Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry


Abstract
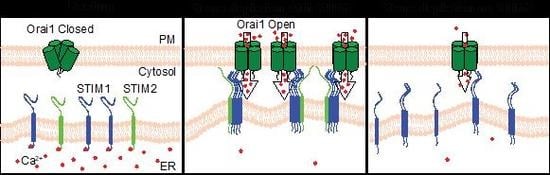
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Store-Operated Ca2+ Entry in NIH 3T3 Cells
2.2. STIM2 Is Expressed and Regulates Intracellular [Ca2+]c in NIH 3T3 Cells
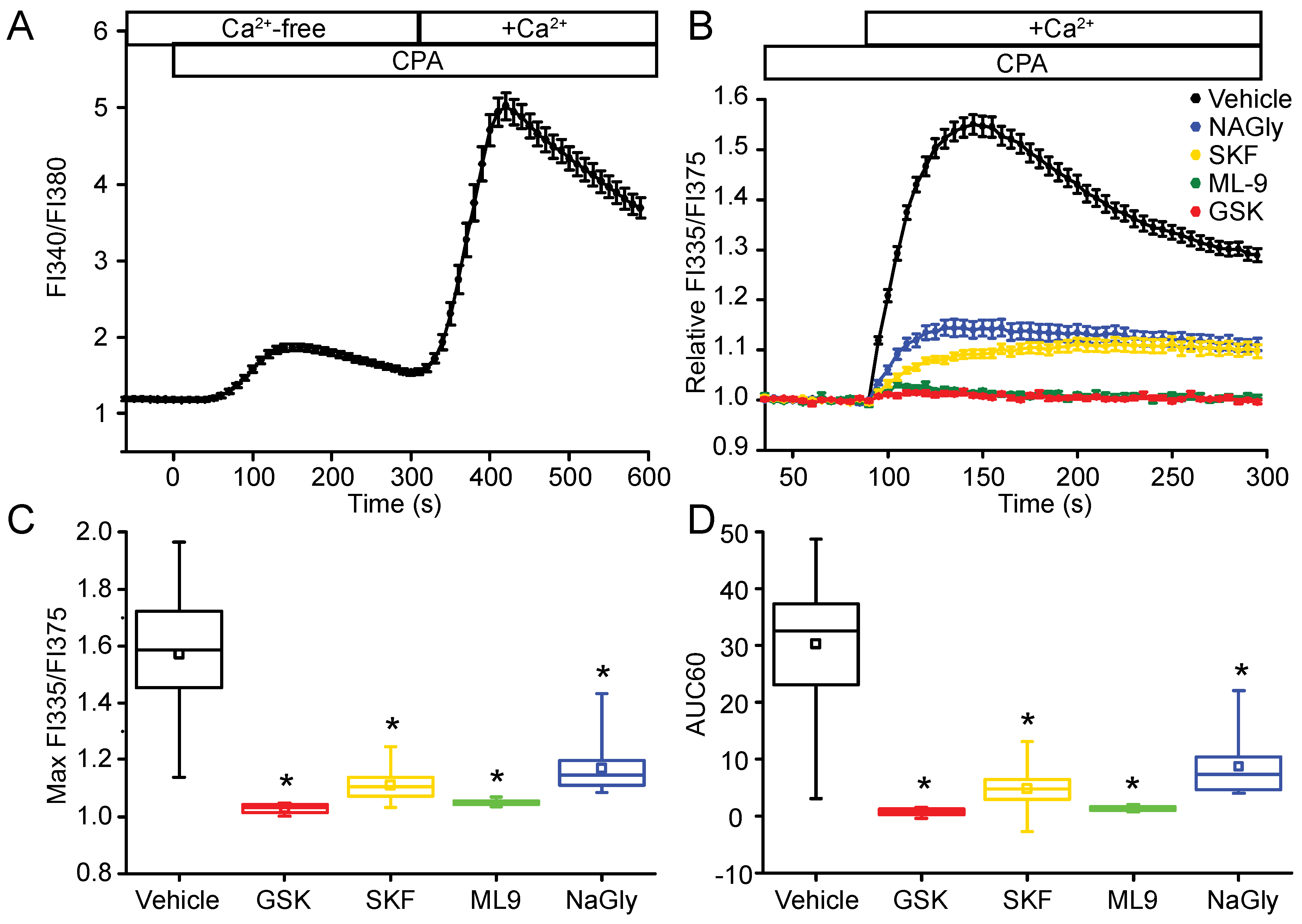
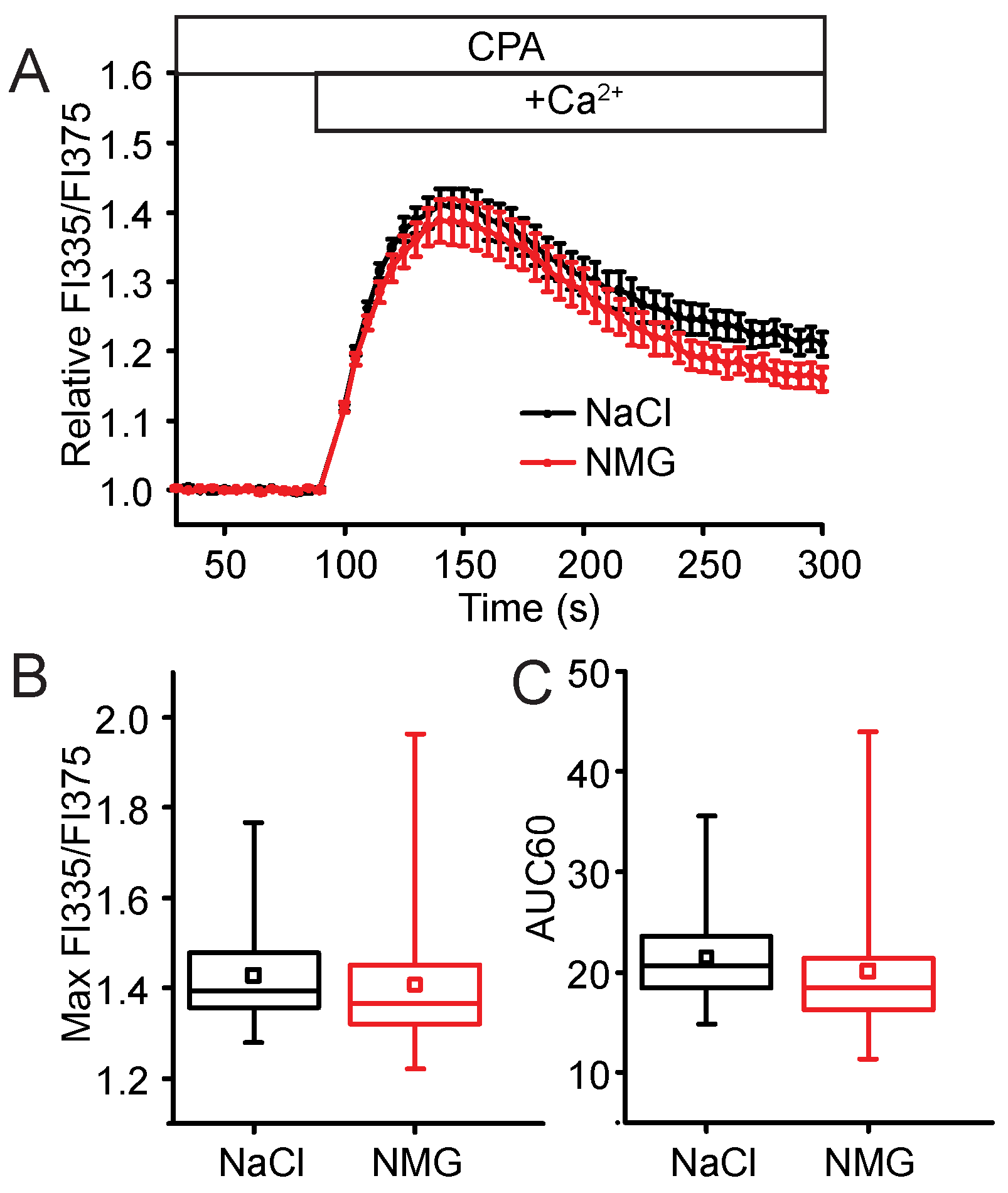
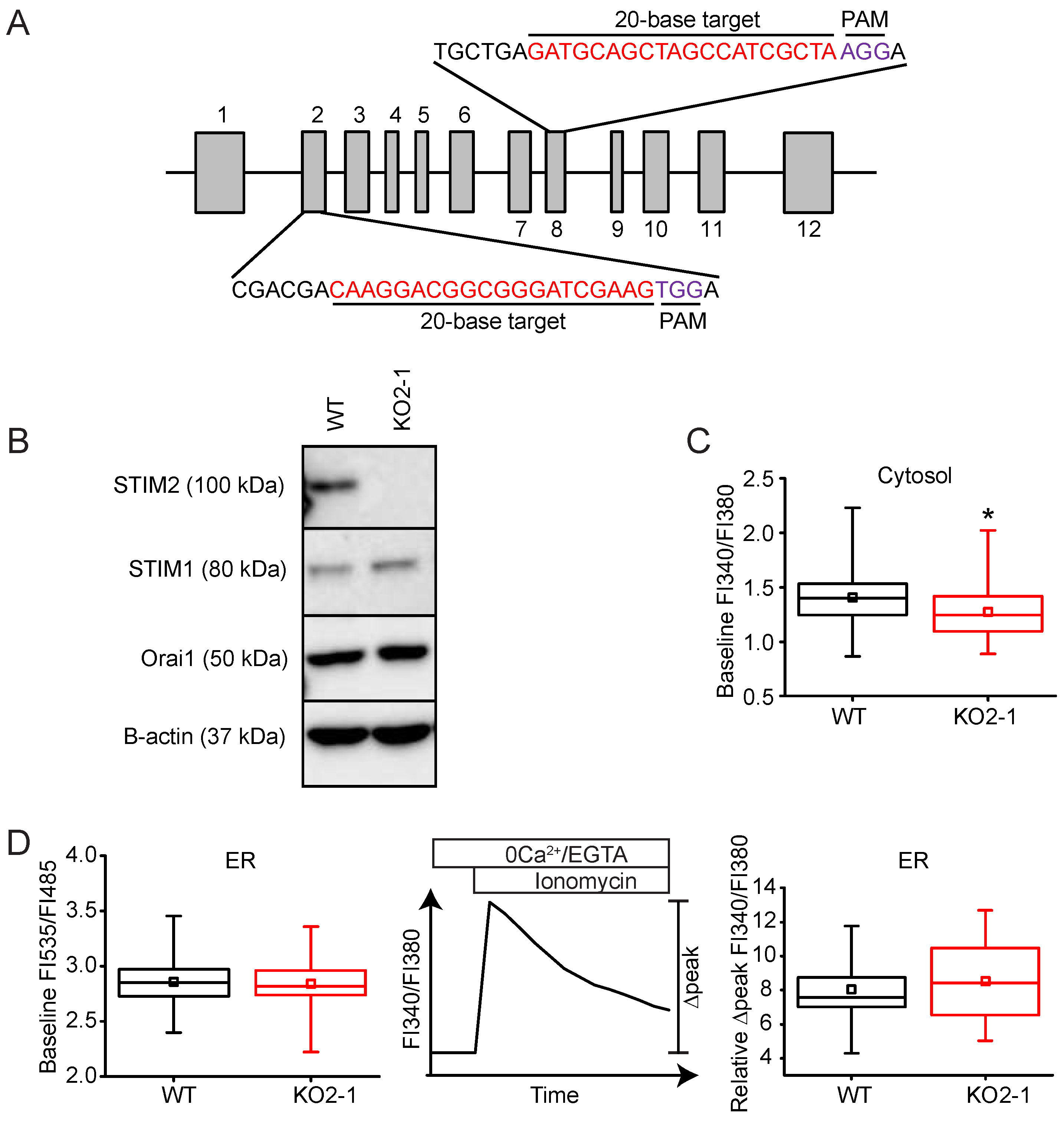
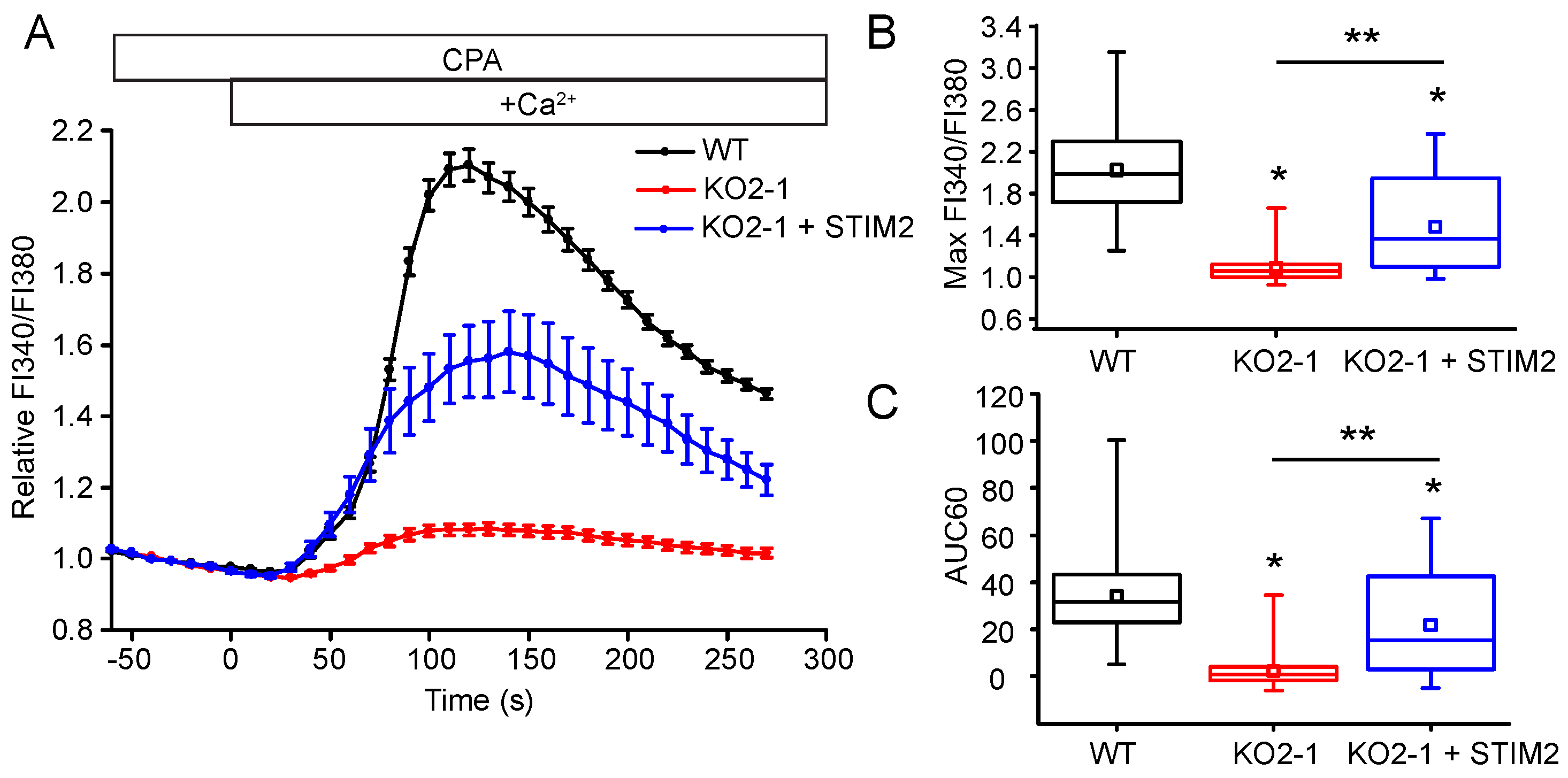
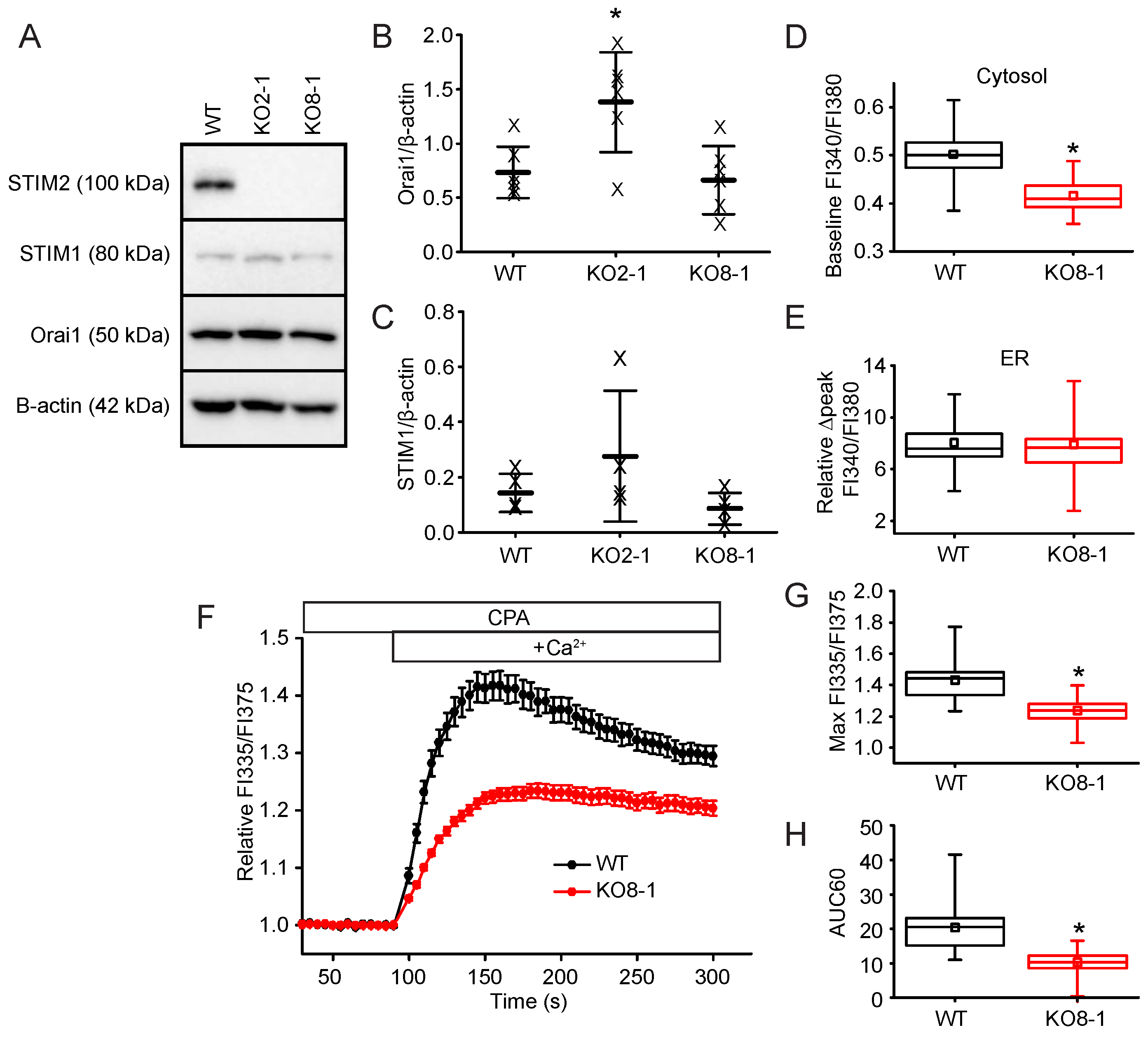
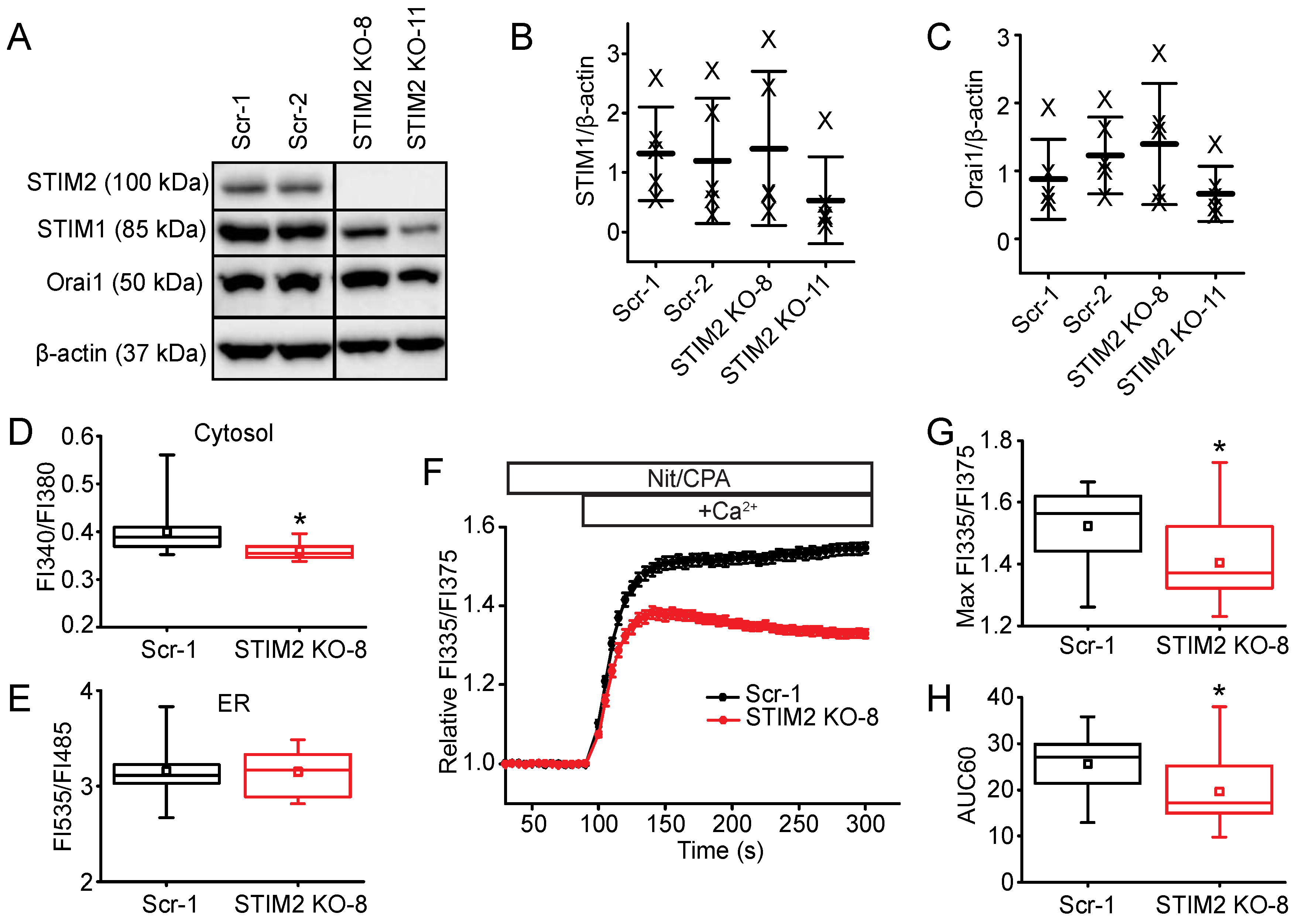
2.3. STIM2 Is an Important Regulator of SOCE
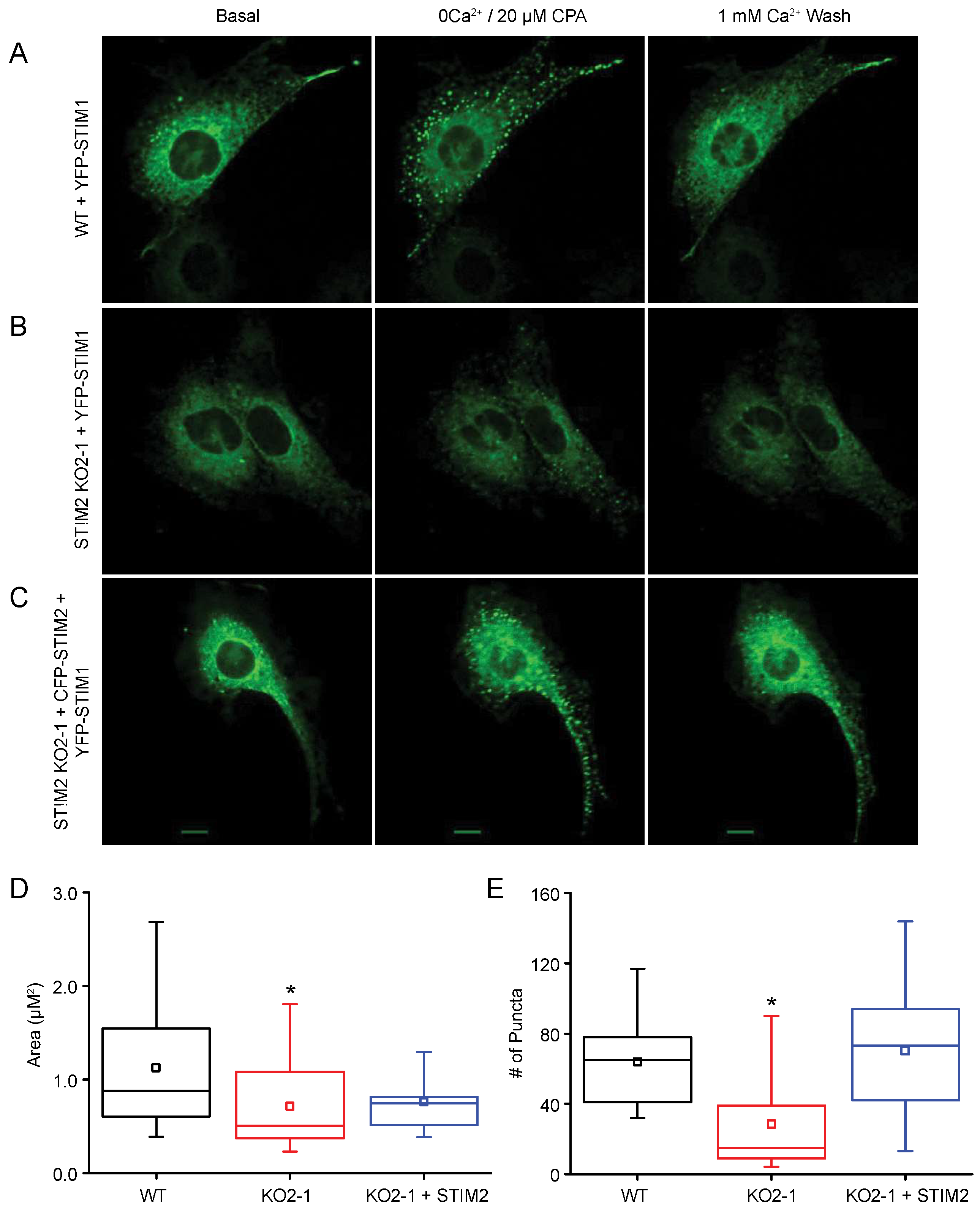
2.4. STIM1 Translocation
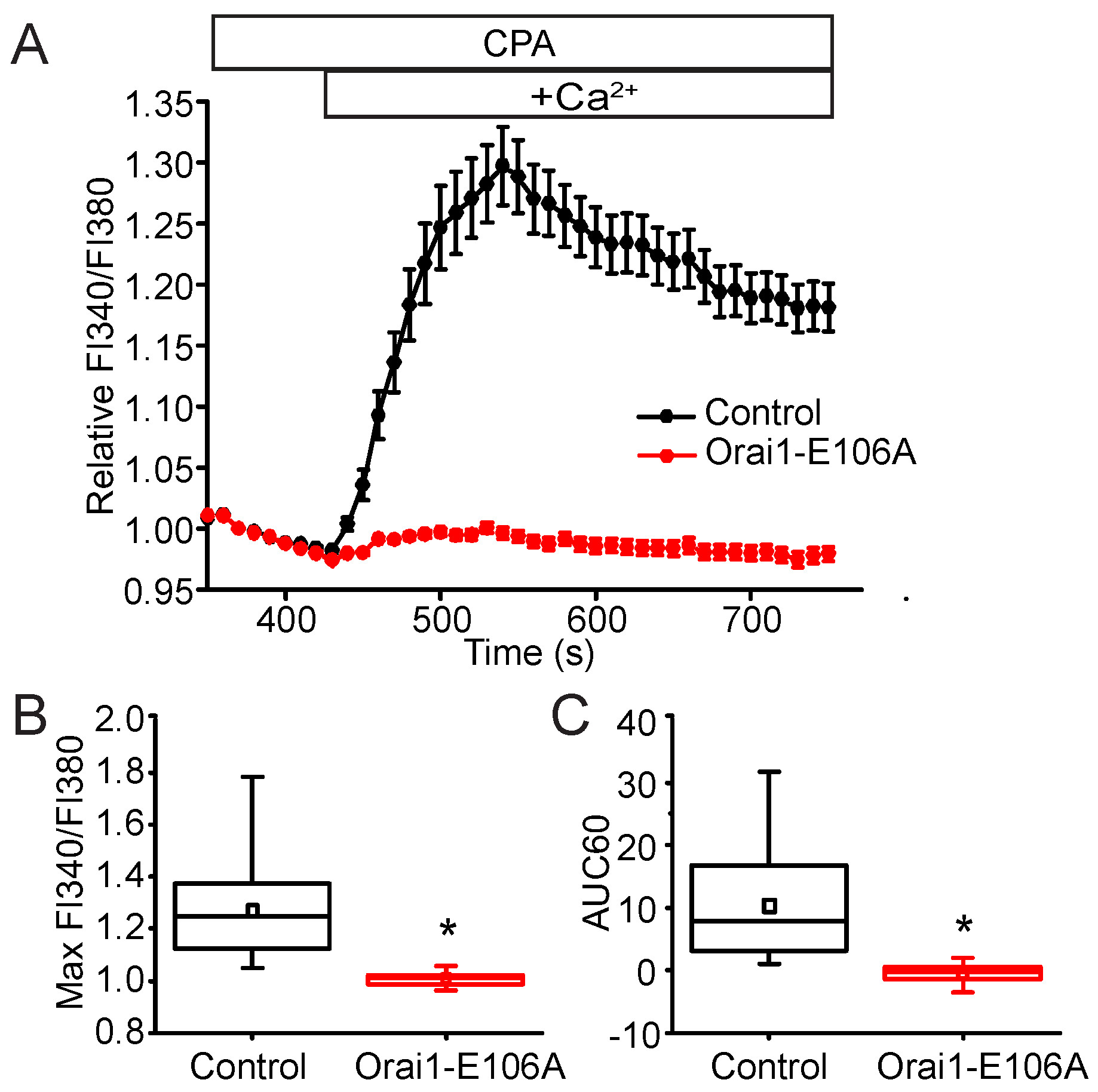
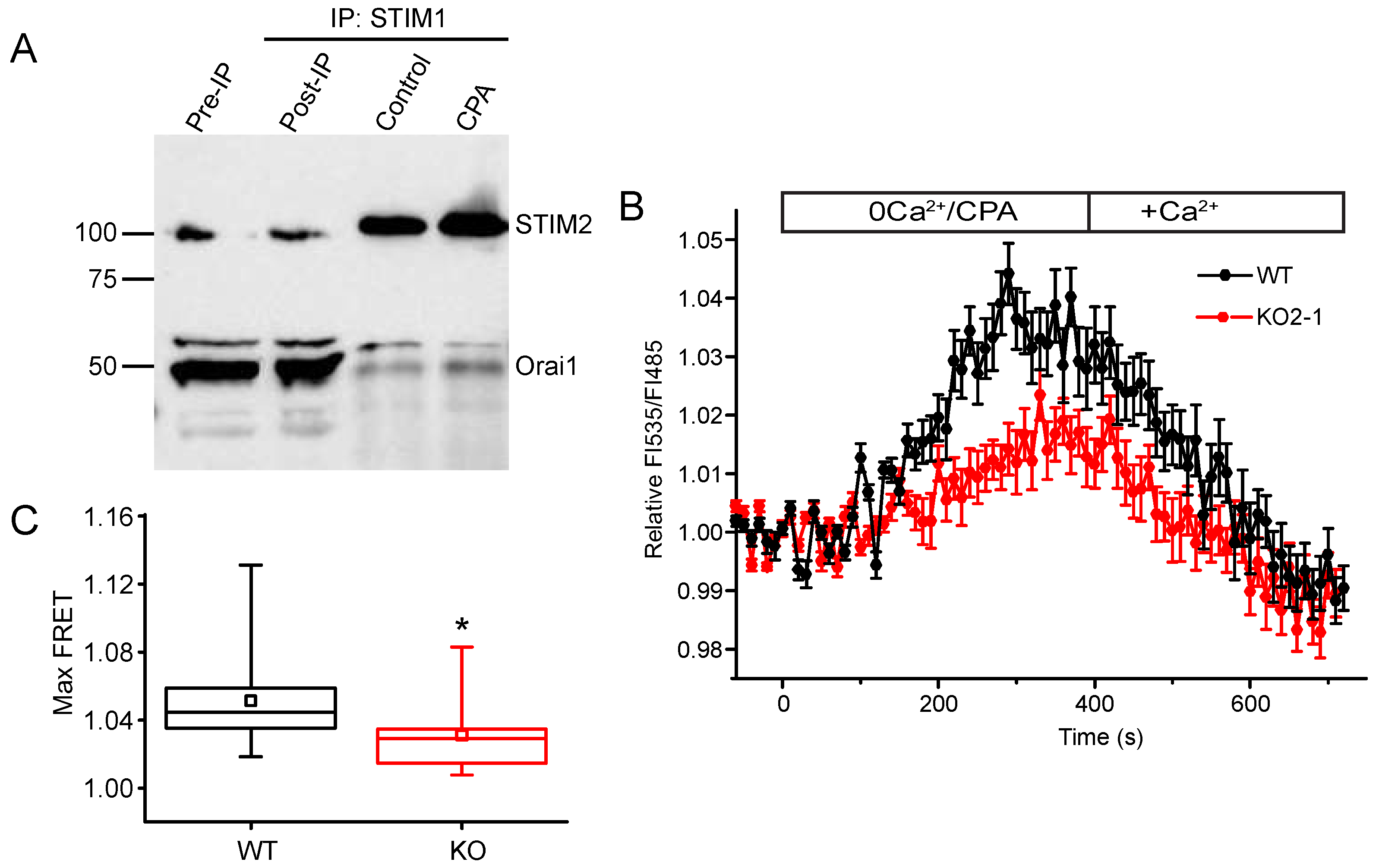
2.5. STIM2 Facilitates STIM1 Interaction with Orai1
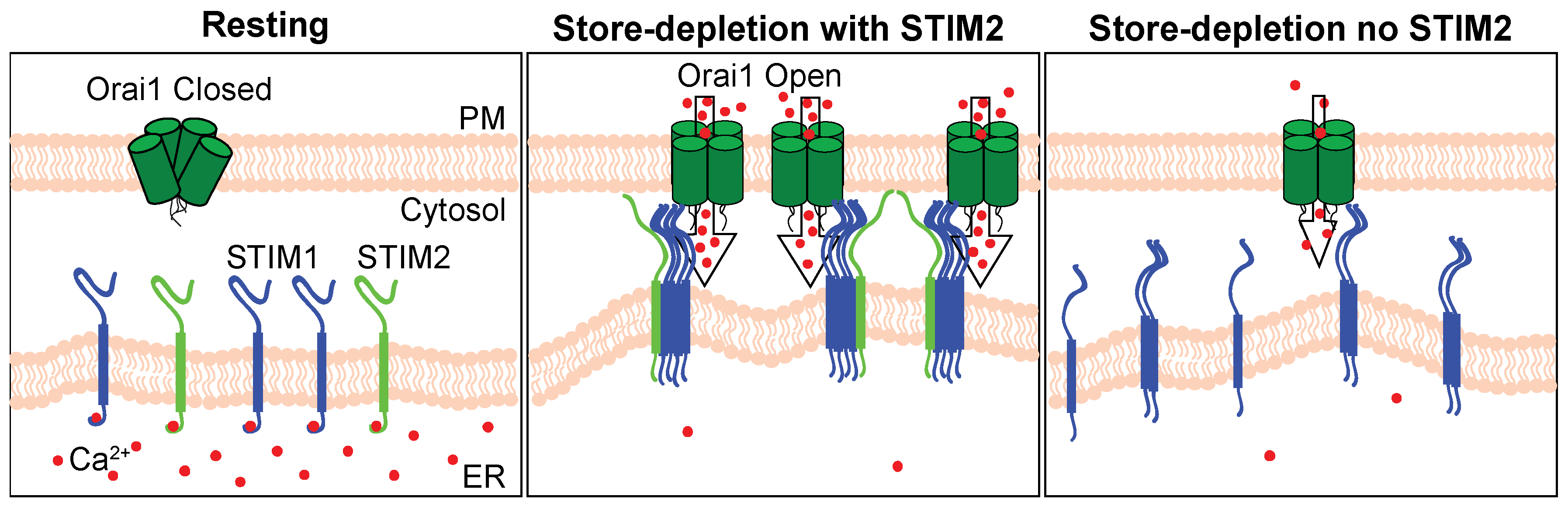
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Lysis, Immunoblotting, and Co-Immunoprecipitation
4.3. Generation and Analysis of STIM2 Knock-Out (KO) Cell Lines
4.4. Cytoplasmic Ca2+ Imaging
4.5. Flex Station
4.6. ER Ca2+ Imaging
4.7. FRET Measurements of Protein-Protein Interactions
