A New Venue of TNF Targeting


1
Barriers in Inflammation, VIB Center for Inflammation Research, Ghent, Department of Biomedical Molecular Biology, Ghent University, 9052 Ghent, Belgium
2
Mouse Genetics in Inflammation, VIB Center for Inflammation Research, Ghent, Department of Biomedical Molecular Biology, Ghent University, 9052 Ghent, Belgium
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(5), 1442; https://doi.org/10.3390/ijms19051442
Submission received: 31 March 2018
/
Revised: 25 April 2018
/
Accepted: 3 May 2018
/
Published: 11 May 2018
(This article belongs to the Special Issue Tumor Necrosis Factor (TNF))
Abstract
:The first Food and Drug Administration-(FDA)-approved drugs were small, chemically-manufactured and highly active molecules with possible off-target effects, followed by protein-based medicines such as antibodies. Conventional antibodies bind a specific protein and are becoming increasingly important in the therapeutic landscape. A very prominent class of biologicals are the anti-tumor necrosis factor (TNF) drugs that are applied in several inflammatory diseases that are characterized by dysregulated TNF levels. Marketing of TNF inhibitors revolutionized the treatment of diseases such as Crohn’s disease. However, these inhibitors also have undesired effects, some of them directly associated with the inherent nature of this drug class, whereas others are linked with their mechanism of action, being pan-TNF inhibition. The effects of TNF can diverge at the level of TNF format or receptor, and we discuss the consequences of this in sepsis, autoimmunity and neurodegeneration. Recently, researchers tried to design drugs with reduced side effects. These include molecules with more specificity targeting one specific TNF format or receptor, or that neutralize TNF in specific cells. Alternatively, TNF-directed biologicals without the typical antibody structure are manufactured. Here, we review the complications related to the use of conventional TNF inhibitors, together with the anti-TNF alternatives and the benefits of selective approaches in different diseases.
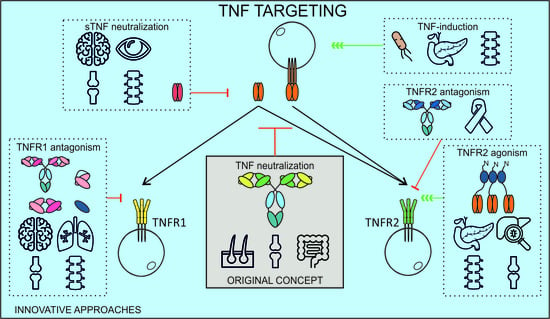
1. Introduction
Since the discovery and identification of tumor necrosis factor (TNF) in the mid 1980s [1], novel techniques allowed the isolation and cloning of the TNF gene for further characterization and TNF became the subject in a lot of studies. Originally, TNF was identified as a factor that necrotizes certain tumors [2,3] and recombinant TNF was useful to discover the biological functions of TNF. As a tumor-necrotizing agent, TNF’s toxicity in animal models was apparently acceptable, thus TNF was quickly launched into clinical trials. Eighteen monotherapy phase I and 10 phase II clinical trials were performed using recombinant human TNF (hTNF) therapy as anti-cancer agent, but none of them was successful as systemic TNF treatment was found to cause dose-dependent toxicities such as fever, hypotension and tachycardia [4,5,6]. Based on these and other studies, it became clear that TNF is a pleiotropic cytokine with major roles in physiology and pathology, amongst others by causing necrotic and apoptotic cell death, cellular regulation, differentiation, inflammation and the regulation of immune cells, and tumorigenesis [7]. The executive functions of TNF exceed multiple disciplines as TNF is important in homeostatic processes as well as in pathological situations ranging from inflammation, neurodegenerative diseases and infections.
Today, 19 members of the TNF superfamily have been identified, based on sequence similarity with TNF. In addition, 29 interacting receptors and several molecules interacting with the cytoplasmic domain of these receptors are recognized [1,7]. All members of the TNF receptor (TNFR) family contain one to six cysteine-rich repeats in their extracellular domains, typically each with three cysteine bridges within their structure [8]. The receptors can be classified in two subgroups: the death domain (DD)-containing receptors and the tumor necrosis factor receptor-associated factor (TRAF)-interacting receptors [9].
2. Biology
TNF is expressed as a 26 kDa transmembrane protein (tmTNF) which can be shed by the metalloproteinase TNF-α-converting enzyme (TACE) or disintegrin and metalloproteinase 17 ADAM17 to release the homotrimeric soluble TNF form (sTNF, monomeric 17 kDa) [10]. TNF is produced by a variety of cell types, such as monocytes and macrophages, T cells, natural killer (NK) cells, neutrophils, and microglia as well as by non-immune cells such as neuronal cells or keratinocytes. Both tmTNF as well as sTNF are biologically active, and the balance between these two forms is influenced by the cell type and its activation status, TACE activity and the expression of the endogenous TACE inhibitor, tissue inhibitor of metalloproteinase (TIMP)-3 [11,12]. TNF binds two homotrimeric transmembrane receptors: the 55 kDa TNF receptor 1 (TNFR1 or CD120a), encoded by the TNFRSF1A gene and the 75 kDa TNF receptor 2 (TNFR2 or CD120b), encoded by TNFRSF1B [9]. Interestingly, instead of only being a ligand, tmTNF can also act as a receptor because tmTNF-bearing cells show biological activity via reverse signaling when activated by mainly TNFR2. However, the biological functions elicited by this “outside-to-inside signaling” have not been completely elucidated [13]. TNFR1 is constitutively and ubiquitously expressed on a broad variety of cells, whereas expression of TNFR2 is inducible and tightly regulated. TNFR2 expression is more restricted and can be typically found on endothelial, immune (including microglia) and neuronal cells [9]. Recently, TNFR2 has also been found to be expressed on tumor cells and has been suggested to function as a tumor oncogene [14,15]. The extracellular domains of the two receptors are conserved and consist of a pre-ligand assembly domain (PLAD) and a ligand-binding domain, which is composed of four cysteine-rich domains and a TACE substrate domain. The PLAD stabilizes the receptors in absence of ligand as homophilic dimers. PLAD-mediated receptor preassembly is necessary for TNF/TNFR signaling and deletion of PLAD completely abrogates ligand binding and signaling [16]. In contrast to their extracellular domains, their intracellular domains are unrelated, explaining the initiation of different signaling cascades [17]. TNFR1 is a DD-containing receptor allowing protein–protein interactions, while TNFR2 does not have such a DD [18,19]. Successful signaling via TNF requires receptor preassembly as trimers prior to ligand binding. Preassembly occurs through the intracellular cytoplasmic tail of the receptors. The DD can recruit two adaptor DD-containing proteins, namely TNFR1-associated death domain (TRADD) or Fas-associated death domain (FADD), whereupon the apoptotic pathway is activated and the caspase cascade is engaged [20]. Importantly, in addition to other ligands such as Fas and TRIAL, TNF via TNFR1 can activate a caspase-independent pro-inflammatory cell death, called necroptosis [21,22,23]. This is a relatively novel programmed necrosis-like inflammatory process.
Upon TNFR2 activation, this receptor recruits TRAF2 and other TRAF2-associated proteins, as well as interacts with other signaling proteins that act independently of TRAF2. Whereas TNFR1 is linked with pro-inflammatory and apoptotic effects, TNFR2 has been associated with a variety of immune regulatory and anti-inflammatory functions [20]. Importantly, a complex interplay between TNFR1 and TNFR2 has been described, and additive, synergistic as well as antagonistic effects have been demonstrated [9].
TNFR1 is activated by either sTNF as well as tmTNF, while TNFR2 can only be activated by tmTNF. Hence, the role of TNFR2 is thought to be underestimated [24]. The membrane-bound forms of both receptors are also a substrate for proteolytical cleavage by TACE, yielding soluble receptor fragments e.g., soluble TNF receptor (sTNFR) [25]. This process is an important self-regulatory mechanism to prevent exaggerated damage and may contribute to the regulation of cellular TNF responsiveness [25]. Increased ectodomain shedding has three consequences: (1) On the one hand, the shed receptors can neutralize the bioactivity of circulating TNF by sequestering it. Hence, sTNFR will act as an intrinsic TNF inhibitor. (2) On the other hand, the process will decrease the number of signaling-competent receptors on the cell surface and cause transient TNF desensitization [26]. Accordingly, mice expressing non-sheddable TNFR1 spontaneously develop liver pathology and autoimmunity, pointing towards the pivotal role of TNFR1 shedding to regulate TNF activity in vivo [27]. The importance of this system is also highlighted in the disease condition of TRAPS (TNF receptor-associated periodic syndrome), an autosomal dominantly inherited disease characterized by unprovoked, often prolonged, attacks of fever and inflammation in multiple organs caused by a mutation in TNFSFR1A [28]. (3) Alternatively, sTNFR1 can form a stable complex with sTNF which can act as a sink in which the circulating TNF levels will be preserved.
3. TNF in Health and Disease
TNF is a pleiotropic cytokine and a master regulator of the immune system, and most cells show at least some TNF responsiveness. Studies revealed two opposite functions of TNF in host defense. At low levels, TNF has beneficial homeostatic functions, such as for host defense mechanisms against for example intracellular pathogens, particularly mycobacteria such as Mycobacterium tuberculosis [29]. However, at high concentrations, TNF can be deleterious and promotes inflammation and organ injury. In disease states, TNF is predominantly secreted by macrophages and monocytes both systemically and locally in the affected tissues, but also many other cells can produce TNF under certain circumstances [30].
3.1. Homeostatic Functions of TNF in Immunity
The use of transgenic mice greatly enhanced our knowledge about the different functions of TNF. TNF deficient mice revealed that TNF has prominent homeostatic functions in addition to its pro-inflammatory roles, and is vital for an optimal functioning of our immune system needed for host defense. Indeed, it has been shown that TNF is required to develop splenic B cell follicles, for the organization of the secondary lymphoid tissue architecture, and for germinal center (GC) and follicular dendritic cell (FDC) formation. Additionally, TNF is indispensable to fight pathogens and prevent further pathogen spreading by the formation and maintenance of granulomas, which are organized accumulations of infected macrophages and lymphocytes [31,32]. Likewise, TNF is highly needed for the resolution of inflammation and to promote tissue repair illustrated by its need for neuronal remyelination, cardiac remodeling and cartilage regeneration. The homeostatic potential of TNF has been proposed to be mainly mediated by TNF/TNFR2 signaling [33,34,35].
As mentioned earlier, the indispensable role of TNF for host defense against (intracellular) bacteria, viruses and fungi is well documented. Indeed, alone, together, or in synergy with interferons (IFNs), TNF is the most potent mediator in this process and it needs both TNF receptors for an efficient defense. Via the induction of chemokines and adhesion molecules [36], TNF is indirectly involved in the recruitment of inflammatory innate immune cells such as neutrophils, monocytes, NK cells and the so-called antigen presenting cells (APCs): immature tissue resident macrophages and dendritic cells (DCs). TNF enhances the pathogen-directed cytotoxicity of the innate immune cells and acts as a stimulatory agent for phagocytes [37]. In addition, TNF-induced NF-κB is required to convert immature tissue resident DCs into functionally mature effector cells. The latter will then stimulate naive T cells in nearby draining lymph nodes to initiate antigen-specific T and B cell responses [38]. For example, TNF is essential for a normal host response to Mycobacterium bovis and, more specifically, TNF derived from hematopoietic cells rather than from stromal origin [39]. To control M. tuberculosis, it has been shown that myeloid and T cells are the primary sources of TNF: myeloid cell-derived TNF is implicated in early immune responses, whereas T cell-derived TNF is essential to sustain protection during chronic infections [40].
Most of the pro-inflammatory functions are mediated by TNFR1, as mice deficient for TNFR1 are highly susceptible to Listeria monocytogenis and M. tuberculosis infection, and TNFR1 is also required for the anti-viral response via induction of apoptotic cell death [38,41]. Moreover, loss-of-function due to TNF knockout is highly mimicked by TNFR1 deficiency [42]. The role of TNF/TNFR1 signaling in M. tuberculosis infection relates to its contribution in granuloma formation that is needed to control the infection. Additionally, it may relate to macrophage activation to produce reactive nitrogen species to destroy intracellular bacteria [43]. Interestingly, mice that express a non-cleavable variant of TNF were partially protected against M. tuberculosis, L. monocytogenes and M. bovis, indicating that tmTNF signaling, which predominantly occurs via TNFR2, is needed for this protection [44]. In addition, mice that express non-cleavable TNFR1 were more resistant to L. monocytogenes infections, suggesting that impaired TNFR1 shedding enhances the antibacterial host defense [27]. Additionally, a recent study indicates that TNFR1 on myeloid and not on T cells is crucial to control M. tuberculosis infection [45].
3.2. Systemic Inflammation
3.2.1. TNF: The Master Regulator of Inflammation
Sepsis is a very complex syndrome that is caused by a dysregulated host response to infection. It is characterized by sustained excessive inflammation and immune suppression, and many studies have shown that TNF is the master mediator of the inflammatory response seen in sepsis and shock, the life-threatening condition caused by circulatory and/or metabolic abnormalities. Indeed, TNF is released from macrophages into the systemic circulation as early as 30 min after an inciting event such as an intraperitoneal (ip) lipopolysaccharide (LPS) injection, with peak concentrations observed after 60–90 min [46]. This primes the activation of other inflammatory mediators such as interleukin (IL-)1. Depending on where TNF is produced and on which cell it acts, TNF not only potently promotes the release of the secondary mediator IL-6, but also drives its own production. The excessive production of pro-inflammatory mediators is followed by a wave of counteracting and anti-inflammatory mediators such as soluble TNFR that sequesters its bioactive ligand [47] (Figure 1). During bacterial infections, these cytokines are the main drivers and are the central mediators of the induced shock after either Gram-positive or Gram-negative bacteremia [38,48]. Thus, dysregulation of the TNF production due to an overreaction of the host may have unforeseeable consequences. In animals, exogenous TNF administration leads to a syndrome that is indistinguishable from septic shock and infusion of TNF into humans results in systemic inflammatory response syndrome (SIRS) [49]. Consequently, sustained elevated endogenous TNF levels can lead to a SIRS, which may evolve to death due to multiple organ failure (MOF). Several studies demonstrate that TNF serum levels in sepsis patients are elevated and associated with mortality [50,51] and they are used as effective markers in the diagnosis of neonatal sepsis [52]. These observations led to the rationale to therapeutically neutralize circulating TNF in septic patients. However, numerous clinical trials failed to demonstrate clear statistical benefits [53,54,55,56,57,58,59]. As sepsis involves the presence of a pathogen, the inability to control the infection when TNF signaling is abrogated might account for the failure seen in these clinical trials. Indeed, in sterile sepsis models (i.e., LPS-induced shock), anti-TNF treated mice show some degree of protection but this is not recapitulated in real infection models such as cecal ligation and puncture (CLP) [60,61]. However, a meta-analysis suggests that immunotherapy with monoclonal antibodies (mAbs) against TNF does reduce the overall mortality in severe septic patients when the drugs is administered before shock. Furthermore, it may improve survival in patients with shock or with high IL6 levels, and thus requires patient stratification [62].
3.2.2. Differential Roles for TNFR1 and TNFR2 in Sepsis
In sepsis, a differential role for TNFR1 and TNFR2 has been uncovered by using transgenic mice in several experimental models, although their exact contributions remain debatable. Mice deficient for TNFR1 were protected against death when they were subjected to endotoxemia, i.e., the injection of a lethal dose of LPS [64,65], whereas TNFR2 knockout (KO) mice were as sensitive as wild type (WT) mice [66]. At the level of the gut, Williams et al. reported that TNFR1 is essential for LPS-induced gut injury by mediating apoptosis of intestinal epithelial cells (IECs) [67], and also in the TNF-induced lethal shock model, the complete survival benefit in TNFR1 KO mice was attributed to the avoidance of TNF-induced gut permeability [41,68,69]. Additionally, the blood–cerebrospinal fluid (CSF) barrier permeability was also less comprised in endotoxic TNFR1 KO mice [70]. This is an important observation, as preservation of the integrity of the brain barriers might overcome the occurrence of sepsis-associated encephalopathy (SAE) [71]. This devastating complication of sepsis is associated with early death in sepsis patients [72]. It has been suggested that TNFR1 is a critical mediator in the onset of SAE because of its stimulating effects on aquaporin-4 and concomitant increase in water content in the brain [73]. Interestingly, TNFR1−/− mice also experienced less sepsis-induced memory deficits [74]. However, despite of these interesting observations, TNFR1 KO mice were not protected against very high LPS doses [42,66], which was also confirmed in our lab [70]. The previous models are sterile models, and observations in CLP, which is considered as the golden standard for human polymicrobial sepsis [75], or in colon ascendens stent peritonitis (CASP) left the scientific community with contradictory results regarding the different roles of TNFR1 and TNFR2 in real sepsis models. Some groups, including ours, found that TNFR1 deficient mice were as sensitive as WT mice in CASP or CLP [70,76,77], whereas others reported that TNFR1 KO mice had prolonged survival in CLP and that TNFR2 KO mice were more sensitive than WT mice [78]. When polymicrobial sepsis was initiated by ip injection of cecal microflora, the levels of TNF were severely elevated and mice deficient for TNFR1 or both TNFRs survived the induced sepsis [61]. Collectively, these studies fail to provide a clear consensus about the exact contribution of each receptor. These data also suggest that lethality does not only depend on TNF, but also other inflammatory mediators contribute to the LPS-induced lethality for instance IL-1β or matrix metalloproteinases (MMPs) [79]. Indeed, former research of the lab reported an important interplay between TNFR1 signaling and MMP8 in sepsis [70]. We found a link between these two mediators in sepsis patients and mice deficient for the both genes were significantly more protected against very lethal endotoxemia and CLP-induced peritonitis than WT or single KO mice. Hence, our research group created a bivalent Camelid-derived heavy-chain only Ab, namely Nanobody (Nb) 70-alb-14, that simultaneously antagonizes TNFR1 and MMP8. Proof-of-concept was provided as treatment with this Nb reduced lethality against CLP [70].
