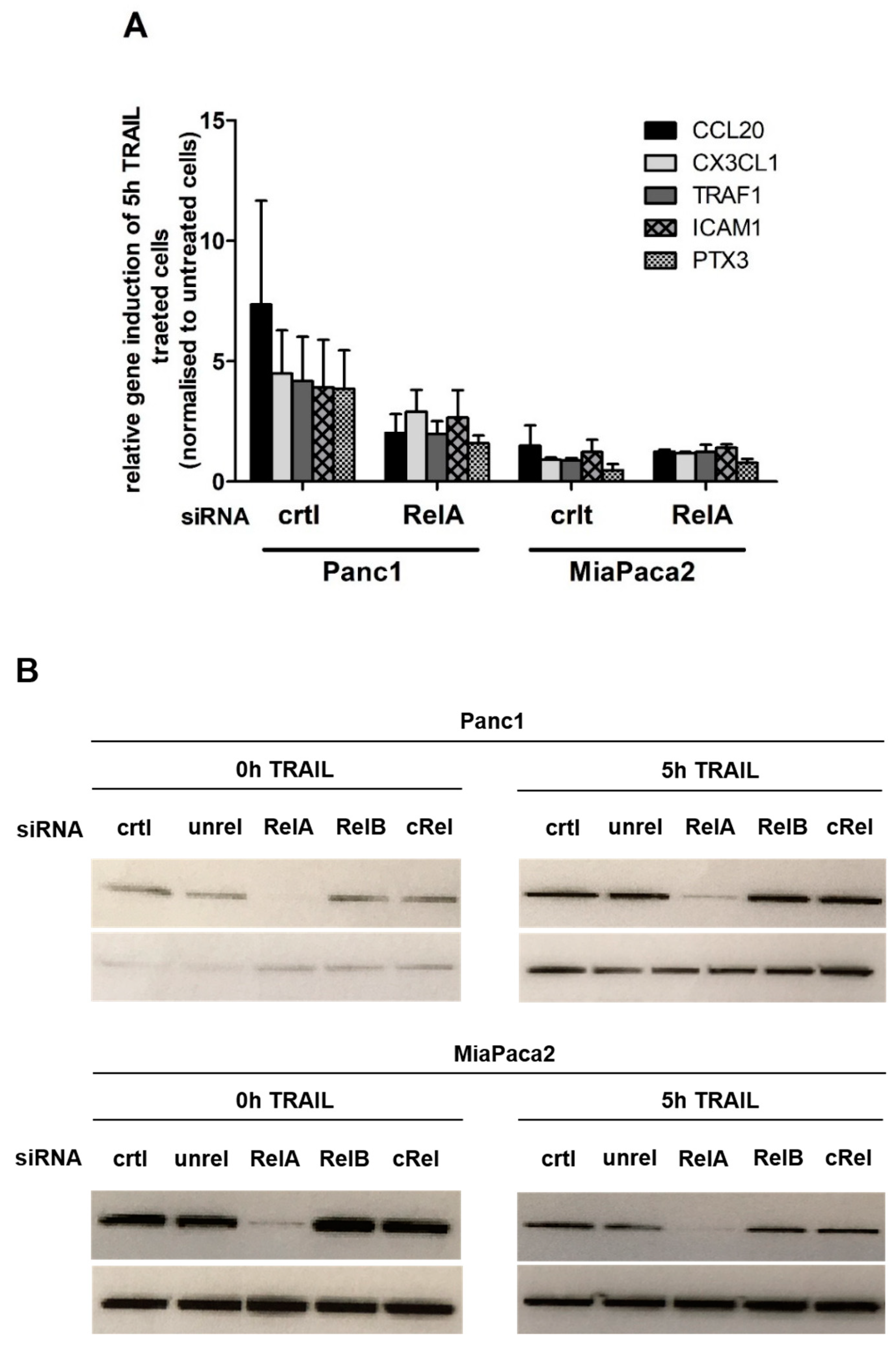
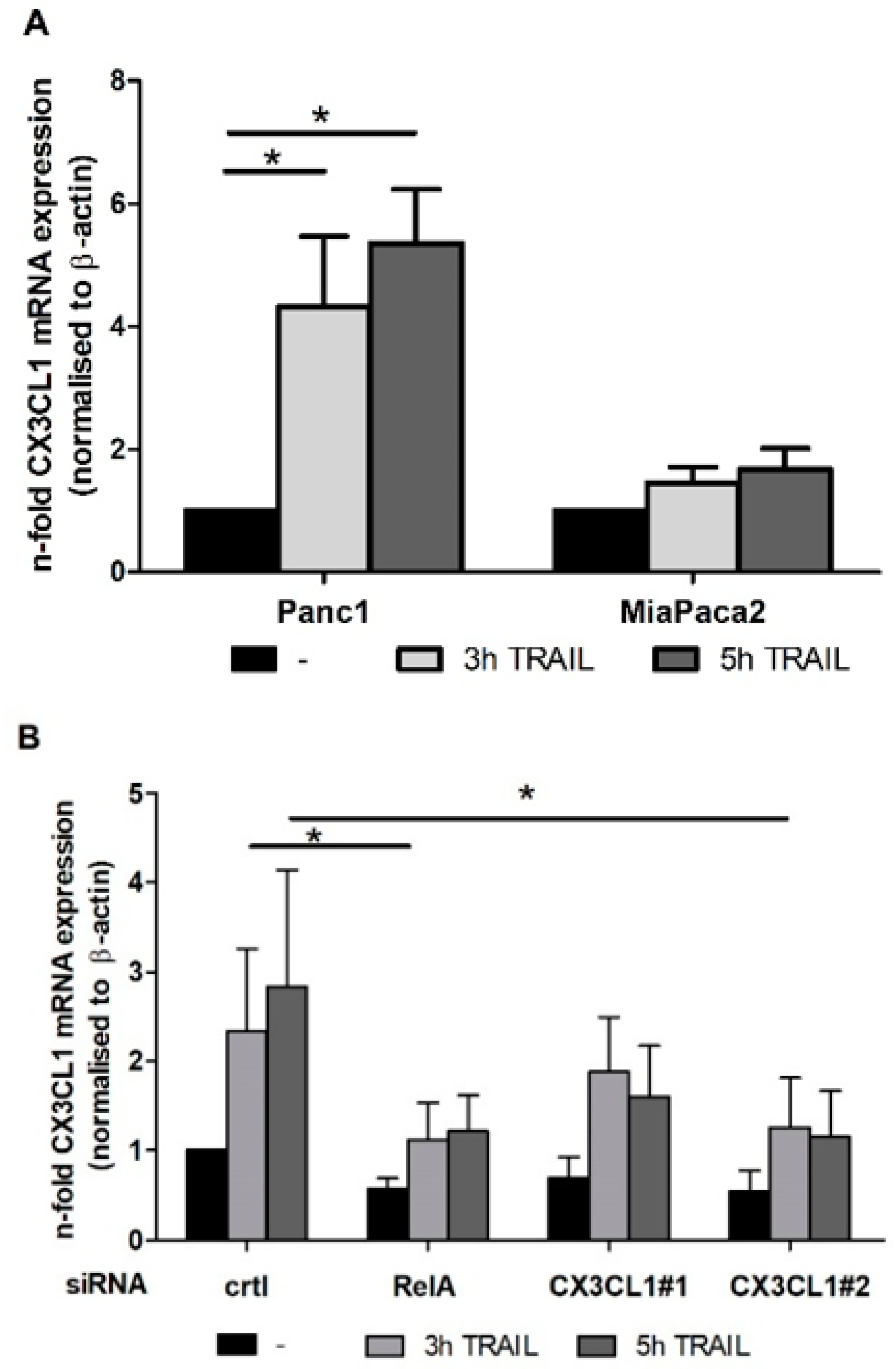
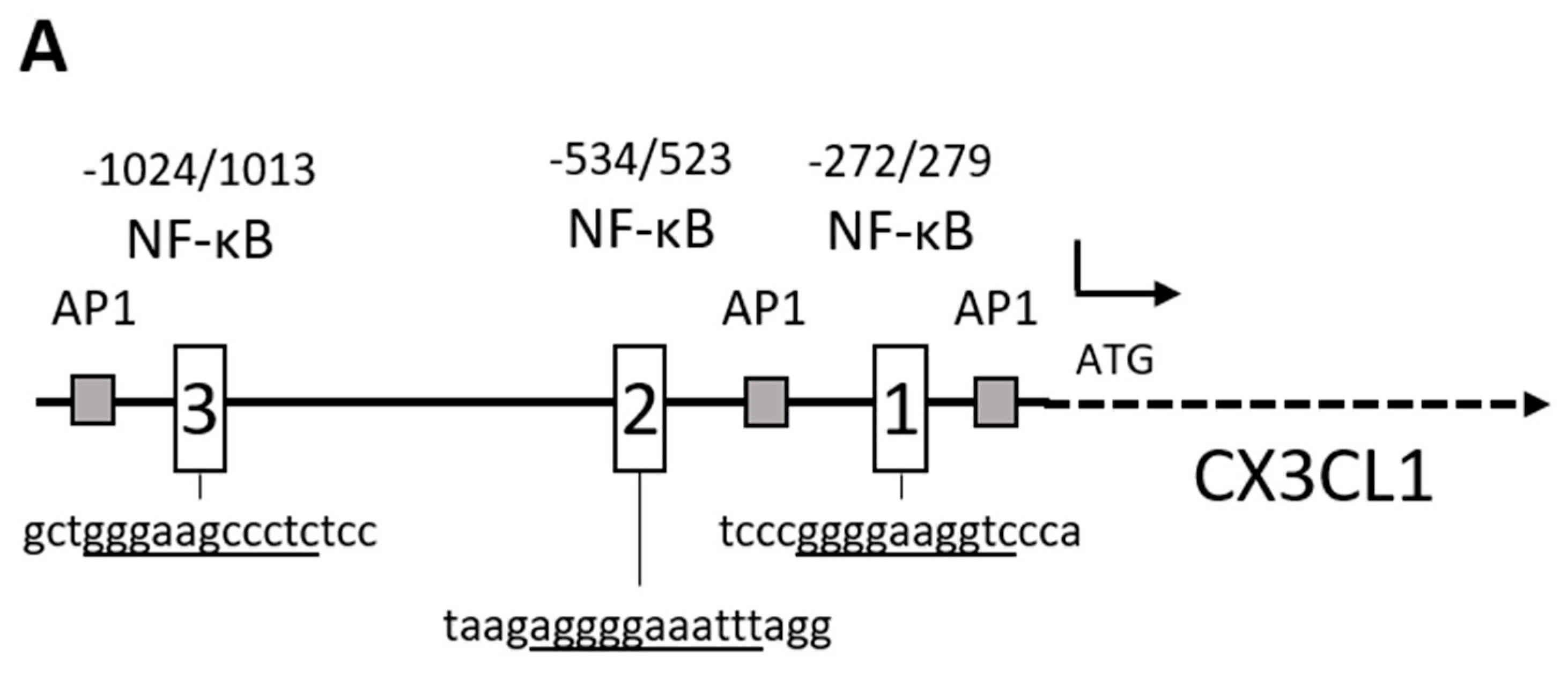
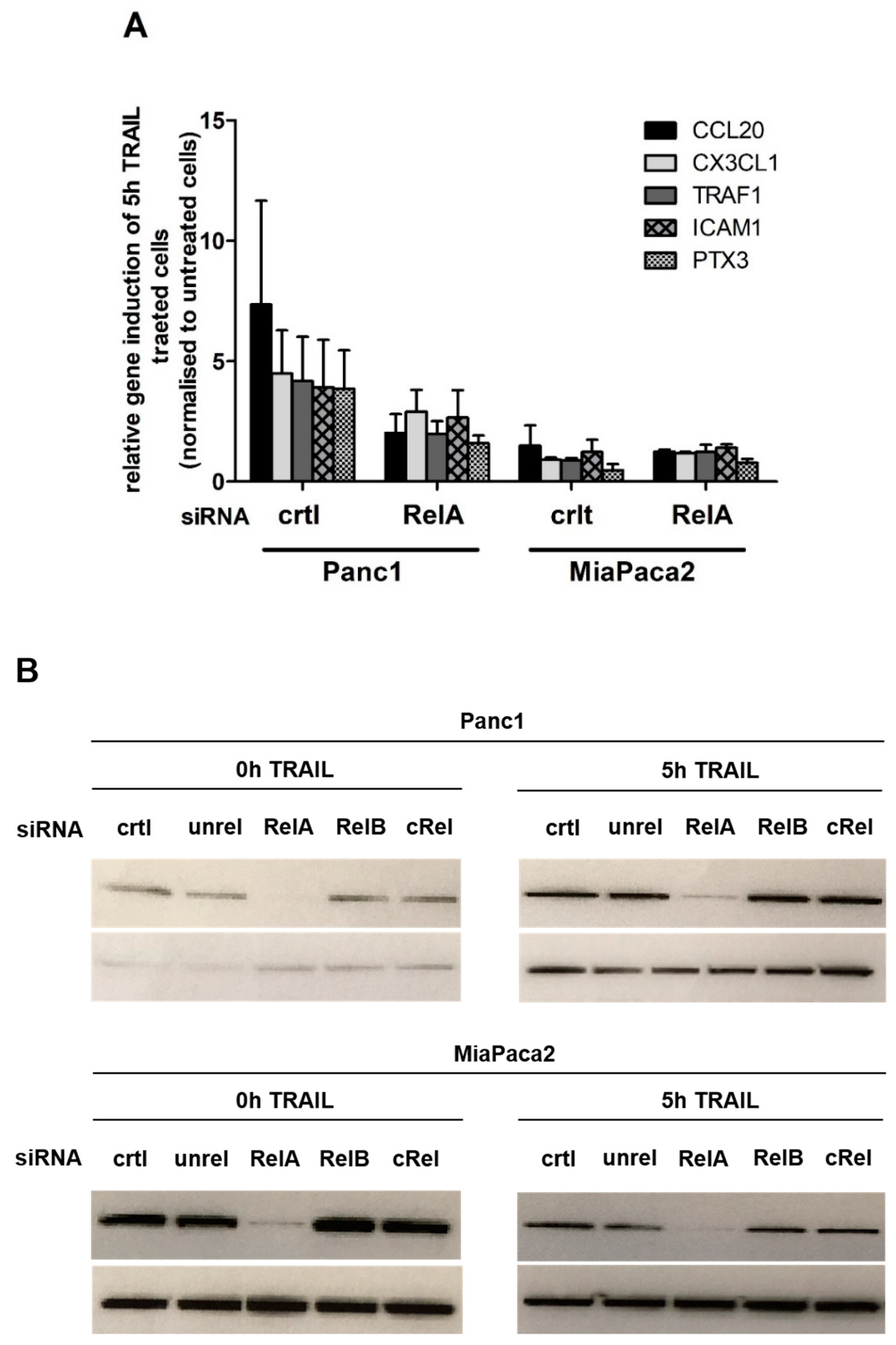
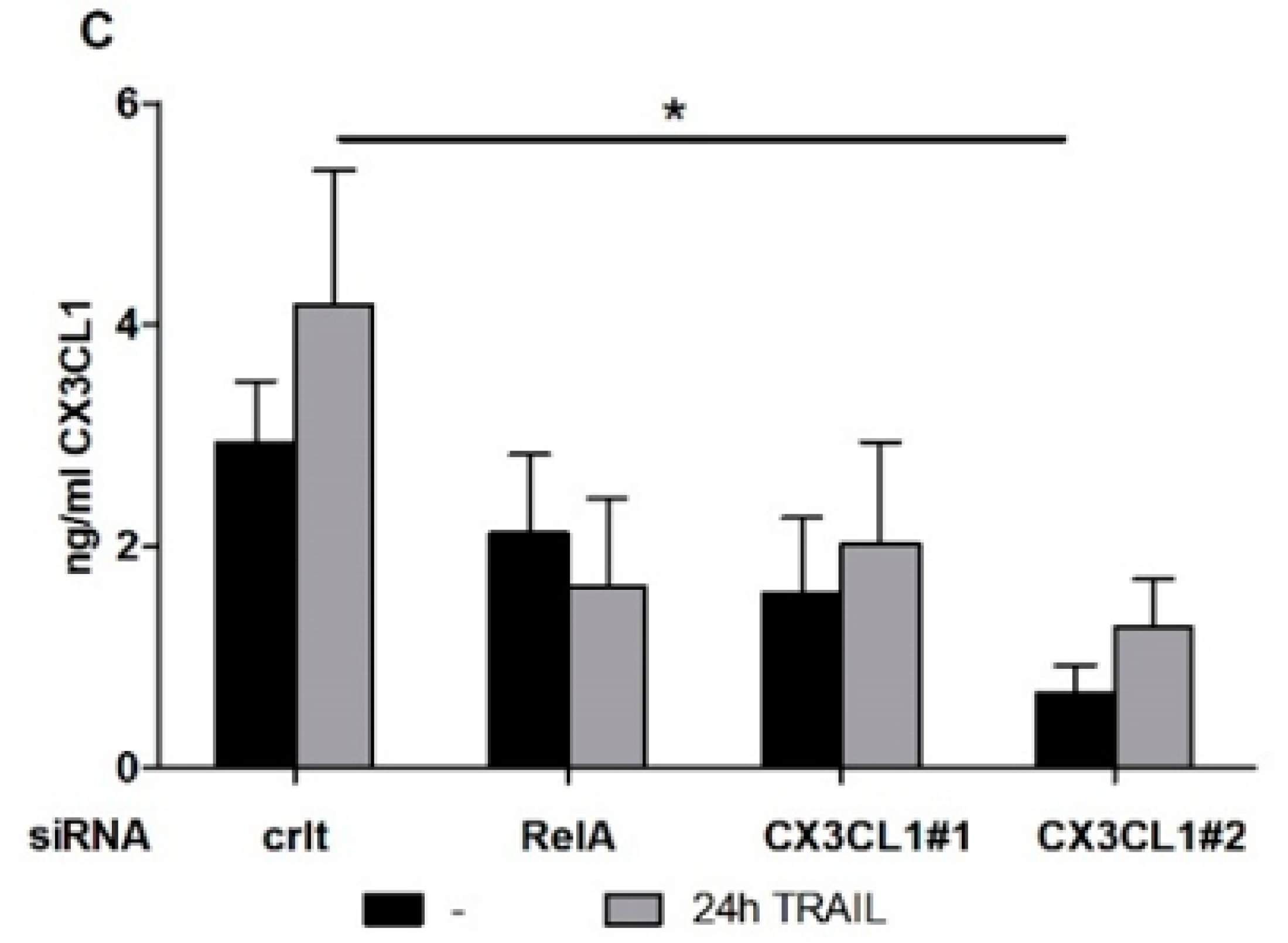
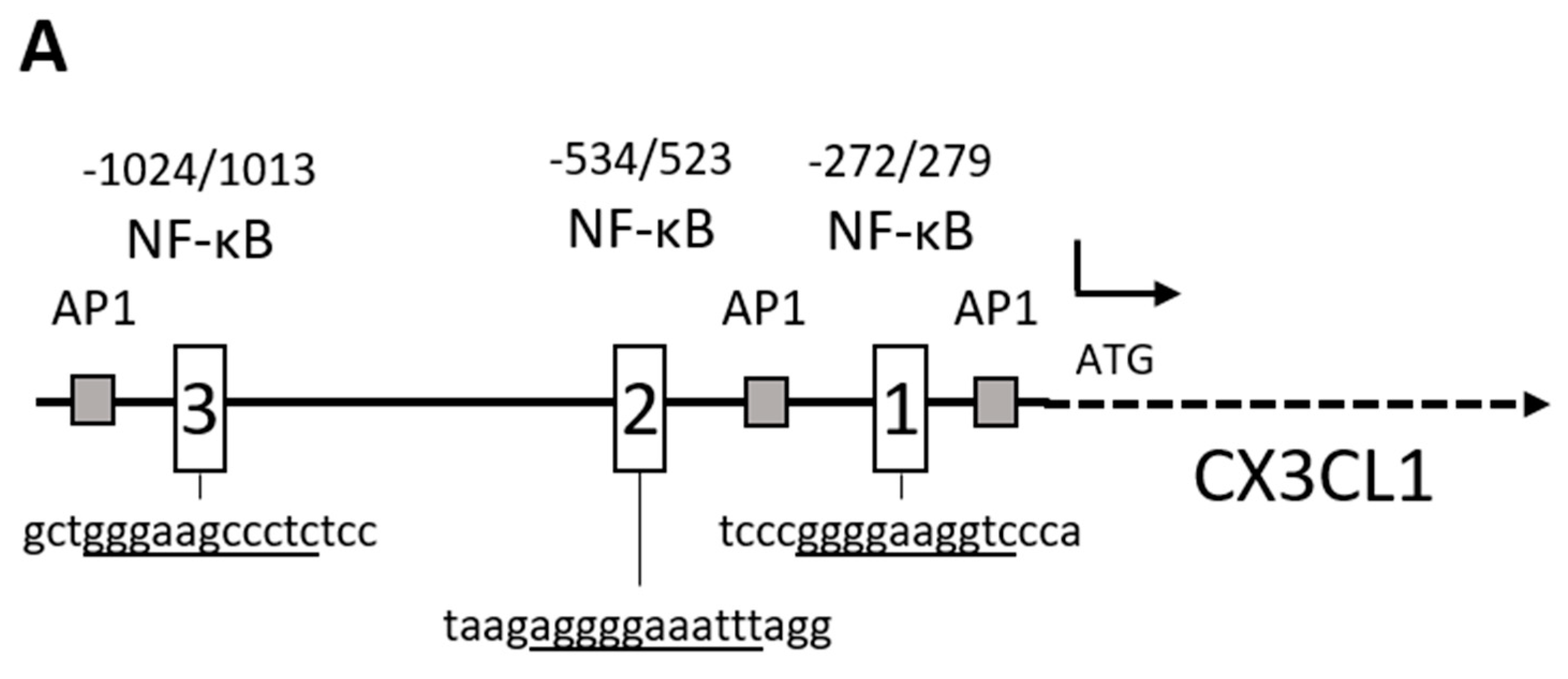
2.1. Chemokine CX3CL1 is a RelA Target Gene
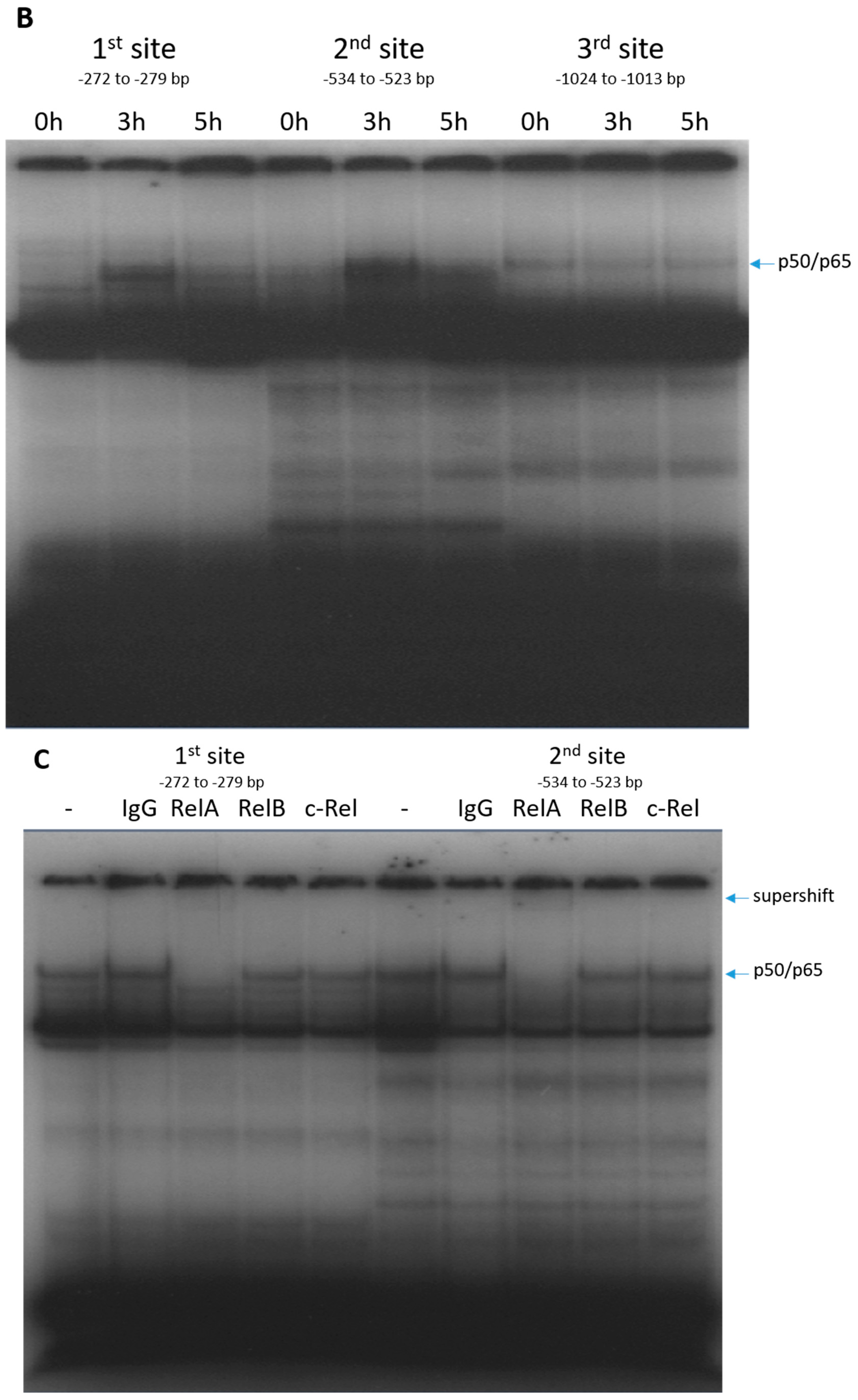
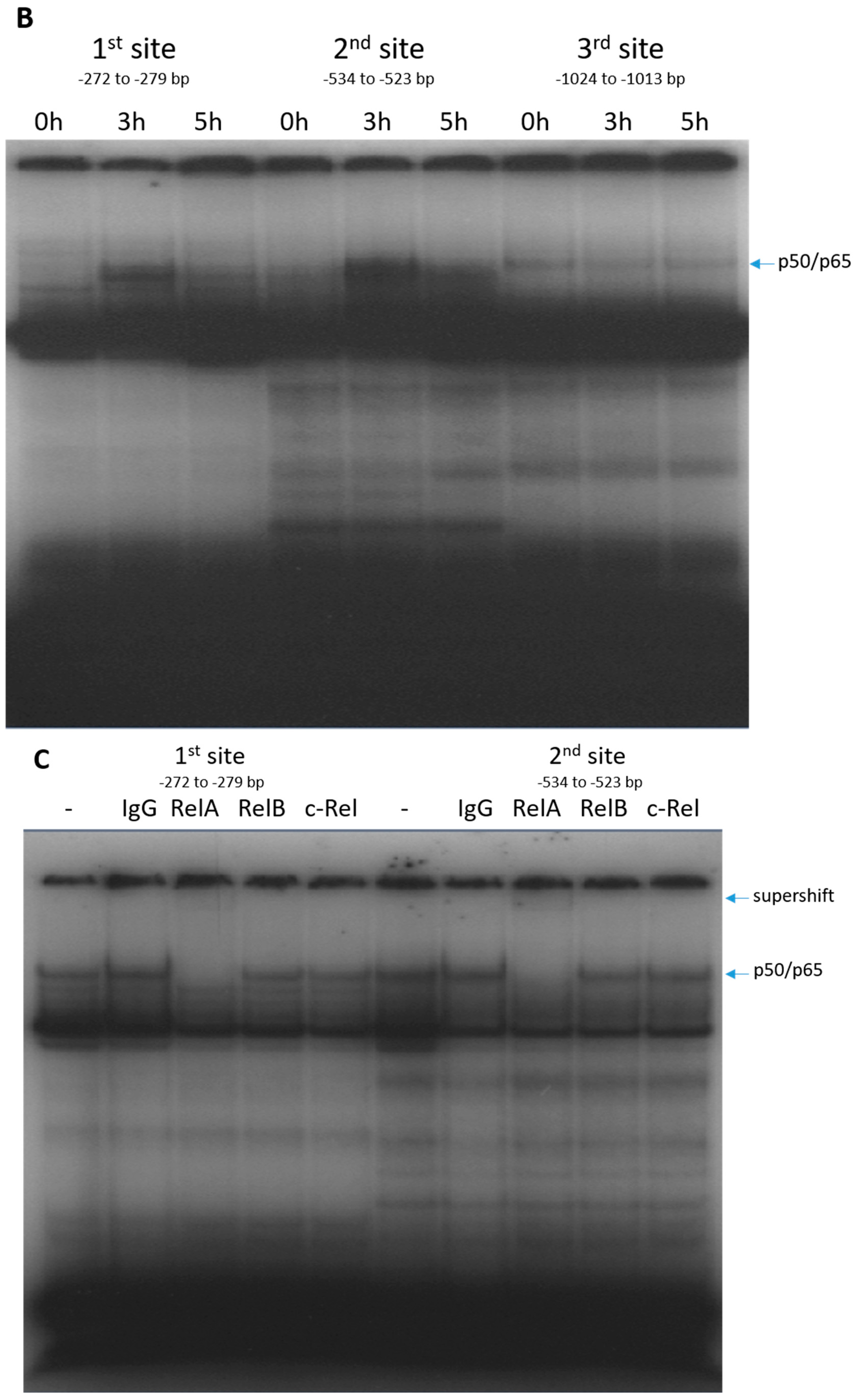
2.2. RelA Binds to CX3CL1 Promoter after TRAIL Treatment
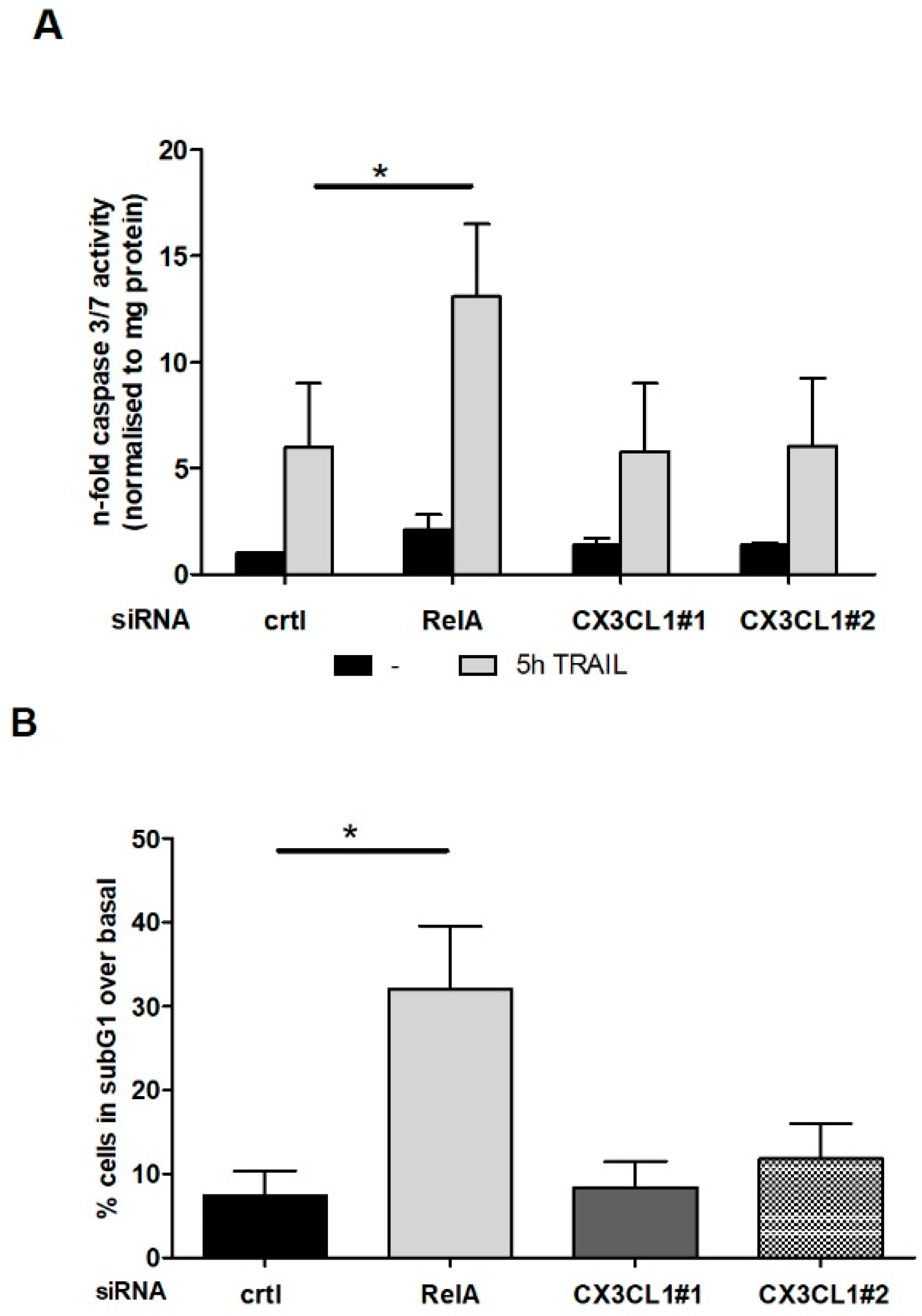
2.3. CX3CL1 Does Not Induce Direct Apoptosis in Panc1 Cells
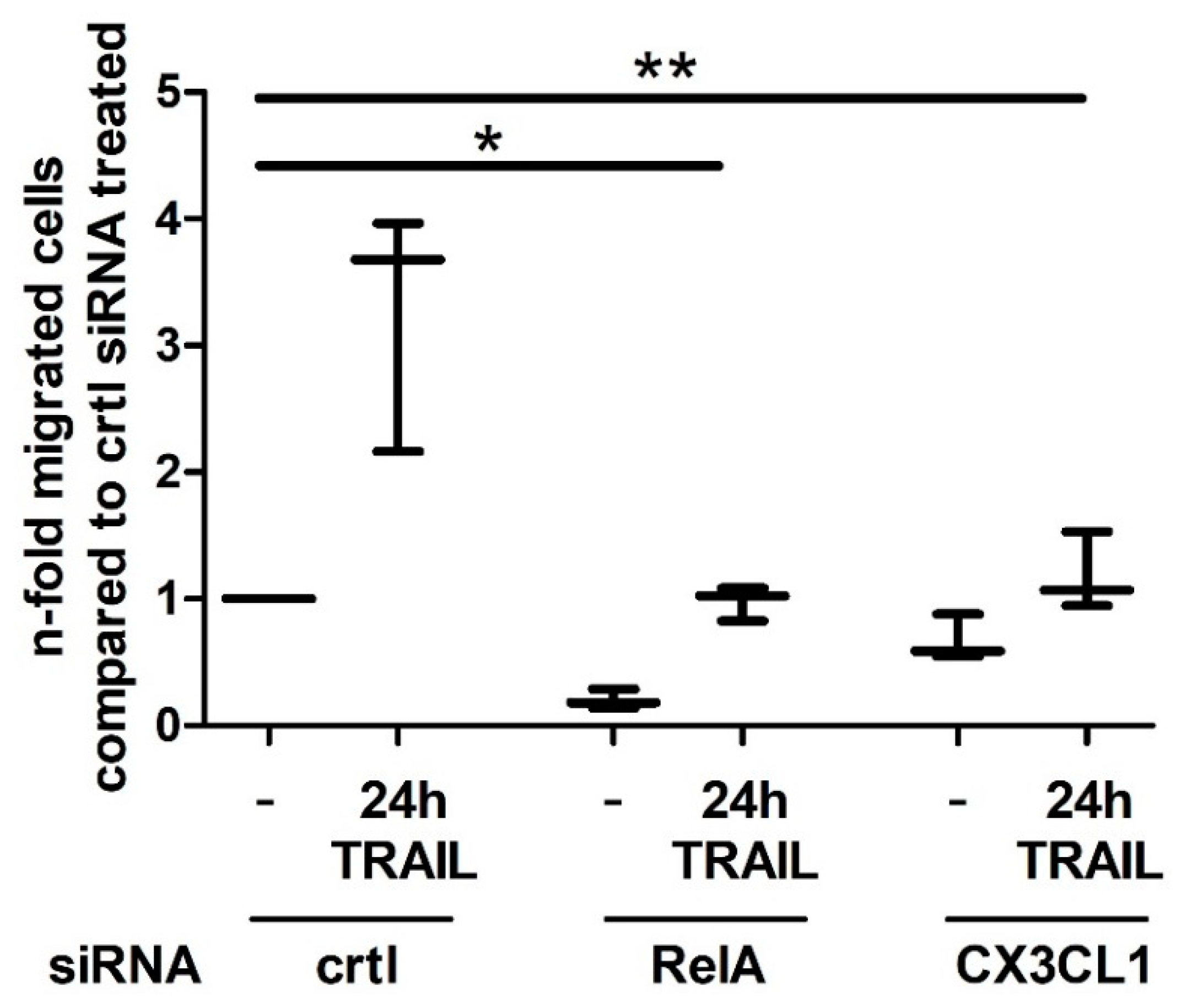
2.4. CX3CL1 Enhances the Migration of PBMCs towards PDAC Cells
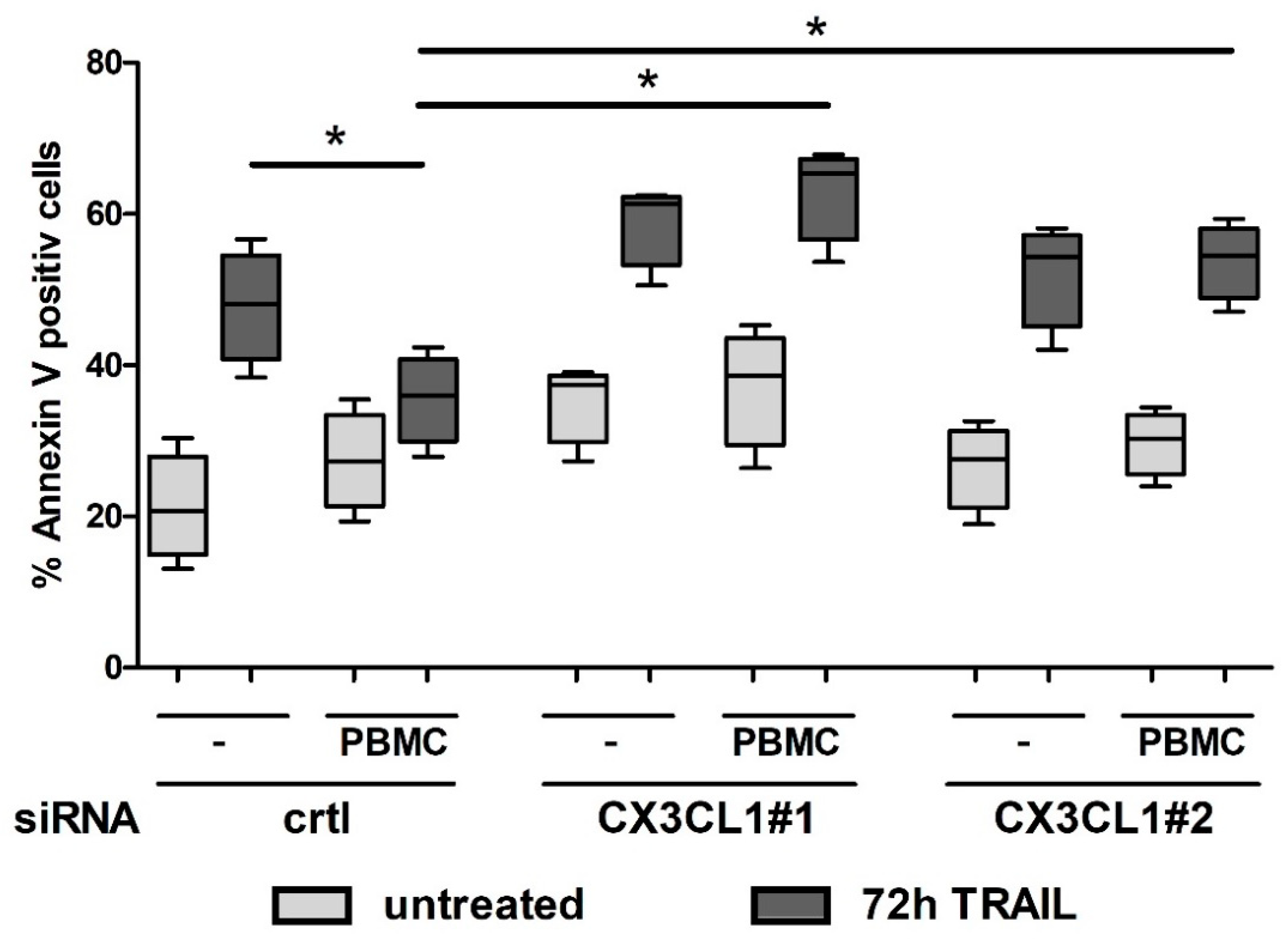
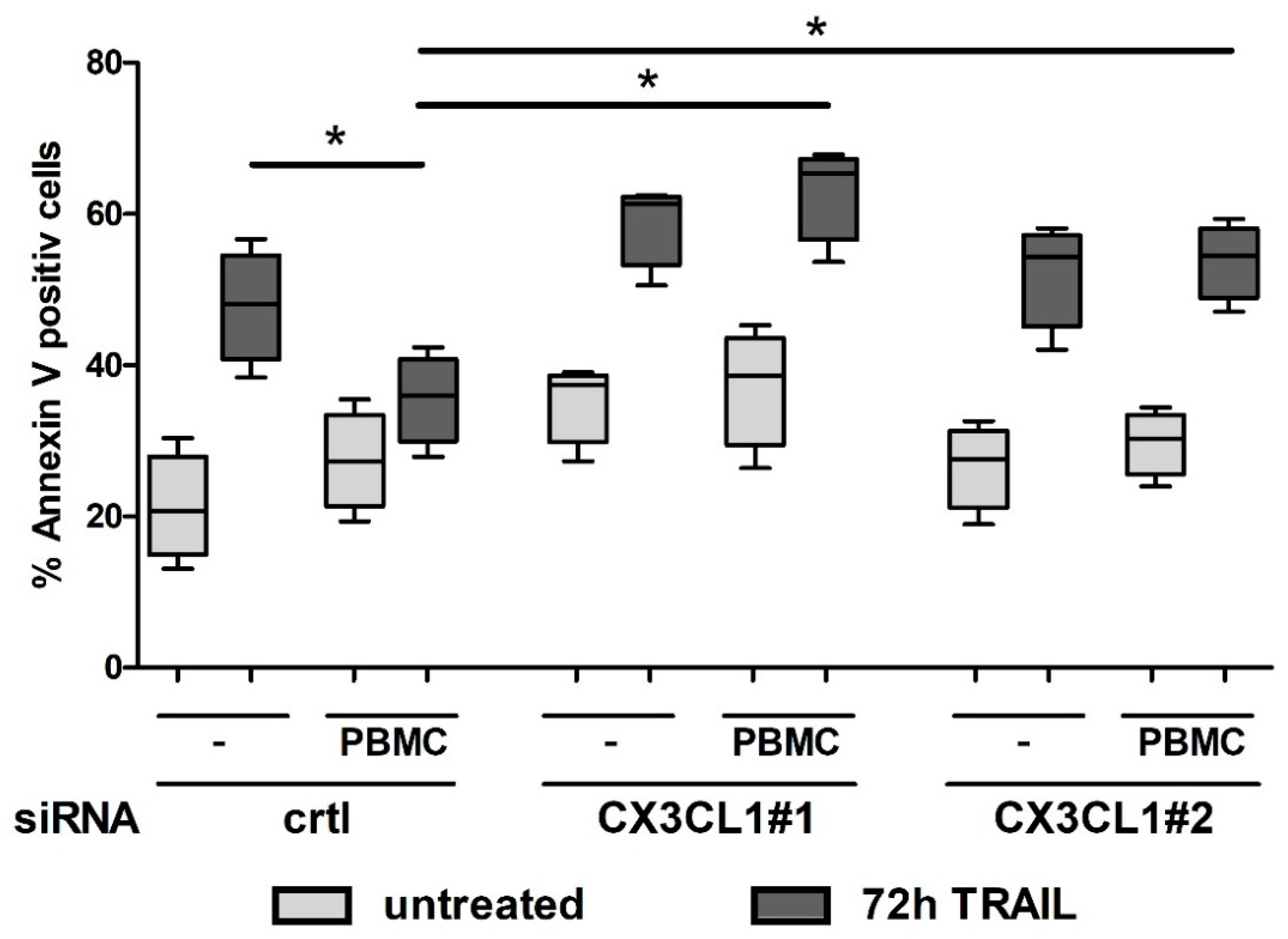
2.5. CX3CL1 Attracted PBMCs Interfere with TRAIL Mediated Apoptosis in Panc1 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. RNA Preparation and Realtime PCR
4.4. siRNA Transfection
4.5. Gel Shift Assays
4.6. Genome Wide Transcriptome Profiling and Cluster Analysis
4.7. PBMC Isolation
4.8. Migration Assay
4.9. Co-Culture Assay
4.10. ELISA
4.11. Western Blotting
4.12. Annexin V/PI Assay
4.13. Caspase-3/-7 Assay
4.14. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | pancreatic ductal adenocarcinoma |
