Quinolones Modulate Ghrelin Receptor Signaling: Potential for a Novel Small Molecule Scaffold in the Treatment of Cachexia

 , ,
, ,
Abstract
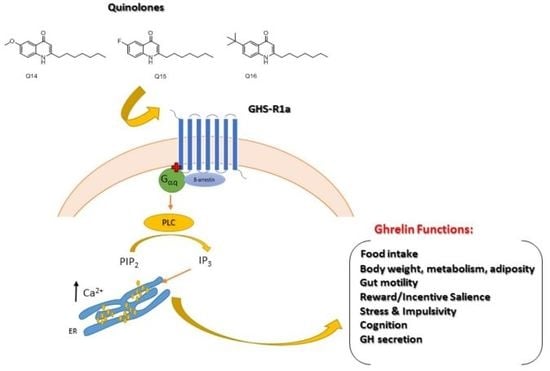
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
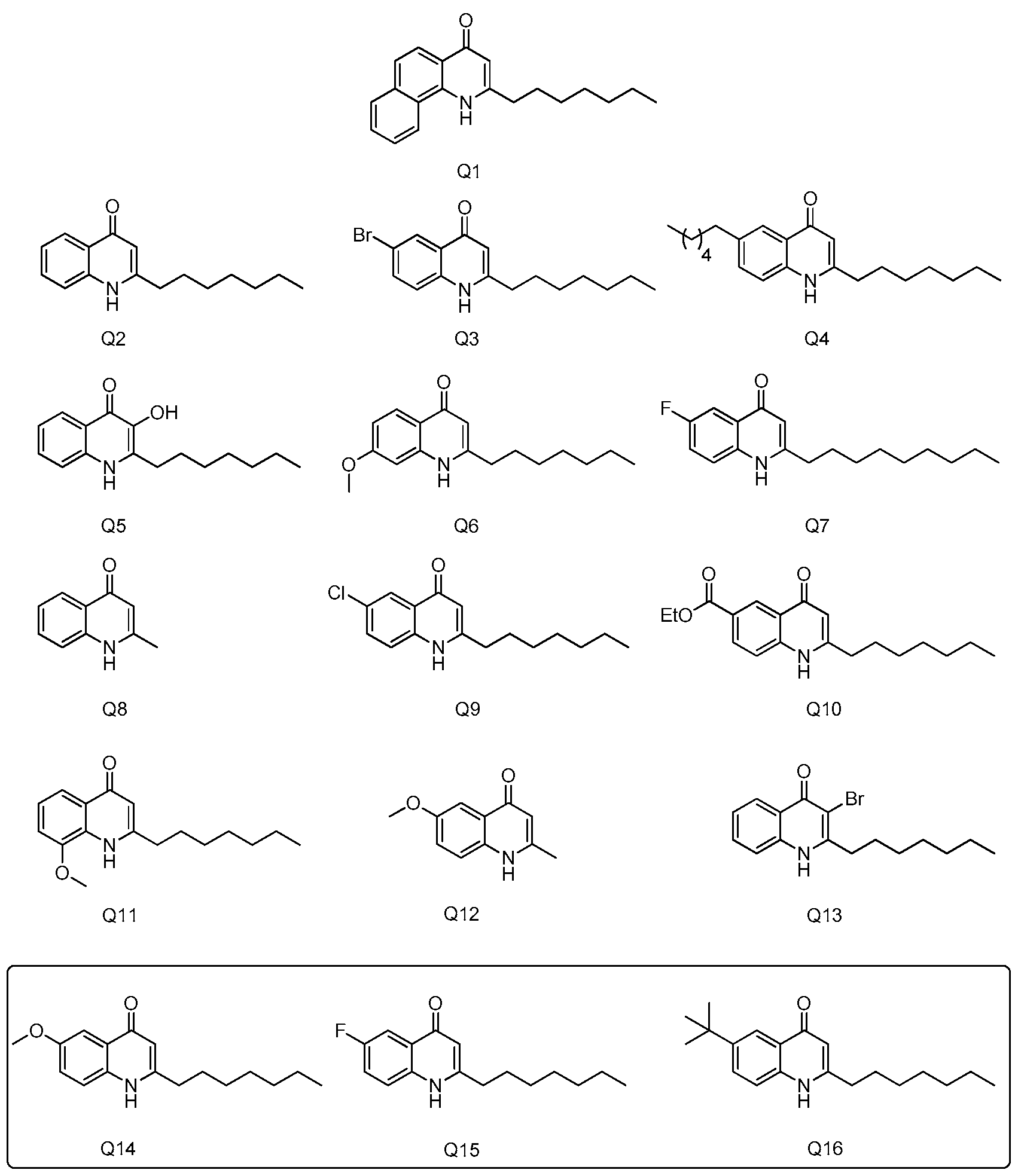
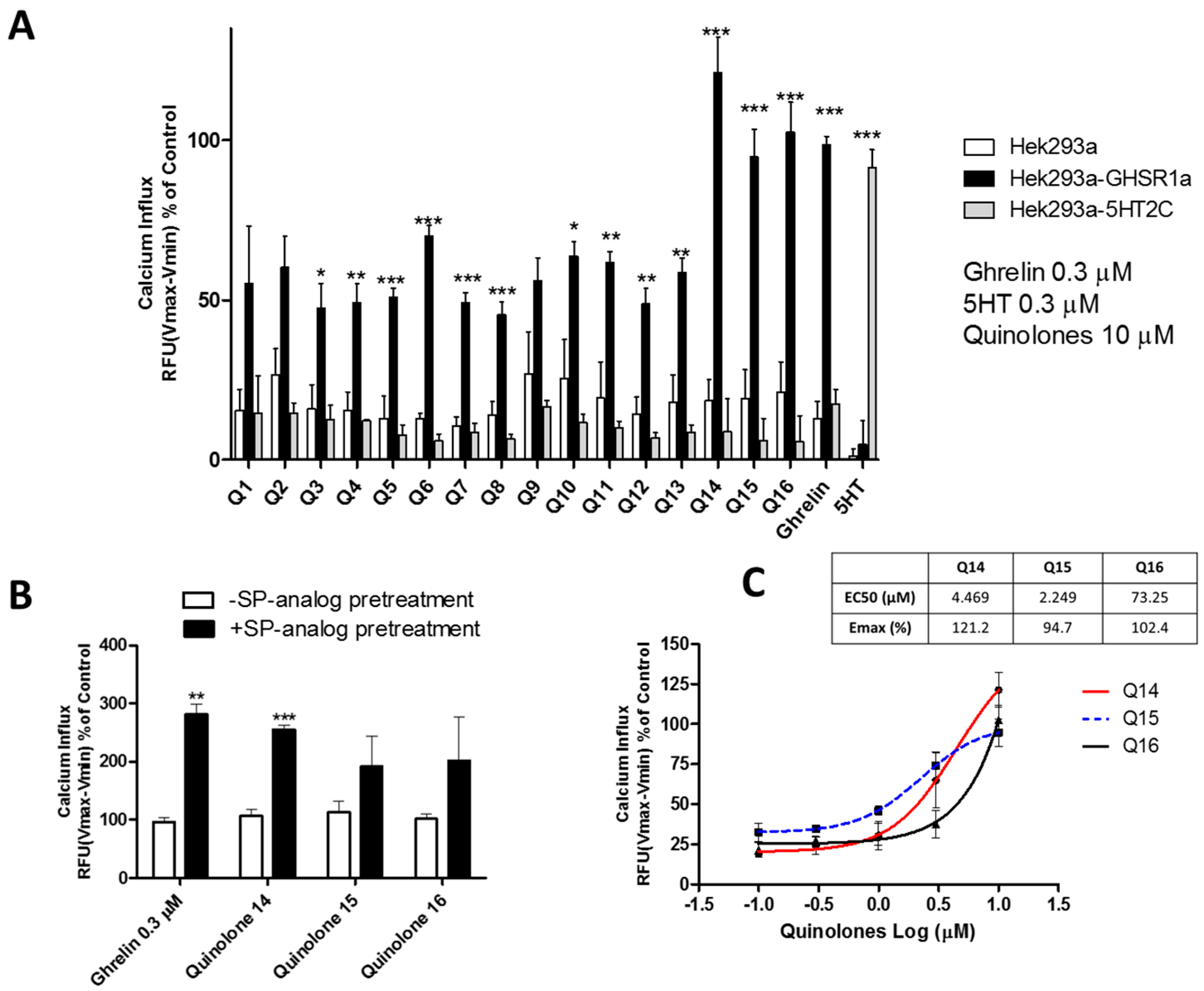
2.1. Quinolones Compounds and Their Effects on GHS-R1a Signaling
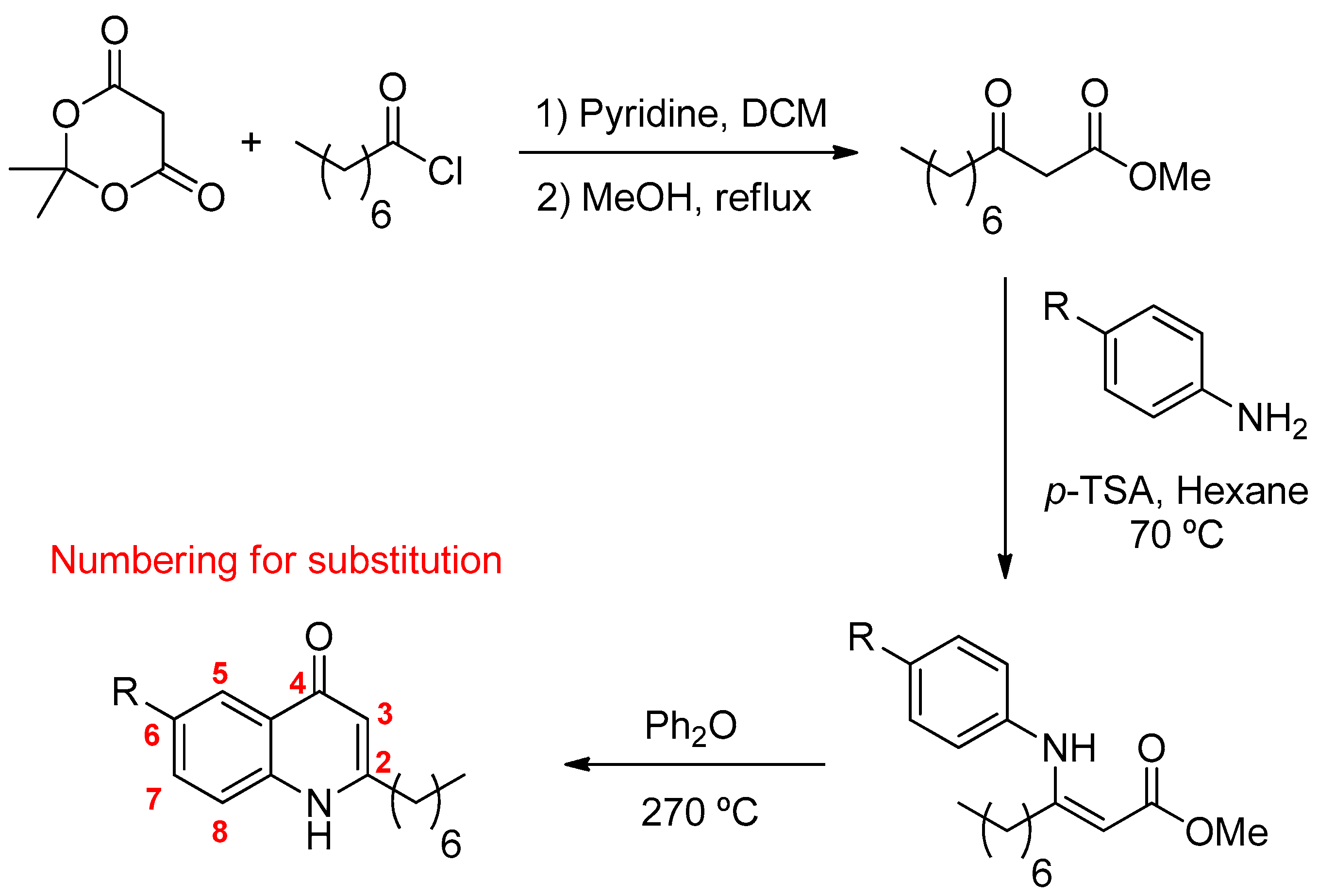
2.1.1. Quinolones Synthesis
2.1.2. Quinolones Compounds Activate GHS-R1a-Mediated Intracellular Calcium Mobilization
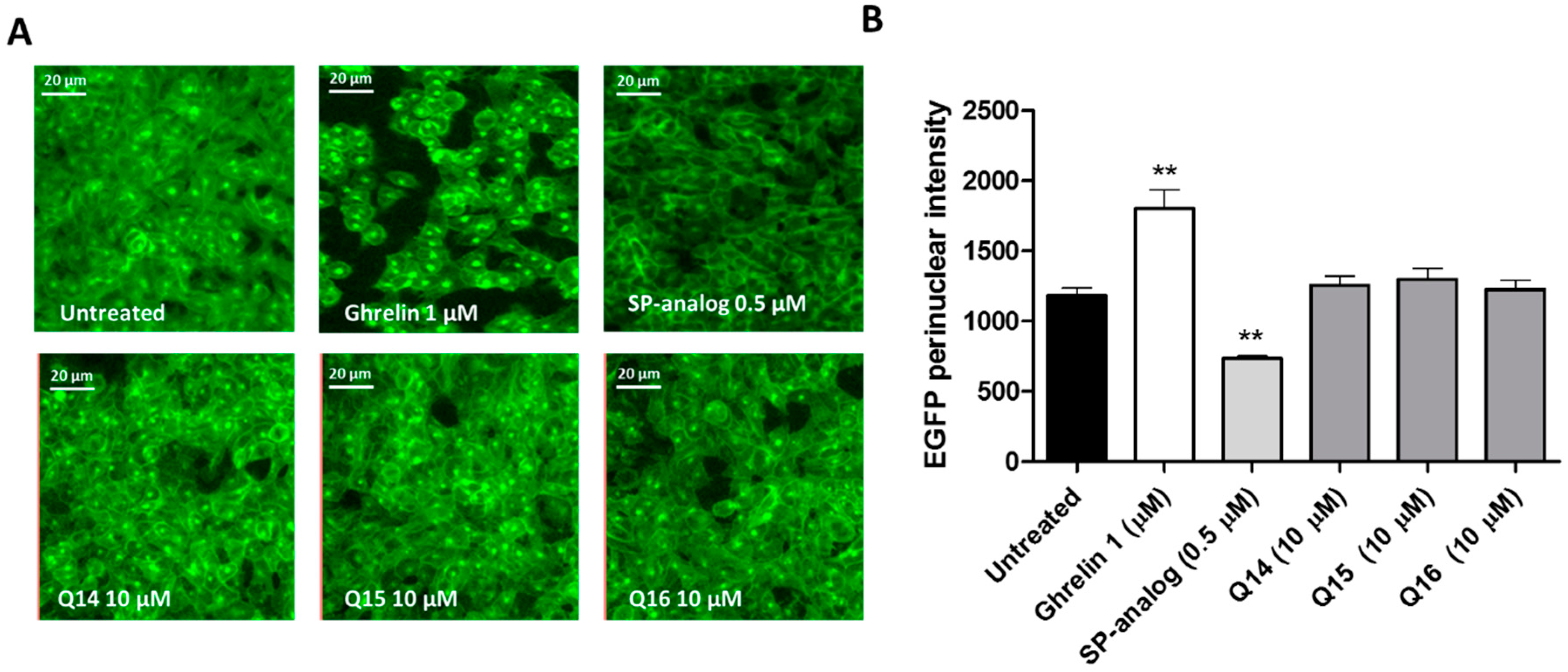
3. Discussion
4. Materials and Methods
4.1. General Procedure for the Preparation of Substituted 2-Alkyl-4-Quinolones
4.1.1. Preparation of the β-Ketoester
4.1.2. Preparation of the Quinolones
4.2. Compounds Preparation
4.3. Cell Culture
4.4. Calcium Mobilization Assay
4.5. Calcium Imaging
4.6. Internalization Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Lok, C. Cachexia: The last illness. Nature 2015, 528, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Voss, A.C.; Hustead, D.S. Definition of cancer cachexia: Effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am. J. Clin. Nutr. 2006, 83, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Morley, J.E.; Argiles, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Drescher, C.; Konishi, M.; Ebner, N.; Springer, J. Loss of muscle mass: Current developments in cachexia and sarcopenia focused on biomarkers and treatment. J. Cachexia Sarcopenia Muscle 2015, 6, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, S.; Sato, T.; Kangawa, K.; Nakazato, M. The Homeostatic Force of Ghrelin. Cell Metab. 2018, 27, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Al Massadi, O.; Lopez, M.; Tschop, M.; Dieguez, C.; Nogueiras, R. Current Understanding of the Hypothalamic Ghrelin Pathways lncucinc Appetite anc Adiposity. Trends Neurosci. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl Hormones of Endocrine Cells Origin in the Gut—Their Discovery and Physiological Relevance. J. Physiol. Pharmacol. 2015, 66, 11–27. [Google Scholar] [PubMed]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. Control of food intake and muscle wasting in cachexia. Int. J. Biochem. Cell Biol. 2013, 45, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.P.; Bonner, T.I.; Foord, S.M.; Harmar, A.J.; Neubig, R.R.; Pin, J.P.; Spedding, M.; Kojima, M.; Kangawa, K. International Union of Pharmacology. LVI. Ghrelin receptor nomenclature, distribution, and function. Pharmacol. Rev. 2005, 57, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Jarkovska, Z.; Krsek, M.; Rosicka, M.; Marek, J. Endocrine and metabolic activities of a recently isolated peptide hormone ghrelin, an endogenous ligand of the growth hormone secretagogue receptor. Endocr. Regul. 2004, 38, 80–86. [Google Scholar] [PubMed]
- Asakawa, A.; Inui, A.; Kaga, T.; Yuzuriha, H.; Nagata, T.; Ueno, N.; Makino, S.; Fujimiya, M.; Niijima, A.; Fujino, M.A.; et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 2001, 120, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Dinan, T.G.; Cryan, J.F. Lean mean fat reducing “ghrelin” machine: Hypothalamic ghrelin and ghrelin receptors as therapeutic targets in obesity. Neuropharmacology 2010, 58, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Hiura, Y.; Takiguchi, S.; Yamamoto, K.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Miyata, H.; Fujiwara, Y.; Mori, M.; Doki, Y. Fall in plasma ghrelin concentrations after cisplatin-based chemotherapy in esophageal cancer patients. Int. J. Clin. Oncol. 2012, 17, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Masaki, T.; Kakuma, T.; Yoshimatsu, H. Centrally administered ghrelin suppresses sympathetic nerve activity in brown adipose tissue of rats. Neurosci. Lett. 2003, 349, 75–78. [Google Scholar] [CrossRef]