3.3. Autoimmunity
3.3.1. Implications of TNF Signaling
Persistently elevated levels of TNF are evident in chronic inflammatory disorders, e.g., rheumatoid arthritis (RA), inflammatory bowel disease (IBD), ankylosing spondylitis (AS) and psoriasis. TNF regulates differentiation, proliferation and apoptosis of several non-immune cells such as IECs, keratinocytes or synovial fibroblast [80,81,82]. In IBD, overexpression of TNF may contribute to increased barrier permeability for luminal pathogens [81]. Importantly, TNF can also initiate necroptosis and this is a common feature in the pathology of several inflammatory diseases and autoimmunity such as in Crohn’s disease (CD) [21,22,23]. The pro-inflammatory role of TNF in these autoimmune diseases is supported by the great clinical effects of TNF-antagonists, which are used in these disorders both in humans as well as in animal models of for instance RA [83,84]. Again, genetically modified mice were an invaluable tool to elucidate the pathogenic role of TNF here. Many valuable insights came from transgenic mice that lack the AU-rich element (ARE) in the 3′-untranslated region of the TNF mRNA. TNF∆ARE mice systemically overexpress TNF due to the increased stability of its mRNA. They spontaneously develop erosive, symmetric polyarthritis and IBD. As the clinical symptoms in the mice resemble the clinical and histopathologic features of RA and IBD in patients, they are an ideal model for these diseases [30,85]. Furthermore, other transgenic mice expressing a hTNF transgene were found to develop a TNF-dependent chronic inflammatory polyarthritis resembling human RA [86,87]. However, this does not necessarily mean that all types of RA and IBD in patients start with a dysregulated TNF expression.
Despite the untoward effect of TNF in the development of autoimmunity, it has also been demonstrated that TNF is sometimes needed to inhibit or control autoimmunity [88]. Indeed, TNF neutralization exacerbates acute injury in the dextran sodium sulfate (DSS) colitis mouse model [89] and exogenous TNF administration could alleviate colitis in oxazolone-treated mice and attenuate the disease severity in RA [90,91]. Furthermore, studies demonstrate that chronic treatment with a low TNF dose or local pancreatic expression of TNF in adult non-obese diabetic (NOD) mice, which is a model for diabetes, delayed spontaneous development of type I diabetes in these mice [92,93]. A similar finding was done in NZB/W mice, a model for systemic lupus erythematosus (SLE), in the development of autoimmunity [94]. Additionally, when these lupus-prone mice were crossed into a TNF deficient background, they experienced aggravated disease [95]. In addition to suppressing systemic autoimmunity, TNF can also suppress organ-specific autoimmunity. The importance of this mechanism is further highlighted in MS mouse models. In the absence of TNF, myelin-specific deleterious T cells remain abnormally prolonged self-reactive to myelin, while in normal conditions they become inactivated [96]. Therefore, TNF may be protective against chronic experimental autoimmune encephalomyelitis (EAE) by downregulation or inactivation of the potentially detrimental autoimmune T cell response against myelin antigens [88]. In conclusion, many models suggest an immunosuppressive and immunoregulatory role for TNF, which may segregate at the level of the two different receptors: the classic pro-inflammatory activities of TNF mediated by TNFR1 may account for chronic inflammatory pathology and tissue damage, especially in situations with persistent and maintained TNF overexpression [88], while immunosuppressive activities might be attributed to TNFR2 [97].
3.3.2. Receptor-Dependent Roles for TNF in Autoimmunity
To analyze the contribution of the two TNF receptors, the aforementioned experimental mouse models were applied in a TNFR1 and/or TNFR2 KO background. In the collagen-induced arthritis (CIA) model for RA, TNFR1 KO mice developed the disease at lower incidence and in a milder form than WT mice [83,98]. In contrast, TNF-driven arthritis was severely aggravated in TNFR2 KO mice [35,99]. The importance of TNFR2 signaling in autoimmune diseases is also illustrated by its prominent role in regulatory T cell (Treg) functioning [100,101,102]. The most potent Tregs express the highest TNFR2 levels and TNF/TNFR2 signaling is required to activate and expand naturally occurring Tregs [101,103,104]. Given that Tregs are essential for immune tolerance and suppress self-reactive T cells [105], their optimal functioning should be considered in new therapies. In RA, selective inhibition of TNFR1 abrogates inflammation by enabling Tregs to suppress IL-17 production, and promotes Treg activity via TNFR2 signaling [106,107]. Likewise, TNFR1 or double TNFR deficient TNF∆ARE mice had a normal histology without any sign of macroscopic illness. In contrast, symptoms were not improved but rather aggravated with more aggressive and destructive arthritis when crossed into the TNFR2 KO background [85]. In addition to these data, mouse and human data in CD point to the importance of the suppressive functions of Tregs which are attributed to TNFR2 [108,109]. Indeed, in mice, Tregs are critical for maintaining intestinal tolerance to luminal antigens and for preventing intestinal inflammation [110]. Genetic data further strengthen the importance to acknowledge the two receptors separately. Indeed, polymorphisms in (the promoter region of) TNFSF1B were associated with increased susceptibility for patients to develop RA, IBD or lupus, suggesting that TNFR2 mutations could lead to increased inflammation due to defective control mechanisms [111,112]. However, the effects of these polymorphisms on the Treg population are not studied and require further examination [113].
3.4. The Role of TNF in Neurodegenerative Diseases
The most under-appreciated role of TNF is its role in neurobiology. In the last decades, TNF has been shown to have several important physiological but also pathological functions in the CNS [12]. In the brain, the duality of TNF is nicely demonstrated at several levels. On the one hand, TNF functions as an essential gliotransmitter, secreted by neurons and glial cells such as microglia and astrocytes. Moreover, TNF regulates synaptic communication between neurons as demonstrated by its involvement in synaptic scaling and plasticity [114], and it orchestrates learning and memory processes via hippocampal neuronal development [115]. Other neurophysiological functions of TNF are listed by Decourt et al. [116]. On the other hand, TNF potentiates excitotoxicity via a microglial and astroglial loop that ultimately results in neuronal death, and inhibition of sTNF ameliorates synaptic dysfunction in aging and improves learning [117]. Conversely, TNF also protects against excitotoxicity via TNFR2 [12,118,119].
Intriguingly, systemic inflammation induces TNF expression in the brain [120] and it has been shown that in mice, TNF can cross the blood–brain barrier (BBB) to reach the brain via a saturable transport system [121]. Microglia which are the tissue-resident macrophages of the CNS are one of the major producers of TNF, participating in numerous pathophysiological conditions in the brain. Indeed, elevated TNF levels are evident in many neurological disorders such as in affected areas in multiple sclerosis (MS, cf. Section 3.4.2), Alzheimer’s disease (AD, cf. Section 3.4.3), Parkinson’s disease (PD, cf. Section 3.4.4), stroke and traumatic brain injury (TBI). Intriguingly, TNF, amongst others, mediates necroptosis which is clearly involved in AD and PD, highlighting one of the possible detrimental effects TNF can exert in these disorders [122]. TNF released in the brain can be both toxic and tropic; however, this is not always clear and depends on the context [12,123,124]. Therefore, unselective targeting of TNF in the brain with therapeutics is not desirable and distinctions should be made based on its function.
3.4.1. Differential Roles of TNFR1 and TNFR2 in Neuronal Health and Disease
In the CNS, microglia secrete TNF and it has been shown that TNF promotes its own release via TNFR1. Hence, it sustains a vicious feedback loop in microglial activation [125]. Additionally, TNFR1 signaling upon TNF interaction could induce the release of glutamate from microglia and astrocytes and also directly potentiates glutamate excitotoxicity through the activation of the glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [125,126,127]. Conversely, TNFR2 activation protects against glutamate-induced excitotoxicity [119,128]. In aging, it has been suggested that TNF/TNFR1 signaling contributes to cognitive decline through its effects on hippocampal long-term depression (LTD). Indeed, in aged rats, the hippocampal levels of the TNF receptors are changed in favor of TNFR1, and inhibition of the TNF/TNFR1 pathway improves behavioral performances and synaptic functioning [117]. Literature also suggests a direct link between the activation of TNFR1 by TNF and neuronal apoptotic death in neurodegenerative disorders [129,130]. Additionally, neurons lacking TNFR2 are more vulnerable to TNF-induced cell death than WT neurons, as TNFR2 overrides the death signals delivered through TNFR1 [131]. In a disease state, TNF robustly stimulates TNFR1 resulting in an apoptotic signal that overweighs the signals through TNFR2. In this respect, hippocampal neurons from TNFR2−/− mice are more vulnerable to a low TNF dose whereas TNF has little effects in TNFR1−/− hippocampal neurons [130]. During neurogenesis, TNF via TNFR1 negatively impacts the hippocampal neurogenesis but is essential for striatal morphology and injury resolution. Conversely, TNFR2 is required for normal hippocampal neurogenesis and morphology in healthy adults, and for hippocampal healing upon injury [132,133,134,135]. In addition, in the context of neuropathic pain, the induced depression and impaired hippocampal plasticity depend on TNFR1 signaling [136]. During stroke, optimal TNF signaling is pivotal for hippocampal neurogenesis, functioning and repair following ischemic insults [137,138]. Table 1 provides a non-exhaustive list of neurological conditions in which different roles for TNFR1/2 are described, and we will deeper dig into MS, AD and PD.
3.4.2. TNF and Its Receptors in Multiple Sclerosis
MS is characterized by immune cell infiltration and upregulation of pro-inflammatory cytokines and chemokines such as IL-1β, IL-17, IL-22, IFN-γ and TNF in CSF [139,140]. An important role for TNF in MS has been described although its exact role remains inconclusive. TNF and its receptors are found in the serum, CSF and lesions of MS patients and TNF levels in serum and CSF are correlated with disease severity [140,141]. Additionally, mouse studies using the established MS mouse model EAE revealed a pathogenic role for TNF in MS [142,143] and CNS-specific overexpression of TNF leads to spontaneous demyelination [144,145]. It has been suggested that TNF-mediated demyelination depends on cellular contacts between TNF-producing cells such as astrocytes or microglia and oligodendrocytes (OLGs). Indeed, human tmTNF expressed by astrocytes is more effective to kill OLGs than sTNF and astrocyte-specific overexpression of tmTNF triggers inflammation and demyelination and this is mediated via TNFR1 [145,146]. It was shown that T cells and myeloid cells are the critical cellular sources of TNF during EAE as T cell-derived TNF determines the severity in EAE by regulating leukocyte influx into the CNS, whereas myeloid-derived TNF controls the early expression of cytokines and determines the onset of EAE [147]. Evidence of its pathogenic role was further provided by anti-TNF treatment that prevented the initiation of clinical symptoms in EAE and ameliorated progression in established disease in mice [148]. To the contrary, although initiation of EAE in TNF KO mice was delayed, the mice eventually developed EAE that was as severe or even more severe with high mortality and extensive inflammation as compared to WT mice [149,150,151]. Furthermore, anti-TNF treatment increased MRI activity and immune activation in several patients with primary progressive MS (PPMS) [152] and in patients with relapsing-remitting MS (RRMS), a phase II clinical study with a TNF inhibitor (Lenercept, a dimeric TNFR1 fused to the immunoglobulin (Ig)-G1 heavy chain) was discontinued because of unexpected exacerbations of the disease [153]. Strikingly, anti-TNF medication can even sporadically induce demyelinating diseases and neuropathies (cf. Section 4.3.4), and several groups found that TNF expressed in lymphoid organs could dampen the immune response by inhibiting the development of encephalitogenic T cells responses [96,147]. Contrasting results have also been described in OLGs as TNF is involved in oligodendrocyte precursor cell (OPC) proliferation and maturation [33] but also causes OLG cell death both in vitro and in vivo [154,155,156,157]. OLGs located at the edge of active lesions express both TNFRs and this could explain the duality of TNF in MS. These results suggest that TNF is again not only destructive but also has essential roles to maintain immune homeostasis in the CNS environment.
Interestingly, the TNFRSF1A locus has been validated as a MS susceptibility locus [158]. The disease genetic variant leads to the expression of a soluble form of TNFR1 that sequesters TNF and thereby abrogates signaling through TNFR2. This suggests that dysregulation of the TNF/TNFR1-pathway has a role in the onset of MS [159]. TNF/TNFR1 signaling has been shown to be involved in OLG apoptosis, demyelination, and initiation of inflammatory processes (Figure 2 and Table 1). First evidence of the harmfulness of TNFR1 has been delivered as EAE disease development was prevented in double TNFR KO as well as in TNFR1 KO mice. This was contrasted by the outcome of TNFR2 null mice which developed a more severe and chronic disease [160,161]. Interestingly, both TNFR1 and TNFR2 KO mice were able to suppress the anti-myelin reactivity, leading to the idea of redundancy in the immunosuppressive functions of the two receptors. However, again, TNFR1 was found to be responsible for the detrimental signals, while mainly TNFR2 was essential for resolving the inflammation and initiating repair [161]. Additionally, in TNFR1 deficient mice that locally express TNF by CNS glial cells, it has been shown that this receptor was able to induce OLG apoptosis, primary inflammatory demyelination and the generation of MS-like plaques [157]. Moreover, EAE mice that only express non-cleavable tmTNF were protected against EAE, suggesting that the interaction between sTNF and TNFR1 mediates the pathology [162]. This concept was further supported by the observation that inhibition of sTNF reduced spinal cord injury [163] and EAE pathology, associated with reduced immunoreactivity, increased expression of neuroprotective and myelin-specific genes and axonal preservation [164,165,166]. Although sTNF inhibition did not prevent OLG loss and demyelination in the cuprizone-induced demyelination model, it induced early remyelination due to improved removal of myelin debris by CNS phagocyting macrophages, indicating that sTNF inhibits the remyelination process [167]. Furthermore, astrocyte-expressed TNF is crucial for blood–brain barrier (BBB) damage and endothelial activation via TNFR1 [114,145,157]. Interestingly, TNFR1 neutralization may indirectly stimulate TNF/TNFR2 signaling in the EAE lesions and promotes remyelination in chemically-induced demyelination [168]. Others successfully applied specific TNFR1 inhibition in EAE using the TNFR1-selective antagonistic mutant TNF protein, PEG-R1antTNF [169] or the commercial monoclonal hamster IgG antibody against mouse TNFR1 [170]. Our research group developed a trivalent TNFR1 Nanobody with very promising characteristics in the EAE pathology [171] (cf. Section 6). MS often presents with memory deficits, and it has been elegantly shown that TNF/TNFR1 signaling in astrocytes is responsible for these cognitive disturbances [172]. However, this symptom has never been therapeutically addressed in mouse studies and the efficacy of TNFR1-inhibiting drugs to overcome memory deficits in MS should definitely be investigated in future research.
TNFR2 is minimally expressed in the CNS in physiological conditions but its expression is boosted in neurological condition in glial cells such as microglia, astrocytes and OLGs [173] but usually not in neurons. In MS, microglia, monocytes and macrophages express TNFR2 and these cell populations also play pivotal roles in the disease [165]. Moreover, TNFR2 null mice subjected to MOG35–55-induced EAE showed exacerbated disease, enhanced Th1 cytokine production, and enhanced CD4+ cell infiltration in the CNS [161]. Conversely, mice only expressing tmTNF (i.e., TNF non-sheddable mice), which mainly signals via TNFR2, were protected during EAE [162]. A neuroprotective role is attributed to TNFR2 because its signaling pathway protects neurons against excitotoxic insults in vitro and in vivo induced by excessive glutamate levels [128,174], and against oxidative stress [168,175]. Furthermore, TNFR2 promotes neuronal survival, OPC maturation and differentiation, OLG proliferation and CNS remyelination [33,168,176,177,178,179]. Given that TNFR2 is also important for Treg expansion and facilitates their suppressive capacity against effector T cells, and since certain Tregs are dysfunctional in MS [180], TNFR2′ optimal signaling should be guaranteed. Intriguingly, it has been shown that also TNFR2 on non-hematopoietic cells is important to modulate the fate of Tregs and their suppressive functions [181]. However, TNFR2 has a dichotomous function: Gao et al. recently showed that peripheral macrophage/monocyte-TNFR2 drives the immune activation via T cell activation and triggers leukocyte transmigration across the BBB, whereas microglial TNFR2 provides protective signals by promoting anti-inflammatory pathways [173,182]. This should be considered for therapeutic purposes. The complexity is even increased as TNF mediates regression of activated myelin-specific T cells. This prevents prolonged primary myelin reactivity and limits the probability for chronic disease. Although it was first thought that this was mediated by TNFR1, more recent studies pointed towards TNFR2, but also redundancy in this immunosuppressive function is described for the two receptors [88,96,183,184].