| TRAIL | TNF alpha related apoptosis inducing ligand |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CX3CR1 | CX3C chemokine receptor 1 |
| RelA | nuclear factor NF-kappa-B p65 subunit |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Van Audenaerde, J.R.M.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E.L.J. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y. Inflammatory Cytokine Signaling during Development of Pancreatic and Prostate Cancers. J. Immunol. Res. 2017, 2017, 7979637. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Bastea, L.; Fleming, A.; Doppler, H.; Edenfield, B.H.; Dawson, D.W.; Zhang, L.; Bardeesy, N.; Storz, P. The Presence of Interleukin-13 at Pancreatic ADM/PanIN Lesions Alters Macrophage Populations and Mediates Pancreatic Tumorigenesis. Cell Rep. 2017, 19, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Grohmann, F.; Dreher, A.; Hasler, R.; Rosenstiel, P.; Legler, K.; Hauser, C.; Egberts, J.H.; Sipos, B.; Schreiber, S.; et al. Role of CCL20 mediated immune cell recruitment in NF-kappaB mediated TRAIL resistance of pancreatic cancer. Biochim. Biophys. Acta 2017, 1864, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Grohmann, F.; Sebens, S.; Wirths, G.; Dreher, A.; Hasler, R.; Rosenstiel, P.; Hauser, C.; Egberts, J.H.; Trauzold, A.; et al. c-Rel is a critical mediator of NF-kappaB-dependent TRAIL resistance of pancreatic cancer cells. Cell Death Dis. 2014, 5, e1455. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Muerkoster, S.S.; Schafer, H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett. 2013, 332, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Sahu, R.P.; Kulkarni-Datar, K.; Srivastava, S.K.; Brown, T.L. BITC Sensitizes Pancreatic Adenocarcinomas to TRAIL-induced Apoptosis. Cancer Growth Metastasis 2010, 2009, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.L.; von Karstedt, S.; Hillenbrand, A.; Henne-Bruns, D.; Knippschild, U.; Trauzold, A.; Lemke, J. Should We Keep Walking along the Trail for Pancreatic Cancer Treatment? Revisiting TNF-Related Apoptosis-Inducing Ligand for Anticancer Therapy. Cancers 2018, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Arlt, A.; Sebens, S.; Schafer, H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco Targets Ther. 2014, 7, 1497–1518. [Google Scholar] [PubMed]
- Zwacka, R.M.; Stark, L.; Dunlop, M.G. NF-kappaB kinetics predetermine TNF-alpha sensitivity of colorectal cancer cells. J. Gene Med. 2000, 2, 334–343. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Schafer, H.; Kalthoff, H. The ‘N-factors’ in pancreatic cancer: Functional relevance of NF-kappaB, NFAT and Nrf2 in pancreatic cancer. Oncogenesis 2012, 1, e35. [Google Scholar] [CrossRef] [PubMed]