- Esposito, A.; Criscitiello, C.; Gelao, L.; Pravettoni, G.; Locatelli, M.; Minchella, I.; Di Leo, M.; Liuzzi, R.; Milani, A.; Massaro, M.; et al. Mechanisms of anorexia-cachexia syndrome and rational for treatment with selective ghrelin receptor agonist. Cancer Treat. Rev. 2015, 41, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Akamizu, T.; Takaya, K.; Irako, T.; Hosoda, H.; Teramukai, S.; Matsuyama, A.; Tada, H.; Miura, K.; Shimizu, A.; Fukushima, M.; et al. Pharmacokinetics, safety, and endocrine and appetite effects of ghrelin administration in young healthy subjects. Eur. J. Endocrinol. 2004, 150, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Currow, D.C.; Abernethy, A.P. Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome. Future Oncol. 2014, 10, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Cavada, E.; Pardo, L.M.; Kandil, D.; Torres-Fuentes, C.; Clarke, S.L.; Shaban, H.; McGlacken, G.P.; Schellekens, H. A Novel Non-Peptidic Agonist of the Ghrelin Receptor with Orexigenic Activity In vivo. Sci. Rep. 2016, 6, 36456. [Google Scholar] [CrossRef] [PubMed]
- Howick, K.; Alam, R.; Chruscicka, B.; Kandil, D.; Fitzpatrick, D.; Ryan, A.M.; Cryan, J.F.; Schellekens, H.; Griffin, B.T. Sustained-release multiparticulates for oral delivery of a novel peptidic ghrelin agonist: Formulation design and in vitro characterization. Int. J. Pharm. 2018, 536, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Uchino, J.; Yokoyama, T.; Naito, T.; Kondo, M.; Yamada, K.; Kitajima, H.; Yoshimori, K.; Sato, K.; Saito, H.; et al. Anamorelin (ONO-7643) for the treatment of patients with non-small cell lung cancer and cachexia: Results from a randomized, double-blind, placebo-controlled, multicenter study of Japanese patients (ONO-7643-04). Cancer 2018, 124, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Currow, D.; Temel, J.S.; Abernethy, A.; Milanowski, J.; Friend, J.; Fearon, K.C. ROMANA 3: A phase 3 safety extension study of anamorelin in advanced non-small-cell lung cancer (NSCLC) patients with cachexia. Ann. Oncol. 