3.4.3. TNF Involvement in Alzheimer’s Disease
As already outlined in the previous sections, TNF has a very important pluripotent role in the brain. The clinical involvement of TNF in AD is evidenced by the observation that TNF serum and CSF levels are correlated with disease severity and that TNF co-localizes with amyloid beta (Aβ) plaques in the brain. In transgenic mouse models of AD, TNF contributes to disease progression and onset [12]. However, TNF is also a known regulator of synaptic communication between cells. Clearly, the role of TNF in the brain is divergent: low levels of TNF are needed in healthy brains, while overexpression of TNF, primary by microglia, is neurodegenerative. It has been proposed that the synaptic effects of TNF are associated with the synaptic dysfunction that has a central role in AD, particularly with respect to cognitive dysfunction [208]. Indeed, TNF can influence synaptic transmission and plasticity such as hippocampal long-term potentiation (LTP) and synaptic scaling that contributes to early memory loss and learning impairment [192,209,210]. Additionally, it has been demonstrated that TNF contributes to amyloidogenesis, although there is no clear consensus where TNF interferes in the amyloidogenesis process. A study in TNF KO AD mice (5×FAD) found that the diminished amount amyloid plaques and Aβ species are a result of reduced Aβ generation and not a consequence of more clearance [211]. Others state that TNF is implicated in both Aβ generation and clearance [211,212]. TNF, whether or not produced by neurons, promotes the expression of astrocyte beta-secretase 1 (BACE1) and suppresses Aβ clearance by inhibiting microglial phagocytosis [212,213,214]. In contrast, transient hippocampal TNF expression decreases Aβ disposition [215]. In further support of the neurotoxicity of TNF in AD, chronic neuronal TNF overexpression promotes brain inflammation and is detrimental for neuronal viability and these inflammatory events coincides with the appearance of cognitive deficits and synaptic dysfunctions [216]. This suggests that TNF participates in multiple stages of AD pathology [216] (Table 1). Clearly, TNF has again both beneficial and detrimental roles in AD, depending on differences in the (spatiotemporal) TNF expression pattern.
Epidemiological studies demonstrate that the relative AD risk was significantly reduced in RA patients that received the anti-TNF drug etanercept [217,218]. To better understand the contribution of peripheral TNF-mediated inflammation to AD pathology, Paouri et al. crossed AD susceptible mice (5xFAD) with Tg197 mice that have a whole-body expression of hTNF. Interestingly, peripheral hTNF which signals only via mouse TNFR1 [219], robustly activates microglial and astrocytes which in turn clear Aβ but also mediate synaptic loss [185]. Despite of this evidence, preclinical studies in mouse AD models with anti-TNF inhibitors left us with conflicting results. In some studies, the use of TNF inhibitors such as etanercept or infliximab demonstrated clinical benefits [220,221], whereas other failed to reproduce that [222,223]. In addition, results from clinical studies are not always clear: a double-blind randomized trial of peripheral administration of etanercept failed to show cognitive or behavioral benefits [224], whereas perispinal administration of etanercept improved cognitive decline in a small short-term pilot study [225,226]. Intrathecal infliximab administration also improved the cognitive behavior, but this was a case report in one woman thus larger studies are imperative [227]. Recently, a phase I open-label crossover study in patients with mild to moderate AD treated with perispinal administration of etanercept together with dietary supplements could provide more insights into the potential effect of etanercept in AD but unfortunately, results were not conclusive about the cognitive benefit compared to patients treated with nutritional supplements alone [228]. These clinical and preclinical data might indicate that the therapy needs to be initiated at very early stages of AD, rather than in advanced disease, or that a more selective TNF neutralizing approach should be implemented.
In AD brains, TNFR1 protein levels and TNFR1 binding affinity were augmented in contrast to TNFR2 levels and binding affinity compared to non-demented patients [229,230]. Interestingly, our research team found that in the choroid plexus of AD patients, TNF is the main inflammatory upstream mediator, providing detrimental signals via TNFR1. The blood–CSF barrier consists of a monolayer of choroid plexus epithelial cells, and we found that TNFR1 contributes to the morphological damage which is typically seen in the choroid plexus of AD patients [186,231]. Additionally, Li et al. reported that Aβ induces neuronal death via TNFR1 in the AD brain [129] and TNFR1 contributes to the amyloidogenesis via the regulation of BACE1 in APP23 transgenic mice [131]. Our recent study reinforces these results in two AD model: the intracerebroventricular (icv) injection of oligomerized Aβ and in APP/PS1 mice [186]. Our results were in line with observations in APP23 mice devoid of TNFR1 that have less memory deficits, neuronal loss and microglia activation compared to normal APP23 mice [131]. Pharmacological evidence was also provided as inhibition of sTNF, signaling through TNFR1, reduced the accumulation of APP fragments in hippocampus and cortex of 3×Tg-AD mice, and restored synaptic dysfunction and LTP impairment in 5xFAD mice [188,189]. These results were in agreement with the study of Paouri et al. [185]. Strikingly, direct TNFR1 blockage with a TNFR1-inhibiting Nanobody TROS alleviated the AβO-induced memory deficits [186] and the Aβ-mediated inhibition of hippocampal LTP is reversed in absence of TNFR1, providing strong evidence that TNFR1 activation is required for the Aβ-mediated inhibition of LTP [192]. Conversely, inhibition of TNFR2 increased Aβ toxicity in vitro [190] and APP23 mice deficient for TNFR2 displayed exacerbated AD pathology compared to APP23 mice with a functional TNFR2 gene [187]. Furthermore, selective inhibition of neuronal TNFR2 enhanced the Aβ and Tau-related pathologic features in AD and diminished microglia activation needed for Aβ clearance [191]. These observations support the idea that TNFR1 has detrimental roles in AD, whereas TNFR2 needs to be spared to counteract the Aβ-mediated pathology and urges more selective targeting of the TNF pathway (see Table 1). Likewise, in seizure models, TNFR2 is important to protect hippocampal neurons against excitotoxicity and pan-TNF inhibitors that do not spare TNFR2 lead to untoward effects in this brain region, again suggesting that TNFR2 is important in hippocampal repair and neurogenesis [205].
3.4.4. TNF in Parkinson’s Disease
Inflammatory processes are described in PD patients and some may argue that they even trigger the disease onset. This also accounts for peripheral inflammation that enhances the degeneration of dopaminergic neurons. This was clear in animal models, but also human data point in that direction [232]. As in several other neurological disease, increased levels of TNF and sTNF(R1) are evident in the CSF and tissues of PD patients, as well as in postmortem brain tissue. The levels found in serum correlated with disease severity according to some researchers [124,233]. One group also found that TNF gene promoter polymorphisms were associated with an earlier age of PD onset [234]. This evidence about the effector role of TNF in PD is strengthened by the observation of very quick fundamental TNF increments in PD mouse models. In addition, TNF was found to be extremely toxic for dopaminergic neurons in vitro. These findings support the idea that the TNF-driven inflammation is essential in the pathogenesis and progression of the disease [12]. Unfortunately, studies in transgenic mice that were subjected to different PD models (injection of parkinsonian neurotoxic agents 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)) have yielded contrasting results. The toxicity of TNF on dopaminergic neurons was demonstrated in TNF KO mice which were less sensitive to MPTP-induced striatal dopaminergic neurotoxicity [235]. As dopaminergic neurons were shown to express TNFR1 and this expression is induced in PD [236], the role of the two receptors was further investigated by several groups. In line with the results obtained in the TNF KO mice, also the double TNFR KO mice were completely protected against MPTP-induced neurotoxicity by suppressing microglial activation [193,194,237]. However, this was rebutted by Leng et al., stating that TNF has protective effects, mediated by TNFR-independent mechanisms [195]. Because the vulnerability of hippocampal neurons to MPTP was increased in mice lacking the two TNFR, a dual and region-dependent role of TNF was proposed and this highlighted the neurotrophic or neuroprotective role of TNF in the hippocampus [194]. This specific role was confirmed by others that observed that TNF does not participate in dopaminergic neuronal cell death in PD but rather alters dopamine metabolism and the survival of dopaminergic terminals [238]. Interestingly, the effects of TNF are not only region-dependent but also dose-dependent: low TNF concentrations in the substantia nigra mediate neuroprotective effects in mice by reducing the nigrostriatal neurodegeneration induced by 6-OHDA although chronic expression of low TNF levels eventually causes dopaminergic cell death, and functionally leads to akinesia. Conversely, high TNF levels induce progressive neuronal cell loss accompanied by gliosis and inflammation [239,240]. In addition, in preclinical studies, contradictory results were obtained as early TNF blockage worsened the outcome after intrastriatal 6-OHDA injection [241] but intranigral infusion of sTNF neutralizing therapeutics attenuated the nigral dopaminergic loss and microglia activation [196,197,198]. This sTNF inhibitor could cross the BBB and had disease-modifying properties upon peripheral administration [198]. Inhibition of TNF synthesis by thalidomide partly protected against MPTP-induced dopamine depletion [235]. Interestingly, a BBB-penetrating Trojan horse has been designed consisting of a TNF decoy receptor fused to a mAb against the mouse transferrin receptor (TfR). This drug was neuroprotective in the 6-OHDA mouse model of PD. In contrast, etanercept that does not penetrate the BBB had no effect on the neurobehavior [242]. To analyze and interpret these incoherent results, one should account for the divergence across all studies, with differences in model, doses and timing of analysis [243]. Furthermore, these models are difficult to extrapolate to human situations as the TNF peak in these mouse models is relatively short, whereas the levels remain elevated along the disease course in PD patients.
4. TNF Inhibitors
4.1. Approved TNF Inhibitors
The initial concept to use recombinant TNF as an anti-tumor agent was quickly followed by the idea to consider TNF as a drug target for inflammatory diseases [244]. Indeed, TNF represents an active and attractive objective for drug development despite the initial skepticism because of the failure of anti-TNF drugs in sepsis patients [245,246]. The rationale to target TNF was first confirmed in a murine RA model [247], and, in 1993, RA patients were successfully treated with mAb cA2, later known as infliximab [248]. This success was the start to further develop anti-TNF drugs in TNF-involving inflammatory diseases. Currently, five anti-TNF biologics and in total 25 drugs that inhibit or modulate the effects of TNF, are approved for clinical use by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of RA, AS, psoriasis and psoriatic arthritis (PsA), juvenile idiopathic arthritis (JIA), CD and ulcerative colitis (UC). Recently, adalimumab was also licensed in some countries to treat uveitis and hidradenitis suppurativa which is a chronic skin disease characterized by recurrent abscesses. Furthermore, there is off-label use in Behcet’s syndrome and amyloidosis [245,249]. The introduction of TNF inhibitors on the market has revolutionized the treatment of these pathologies and anti-TNF therapy is now standard of care for RA. Moreover, these blockbusters currently belong to the top-10 best-selling drugs in the world, with adalimumab being the world’s best-selling medicine, counting for US $10 billion per year and the total sale of the various anti-TNF drugs exceeds US $25 billion [245]. Currently, another 151 TNF inhibitors are in the clinical pipeline.
Three of the five approved TNF-inhibitors are full-length monoclonal antibodies (mAbs): infliximab (Remicade® and biosimilars Remsima®, Inflectra®, Flixabi®, and Ixifi®), adalimumab (Humira® and Cyltezo®, Imraldi®, Amgevita®, and Solymbic®) and golimumab (Simponi®). Next to these, certolizumab (Cimzia®) and etanercept (Enbrel® and biosimilars Erelzi® and Benepali®) are also approved. Although they all neutralize the TNF activity, they each have different characteristics and routes of administration. Furthermore, all of them are equally effective against RA, but not against CD. These discrepancies are attributable to different mechanisms of actions that are not completely understood [38,250,251] (Table 2 and Figure 3).
In 1998, infliximab was the first TNF-targeting antibody approved in the US to treat CD and later UC. It is a chimeric monoclonal IgG1 Ab that comprises a human constant domain and murine variable regions. The infliximab biosimilar CT-P13 (Remisma® or Inflectra®) is highly similar to its originator and therefore clinically used in the same way [252]. Golimumab and adalimumab are full human Abs that were produced by recombinant DNA technology and certolizumab is a humanized Fab’ fragment that is conjugated to polyethylene glycol (PEG) to increase the serum half-life. This reduces the requirement for frequent dosing and possibly reduces the immunogenic nature. Finally, etanercept is a fusion protein of the extracellular domain of human TNFR2 receptor coupled to the Fc region of human IgG1. Etanercept binds circulating sTNF and acts as a decoy receptor that prevents TNF-interaction with the cell surface receptors.
4.2. Mechanisms of Action of TNF Inhibitors
All anti-TNF agents have the same target but not all of them are equally efficacious in all considered diseases, suggesting that different working mechanisms are inherit to certain antibody structures. It is clear that particularly in CD alternative effector mechanisms rather than pure TNF neutralization account for their efficacy whereas this is less the case in RA in which all marketed anti-TNF drugs are indicated. First, their affinities for TNF are different and generally tmTNF is neutralized with lower affinity than sTNF [252]. Etanercept is the only one that is capable of neutralizing lymphotoxin-α (LT-α) and it only neutralizes trimeric TNF. However, TNF inhibitors show equal sTNF neutralizing potency [276]. Neutralization of sTNF or tmTNF blocks the TNF-mediated activation of TNFRs and this results in suppression of inflammatory mediators. Indeed, TNF recruits pro-inflammatory immune cells (cf. Section 3.2) and this process is thus abrogated by the drugs. Use of the TNF inhibitor also reduces intestinal permeability in CD through decreased intestinal epithelial cell apoptosis [276].
The TNF blockers’ ability to crosslink tmTNF can be different, e.g., infliximab forms more stable complexes with tmTNF than etanercept. Consequently, binding of infliximab to tmTNF can activate the “outside-to-inside signaling” or reverse signaling, and, in that case, TNF is considered as a receptor rather than a ligand (Figure 3). As a direct consequence of this interaction, apoptosis is induced in the tmTNF-expressing immune cells and this was proposed as one of the mechanisms of action in CD. This mechanism also impairs their production of pro-inflammatory mediators. Interestingly, apoptosis can also indirectly be induced in immune cells upon anti-TNF treatment. In CD, there is an anti-apoptotic signal induced by the interaction between monocytic tmTNF and TNFR2 expressed by CD4+ T cells. This mechanism is critical for granulomatous inflammation seen in CD, but this interaction is inhibited by anti-TNF resulting in lamina propria T cell apoptosis [277,278]. As the affinity for tmTNF is not similar among the different anti-TNF drugs, clinical features against this type of inflammation are not equal as well. Indeed, etanercept cannot activate reverse signaling via tmTNF which might explain its inefficiency in CD [13,279]. Thus, these drugs might stimulate apoptosis by reverse signaling or impairing apoptosis by abrogation of the TNF/TNFR signaling pathway. Importantly, TNF can also mediate other forms of cell death such as necroptosis [22,122] (cf. Section 2). Consequently, blockage of TNF might also impair this inflammatory process which might also account for the efficiency of the drugs. However, to our knowledge, not much research has been done about this topic.
In addition to their direct TNF-related capacities, the TNF-inhibitors have a panoply of other effects although currently not all their molecular mechanisms of action are completely understood [252,276]. Infliximab, adalimumab and golimumab are the only full-length mAbs and thus they also possess Fc-effector activity in addition to their general TNF-blockage properties. As a result, they can induce antibody-dependent cellular cytotoxicity (ADCC) and activate the complement pathway leading to cell-dependent cytotoxicity (CDC) and apoptosis (Figure 3). Etanercept contains a truncated Fc-domain without the CH1 domain of IgG1, therefore it induces ADCC and CDC but to a lower extent than the mAbs [252]. Certolizumab pegol, being a Fab’ fragment, is due to its structure incapable of inducing ADCC and CDC and therefore its working mechanism does not rely on the complement pathway [280] (Figure 3).