4.8. Morphometric Analysis of STIM1 Localization
4.9. Reagents
4.10. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| STIM | Stromal Interaction Molecule |
| Ca2+ | Calcium |
| ER | Endoplasmic Reticulum |
| PM | Plasma Membrane |
References
- Putney, J. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Won, D.H.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store- depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Zheng, L.; Li, G.-Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 2010, 21, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Srinivasan, P.; Razavi, S.; Seymour, S.; Meraner, P.; Gudlur, A.; Stathopulos, P.B.; Ikura, M.; Rao, A.; Hogan, P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013, 20, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357 Pt 3, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 Is a Feedback Regulator that Stabilizes Basal Cytosolic and Endoplasmic Reticulum Ca2+ Levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.-Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Stathopulos, P.B.; Li, G.-Y.; Ikura, M. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem. Biophys. Res. Commun. 2008, 369, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Thiel, M.; Lis, A.; Penner, R. STIM2 drives Ca2+ oscillations through store-operated Ca2+ entry caused by mild store depletion. J. Physiol. 2013, 591 Pt 6, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.S.; Hwang, S.-Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009, 19, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Bakowski, D.; di Capite, J.; Nelson, C.; Parekh, A.B. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 6969–6974. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Spassova, M.A.; Hewavitharana, T.; He, L.P.; Xu, W.; Johnstone, L.S.; Dziadek, M.A.; Gill, D.L. STIM2 Is an Inhibitor of STIM1-Mediated Store-Operated Ca2+ Entry. Curr. Biol. 2006, 16, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I crac and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.T.; Dehaven, W.I.; Bird, G.S.; Putney, J.W., Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J. Cell