- Plantivaux, A.; Szegezdi, E.; Samali, A.; Egan, L. Is there a role for nuclear factor kappaB in tumor necrosis factor-related apoptosis-inducing ligand resistance? Ann. N. Y. Acad. Sci. 2009, 1171, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.P.; Farrow, B.J.; Kim, S.; May, M.J.; Hellmich, M.R.; Evers, B.M. Selective targeting of the nuclear factor-kappaB pathway enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated pancreatic cancer cell death. Surgery 2002, 132, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.H.; Trauzold, A.; Roder, C.; Pan, G.; Zheng, C.; Kalthoff, H. The potential molecular mechanism of overexpression of uPA, IL-8, MMP-7 and MMP-9 induced by TRAIL in pancreatic cancer cell. Hepatobiliary Pancreat Dis. Int. 2008, 7, 201–209. [Google Scholar] [PubMed]
- Greten, F.R.; Weber, C.K.; Greten, T.F.; Schneider, G.; Wagner, M.; Adler, G.; Schmid, R.M. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology 2002, 123, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Sun, B.; Jiang, H.; Pan, S.; Chen, H.; Wang, S.; Krissansen, G.W.; Sun, X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010, 291, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Gehrz, A.; Muerkoster, S.; Vorndamm, J.; Kruse, M.L.; Folsch, U.R.; Schafer, H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003, 22, 3243–3251. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Y.; Chen, J.; Ma, H.; Shao, Z.; Chen, H.; Jin, G. High expression of CX3CL1/CX3CR1 axis predicts a poor prognosis of pancreatic ductal adenocarcinoma. J. Gastrointest. Surg. 2012, 16, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Gan, A.M.; Butoi, E.; Manea, A.; Pirvulescu, M.M.; Stan, D.; Simion, V.; Calin, M.; Simionescu, M.; Manduteanu, I. Functional analysis of the fractalkine gene promoter in human aortic smooth muscle cells exposed to proinflammatory conditions. FEBS J. 2014, 281, 3869–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, F.; Piemonti, L.; Fedele, G.; Destro, A.; Roncalli, M.; Albarello, L.; Doglioni, C.; Anselmo, A.; Doni, A.; Bianchi, P.; et al. The chemokine receptor CX3CR1 is involved in the neural tropism and malignant behavior of pancreatic ductal adenocarcinoma. Cancer Res. 2008, 68, 9060–9069. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Yasuda, N. Therapeutic intervention of inflammatory/immune diseases by inhibition of the fractalkine (CX3CL1)-CX3CR1 pathway. Inflamm. Regen. 