In IBD, also the interplay between the IgG1-Fc domain of the anti-TNF antibodies and the Fcγ-receptors (FcγR) on macrophages accounts for the efficacy of the anti-TNF antibodies by increasing the number of regulatory CD206+ macrophages upon activation. This M2-type macrophage subset expresses specific membrane markers and inhibits T cell proliferation [281]. Alternatively, adalimumab promotes the interaction between monocytes and Tregs via TNFR2 in RA. Adalimumab enhances the expression of tmTNF in monocytes upon binding which improves the interaction between tmTNF and TNFR2 on Tregs and boosts their suppressive activities [282]. In addition, infliximab gives rise to a CD4+CD25hiFoxP3+ Treg population that restrains pro-inflammatory cytokine production. This newly generated Treg population compensates for the natural Treg pool that is defective in autoimmune diseases such as RA [283].
The anti-TNF inhibitors were investigated in many observational studies as well as in open-label extensions of the original double-blind trials and in post-marketing observational studies. These studies provided data about the long-term efficacy and safety of the drugs. Generally, the anti-TNF inhibitors were found to be well-tolerated and to improve health-related quality-of-life (QoL) outcomes in the aforementioned diseases [284]. For RA, anti-TNF drugs are now standard-of-care, initiated after failure of treatment with the immunomodulator methotrexate (MTX) in patients. In most of the RA cases (70–80%) TNF-inhibitors are used as combination therapy with MTX. Systematic reviews of clinical trials demonstrated an additional effect of this combination in RA [245,285], whereas for CD the results from clinical trials comparing monotherapy with combination therapy were conflicting [286]. It should be noted that the risk for adverse outcomes is possibly increased with combination therapy. The combination of etanercept with MTX has similar efficacy in the therapy of RA as infliximab and adalimumab, while it is not active against CD.
4.3. Pitfalls of TNF Inhibitors
The introduction of TNF-antagonists for treatment of inflammatory disorders substantially improved the QoL of the patients, and in IBD it also reduced the number of surgeries and hospitalizations. However, the long-lasting use of these drugs coincides with a number of important adverse events (Figure 4) [287].
4.3.1. High Costs
Anti-TNF drugs are real blockbusters for the pharmaceutical companies because this is the best-selling pharmaceutical drug class with sales over US $25 billion. This puts a monumental pressure on health care systems, meaning that many countries even cannot afford a decent policy around these drugs. A retrospective study performed in 2016 estimated the annual cost of the use of biologics per patients in the USA. The most used biological was etanercept (48%), followed by adalimumab (29%) and infliximab (12%) and the annual costs per treated patient were US $24,859 for etanercept, US $26,537 for adalimumab and US $26,468 infliximab [288].
4.3.2. Clinical Response
Although many patients benefit from treatment with anti-TNF drugs, a big problem in the clinic remains the high number of patients that do not respond to the therapy: 13–40% of patients fail to respond to initial anti-TNF therapy (primary non-responders) and up to 50% of patients lose responsiveness during therapy (secondary non-responders) [289]. Primary non-response is defined by the lack of improvement of clinical signs and symptoms with induction therapy. Loss of clinical remission frequently occurs in CD patients treated with anti-TNF drugs and when the treatment fails the therapeutic options are often limited [290]. Therefore, early identification of patients at risk is of major clinical importance. Whether a patient responds well to the initiated therapy depends on multiple clinical (e.g., disease phenotype and response to previous therapies), genetic and immuno-pharmacological variables [291]. In IBD patients, mucosal healing is not obtained in 50% of the patients treated with anti-TNF biologics, and therapeutic efficacy is shown to be dependent on the interaction between the Fc region of the anti-TNF IgG and the cellular FcγR. Recently, a hypo-fucosylated form of adalimumab was designed and was found to have improved mucosal healing properties thanks to its higher affinity to FcγRIII and induction of CD206+ macrophages [281]. Consequently, some studies correlated the low-affinity FcγRIIIa allotype in IBD patients with lower changes to respond to therapies with IgG1 Ab infliximab and reduced mucosal healing [292]. Currently, elaborate studies are ongoing to identify inflammatory biomarkers that allow the stratification of patients into responders and non-responders. Some biomarkers seem promising, although they still have to be validated in large patient cohorts to verify their specificity and reliability to predict the response in the clinic [252,291,293].
Secondary non-responsiveness can be explained by the formation of anti-drug antibodies (ADAs) (e.g., anti-anti-TNF antibodies) in a subset of patients and the risk of loss-of-response is increased by at least threefold when ADAs are present [291] (Figure 4). ADAs were not only found against the chimeric mAb infliximab, but also against the fully humanized mAbs adalimumab and golimumab. This is possibly a consequence of the interaction between anti-TNF and tmTNF on antigen presenting cells and the subsequent rapid internalization. The internalized anti-TNF will be processed and its peptides displayed on the surface of the APCs will mount a T cell proliferation response [294]. Unfortunately, technical factors, standardization of the assays used to determine ADAs, and the timing of the measurements makes this a complex subject to investigate. The neutralizing ADAs that inhibit the functionality of the biologics are only a subset of the ADAs that can be found in patients. Neutralizing ADAs are generally directed against the biological active site of the drug, e.g., antigen binding part of the drug. The loss-of-response elicited by neutralizing ADAs can be countered by dose-escalation or by switching to another anti-TNF drug, but sometimes the therapy needs to be discontinued [295]. A good drug compliance and concomitant treatment with immunosuppressive agents demonstrated a reduction in the occurrence of ADAs in multiple clinical trials and improved clinical outcomes [276,296]. In addition, co-treatment with MTX was found to be beneficial in that regard [286]. ADAs may form immune complexes that are rapidly cleared by the reticulo-endothelial system and are associated with decreased drug levels, short duration of response and higher risk of infusion reactions and even acute hypersensitivity (anaphylaxis) [297,298]. Luckily, the impact of the ADA is inversely correlated with their frequency of occurrence, meaning that binding ADAs are generally more common than neutralizing ADAs or ADAs that form immune complexes. Surprisingly, the presence of ADAs may be permanent but may also be transient, appearing in one single measurement without recurrence [299].
To reduce the risk of loss-of-response, therapeutic drug monitoring (TDM) has become standard of care in the clinical setting for many clinicians, because there is a well-established correlation between serum trough levels of the drugs and clinical response [291]. Indeed, adaption of the dose based on the trough levels were more effective at inducing remission in IBD patients than clinic-based dosing [300]. TDM allows increasing the dose in patients with sub-optimal drug levels and this leads to better clinical effects. Moreover, it is widely believed that sub-therapeutic doses contribute to the development of ADAs, further highlighting the importance of TDM. Dose de-escalation is done in patients with supra-optimal levels, leading to lower drug exposure and reduced costs without impact on the clinical response [301,302].
4.3.3. Increased Susceptibility to Infection and Malignancies
The beneficial effects of anti-TNF medication are undeniable but there are serious concerns considering their safety. In addition to acute problems, such as infusion reactions, other severe adverse events might occur (Figure 4). Because TNF has an important function in host defense and in the protection against (intracellular) bacteria, as discussed in Section 3.2, infectious complications due to shutdown of this arm of the immune system are a big concern in patients treated with anti-TNF drugs. Post-marketing data have revealed a rate of 0.69% serious infections and also a drastic increase of activated tuberculosis with aberrant granuloma formation was reported [303,304]. These unwanted effects are considered as a class effect, because all anti-TNF drugs appear to have an equally high risk in acquiring new tuberculosis infections, although the mAbs seem to cause more infections with reactive latent tuberculosis [304]. This serious health issue initiated the recommendation to screen for tuberculosis with the QuantiFERON Gold Test and to treat infections, even when they are latent, before initiating the anti-TNF treatment [305]. Since then, the number of reports on tuberculosis infections has decreased, but other untypical opportunistic viral and fungal infections have popped up, including cytomegolavirus infection, Pneumocystis jirovecii pneumonia, histoplasmosis and aspergillosis. Risk factors to develop these infections are age and concomitant treatment with corticosteroids, and the overall risk of these infections should be considered before the treatment is started [306].
Regarding the risk for malignancies, studies in mice attributed an important role to TNF in the process of tumor immune surveillance [307]. However, TNF KO mice do not spontaneously develop tumors, not even in a susceptible background [38]. Notwithstanding this observation, serious concerns about anti-TNF drugs in human patients remained. Earlier studies reported an increased risk for lymphomas (Hodgkin’s lymphoma, B cell lymphoma, etc.) and other malignancies, but more recent studies and registry databases found no association between anti-TNF treatment and the occurrence of solid or hematologic cancers [308]. However, it is possible that significances are unclear because the studies are underpowered due to the low incidence of these adverse effects. In addition, because the relatively short period of clinical use of the anti-TNF drugs and the long time it may take for tumors to develop, it may be too early to make good and relevant association and risk studies. Anyway, in the case cancer is found during anti-TNF treatment, it is advisable to interrupt the treatment until the cancer is under control [309]. Noteworthy to conclude, patients that use anti-TNF medication for chronic inflammatory disease are already at higher risk for infections and malignancies for several disease-related reasons, regardless of their treatment [308,310].
4.3.4. Demyelinating Disease and Other Neurological Side Effects
Neurological side effects have been reported and recent data suggest a role for anti-TNF drugs in the induction of neurological disease, especially demyelination of the CNS as well as implications at the level of the peripheral nervous system (Figure 4). The prevalence of these side effects has been estimated to range between 0.05% and 0.2% for infliximab, etanercept and adalimumab [311]. Eighty percent of the reports about CNS demyelination are about optic neuritis, but also cases of MS or MS-like diseases have been reported. In addition, peripheral nervous system disorders were documented, such as Miller Fisher syndrome, Guillain-Barré syndrome and other neuropathies [312]. There are several hypotheses to explain the possible relationship between TNF-antagonists and demyelination but none of them is believed to be adequate [313]: (1) The occurrence of demyelinating disease could be attributed to the unmasking of a latent pre-existing form of MS, to the emergence of a new demyelination episode or to incidental coexistence of the two disorders. (2) The administration of anti-TNF agents could unmask a latent infection that is critical for the development of MS [311]. (3) Local TNF production in the CNS by pathogenic T cells induces demyelination and, therefore, the demyelination seen with TNF-antagonists may look paradoxical. However, the presence of CNS barriers, e.g., the blood–CSF and BBB, renders CNS access almost impossible as the biologics have a size of approximately 150 kDa. Even though the permeability of the barriers is increased in inflammatory conditions, this does not lead to significant blood-derived protein increment, and by inference anti-TNF biologics, in the CSF [313]. In patients, infliximab was not detected in the CSF, even in presence of active MS and BBB impairment [152]. (4) Prolonged blockage of peripheral TNF increases the T cell response to a specific antigen. This may lead to a significantly increased amount of highly activated myelin-specific autoreactive T cells, ultimately exacerbating autoimmune demyelinating diseases [313]. (5) Anti-TNF drugs can neutralize TNF systemically but not within the CNS. This results in an overall reduction in TNF in the body but relatively unchanged TNF levels in the brain. This creates an artificially high local concentration of brain (the “sponge” effect) leading to local tissue injury. Either way, once unexplained neurologic symptoms appear, the anti-TNF treatment should be discontinued [312,313].
In addition to these neurological disorders, CD patients treated with anti-TNF drugs reported fatigue which was significantly associated with the use of these drugs. Subgroup analyses also indicated that long-term therapy duration and combination without azathioprine were risk factors for the occurrence of fatigue [314,315].
4.3.5. Paradoxical Side Effects
In addition to the “common” adverse effects, there are also paradoxical side effects described in patients treated with anti-TNF drugs (Figure 4). They represent the unexpected onset or exacerbation of an autoimmune disease for which TNF blockers are indicated other than the one the patient is treated for. These disorders are mainly reported in patients with rheumatic diseases and IBD [316]. Psoriasiform skin reactions are the most frequently observed dermatological adverse effects seen in patients treated with anti-TNF therapy, but also uveitis, vasculitis, Graves’ disease and granulomatous diseases such as sarcoidosis have been reported [316,317,318,319,320]. Psoriatic skin reactions mostly occur about five months after first exposure to TNF blockers. Large cohorts also reported that RA patients treated with etanercept for juvenile idiopathic arthritis, AS or RE developed new-onset CD. Another common paradoxical side effect is the appearance of autoantibodies and a subset of patients developed drug-induced lupus erythematosus (DILE) [321]. SLE is a heterogeneous disease characterized by the production of autoantibodies that form immune complexes leading to inflammation in various organs. TNF is involved in the pathology of SLE but an open-label study showed that TNF inhibition by itself led to the paradoxical formation of autoantibodies [322]. Unfortunately, these adverse effects are probably underreported and the mechanisms unclear. There are indications that an imbalance of cytokines towards IFNs, chemokines and probably IL-17 is implicated in the pathogenesis [323,324]. The first hypothesis attributes a central role to type I IFN-α, which is highly implicated in psoriasis [320]. TNF downregulates the production of IFNs by the plasmacytoid DCs, thus TNF inhibition would enhance IFN production thereby favoring psoriasis development [320]. In addition, an imbalance in cytokines of the IL-12/IL-23 pathway via activation of the Th17 pathway is proposed as a possible mechanism [325]. A third hypothesis accounts for the impairment of the TNF-induced apoptosis of autoreactive T cells by the anti-TNF drugs. This lack of autoreactive T cell destruction induces new or aggravated forms of autoimmunity. In these cases, boosting or restoring TNF activity might be therapeutic [326]. These paradoxical side effects appeared between one month and one year after initiation of the therapy. When these symptoms appear in patients, withdrawal of the treatment reverses this unwanted effect in nearly 75% of the cases [316]. As this side effect is considered as a class effect that is seen with all TNF inhibitors, switching to another inhibitor is mostly not helpful.