2017, 28, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Katakami, N.; Yokoyama, T.; Atagi, S.; Yoshimori, K.; Kagamu, H.; Saito, H.; Takiguchi, Y.; Aoe, K.; Koyama, A.; et al. Anamorelin (ONO-7643) in Japanese patients with non-small cell lung cancer and cachexia: Results of a randomized phase 2 trial. Support. Care Cancer 2016, 24, 3495–3505. [Google Scholar] [CrossRef] [PubMed]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Pietra, C.; Takeda, Y.; Tazawa-Ogata, N.; Minami, M.; Yuanfeng, X.; Duus, E.M.; Northrup, R. Anamorelin HCl (ONO-7643), a novel ghrelin receptor agonist, for the treatment of cancer anorexia-cachexia syndrome: Preclinical profile. J. Cachexia Sarcopenia Muscle 2014, 5, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, S.W.; Oh, J.; Hong, G.S.; Seo, E.K.; Oh, U.; Shim, W.S. Ghrelin receptor is activated by naringin and naringenin, constituents of a prokinetic agent Poncirus fructus. J. Ethnopharmacol. 2013, 148, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.K.; Lo, Y.H.; Wu, C.C.; Chung, T.Y.; Tzen, J.T.C. Identification of biosynthetic intermediates of teaghrelins and teaghrelin-like compounds in oolong teas, and their molecular docking to the ghrelin receptor. J. Food Drug Anal. 2015, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; McNamara, O.; Dinan, T.G.; McCarthy, J.V.; McGlacken, G.P.; Cryan, J.F. Semagacestat, a gamma-secretase inhibitor, activates the growth hormone secretagogue (GHS-R1a) receptor. J. Pharm. Pharmacol. 2013, 65, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; McGlacken, G.P. Access to trifluoromethylated 4-alkoxy-2-pyrones, pyridones and quinolones. Tetrahedron 2015, 71, 2906–2913. [Google Scholar] [CrossRef]