Sci. 2008, 121 Pt 6, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Deak, A.T.; Groschner, L.N.; Alam, M.R.; Seles, E.; Bondarenko, A.I.; Graier, W.F.; Malli, R. The endocannabinoid N-arachidonoyl glycine (NAGly) inhibits store-operated Ca2+ entry by preventing STIM1–Orai1 interaction. J. Cell Sci. 2013, 126, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release–activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Ambudkar, I.S. The dynamic complexity of the TRPC1 channelosome. Channels (Austin) 2011, 5, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Sobradillo, D.; Hernández-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S.; Ong, H.L.; Singh, B.B. Molecular and functional determinants of Ca2+ signaling microdomains. In Signal Transduction: Pathways, Mechanisms and Diseases; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2010; pp. 237–253. [Google Scholar]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.E.; Jin, C.; Reed, J.C.; Tsien, R.Y. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 2004, 101, 17404–17409. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Peel, S.E.; Liu, B.; Hall, I.P. A key role for STIM1 in store operated calcium channel activation in airway smooth muscle. Respir. Res. 2006, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wang, J.; Peng, G.; Shimoda, L.A.; Sylvester, J.T. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L17–L25. [Google Scholar] [CrossRef] [PubMed]
- Bréchard, S.; Plançon, S.; Melchior, C.; Tschirhart, E.J. STIM1 but not STIM2 is an essential regulator of Ca2+ influx-mediated NADPH oxidase activity in neutrophil-like HL-60 cells. Biochem. Pharmacol. 2009, 78, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Fujii, Y.; Baba, A.; Hikida, M.; Kurosaki, T.; Baba, Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity 2011, 34, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Flores, B.; Garay, E.; Vázquez-Cuevas, F.G.; Arellano, R.O. Differential role of STIM1 and STIM2 during transient inward (Tin) current generation and the maturation process in the Xenopus oocyte. BMC Physiol. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef] [PubMed]