5. Other Anti-TNF and TNF-Modulating Drugs
5.1. TNF Inhibitors
In addition to the well-known approved anti-TNF inhibitors, there are several other anti-TNF drugs developed or under development (Table 2). In China, a phase I clinical trial is completed with a humanized anti-TNF mAb SSS-07 against RA, but no results are provided yet (NCT02460393). The bovine polyclonal milk-derived anti-TNF Ab AVX-470 can be administered orally thanks to the stability of bovine Igs in human intestinal secretions. After oral administration, this Ab remained localized in the gut, and in a double-blind, placebo controlled study AVX-470 appeared to be safe and well tolerated, and was associated with dose-dependent increases in clinical and endoscopic remission in patients with active UC (NCT01759056) [262,263]. The involved company Avaxia Biologics has created orally administered anti-TNF mAbs, Avaximabs®, that are stable in the gastro-intestinal tract. These will be explored for the treatment of necrotizing enterocolitis (NEC). Unfortunately, clinical drug development goes along with many failures too: CDP571 (Humicade®), a humanized mAb against TNF failed to demonstrate clinical efficacy for sparing steroids in CD patients and further development was discontinued [264]. Other approaches than Abs are also considered, as illustrated with the clinical success of the Nanobodies (Nbs) [327]. A highly promising Nb is generated by Ablynx against TNF and is called ozoralizumab [328]. This drug is now under clinical investigation to treat autoimmune diseases and proof-of-concept was already obtained in a phase II RA study [265]. Another bivalent Nb was also engineered consisting of two monomeric variable domains of heavy-chain only Abs (VHHs). The construct VHH#1-3 antagonizes the binding of TNF to its receptors with picomolar potencies. As this drug has a different mode of binding, i.e., it can bind a single trimeric TNF and blocks two of the three receptor binding sites of TNF, it distinguishes itself from other TNF neutralizing drugs [266]. The ease of cloning and production allows Nbs to be locally secreted by the genetically modified probiotic Lactococcus lactis after oral administration, as was done with an anti-TNF Nb [329]. Preclinically, this innovative approach was efficacious in colitis without causing immunogenicity and is under clinical investigation. In addition, variable new antigen receptor (VNAR) domains against TNF were developed, originated from immunized sharks. Multivalent VNARs neutralize TNF at picomolar concentrations and were as efficacious as adalimumab in in vitro models of intestinal epithelial barrier dysfunction. Therefore, these drugs could be considered as a novel alternative class of biological agents [330]. Similar to etanercept, Lenercept is a soluble fusion protein consisting of TNFR1 fused to the hinge region of the IgG1 Fc region. It entered clinical trials for indications such as sepsis, RA and MS. However, the clinical trial for treatment of RRMS had to be terminated due to unforeseen exacerbations of the symptoms [53,153]. Hitanercept is a variant of etanercept that carries a mutation in the TNFR2 domain of the fusion protein and exhibits higher affinity to sTNF and tmTNF than etanercept [269,331]. In the CIA model for RA, hitanercept is more efficacious compared to etanercept. Interestingly, hitanercept is also more potent to induce reverse signaling via tmTNF and to mediate CDC and ADCC. Therefore, the drug also has therapeutic potential in CD and UC. Currently, the tolerance, pharmacokinetics and preliminary efficacy of hitanercept in RA are assessed in a phase I clinical trial in China (NCT02481180) [269]. HL036 is a small TNFR1 fragment (19 kDa) with enhanced ocular tissue penetration. This drug is formulated as an ophthalmic solution and is currently under clinical investigation (phase II) for dry eyes disease (NCT03334539) after it was shown to be safe in healthy volunteers. In addition, other formulations for inflammatory ocular diseases are currently considered. Onercept is a PEGylated form of soluble human TNFR1 that was tested in psoriasis and CD. Despite promising early clinical results, onercept proved not to have an exceptional efficacy and safety profile in both diseases and therefore further development was stopped [267,268,332,333]. Another potentially interesting candidate that is based on the same rationale as onercept is pegsunercept, a PEGylated soluble TNFR1. It has been tested for RA, but also here, development was discontinued and this decision was based on recommendations of two separate independent Data and Safety Monitoring Boards [271]. Likewise, the development of the polyclonal anti-TNF Fab fragment Azd9773 (CytoFab®) was suspended as the drug failed to show efficacy in severe sepsis and septic shock [270]. Another new therapeutic approach relies on the active immunization with TNF-kinoid or with CYT007-TNFQb, inducing endogenous polyclonal anti-TNF antibodies that neutralize circulating TNF in inflammatory immune-mediated diseases. With TNF-kinoid, proof-of-concept was obtained in a mouse RA model and these findings were translated into the clinic in patients that experience secondary non-responsiveness of TNF-antagonists. A phase Ia clinical trial showed that therapeutic vaccination induced dose- and schedule-dependent anti-TNF Abs in RA patients and was well tolerated. Moreover, patients with anti-TNF Abs showed a trend towards clinical improvement [272]. The drug was also investigated to treat CD patients, and a high clinical response was reported with remission rates in half of the patients. However, the clinical efficacy needs to be weighed against the potential harmful consequences of life-long ablation of TNF and probably for that reason further development was suspended [273]. The same holds true for CYT007-TNFQb of which the phase I/II clinical trial was discontinued in psoriasis patients [274]. Progranulin is an endogenous glycoprotein expressed in neurons and glia cells that directly interacts with TNFR1 and TNFR2. Proganulin has anti-inflammatory activities by the inhibition of the TNF activity, and Atsttrin is a progranulin-derived engineered protein that showed efficacy against RA and osteoarthritis in preclinical models [334,335]. Up until now, no clinical trials are reported, but also this candidate may be an interesting alternative to the generally used anti-TNF biologics. The small chemical triazoloquinoxaline inhibitor R-7050 is a TNFR complex inhibitor that improves the outcome upon intracerebral hemorrhage, suggesting its use as adjunct therapy in the treatment of neurological injury [336]. The drug does not interfere with TNF-TNFR1 binding, but acts via the inhibition of receptor-adaptor molecules complex formation and subsequent receptor internalization [336]. In addition, interesting is the approach exploited by the group of Pardridge. They engineered a BBB-penetrating TNF inhibitor by fusion of the extracellular domain of TNFR2 to a chimeric monoclonal antibody against the mouse TfR. This Trojan horse approach led to rapid therapeutically relevant amounts of drug in the brain following intravenous, subcutaneous and ip administration, and was protective in mouse models of PD, AD and ischemic stroke [223,242,275,337]. Finally, a novel chemically synthesized anti-TNF compound is described. C87 is an TNF-TNFR interaction modulator as it directly binds to TNF and prevents TNFR signaling and subsequent Casp-8 and NF-κB activation. It was found from an initial screen of ~90,000 compounds and has in vivo potency. The only remaining challenge is to determine toxicity and stability with longer-term use [338].
5.2. TNF Modulators
Other less specific anti-TNF agents are thalidomide and its derivatives lenalidomide and pomalidomide, curcumin and minocycline. Initially, thalidomide was indicated as an effective tranquilizer and painkiller associated with enormous teratogenic side effects in human. Thalidomide is now recently re-introduced as well-known (non-specific) TNF-inhibitor as it reduces the rate of TNF synthesis by enhancing the degradation of the transcript. Currently, thalidomide is under investigation to treat neurodegenerative disorders that implicate TNF-signaling such as AD, PD and amyotrophic lateral sclerosis (ALS) as this small-drug molecule can penetrate into the brain [235,339,340,341,342]. The broad-spectrum tetracycline antibiotic drug minocycline decreases TNF synthesis in addition to its bacteriostatic and anti-inflammatory actions. Additionally, it also inhibits MMPs, reduces cyclooxygenase 2 (COX-2) activity and prostaglandin E2 production, and attenuates apoptosis [12]. In PD models, minocycline attenuated MPTP-induced microglia activation, but could not abolish the neurotoxicity [237]. By contrast, curcumin (diferuloymethane) is a natural anti-inflammatory agent that inhibits TNF transcription at several levels, but mostly via inhibition of NF-κB. Consequently, curcumin also antagonizes other pro-inflammatory cytokines including IL-1β and IL-6. It is a broad-acting anti-TNF that can be orally consumed via natural food spices. Unfortunately, it is poorly soluble in water and has a poor bio-availability [343]. Currently, neuroprotective characteristics are attributed to curcumin and therefore it is under active investigation for AD amongst others [344]. In addition, xanthine derivate pentoxifylline and bupropion have shown to decrease TNF synthesis [345]. By increasing the signaling at beta-adrenoreceptors and D1 receptors, bupropion increases cyclic AMP (cAMP) which subsequently inhibits TNF synthesis [346]. Unexpectedly, a novel crosstalk pathway between neural and immune receptors was found as several 5-hydroxytryptamine (HT) agonist hallucinogens (such as (R)-DOI, TCB-2, LSD and LA-SS-Az) are potent TNF inhibitors, with DOI being the most potent one. This indicates that activation of the serotonin 5-HT(2A) receptors represents a novel potential therapeutic avenue for TNF-involving disorders [347,348].
6. A New Chapter of Inventive TNF Manipulating Approaches
The anti-TNF drugs on the market were based on the wide perception that TNF is a pathological factor, ignoring the fact that TNF can also have beneficial and unique indispensable properties e.g., in immune regulation and tissue regeneration, as discussed above [33,93,102,114,349,350]. This is also illustrated by the numerous side-effects that are inherent to long-lasting TNF blockage. Therefore, more discriminative approaches hold the potential to increase the safety and efficacy of the drugs. Indeed, as outlined in previous sections, especially for neurological diseases more selective approaches are warranted as there is a clear discrepancy between TNFR1 and TNFR2 signaling in the brain typified by the aggravated disease symptoms that are induced by anti-TNF drugs in MS patients. Nonetheless, also non-neurological diseases will benefit from receptor-discriminatory drugs as pan-TNF neutralization induces neurological phenomena in some patients.
6.1. Selective TNFR1 Targeting
Given that TNFR1 and TNFR2 mediate different cellular effects, it is interesting to selectively target one of the two. This field is currently actively explored via several approaches [97]. ATROSAB is a human TNFR1-specific antibody that demonstrated to efficiently block the activity of TNF and LT-α in vitro [351,352]. Williams et al. described a monoclonal hamster IgG against mouse TNFR1 that was effective to reduce symptoms in EAE [170], and also a TNFR1 selective antagonistic mutant TNF protein, PEG-R1ant-TNF has been described to have this property [169]. PEG-R1ant-TNF was also effective in the CIA model for RA and against arterial inflammation [353,354]. The efficacy of a single TNFR1-binding domain bispecific antibody, MDS5541, was evaluated against RA in vitro in synovial membrane cell cultures from RA patients and in vivo in the CIA model of RA [107,355]. Selective TNFR1 inhibition established with this drug led to the expansion and activation of Tregs with upregulation of FoxP3-dependent genes. This effect again highlights the importance of preserving the TNF/TNFR2-mediated signaling pathway. In addition, two fully human anti-TNFR1 single domain antibodies have been developed and were investigated by GlaxoSmithKline [356]. The first, GSK1995057 attenuated lung injury in different preclinical models of acute respiratory distress syndrome [356,357]. However, in a phase I clinical trial with this small inhibitor, infusion reactions arose in healthy volunteers because of the presence of naturally occurring pre-existing ADAs [358,359]. Consequently, a new trial was initiated in which only healthy subjects prospectively demonstrated to be seronegative for the pre-existing ADAs were eligible for participation (TFR116343). In these subjects, nebulized GSK1995057 prevented acute lung injury in an LPS-induced model. The drug will now be evaluated in a phase IIa clinical trial [360]. To deal with the problem of pre-existing ADAs, a second single domain was designed (GSK2862277). In a phase I trial, this drug was well tolerated by both the inhaled and iv route [359]. A placebo-controlled randomized phase II trial was set up in patients that undergo oesophagectomy surgery and that were at risk to develop acute respiratory distress syndrome. The drug GSK2862277 was administered as an orally inhaled aerosol pre-operative, but the trial was terminated earlier as the study met the designed stopping criteria (NCT02221037). However, GSK still concludes that selective antagonisms of TNFR1 using inhaled drugs might offer therapeutic benefit in patients with acute respiratory distress syndrome. Another interesting approach to selectively target TNFR1 is by interfering with the PLAD association, necessary for TNF/TNFR1 signaling [16]. Targeting the TNFR1 PLAD domain has already been proposed by several groups as a promising strategy in autoimmune diseases such as diabetes and RA [361,362]. The marketed anti-asthma drug zafirlukast also disrupts the interaction between the TNFR1 PLAD domain and is thus considered as a selective TNFR inhibitor [363].
Importantly, our research group also generated a trivalent human TNFR1 inhibiting Nanobody consisting of two paratopic TNFR1 binding Nbs linked to an anti-albumin Nb. This Nb only binds to human TNFR1 without being cross reactive for the mouse homologue. One of these Nbs also competitively inhibited the TNF/TNFR1 signaling, but the potency of this Nb was improved after incorporation in the TROS construct. This construct effectively inhibited TNFR1 signaling in vitro and ex vivo on isolated colon biopsies from CD patients [328]. Proof-of-concept of this promising molecule has been first delivered in humanized mice carrying a human TNFRSF1A gene, subjected to the mouse EAE model of MS. In these transgenic mice Prophylactic as well as therapeutic ip administration of the drug prevented or halted disease development, respectively [171]. The drug prevented demyelination and treatment with TROS maintained the expression of several important neuroprotective genes that are downregulated in MS patients. Because choroid plexus TNFR1 is also an important detrimental mediator in AD as was recently shown by the group, also the possibility to inhibit TNFR1 therapeutically with TROS was investigated in the acute AD model of icv oligomerized amyloid beta (AβO) injection. Strikingly, TROS prevented the AβO-induced memory decline in these mice after icv injection, confirming the therapeutic possibilities of this molecule [186].
6.2. Selective TNFR2 Targeting
The opposite approach that is currently investigated implies TNFR2 activation to stimulate the TNFR2-mediated protective pathways in autoimmunity and neurodegenerative diseases [364]. It has been suggested that this strategy might be superior to TNFR1 antagonism because of the restricted cellular expression of TNFR2. TNFR2 is not only considered as a costimulatory receptor for T cells and critically involved in the development of Tregs, recent studies also provide new insights into the role of tmTNF/TNFR2 signaling in the suppressive activity of myeloid-derived suppressor cells (MDSCs). Indeed, MDSCs require membrane TNFR2 expression to exert optimal suppressive activity [365,366]. Interestingly, chronic inflammation increases the sensitivity of these suppressive cells for TNFR2 costimulation [367]. Efficient TNFR2 activation requires oligomerization of TNFR2 by tmTNF. Alternatively, oligomerized soluble forms of tmTNF should mimic receptor activation via tmTNF [168,368]. Several TNFR2 agonists have already been developed such as the TNFR2-specific variant of mouse TNF that is trimerized using the trimerization domain of chicken tenascin C as has been done in TNCscTNF80. This drug protected against graft-versus-host disease (GVHD) via host Treg expansion [369]. Lamontain and colleagues recently confirmed that stimulation with TNCscTNF80 effectively leads to expansion of Tregs and ameliorates established CIA in mice [370]. Another TNFR2-selective TNF mutein EHD2-scTNFR2 consists of a covalently stabilized human TNFR2-selective single-chain TNF fused to the dimerization domain EHD2 derived from the heavy chain domain of IgEs [174]. TNFR2 agonists could rescue human neurons from death induced by oxidative stress or stimulate Tregs in type I diabetes. These could be useful in patients with TNF-mediated neurodegeneration or as a correctional therapy in diabetes patients [371]. Additionally, TNFR2 is expressed by all diseased CD8+ T cells and TNFR2 agonism has been shown to selectively kill insulin-autoreactive CD8+ T cells in blood of diabetic patients by an altered signaling pathway [184]. This approach is very attractive and is preclinically assessed for type I diabetes and other autoimmune diseases. However, despite the promising features of TNFR2 agonism, it can also be a risky approach as it might enhance the accumulation of pathogenic T cells [372]. In type I diabetes, it has been suggested that tmTNF signaling via TNFR2 is responsible for islet destruction, arguing for TNFR2 antagonism instead [373]. Interestingly, Dong et al. combined TNFR1 antagonism and TNFR2 agonism in a model of N-methyl-d-aspartate (NMDA)-induced acute neurodegeneration. Administration of ATROSAB or of a TNFR2-selective TNF mutant EHD2-scTNFRR2 reverted neurodegeneration-associated memory impairment and protected cholinergic neurons against cell death [174]. In addition of the use of TNFR2-agonizing TNF muteins, one may also consider co-administration of the cholesterol lowering drug lovastatin. Indeed, in further support of the importance to preserve TNFR2 signaling in AD, it was shown that statins reduced progression of AD in clinical trials or even prevented the onset of it [374,375], and lovastatin established this by increasing TNFR2 expression. Additionally, the drug protected primary cortical neurons against glutamate-induced excitotoxicity [376].
In contrast to TNFR2 agonism, TNFR2 inhibition was proposed as an effective anti-cancer strategy as TNFR2 has been identified as a human cancer oncogene. Indeed, many cancer cells are characterized by TNFR2 expression that promotes the expansion of tumor cells [14]. Recently, TNFR2 has also been found on the surface of a highly immuno-suppressive tumor-infiltrating subset of Tregs [109], and therapies that target and eliminate Tregs are currently investigated as cancer treatment [377]. TNFR2 antagonism could therefore act as a double-edged sword as the group of Faustman elegantly showed. TNFR2 antibodies directly blocked the tumor growth, inhibited Treg proliferation and enabled T effector cell expansion which could help amplify effective anti-tumor immune responses [15]. Furthermore, the combination of TNFR2 antagonists with immunotherapeutic stimulants synergistically improves the therapeutic efficacy in colon cancer mouse models [378].
6.3. TNF-Inducing Vaccines
Interestingly, also vaccines that stimulate endogenous TNF release are currently evaluated as a long-term modulating immuno-intervention in clinical trials in type 1 diabetes and have been investigated in MS (NCT02081326; NCT00607230; NCT00202410) [349,379]. These approaches are less toxic than recombinant TNF treatment, and can be established via immunization with the Bacillus Calmette-Guérin (BCG) vaccine. This live tuberculosis vaccine contains Mycobacterium bovis, known to stimulate the innate immune system by inducing the host to produce TNF that subsequently kills the autoreactive T cells. BCG vaccination of longstanding type I diabetic subjects led to more death insulin-autoreactive T cells and transiently induced beneficial Tregs for 4–6 weeks after vaccination [379]. This vaccine is now evaluated in a phase II trial with a duration of five years to establish the long term consequences [349]. In addition, in MS, the clinical benefit was shown, consistent with the outcome in diabetic patients [380]. Vaccinated RRMS patients had a reduction in disease activity and the progression of brain lesions was prevented. The effects of BCG vaccination were also assessed in subjects with clinically isolated syndromes, and at the end of the five-year trial, 58% of the subjects did not progress into MS whereas this was only 30% of the placebo-treated group, and no adverse events were reported [381].
6.4. Selective Targeting of sTNF
Instead of targeting the receptors, approaches that selectively target sTNF have also been proposed. One of the most promising drugs is the dominant negative peptide Xpro1595 [382,383]. Xpro1595 selectively binds to soluble TNF monomers without interfering with tmTNF and forms inactive heterotrimers that are unable to interact with the TNFR [382]. Interestingly, XPro1595 suppressed inflammation in both the CIA and the mouse collagen antibody-induced arthritis (CAIA) model for RA without compromising the innate immunity to L. monocytogenes infection [382]. The efficacy of XPro1595 has also been tested in other preclinical models. Indeed, in two mouse MS model, EAE and the cuprizone model, subcutaneous injection of XPro1595 was therapeutic and promoted axon preservation and remyelination [164,165,167] and in AD 5xFAD mice it decreased Aβ plaque load and rescued impaired long-term potentiation [189]. Locomotor functioning was improved after spinal cord injury, but only after central administration [163] and intraocular administration of XPro1595 promoted retinal ganglion cell survival in a rat model of ocular hypertension glaucoma [384]. Icv infusion of XPro1595 also prevented the development of depressive-like symptoms induced by exposure to artificial light at night [385]. Peripheral administration of XENP345, a PEGylated variant of XPro1595, in the 6-OHDA model for PD attenuated nigral and dopaminergic cell loss [196,198] (cf. Section 3.4.4). Finally, in the established mouse model of Huntington’s disease (R6/2 mice), particularly icv injection of XPro1595 improved the functional outcome of these mice [386]. Vaccination with virus-like particles of the bacteriophage Qbeta that was covalently linked to sTNF led to Abs specifically neutralizing sTNF. These endogenously raised Abs protected mice from inflammation in RA mouse models [274].