- Clarke, S.L.; McGlacken, G.P. Methyl fluorosulfonyldifluoroacetate (MFSDA): An Underutilised Reagent for Trifluoromethylation. Chem.-Eur. J. 2017, 23, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Heeb, S.; Fletcher, M.P.; Chhabra, S.R.; Diggle, S.P.; Williams, P.; Camara, M. Quinolones: From antibiotics to autoinducers. FEMS Microbiol. Rev. 2011, 35, 247–274. [Google Scholar] [CrossRef] [PubMed]
- Ó Muimhneacháin, E.; Reen, F.J.; O’Gara, F.; McGlacken, G.P. Analogues of Pseudomonas aeruginosa signalling molecules to tackle infections. Org. Biomol. Chem. 2018, 16, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Phelan, J.P.; Gallagher, L.; Shanahan, R.M.; Cano, R.; Ó Muimhneacháin, E.; O’Gara, F.; McGlacken, G.P. Exploiting Interkingdom Interactions for Development of Small-Molecule Inhibitors of Candida albicans Biofilm Formation. Antimicrob. Agents Chemother. 2016, 60, 5894–5905. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Clarke, S.L.; Legendre, C.; McSweeney, C.M.; Eccles, K.S.; Lawrence, S.E.; O’Gara, F.; McGlacken, G.P. Structure–function analysis of the C-3 position in analogues of microbial behavioural modulators HHQ and PQS. Org. Biomol. Chem. 2012, 10, 8903–8910. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Mooij, M.J.; Holcombe, L.J.; McSweeney, C.M.; McGlacken, G.P.; Morrissey, J.P.; O’Gara, F. The Pseudomonas Quinolone Signal (PQS), and its precursor HHQ, modulate interspecies and interkingdom behaviour. FEMS Microbiol. Ecol. 2011, 77, 413–428. [Google Scholar] [CrossRef] [PubMed]
- McGlacken, G.P.; McSweeney, C.M.; O’Brien, T.; Lawrence, S.E.; Elcoate, C.J.; Reen, F.J.; O’Gara, F. Synthesis of 3-halo-analogues of HHQ, subsequent cross-coupling and first crystal structure of Pseudomonas quinolone signal (PQS). Tetrahedron Lett. 2010, 51, 5919–5921. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Theeuwes, W.F.; McMullen, M.K.; McMullen, A.K.; Dinan, T.G.; Cryan, J.F.; Schellekens, H. Devil’s Claw to suppress appetite–ghrelin receptor modulation potential of a Harpagophytum procumbens root extract. PLoS ONE 2014, 9, e103118. [Google Scholar] [CrossRef] [PubMed]