- Tamarina, N.A.; Kuznetsov, A.; Philipson, L.H. Reversible translocation of EYFP-tagged STIM1 is coupled to calcium influx in insulin secreting &beta-cells. Cell Calcium 2008, 44, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Zbidi, H.; Jardin, I.; Woodard, G.E.; Lopez, J.J.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A. STIM1 and STIM2 are located in the acidic Ca2+ stores and associates with Orai1 upon depletion of the acidic stores in human platelets. J. Biol. Chem. 2011, 286, 12257–12270. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; De Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum—Plasma membrane junctions. Sci. Signal. 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, B.C.; Pingle, S.C.; Ahern, G.P. Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J. Leukocyte Biol. 2011, 89, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Borelly, L.; Somasundaram, A.; Yamashita, M.; Ren, D.; Miller, R.J.; Prakriya, M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 2008, 586 Pt 22, 5383–5401. [Google Scholar] [CrossRef] [PubMed]
- Courjaret, R.; Machaca, K. Mid-range Ca2+ signalling mediated by functional coupling between store-operated Ca2+ entry and IP3-dependent Ca2+ release. Nat. Commun. 2014, 5, 3916. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Hewavitharana, T.; Deng, X.; Soboloff, J.; Gill, D.L. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 2007, 42, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, M.K.; Stegner, D.; Berna-Erro, A.; Bittner, S.; Braun, A.; Kleinschnitz, C.; Stoll, G.; Wiendl, H.; Meuth, S.G.; Nieswandt, B. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J. Immunol. 2010, 184, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Oni-Biton, E.; Segal, M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 2017, 595, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Müller, H.-M.; Nickel, W.; Seedorf, M. Oligomerization and Ca2+/calmodulin control binding of the ER Ca2+ -sensors STIM1 and STIM2 to plasma membrane lipids. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Chiu, W.-T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [PubMed]
- Ruano, Y.; Mollejo, M.; Ribalta, T.; Fiaño, C.; Camacho, F.I.; Gómez, E.; de Lope, A.R.; Hernández-Moneo, J.-L.; Martínez, P.; Meléndez, B. Identification of novel candidate target genes in amplicons of Glioblastoma multiforme tumors detected by expression and CGH microarray profiling. Mol. Cancer 2006, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, H.A.; Leech, C.A.; Kopp, R.F.; Roe, M.W. Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry. Int. J. Mol. Sci. 2018, 19, 1522. https://doi.org/10.3390/ijms19051522
Nelson HA, Leech CA, Kopp RF, Roe MW. Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry. International Journal of Molecular Sciences. 2018; 19(5):1522. https://doi.org/10.3390/ijms19051522
Chicago/Turabian StyleNelson, Heather A., Colin A. Leech, Richard F. Kopp, and Michael W. Roe. 2018. "Interplay between ER Ca2+ Binding Proteins, STIM1 and STIM2, Is Required for Store-Operated Ca2+ Entry" International Journal of Molecular Sciences 19, no. 5: 1522. https://doi.org/10.3390/ijms19051522