6.5. Cell-Type Restricted TNF(R) Targeting
Cell-type restricted targeting of TNF is a new innovative approach that has been suggested by the group of Nedospasov [387]. TNF signaling represents a complex network, and the deleterious or beneficial outcome of TNF depends not only on the receptor via which it signals, but also on the physiological circumstances and the cell type. Indeed, cell-type specific conditional TNF knockout mice point to differential roles for myeloid and T cell-derived TNF in host defense and in several inflammatory mouse models (Table 3). For the immune response against L. monocytogenes, both myeloid and T cell-derived TNF have a similar contribution depending on the bacterial load [387]. The situation in different during M. tuberculosis infection in which T cell-derived TNF is needed to control the infection and can better not be blocked, whereas myeloid cell-derived TNF is dispensable [40]. For the formation of GCs, another source of TNF, namely B cells, is important. However, to maintain the formation of the GCs and FDC networks in lymph nodes, B cell-TNF synergizes with T cell-TNF [388]. In disease settings, TNF derived from myeloid cells was generally found to have detrimental functions in LPS/d-galactosamine induced hepatotoxicity, in experimental arthritis and in EAE [147]. Conditional ablation of macrophage TNF led to protection against diabetic nephropathy in streptozotocin-induced diabetes [389]. Conversely, T cell-derived TNF demonstrated nonredundant protective roles in these diseases, although in EAE also pathogenic effects of T cell-derived TNF are described [147]. Interestingly, intestinal inflammation could be induced by chronic TNF expression by the IECs whereas in the T cell transfer model of murine colitis, TNF from non-T cells seems to be responsible for colitis induction [390]. Intestinal pathology in TNF∆ARE mice is induced by TNF derived from innate (myeloid cells) and adaptive (CD8+ T cells) effector cells as well as IECs via interacting with TNFR1 on mesenchymal cells [85,391,392]. These examples led to the idea of selective inhibition of TNF produced by myeloid cells thereby sparing the protective effects of TNF that is produced by other cells such as T cells [393]. To establish site-directed TNF neutralization myeloid cell-specific TNF inhibitors (MYSTIs) were designed that locally enrich at the cell membrane. These VHH-based bispecifics consist of one arm that binds TNF and another arm directed against the myeloid surface markers F4/80 or CD11b. These drugs retain endogenously generated TNF in vitro, and in vivo the F4/80-directed MYSTI protects against LPS/D-Gal lethal toxicity and was also active in the anti-collagen antibody transfer arthritis model [394,395]. Additionally, treatment with these drugs led to beneficial outcomes in an in vivo model of acute hepatotoxicity as macrophage-derived TNF is directly captured at the source of production [394,396].
Interestingly, in the TNF-induced shock model IEC-restricted TNFR1 expression is sufficient to induce IEC-apoptosis, but chronic targeting of IECs by endogenous TNF is not enough to induce IBD pathology indicating that TNFR1 expressed on other cell types is required for effective pathology [68,69,397]. This means that selective IEC targeting of TNFR1 is not interesting to consider as IBD therapy. In TNF∆ARE mice, the RA pathology is independent of TNF-mediated adaptive immune responses (T and B cells), but it is mediated by TNFR1 on joint (synovial) fibroblasts [85,391,398]. In addition, astrocyte-specific TNFR1 blockage might be considered to prevent memory deficits in MS [172] or CD8 T cell-TNFR2 antagonism for induction of apoptosis in autoreactive lymphocytes in diabetes [184].
Instead of targeted TNF neutralization, there are also several indications that might benefit from targeted TNF supply, for example to exploit its anti-cancer properties. Unfortunately, TNF’s intrinsic and unacceptable toxicity hampers its use as immunotherapeutic. This obstacle can be circumvented by the creation of Activity-on-Target cytokines (AcTakines). These immunocytokines consist of mutated cytokines with reduced binding affinity coupled to a targeting moiety that guides the cytokine to the desired cell target. The activity of the mutated cytokine is only restored after local enrichment at the targeted cell types. This strategy will greatly reduce the off-target adverse effects and improve the desired efficacy [399].
6.6. Multispecific Approaches
Simultaneous blockage of multiple pathways might directly cope possible compensation mechanisms that neutralize the initial effect of the drug. Alternatively, this approach can also directly increase the potency of drugs. In the pathogenesis of psoriasis, a prominent role for TNF has been described and this is illustrated by the success of the TNF inhibitors in this disease. In addition to TNF, recent data also suggest an important role for type I IFN in psoriasis. Interestingly, blockage of TNFR1 or the IFN receptor 1 (IFNAR1) only partially protects mice against imiquimod-induced psoriasis, whereas double KO mice lacking both receptors showed superior protection in this model. This was explained by the presence of a sustained type I IFN production in TNFR1 single KO mice [324]. As there is also a clear synergy between TNF and IL17A or IL17F, bispecific antibodies that bind both TNF and IL-17 were designed with the intend to be superior for the treatment of RA, PsA and AS, and to overcome limited therapeutic responses obtained with single cytokine neutralization [401]. Two different bispecific biologics (e.g., COVA322 and ABT-122) are currently under clinical investigation, but the clinical development of COVA322 was terminated due to safety issues (NCT022437870). ABT-122 is a dual-variable domain IgG under clinical development by AbbVie and seems promising after evaluation in several phase I and II trials in healthy volunteers and in subjects with RA and PsA [402,403]. Similar approaches were inquired with the TNF/IL-6 and TNF/IL-23 neutralizing Abs that were patented [404,405]. The first approach could ameliorate the clinical progress in CIA more than the single treatments did [406]. Additionally, RA patients were treated with the combination of IL1 and TNF neutralizing drugs but this therapy did not have added value [407], although it seemed promising in preclinical sepsis models and in acute myeloid leukemia [79,408]. However, the complete blockage of host defense mechanisms should be kept in mind as a plausible destructive side effect. In the context of sepsis, our group developed a bispecific Nb that targets MMP8 and TNFR1, as discussed in Section 3.2.2. In addition to these double cytokine-hitters, the combination between anti-TNF with anti-angiogenic agents were studied in the context of RA. The bispecific Zybody that was generated by the genetic fusion of Ang2-targeting peptides to the heavy chain of an anti-TNF Ab showed superior efficacy compared to the single hit strategy [409].
The establishment of a bispecific therapeutic also allows targeting of cytokine-neutralization on cytokine-producing cells (cf. Section 6.5) or at particular anatomical sites such as regions of inflammation. For instance, anti-TNF Nbs that are coupled to anti-albumin do not only have improved pharmacokinetic properties, but also accumulate in the inflamed joints of CIA mice [410]. Alternatively, in an attempt for improved local delivery to the inflamed joint, TNF-inhibitors were coupled to a single-chain variable fragment (scFv) that recognize collagen type II that is post-translationally modified by reactive oxygen species (ROS) [411], and the so-called MYSTIs introduced in the previous section, also aim for site-directed TNF neutralization. Others also investigated the possibility of simultaneous targeting of MMP14 or cadherin-11 together with TNF in an attempt to direct the pannus-cartilage junction in RA-affected joints [393].
7. Concluding Remarks
It is clear from the increasing amount of studies regarding the differential roles of the TNF receptors, the TNF format, i.e., tmTNF or sTNF, and the function of TNF derived from specific cell subsets, that the use of anti-TNF drugs can be improved or adjusted to the specific disease context. Given the relatively long-time experience with the approved anti-TNF drugs, they can be considered as relatively safe; however, improvements are warranted. In addition to the disease settings for which a market authorization is settled, many other diseases in which a specific TNF subtype/receptor is involved can be approached. The examples are numerous and exceed the field of neurology although this was mainly the focus in this review. Selective TNFR1 targeting is described for dry-skin induced chronic itching [412], neuropathic pain [136], ventilator-induced acute lung injury [356], eye disorders [203], and cardiomyopathy and myocardial ischemic injury [413,414,415,416], and this list is not limited to these examples.
Although the expression of TNFR2 is limited in healthy conditions, in (autoimmune) diseases, the expression of TNFR2 is significantly elevated. Importantly, TNFR2 has a superior toxicity profile because of its limited tissue expression compared to the ubiquitous expression of TNFR1 [349]. As discussed in the manuscript, TNFR2 agonism is an attractive checkpoint therapy to modulate the immune regulation through Treg activation, which can be very interesting in, for instance, graft-versus-host disease [369]. Therapies that combine the two strategies in diseases, in which TNFR1 signaling leads to devastating outcomes and stimulation of TNFR2 is needed to promote proliferation or regeneration, to achieve replenishment of the nonfunctional Treg population are attractive.
Additionally, as discussed, cell-specific drugs that are directed against the intended TNFR expressed by a specific cell type are interesting for future research. This would lead to local accumulation, less undesired side effects and thus safer therapies. Collectively, during the development of new innovative therapies, one can not only consider the removal of the detrimental cells or signals that are present, e.g., blockage of the inflammatory TNFR1 pathway or removal of autoreactive T cells, but also think about therapies that stimulate the cells or cellular properties that are defective such as suppressive cell types. Therefore, therapeutic treatments should always find the ideal balance between doing enough good and preventing bad.
In addition, critical notes are needed, and considerable attention should be paid to adverse effects that might pop-up with TNFR1 antagonizing treatments, as the sensitivity to several infectious diseases is clearly increased in mice lacking TNFR1 [417]. However, cell-specific TNFR1 targeting might again offer a solution to increase the safety. Besides, antagonizing a receptor always carries the risk that the receptor is not blocked but instead becomes activated. This event has already previously been described by others, and even led to early termination of a clinical trial [358]. Seemingly, TNFR1 antagonists could not only be converted into potent receptor activators by the induction of TNFR1-oligomerization, but agonistic activities can also be induced upon cross-linking by secondary Abs such as by drug-induced antibodies or pre-existing antibodies that cluster the drug/receptor complex and consequently activate the downstream pathway [358,418,419,420].
To conclude, therapeutic manipulation of TNF remains a very attractive field, and although we know already a lot about the biology of TNF, there is a lot to uncover. This will allow us to select the appropriate treatment for a specific patient population. In addition, improved patient empowerment will drive the development of innovative medicines that deliver more relevant and impactful patient outcomes [421]. Indeed, precision and personalized medicine are currently booming and the needs of the patients should be considered when developing new drugs. Therefore, not only a profound molecular understanding of the considered diseases is indispensable, but also the challenges faced by the patients during their everyday living and their QoL should be accounted for during drug development. Hence, a collaborative approach is essential and will facilitate the introduction of real personalized medicine into clinical practice.
Author Contributions
S.S. drafted the manuscript; and C.L. and R.E.V. edited the manuscript and approved the final version.
Funding
S.S. is supported by Horizon2020 (B-Smart). Research in the authors’ lab is sponsored by the Research Foundation-Flanders (FWO), the Concerted Research Actions of Ghent University, The Foundation for Alzheimer’s Research Belgium (SAO-FRA), the EU Cost action MouseAge (BM1402) and the Baillet Latour Fund.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| ADA | Anti-drug antibody |
| ADAM | A disintegrin and metalloproteinase |
| ADCC | Antibody-dependent cellular cytotoxicity |
| ALS | Amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APC | Antigen-presenting cells |
| AS | Ankylosing spondylitis |
| Aβ(O) | (oligomerized) Amyloid beta |
| BACE1 | Beta-secretase 1 |
| BBB | Blood–brain barrier |
| BCG | Bacillus Calmette-Guérin |
| CAIA | Collagen antibody-induced arthritis |
| cAMP | Cyclic AMP |
| CASP | Colon ascendens stent peritonitis |
| CD | Crohn’s disease |
| CDC | Cell-dependent cytotoxicity |
| CIA | Collagen-induced arthritis |
| CLP | Cecal ligation and puncture |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CSF | Cerebrospinal fluid |
| DC | Dendritic cell |
| DD | Death domain |
| DILE | Drug-induced lupus erythematosus |
| DSS | Dextran sodium sulfate |
| EAE | Experimental autoimmune encephalomyelitis |
| EMA | European Medicine Agency |
| FADD | Fas-associated death domain |
| FcγR | Fcγ-receptor |
| FDA | Food and Drug Administration |
| FDC | Follicular dendritic cell |
| GC | Germinal center |
| GVHD | Graft-versus-host disease |
| HT | 5-hydroxytryptamine |
| hTNF | Human TNF |
| ip | Intraperitoneal |
| IBD | Inflammatory bowel disease |
| icv | Intracerebroventricular |
| IEC | Intestinal epithelial cells |
| IFN | Interferon |
| IFNAR | Interferon-α receptor |
| Ig | Immunoglobulin |
| IL | Interleukin |
| JIA | Juvenile idiopathic arthritis |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| LT-α | Lymphotoxin-α |
| mAb | Monoclonal antibody |
| MDSC | Myeloid-derived suppressor cells |
| MMP | Matrix metalloproteinase |
| MOF | Multiple organ failure |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | Multiple sclerosis |
| MTX | Methotrexate |
| Nb | Nanobody |
| NEC | Necrotizing enterocolitis |
| NK | Natural killer |
| NOD | Non-obese diabetic |
| OLG | Oligodendrocytes |
| OPC | Oligodendrocyte precursor cell |
| PD | Parkinson’s disease |
| PEG | Polyethylene glycol |
| PLAD | Pre-ligand assembly domain |
| PPMS | Primary progressive MS |
| PsA | Psoriatic arthritis |
| QoL | Quality-of-life |
| RA | Rheumatoid arthritis |
| ROS | Reactive oxygen species |
| RRMS | Relapsing-remitting MS |
| scFv | Single-chain variable fragment |
| SIRS | Systemic inflammatory response syndrome |
| SLE | Systemic lupus erythematosus |
| sTNF(R) | Soluble TNF(R) |
| TACE | TNF-α converting enzyme |
| TDM | Therapeutic drug monitoring |
| TIMP | Tissue inhibitor of metalloproteinase |
| tmTNF | Transmembrane TNF |
| TNF | Tumor necrosis factor |
| TNFR | Tumor necrosis factor receptor |
| TRADD | TNFR1-associated death domain |
| TRAF(2) | TNF receptor-associated factor (2) |
| TRAPS | TNF receptor-associated periodic syndrome |
| Tregs | Regulatory T cells |
| UC | Ulcerative colitis |
| VHH | Variable domain of heavy-chain only Abs |
| VNAR | Variable new antigen receptor |
| WT | Wild type |
References
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 Years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Tumor necrosis factor (TNF). Science 1985, 230, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Creaven, P.J.; Plager, J.E.; Dupere, S.; Huben, R.P.; Takita, H.; Mittelman, A.; Proefrock, A. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol. 1987, 20, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, B.; Kurzrock, R.; Talpaz, M.; Blick, M.; Saks, S.; Gutterman, J.U. A phase I trial of intravenously-administered recombinant tumor necrosis factor-alpha in cancer patients. J. Clin. Oncol. 1988, 6, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Zhou, S.; Diaz, L.A., Jr.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Naismith, J.H.; Sprang, S.R. Modularity in the TNF-receptor family. Trends Biochem. Sci. 1998, 23, 74–79. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L.; et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Targeting TNFR2, an immune checkpoint stimulator and oncoprotein, is a promising treatment for cancer. Sci. Signal. 2017, 10, eaal2328. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal. 2017, 10, eaaf8608. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74, 845–853. [Google Scholar] [CrossRef]
- Faustman, D.L.; Davis, M. TNF receptor 2 and disease: Autoimmunity and regenerative medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Bernardazzi, C.; de Souza, H.S. Cell death and inflammatory bowel diseases: Apoptosis, necrosis, and autophagy in the intestinal epithelium. BioMed Res. Int. 2014, 2014, 218493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.A.; Lehman, J.; Roderick, J.E.; Martinez-Marin, D.; Zelic, M.; Doran, C.; Hermance, N.; Lyle, S.; Pasparakis, M.; Fitzgerald, K.A.; et al. Dendritic cell RIPK1 maintains immune homeostasis by preventing inflammation and autoimmunity. J. Immunol. 2018, 200, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Wallach, D.; Engelmann, H.; Nophar, Y.; Aderka, D.; Kemper, O.; Hornik, V.; Holtmann, H.; Brakebusch, C. Soluble and cell surface receptors for tumor necrosis factor. Agents Actions Suppl. 1991, 35, 51–57. [Google Scholar] [PubMed]
- Aderka, D. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 1996, 7, 231–240. [Google Scholar] [CrossRef]