- Camina, J.P.; Lodeiro, M.; Ischenko, O.; Martini, A.C.; Casanueva, F.F. Stimulation by ghrelin of p42/p44 mitogen-activated protein kinase through the GHS-R1a receptor: Role of G-proteins and beta-arrestins. J. Cell. Physiol. 2007, 213, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Sivertsen, B.; Holliday, N.; Madsen, A.N.; Holst, B. Functionally biased signalling properties of 7TM receptors–opportunities for drug development for the ghrelin receptor. Br. J. Pharmacol. 2013, 170, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Els, S.; Beck-Sickinger, A.G.; Chollet, C. Ghrelin Receptor: High Constitutive Activity and Methods for Developing Inverse Agonists. Methods Enzymol. 2010, 485, 103–121. [Google Scholar] [PubMed]
- Holst, B.; Cygankiewicz, A.; Jensen, T.H.; Ankersen, M.; Schwartz, T.W. High constitutive signaling of the ghrelin receptor—Identification of a potent inverse agonist. Mol. Endocrinol. 2003, 17, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Fortin, J.P.; Beinborn, M.; Kopin, A.S. Four missense mutations in the ghrelin receptor result in distinct pharmacological abnormalities. J. Pharmacol. Exp. Ther. 2007, 322, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Smith, R.; Judith, F.; Schleim, K.; Frazier, E.; Chen, H.; Krupa, D.; Hora, D., Jr.; Nargund, R.; Patchett, A.; et al. MK-0677, a potent, novel, orally active growth hormone (GH) secretagogue: GH, insulin-like growth factor I, and other hormonal responses in beagles. Endocrinology 1996, 137, 5284–5289. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Fearon, K.C. Cachexia: Prevalence and impact in medicine. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mansson, J.V.; Alves, F.D.; Biolo, A.; Souza, G.C. Use of ghrelin in cachexia syndrome: A systematic review of clinical trials. Nutr. Rev. 2016, 74, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Shanahan, R.; Cano, R.; O’Gara, F.; McGlacken, G.P. A structure activity-relationship study of the bacterial signal molecule HHQ reveals swarming motility in Bacillus atrophaeus. Org. Biomol. Chem. 2015, 13, 5537–5541. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, R.; Reen, F.J.; Cano, R.; O’Gara, F.; McGlacken, G.P. The requirements at the C-3 position of alkylquinolones for signalling in Pseudomonas aeruginosa. Org. Biomol. Chem. 2017, 15, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Ritter, S.L.; Hall, R.A. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 2009, 10, 819–830. [Google Scholar] [CrossRef] [PubMed]
- M’Kadmi, C.; Leyris, J.P.; Onfroy, L.; Gales, C.; Sauliere, A.; Gagne, D.; Damian, M.; Mary, S.; Maingot, M.; Denoyelle, S.; et al. Agonism, Antagonism, and Inverse Agonism Bias at the Ghrelin Receptor Signaling. J. Biol. Chem. 2015, 290, 27021–27039. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Mary, S.; Damian, M.; Louet, M.; Floquet, N.; Fehrentz, J.A.; Marie, J.; Martinez, J.; Baneres, J.L. Ligands and signaling proteins govern the conformational landscape explored by a G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 8304–8309. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, V.T.; van Oeffelen, W.E.P.A.; Torres-Fuentes, C.; Chruścicka, B.; Druelle, C.; Golubeva, A.V.; van de Wouw, M.; Dinan, T.G.; Cryan, J.F.; Schellekens, H. Differential Functional Selectivity and Downstream Signalling Bias of Ghrelin Receptor Antagonists and Inverse Agonists. FASEB J. 2018, in press. [Google Scholar]
- Sanger, G.J. Ghrelin and motilin receptor agonists: Time to introduce bias into drug design. Neurogastroenterol. Motil. 2014, 26, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Evron, T.; Peterson, S.M.; Urs, N.M.; Bai, Y.; Rochelle, L.K.; Caron, M.G.; Barak, L.S. G Protein and beta-arrestin signaling bias at the ghrelin receptor. J. Biol. Chem. 2014, 289, 33442–33455. [Google Scholar] [CrossRef] [PubMed]