2016, 36, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A potential new target for inflammatory diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Erreni, M.; van Brakel, M.; Debets, R.; Allavena, P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J. Immunother. Cancer 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, B.; Mummidi, S.; Perla, R.P.; Bysani, S.; Dulin, N.O.; Liu, F.; Melby, P.C. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem. J. 2003, 373, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, E.; Pistoia, V.; Corcione, A. Role of fractalkine/CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediat. Inflamm. 2014, 2014, 480941. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Hu, H.D.; Hu, P.; Lan, Y.H.; Peng, M.L.; Chen, M.; Ren, H. Gene therapy with CX3CL1/Fractalkine induces antitumor immunity to regress effectively mouse hepatocellular carcinoma. Gene Ther. 2007, 14, 1226–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Huebener, N.; Fest, S.; Weixler, S.; Schroeder, U.; Gaedicke, G.; Xiang, R.; Schramm, A.; Eggert, A.; Reisfeld, R.A.; et al. Fractalkine (CX3CL1)- and interleukin-2-enriched neuroblastoma microenvironment induces eradication of metastases mediated by T cells and natural killer cells. Cancer Res. 2007, 67, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Ono, T.; Yamanoi, A.; Tachibana, M.; Nagasue, N. Fractalkine-CX3CR1 axis regulates tumor cell cycle and deteriorates prognosis after radical resection for hepatocellular carcinoma. J. Surg. Oncol. 2007, 95, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Hyakudomi, M.; Matsubara, T.; Hyakudomi, R.; Yamamoto, T.; Kinugasa, S.; Yamanoi, A.; Maruyama, R.; Tanaka, T. Increased expression of fractalkine is correlated with a better prognosis and an increased number of both CD8+ T cells and natural killer cells in gastric adenocarcinoma. Ann. Surg. Oncol. 2008, 15, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Tanaka, F.; Yamaguchi, H.; Sadanaga, N.; Inoue, H.; Mori, M. The high expression of Fractalkine results in a better prognosis for colorectal cancer patients. Int. J. Oncol. 2005, 26, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Cabioglu, N.; Assi, H.; Sabourin, J.C.; Delaloge, S.; Sahin, A.; Broglio, K.; Spano, J.P.; Combadiere, C.; Bucana, C.; et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann. Oncol. 