- Xanthoulea, S.; Pasparakis, M.; Kousteni, S.; Brakebusch, C.; Wallach, D.; Bauer, J.; Lassmann, H.; Kollias, G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 2004, 200, 367–376. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kda TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Mootoo, A.; Stylianou, E.; Arias, M.A.; Reljic, R. TNF-alpha in tuberculosis: A cytokine with a split personality. Inflamm. Allergy Drug Targets 2009, 8, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.; Douni, E.; Kassiotis, G.; Kontoyiannis, D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann. Rheum. Dis. 1999, 58 (Suppl. 1), I32–I39. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNF alpha-deficient mice: A critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.W.; Dunn, A.; Grail, D.; Inglese, M.; Noguchi, Y.; Richards, E.; Jungbluth, A.; Wada, H.; Moore, M.; Williamson, B.; et al. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8093–8098. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, S.; Rickelt, S.; Soriano, M.E.; Schips, T.G.; Maier, H.J.; Davos, C.H.; Varela, A.; Kaklamanis, L.; Mann, D.L.; Capetanaki, Y. Tumor necrosis factor-alpha confers cardioprotection through ectopic expression of keratins K8 and K18. Nat. Med. 2015, 21, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Bluml, S.; Binder, N.B.; Niederreiter, B.; Polzer, K.; Hayer, S.; Tauber, S.; Schett, G.; Scheinecker, C.; Kollias, G.; Selzer, E.; et al. Antiinflammatory effects of tumor necrosis factor on hematopoietic cells in a murine model of erosive arthritis. Arthritis Rheum. 2010, 62, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.; Maier, O. Targeting sTNF/TNFR1 signaling as a new therapeutic strategy. Antibodies 2015, 4, 48–70. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.; Marino, M.W.; Brown, N.; Abel, B.; Bekker, L.G.; Quesniaux, V.J.; Fick, L.; Ryffel, B. Correction of defective host response to Mycobacterium Bovis BCG infection in TNF-deficient mice by bone marrow transplantation. Lab. Investig. 2000, 80, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Allie, N.; Grivennikov, S.I.; Keeton, R.; Hsu, N.J.; Bourigault, M.L.; Court, N.; Fremond, C.; Yeremeev, V.; Shebzukhov, Y.; Ryffel, B.; et al. Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci. Rep. 2013, 3, 1809. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Lesslauer, W.; Lotscher, H.; Lang, Y.; Koebel, P.; Kontgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Mackay, F.; Bluethmann, H.; Zinkernagel, R. Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Circ. Shock 1994, 44, 51–56. [Google Scholar] [PubMed]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef]
- Garcia, I.; Olleros, M.L.; Quesniaux, V.F.; Jacobs, M.; Allie, N.; Nedospasov, S.A.; Szymkowski, D.E.; Ryffel, B. Roles of soluble and membrane TNF and related ligands in mycobacterial infections: Effects of selective and non-selective TNF inhibitors during infection. Adv. Exp. Med. Biol. 2011, 691, 187–201. [Google Scholar] [PubMed]
- Segueni, N.; Benmerzoug, S.; Rose, S.; Gauthier, A.; Bourigault, M.L.; Reverchon, F.; Philippeau, A.; Erard, F.; Le Bert, M.; Bouscayrol, H.; et al. Innate myeloid cell TNFR1 mediates first line defence against primary Mycobacterium tuberculosis infection. Sci. Rep. 2016, 6, 22454. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.G.; Tompkins, R.G.; Gelfand, J.A.; Michie, H.R.; Stanford, G.G.; van der Meer, J.W.; Endres, S.; Lonnemann, G.; Corsetti, J.; Chernow, B.; et al. Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J. Infect. Dis. 1990, 161, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Gogos, C.A.; Drosou, E.; Bassaris, H.P.; Skoutelis, A. Pro-versus anti-inflammatory cytokine profile in patients with severe sepsis: A marker for prognosis and future therapeutic options. J. Infect. Dis. 2000, 181, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Feezor, R.J.; Oberholzer, C.; Baker, H.V.; Novick, D.; Rubinstein, M.; Moldawer, L.L.; Pribble, J.; Souza, S.; Dinarello, C.A.; Ertel, W.; et al. Molecular characterization of the acute inflammatory response to infections with gram-negative versus gram-positive bacteria. Infect. Immun. 2003, 71, 5803–5813. [Google Scholar] [CrossRef] [PubMed]
- Schulte, W.; Bernhagen, J.; Bucala, R. Cytokines in sepsis: Potent immunoregulators and potential therapeutic targets—An updated view. Mediat. Inflamm. 2013, 2013, 165974. [Google Scholar] [CrossRef] [PubMed]
- Rigato, O.; Ujvari, S.; Castelo, A.; Salomao, R. Tumor necrosis factor alpha (TNF-alpha) and sepsis: Evidence for a role in host defense. Infection 1996, 24, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Kothari, N.; Bogra, J.; Abbas, H.; Kohli, M.; Malik, A.; Kothari, D.; Srivastava, S.; Singh, P.K. Tumor necrosis factor gene polymorphism results in high TNF level in sepsis and septic shock. Cytokine 2013, 61, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, E.; Sarikcioglu, A.; Aksaray, N.; Seydaoglu, G.; Seyhun, Y.; Yaman, A. Role of procalcitonin, C-reactive protein, interleukin-6, interleukin-8 and tumor necrosis factor-alpha in the diagnosis of neonatal sepsis. Turk. J. Pediatr. 2007, 49, 7–20. [Google Scholar] [PubMed]
- Abraham, E.; Laterre, P.F.; Garbino, J.; Pingleton, S.; Butler, T.; Dugernier, T.; Margolis, B.; Kudsk, K.; Zimmerli, W.; Anderson, P.; et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: A randomized, double-blind, placebo-controlled, multicenter phase iii trial with 1342 patients. Crit. Care Med. 2001, 29, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Glauser, M.P.; Butler, T.; Garbino, J.; Gelmont, D.; Laterre, P.F.; Kudsk, K.; Bruining, H.A.; Otto, C.; Tobin, E.; et al. P55 tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 study group. JAMA 1997, 277, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Wunderink, R.; Silverman, H.; Perl, T.M.; Nasraway, S.; Levy, H.; Bone, R.; Wenzel, R.P.; Balk, R.; Allred, R.; et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAB sepsis study group. JAMA 1995, 273, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 1985, 229, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Fong, Y.; Hesse, D.G.; Manogue, K.R.; Lee, A.T.; Kuo, G.C.; Lowry, S.F.; Cerami, A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987, 330, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Newham, P.; Ross, D.; Ceuppens, P.; Das, S.; Yates, J.W.; Betts, C.; Reens, J.; Randall, K.J.; Knight, R.; McKay, J.S. Determination of the safety and efficacy of therapeutic neutralization of tumor necrosis factor-α (TNF-α) using azd9773, an anti-TNF-α immune fab, in murine clp sepsis. Inflamm. Res. 2014, 63, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, K.; Menges, T.; Gardlund, B.; Harm Zwaveling, J.; Smithes, M.; Vincent, J.L.; Tellado, J.M.; Salgado-Remigio, A.; Zimlichman, R.; Withington, S.; et al. Randomized, placebo-controlled trial of the anti-tumor necrosis factor antibody fragment afelimomab in hyperinflammatory response during severe sepsis: The ramses study. Crit. Care Med. 2001, 29, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.K.; Bolgos, G.; Miller, C.; Nguyen, D.T.; DeForge, L.E.; Remick, D.G. Anti-tumor necrosis factor antibody therapy fails to prevent lethality after cecal ligation and puncture or endotoxemia. J. Immunol. 1992, 148, 2724–2730. [Google Scholar] [PubMed]
- Secher, T.; Vasseur, V.; Poisson, D.M.; Mitchell, J.A.; Cunha, F.Q.; Alves-Filho, J.C.; Ryffel, B. Crucial role of TNF receptors 1 and 2 in the control of polymicrobial sepsis. J. Immunol. 2009, 182, 7855–7864. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Han, M.; Yi, R.; Kwon, S.; Dai, C.; Wang, R. Anti-TNF-α therapy for patients with sepsis: A systematic meta-analysis. Int. J. Clin. Pract. 2014, 68, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. The anti-inflammatory effect of exercise: Its role in diabetes and cardiovascular disease control. Essays Biochem. 2006, 42, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, K.; Matsuyama, T.; Kündig, T.M.; Wakeham, A.; Kishihara, K.; Shahinian, A.; Wiegmann, K.; Ohashi, P.S.; Krönke, M.; Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. Monocytogenes infection. Cell 1993, 73, 457–467. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Hauwermeiren, F.; Lodens, S.; De Rycke, R.; Van Wonterghem, E.; Staes, A.; Gevaert, K.; Lopez-Otin, C.; Libert, C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 2013, 5, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J.; Torrance, D.S.; Stocking, K.L.; Glaccum, M.B.; Otten, C.; Willis, C.R.; Charrier, K.; Morrissey, P.J.; Ware, C.B.; Mohler, K.M. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 1998, 160, 943–952. [Google Scholar] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Watson, A.J.; Frey, M.R.; Miguel, J.C.; Burkitt, M.D.; Sutton, R.; Hughes, K.R.; Hall, L.J.; Caamaño, J.H.; et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis. Models Mech. 2013, 6, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Armaka, M.; Karagianni, N.; Kranidioti, K.; Vandenbroucke, R.E.; Loges, S.; Van Roy, M.; Staelens, J.; Puimege, L.; Palagani, A.; et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J. Clin. Investig. 2013, 123, 2590–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Grine, L.; Lodens, S.; Van Wonterghem, E.; De Rycke, R.; De Geest, N.; Hassan, B.; Libert, C. TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol. 2015, 8, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Van Ryckeghem, S.; Vandewalle, J.; Ballegeer, M.; Van Wonterghem, E.; Eggermont, M.; Decruyenaere, J.; De Bus, L.; Libert, C.; Vandenbroucke, R.E. Simultaneous inhibition of tumor necrosis factor receptor 1 and matrix metalloproteinase 8 completely protects against acute inflammation and sepsis. Crit. Care Med. 2018, 46, e67–e75. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Lint, P.; Demeestere, D.; Van Wonterghem, E.; Vanlaere, I.; Puimege, L.; Van Hauwermeiren, F.; De Rycke, R.; Mc Guire, C.; et al. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-csf barrier contributes to lethality during systemic inflammatory diseases. J. Neurosci. 2012, 32, 9805–9816. [Google Scholar] [CrossRef] [PubMed]
- Tauber, S.C.; Eiffert, H.; Bruck, W.; Nau, R. Septic encephalopathy and septic encephalitis. Expert Rev. Anti Infect. Ther. 2017, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Jacob, A.; Cunningham, P.; Hensley, L.; Quigg, R.J. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem. Int. 2008, 52, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Calsavara, A.C.; Soriani, F.M.; Vieira, L.Q.; Costa, P.A.; Rachid, M.A.; Teixeira, A.L. TNFR1 absence protects against memory deficit induced by sepsis possibly through over-expression of hippocampal BDNF. Metab. Brain Dis. 2015, 30, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Zantl, N.; Uebe, A.; Neumann, B.; Wagner, H.; Siewert, J.R.; Holzmann, B.; Heidecke, C.D.; Pfeffer, K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect. Immun. 1998, 66, 2300–2309. [Google Scholar] [PubMed]
- Hildebrand, F.; Pape, H.C.; Hoevel, P.; Krettek, C.; van Griensven, M. The importance of systemic cytokines in the pathogenesis of polymicrobial sepsis and dehydroepiandrosterone treatment in a rodent model. Shock 2003, 20, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Ebach, D.R.; Riehl, T.E.; Stenson, W.F. Opposing effects of tumor necrosis factor receptor 1 and 2 in sepsis due to cecal ligation and puncture. Shock 2005, 23, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G.; Call, D.R.; Ebong, S.J.; Newcomb, D.E.; Nybom, P.; Nemzek, J.A.; Bolgos, G.E. Combination immunotherapy with soluble tumor necrosis factor receptors plus interleukin 1 receptor antagonist decreases sepsis mortality. Crit. Care Med. 2001, 29, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.C.; Kollias, G.; Toftgard, R.; et al. Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Reinecker, H.C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MacDermott, R.P.; Raedler, A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 1993, 94, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Kollias, G. Fibroblast biology. Synovial fibroblasts in rheumatoid arthritis: Leading role or chorus line? Arthritis Res. 2000, 2, 342–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, L.; Iselin, S.; De Libero, G.; Lesslauer, W. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1-treated and TNFR1-deficient mice. J. Immunol. 1996, 157, 3178–3182. [Google Scholar] [PubMed]
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s disease cA2 study group. N. Engl. J. Med. 1997, 337, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF Au-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [PubMed]
- Li, G.; Wu, Y.; Jia, H.; Tang, L.; Huang, R.; Peng, Y.; Zhang, Y. Establishment and evaluation of a transgenic mouse model of arthritis induced by overexpressing human tumor necrosis factor alpha. Biol. Open 2016, 5, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002, 13, 315–321. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Noti, M.; Corazza, N.; Mueller, C.; Berger, B.; Brunner, T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J. Exp. Med. 2010, 207, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Chi, H.H.; Rajaiah, R.; Moudgil, K.D. Exogenous tumour necrosis factor alpha induces suppression of autoimmune arthritis. Arthritis Res. Ther. 2008, 10, R38. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Seino, H.; Abo, T.; Tanaka, S.; Shintani, S.; Ohta, S.; Tamura, K.; Sawai, T.; Nobunaga, T.; Oteki, T.; et al. Recombinant human tumor necrosis factor alpha suppresses autoimmune diabetes in nonobese diabetic mice. J. Clin. Investig. 1989, 84, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Grewal, K.D.; Wong, F.S.; Picarella, D.E.; Janeway, C.A., Jr.; Flavell, R.A. Local expression of transgene encoded TNF alpha in islets prevents autoimmune diabetes in nonobese diabetic (NOD) mice by preventing the development of auto-reactive islet-specific T cells. J. Exp. Med. 1996, 184, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.O.; McDevitt, H.O. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature 1988, 331, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Kollias, G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur. J. Immunol. 2000, 30, 2038–2047. [Google Scholar] [CrossRef]
- Kassiotis, G.; Kollias, G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: Implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 2001, 193, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Libert, C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev. 2011, 22, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Ho, A.; Koarada, S.; Morito, F.; Ushiyama, O.; Suzuki, N.; Kikuchi, Y.; Ohta, A.; Mak, T.W.; Nagasawa, K. Collagen-induced arthritis in TNF receptor-1-deficient mice: TNF receptor-2 can modulate arthritis in the absence of TNF receptor-1. Clin. Immunol. 2001, 99, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y.; Erdmann, J.; Alexopoulou, L.; Kollias, G.; Ross, F.P.; Teitelbaum, S.L. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 2000, 275, 27307–27310. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Baumel, M.; Mannel, D.N.; Howard, O.M.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FoxP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011, 585, 3611–3618. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, B.; Chen, X.; Oppenheim, J.J.; Baeyens, A.; Gregoire, S.; Chader, D.; Gorochov, G.; Miyara, M.; Salomon, B.L. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 2016, 22, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3+ conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Balancing TNFR1 and TNFR2 jointly for joint inflammation. Arthritis Rheumatol. 2014, 66, 2657–2660. [Google Scholar] [CrossRef] [PubMed]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.; Stoop, A.A.; Williams, R.O. Selective TNFR1 blockade is anti-inflammatory and reveals an immunoregulatory role for TNFR2. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Nichols, F.C.; Puddington, L.; Lingenheld, E.G.; Zhu, L.; Rajan, T.V.; Clark, R.B. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J. Immunol. 2011, 186, 6779–6787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, X.; Zhou, Q.; Howard, O.M.; Netea, M.G.; Oppenheim, J.J. TNFR2 is critical for the stabilization of the CD4+FoxP3+ regulatory t. Cell phenotype in the inflammatory environment. J. Immunol. 2013, 190, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Boden, E.K.; Snapper, S.B. Regulatory T cells in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2008, 24, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Pierik, M.; Vermeire, S.; Steen, K.V.; Joossens, S.; Claessens, G.; Vlietinck, R.; Rutgeerts, P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment. Pharmacol. Ther. 2004, 20, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Bae, S.C.; Lee, Y.H. Associations between functional TNFR2 196 m/r polymorphisms and susceptibility to rheumatoid arthritis: A meta-analysis. Rheumatol. Int. 2014, 34, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Anderson, S.K. Association of TNFRSF1B promoter polymorphisms with human disease: Further studies examining T-regulatory cells are required. Front. Immunol. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and cA2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; Merrill, J.E. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989, 170, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Kuno, R.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Autocrine activation of microglia by tumor necrosis factor-alpha. J. Neuroimmunol. 2005, 162, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNF-alpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-d-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa b pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, L.; Lindholm, K.; Konishi, Y.; Yue, X.; Hampel, H.; Zhang, D.; Shen, Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J. Neurosci. 2004, 24, 1760–1771. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lindholm, K.; Konishi, Y.; Li, R.; Shen, Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J. Neurosci. 2002, 22, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.; Redlich, R.; Buschel, A.; Opel, N.; Grotegerd, D.; Zaremba, D.; Dohm, K.; Burger, C.; Meinert, S.L.; Forster, K.; et al. TNF receptors 1 and 2 exert distinct region-specific effects on striatal and hippocampal grey matter volumes (VBM) in healthy adults. Genes Brain Behav. 2017, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Palmer, T.D. Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Zoecklein, L.; Papke, L.; Gamez, J.; Denic, A.; Macura, S.; Howe, C. Tumor necrosis factor alpha is reparative via TNFR2 in the hippocampus and via TNFR1 in the striatum after virus-induced encephalitis. Brain Pathol. 2009, 19, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Heldmann, U.; Thored, P.; Claasen, J.H.; Arvidsson, A.; Kokaia, Z.; Lindvall, O. TNF-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp. Neurol. 2005, 196, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Bruce, A.J.; Boling, W.; Kindy, M.S.; Peschon, J.; Kraemer, P.J.; Carpenter, M.K.; Holtsberg, F.W.; Mattson, M.P. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996, 2, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, P.B.; Provitera, V.; De Rosa, T.; Tartaglia, G.; Gorga, F.; Perrella, O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: A correlation with clinical activity. Immunopharmacol. Immunotoxicol. 1998, 20, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Doolittle, T.H.; Lincoln, R.; Brown, R.H.; Dinarello, C.A. Cytokine accumulations in CSF of multiple sclerosis patients: Frequent detection of interleukin-1 and tumor necrosis factor but not interleukin-6. Neurology 1990, 40, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Almoallim, H.; Al-Ghamdi, Y.; Almaghrabi, H.; Alyasi, O. Anti-tumor necrosis factor-α induced systemic lupus erythematosus. Open Rheumatol. J. 2012, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Tack, C.J.; Kleijwegt, F.S.; Van Riel, P.L.; Roep, B.O. Development of type 1 diabetes in a patient treated with anti-TNF-alpha therapy for active rheumatoid arthritis. Diabetologia 2009, 52, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef] [PubMed]