- Blayo, A.L.; Maingot, M.; Aicher, B.; M’Kadmi, C.; Schmidt, P.; Muller, G.; Teifel, M.; Gunther, E.; Gagne, D.; Denoyelle, S.; et al. New trisubstituted 1,2,4-triazoles as ghrelin receptor antagonists. Bioorg. Med. Chem. Lett. 2015, 25, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Slosky, L.M.; Pack, T.F.; Urs, N.M.; Boone, P.; Mao, L.; Abraham, D.; Caron, M.G.; Barak, L.S. Ghrelin receptor antagonism of hyperlocomotion in cocaine-sensitized mice requires betaarrestin-2. Synapse 2018, 72. [Google Scholar] [CrossRef] [PubMed]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, Y.; Sugano, K.; Yonemitsu, O. Meldrum’s acid in organic synthesis. 2. A general and versatile synthesis of beta-keto esters. J. Org. Chem. 1978, 43, 2087–2088. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Fuentes, C.; Pastor-Cavada, E.; Cano, R.; Kandil, D.; Shanahan, R.; Juan, R.; Shaban, H.; McGlacken, G.P.; Schellekens, H. Quinolones Modulate Ghrelin Receptor Signaling: Potential for a Novel Small Molecule Scaffold in the Treatment of Cachexia. Int. J. Mol. Sci. 2018, 19, 1605. https://doi.org/10.3390/ijms19061605
Torres-Fuentes C, Pastor-Cavada E, Cano R, Kandil D, Shanahan R, Juan R, Shaban H, McGlacken GP, Schellekens H. Quinolones Modulate Ghrelin Receptor Signaling: Potential for a Novel Small Molecule Scaffold in the Treatment of Cachexia. International Journal of Molecular Sciences. 2018; 19(6):1605. https://doi.org/10.3390/ijms19061605
Chicago/Turabian StyleTorres-Fuentes, Cristina, Elena Pastor-Cavada, Rafael Cano, Dalia Kandil, Rachel Shanahan, Rocio Juan, Hamdy Shaban, Gerard P. McGlacken, and Harriët Schellekens. 2018. "Quinolones Modulate Ghrelin Receptor Signaling: Potential for a Novel Small Molecule Scaffold in the Treatment of Cachexia" International Journal of Molecular Sciences 19, no. 6: 1605. https://doi.org/10.3390/ijms19061605