2006, 17, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, J.Y.; Ni, Y.B.; Chan, S.K.; Shao, M.M.; Kwok, Y.K.; Chan, K.W.; Tan, P.H.; Tse, G.M. CX3CL1 expression is associated with poor outcome in breast cancer patients. Breast Cancer Res. Treat. 2013, 140, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, F.; Nasreddine, S.; Donnadieu, A.C.; Emilie, D.; Combadiere, C.; Prevot, S.; Machelon, V.; Balabanian, K. Identification of the chemokine CX3CL1 as a new regulator of malignant cell proliferation in epithelial ovarian cancer. PLoS ONE 2011, 6, e21546. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schäfer, H.; Arlt, A. TRAIL/NF-κB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 1661. https://doi.org/10.3390/ijms19061661
Geismann C, Erhart W, Grohmann F, Schreiber S, Schneider G, Schäfer H, Arlt A. TRAIL/NF-κB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. International Journal of Molecular Sciences. 2018; 19(6):1661. https://doi.org/10.3390/ijms19061661
Chicago/Turabian StyleGeismann, Claudia, Wiebke Erhart, Frauke Grohmann, Stefan Schreiber, Günter Schneider, Heiner Schäfer, and Alexander Arlt. 2018. "TRAIL/NF-κB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines" International Journal of Molecular Sciences 19, no. 6: 1661. https://doi.org/10.3390/ijms19061661