- Akassoglou, K.; Probert, L.; Kontogeorgos, G.; Kollias, G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J. Immunol. 1997, 158, 438–445. [Google Scholar] [PubMed]
- Zajicek, J.P.; Wing, M.; Scolding, N.J.; Compston, D.A. Interactions between oligodendrocytes and microglia. A major role for complement and tumour necrosis factor in oligodendrocyte adherence and killing. Brain J. Neurol. 1992, 115 Pt 6, 1611–1631. [Google Scholar] [CrossRef]
- Kruglov, A.A.; Lampropoulou, V.; Fillatreau, S.; Nedospasov, S.A. Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J. Immunol. 2011, 187, 5660–5670. [Google Scholar] [CrossRef] [PubMed]
- Ruddle, N.H.; Bergman, C.M.; McGrath, K.M.; Lingenheld, E.G.; Grunnet, M.L.; Padula, S.J.; Clark, R.B. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 1990, 172, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Bauer, J.; Akassoglou, K.; Lassmann, H.; Kollias, G.; Probert, L. A tumor necrosis factor-induced model of human primary demyelinating diseases develops in immunodeficient mice. Eur. J. Immunol. 1999, 29, 912–917. [Google Scholar] [CrossRef]
- Frei, K.; Eugster, H.P.; Bopst, M.; Constantinescu, C.S.; Lavi, E.; Fontana, A. Tumor necrosis factor alpha and lymphotoxin alpha are not required for induction of acute experimental autoimmune encephalomyelitis. J. Exp. Med. 1997, 185, 2177–2182. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.; Woody, J.N.; Hartung, H.P.; Polman, C.H. Increased mri activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Arnason, B.G.W. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The lenercept multiple sclerosis study group and the university of british columbia ms/mri analysis group. Neurology 1999, 53, 457–465. [Google Scholar]
- D’Souza, S.D.; Bonetti, B.; Balasingam, V.; Cashman, N.R.; Barker, P.A.; Troutt, A.B.; Raine, C.S.; Antel, J.P. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J. Exp. Med. 1996, 184, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.W.; Raine, C.S. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann. Neurol. 1988, 23, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Raine, C.S.; Farooq, M.; Norton, W.T.; Brosnan, C.F. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J. Immunol. 1991, 147, 1522–1529. [Google Scholar] [PubMed]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1a as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Eugster, H.P.; Frei, K.; Bachmann, R.; Bluethmann, H.; Lassmann, H.; Fontana, A. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur. J. Immunol. 1999, 29, 626–632. [Google Scholar] [CrossRef]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Kranidioti, K.; Xanthoulea, S.; Denis, M.; Kotanidou, A.; Douni, E.; Blackshear, P.J.; Kontoyiannis, D.L.; Kollias, G. Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity. Eur. J. Immunol. 2006, 36, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Novrup, H.G.; Bracchi-Ricard, V.; Ellman, D.G.; Ricard, J.; Jain, A.; Runko, E.; Lyck, L.; Yli-Karjanmaa, M.; Szymkowski, D.E.; Pearse, D.D.; et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J. Neuroinflamm. 2014, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-κB. Brain J. Neurol. 2011, 134, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain J. Neurol. 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Evangelidou, M.; Karamita, M.; Vamvakas, S.S.; Szymkowski, D.E.; Probert, L. Altered expression of oligodendrocyte and neuronal marker genes predicts the clinical onset of autoimmune encephalomyelitis and indicates the effectiveness of multiple sclerosis-directed therapeutics. J. Immunol. 2014, 192, 4122–4133. [Google Scholar] [CrossRef] [PubMed]
- Karamita, M.; Barnum, C.; Mobius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight 2017, 2, e87455. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; Kawara, T.; Arita, S.; Furuya, T.; Yamashita, T.; et al. Therapeutic effect of pegylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J. Control. Release 2011, 149, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, e90117. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; De Rycke, R.; Toussaint, W.; Vanhoutte, L.; Vanhove, C.; De Vos, F.; Vandenbroucke, R.E.; Libert, C. TNFR1 inhibition with a nanobody protects against EAE development in mice. Sci. Rep. 2017, 7, 13646. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNF-alpha impairs memory via astrocyte signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Dalby-Hansen, C.; Ellman, D.G.; Madsen, P.M.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fischer, R.; Naude, P.J.; Maier, O.; Nyakas, C.; Duffey, M.; Van der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Fischer, R.; Agresti, C.; Pfizenmaier, K. TNF receptor 2 protects oligodendrocyte progenitor cells against oxidative stress. Biochem. Biophys. Res. Commun. 2013, 440, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Motti, D.; Karmally, S.; Szymkowski, D.E.; Lambertsen, K.L.; Bethea, J.R.; Brambilla, R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J. Neurosci. 2016, 36, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Wajant, H.; Kontermann, R.; Pfizenmaier, K.; Maier, O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia 2014, 62, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.R.; Williams, J.L.; Muccigrosso, M.M.; Liu, L.; Sun, T.; Rubin, J.B.; Klein, R.S. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 2012, 124, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Buc, M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediat. Inflamm. 2013, 2013, 963748. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, N.; Papadopoulos, D.; Denis, M.C.; Mitsikostas, D.D.; Kollias, G. TNFR2 on non-haematopoietic cells is required for FoxP3+ treg-cell function and disease suppression in eae. Eur. J. Immunol. 2012, 42, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Arico, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, R.; Eugster, H.P.; Frei, K.; Fontana, A.; Lassmann, H. Impairment of TNF-receptor-1 signaling but not fas signaling diminishes T-cell apoptosis in myelin oligodendrocyte glycoprotein peptide-induced chronic demyelinating autoimmune encephalomyelitis in mice. Am. J. Pathol. 1999, 154, 1417–1422. [Google Scholar] [CrossRef]
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Kartalou, G.I.; Zenelak, S.; Georgopoulos, S. Peripheral tumor necrosis factor-alpha (TNF-alpha) modulates amyloid pathology by regulating blood-derived immune cells and glial response in the brain of AD/TNF transgenic mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Gorle, N.; Vandendriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, P.; Xie, J.; Staufenbiel, M.; Li, R.; Shen, Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα. Hum. Mol. Genet. 2014, 23, 4906–4918. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, R.; Shiosaki, K. Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and beta-amyloid toxicity in human neuronal cell line. J. Biol. Chem. 1997, 272, 3550–3553. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.L.; Narrow, W.C.; Mastrangelo, M.A.; Olschowka, J.A.; O’Banion, M.K.; Bowers, W.J. Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic Alzheimer disease mice leads to significant modulation of amyloid- and tau-related pathologies. Am. J. Pathol. 2013, 182, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, J.; Rowan, M.J.; Anwyl, R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur. J. Neurosci. 2005, 22, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Leng, A.; Mura, A.; Feldon, J.; Ferger, B. Tumor necrosis factor-alpha receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci. Lett. 2005, 375, 107–111. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Ruhn, K.A.; Martinez, T.N.; McAlpine, F.E.; Blesch, A.; Tansey, M.G. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiParkinsonian rats. Mol. Ther. 2008, 16, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiParkinsonian rats. J. Parkinsons Dis. 2014, 4, 349–360. [Google Scholar] [PubMed]
- Sullivan, P.G.; Bruce-Keller, A.J.; Rabchevsky, A.G.; Christakos, S.; Clair, D.K.; Mattson, M.P.; Scheff, S.W. Exacerbation of damage and altered NF-κB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci. 1999, 19, 6248–6256. [Google Scholar] [CrossRef] [PubMed]
- Longhi, L.; Ortolano, F.; Zanier, E.R.; Perego, C.; Stocchetti, N.; De Simoni, M.G. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir. Suppl. 2008, 102, 409–413. [Google Scholar] [PubMed]
- Longhi, L.; Perego, C.; Ortolano, F.; Aresi, S.; Fuma galli, S.; Zanier, E.R.; Stocchetti, N.; De Simoni, M.G. Tumor necrosis factor in traumatic brain injury: Effects of genetic deletion of p55 or p75 receptor. J. Cereb. Blood Flow Metab. 2013, 33, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.S.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998, 18, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Balosso, S.; Ravizza, T.; Perego, C.; Peschon, J.; Campbell, I.L.; De Simoni, M.G.; Vezzani, A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann. Neurol. 2005, 57, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Balosso, S.; Ravizza, T.; Pierucci, M.; Calcagno, E.; Invernizzi, R.; Di Giovanni, G.; Esposito, E.; Vezzani, A. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: Implications for seizure susceptibility. Neuroscience 2009, 161, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Schafers, M.; Schmidt, C.; Vogel, C.; Toyka, K.V.; Sommer, C. Tumor necrosis factor-alpha (TNF) regulates the expression of ICAM-1 predominantly through TNF receptor 1 after chronic constriction injury of mouse sciatic nerve. Acta Neuropathol. 2002, 104, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Stallforth, S.; Sommer, C. Altered pain behavior and regeneration after nerve injury in TNF receptor deficient mice. J. Peripher. Nerv. Syst. JPNS 2006, 11, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease: Rationale and current evidence. CNS Drugs 2009, 23, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial tnfalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Zenelak, S.; Georgopoulos, S. Genetic deletion of tumor necrosis factor-alpha attenuates amyloid-beta production and decreases amyloid plaque formation and glial response in the 5xfad model of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2017, 60, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Marx, F.; Steiner, E.; Hartmann, T.; Grubeck-Loebenstein, B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999, 13, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in swedish mutant APP transgenic mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 2011, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3XTG-AD mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.C.; Kane, M.; Ghimire, S.; Gautam, S.; Gui, J. Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: A nested case-control analysis. CNS Drugs 2016, 30, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.M.; Lin, M.T.; Larson, E.B.; Maezawa, I.; Tseng, L.H.; Edwards, K.L.; Schellenberg, G.D.; Hansen, J.A.; Kukull, W.A.; Jin, L.W. Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch. Neurol. 2006, 63, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.; Tartaglia, L.A.; Lee, A.; Bennett, G.L.; Rice, G.C.; Wong, G.H.; Chen, E.Y.; Goeddel, D.V. Cloning and expression of cdnas for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA 1991, 88, 2830–2834. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.Q.; Zhang, Q.Q.; Zhang, Y.D.; Xu, J. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11C-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Choi, S.M.; Jho, J.; Park, M.S.; Kang, J.; Park, S.J.; Ryu, J.H.; Jo, J.; Kim, H.H.; Kim, B.C. Infliximab ameliorates AD-associated object recognition memory impairment. Behav. Brain Res. 2016, 311, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, F.; Vernay, A.; Leuba, G.; Schenk, F. Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolism. Brain Res. Bull. 2009, 80, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Knox, J.; Chang, J.; Derbedrossian, A.; Vasilevko, V.; Cribbs, D.; Boado, R.J.; Pardridge, W.M.; Sumbria, R.K. Blood-brain barrier penetrating biologic TNF-alpha inhibitor for Alzheimer’s disease. Mol. Pharm. 2017, 14, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Puntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E.; Gross, H.; Weinberger, A.; Cohen, H. TNF-alpha modulation for treatment of Alzheimer’s disease: A 6-month pilot study. MedGenMed 2006, 8, 25. [Google Scholar] [PubMed]
- Tobinick, E.L.; Gross, H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J. Neuroinflamm. 2008, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Wang, B.R.; Jiang, W.W.; Chen, J.; Zhu, Y.W.; Zhong, L.L.; Zhang, Y.D.; Xu, J. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J. Am. Geriatr. Soc. 2011, 59, 1142–1144. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, S.; Huber, L.; Stein, R.; Schmid, K.; Swick, A.; Wand, P.; Brody, M.; Strum, S.; Joyal, S.V. Open label, crossover, pilot study to assess the efficacy and safety of perispinal administration of etanercept (enbrel®) in combination with nutritional supplements versus nutritional supplements alone in subjects with mild to moderate Alzheimer’s disease receiving standard care. FASEB J. 2016, 30 (Suppl. 1), Ib296. [Google Scholar]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimer’s Dis. JAD 2010, 19, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Cribbs, D.H.; Anderson, A.J.; Cummings, B.J.; Su, J.H.; Wasserman, A.J.; Cotman, C.W. The induction of the TNF-alpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res. 2003, 28, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorle, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid beta oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.; Herrera, A.J.; Venero, J.L.; Santiago, M.; De Pablos, R.M.; Villaran, R.F.; Espinosa-Oliva, A.M.; Arguelles, S.; Sarmiento, M.; Delgado-Cortes, M.J.; et al. Peripheral inflammation increases the damage in animal models of nigrostriatal dopaminergic neurodegeneration: Possible implication in Parkinson’s disease incidence. Parkinson’s Dis. 2011, 2011, 393769. [Google Scholar] [CrossRef] [PubMed]
- Kouchaki, E.; Kakhaki, R.D.; Tamtaji, O.R.; Dadgostar, E.; Behnam, M.; Nikoueinejad, H.; Akbari, H. Increased serum levels of TNF-alpha and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin. Neurol. Neurosurg. 2018, 166, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Lindenau, J.D.; Altmann, V.; Schumacher-Schuh, A.F.; Rieder, C.R.; Hutz, M.H. Tumor necrosis factor alpha polymorphisms are associated with Parkinson’s disease age at onset. Neurosci. Lett. 2017, 658, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from Parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Rousselet, E.; Callebert, J.; Parain, K.; Joubert, C.; Hunot, S.; Hartmann, A.; Jacque, C.; Perez-Diaz, F.; Cohen-Salmon, C.; Launay, J.M.; et al. Role of TNF-alpha receptors in mice intoxicated with the Parkinsonian toxin MPTP. Exp. Neurol. 2002, 177, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Chertoff, M.; Di Paolo, N.; Schoeneberg, A.; Depino, A.; Ferrari, C.; Wurst, W.; Pfizenmaier, K.; Eisel, U.; Pitossi, F. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor alpha in the nigrostriatal dopaminergic circuit of adult mice. Exp. Neurol. 2011, 227, 237–251. [Google Scholar] [CrossRef] [PubMed]
- De Lella Ezcurra, A.L.; Chertoff, M.; Ferrari, C.; Graciarena, M.; Pitossi, F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol. Dis. 2010, 37, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Gemma, C.; Catlow, B.; Cole, M.; Hudson, C.; Samec, A.; Shah, N.; Vila, J.; Bachstetter, A.; Bickford, P.C. Early inhibition of tnfalpha increases 6-hydroxydopamine-induced striatal degeneration. Brain Res. 2007, 1147, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.H.; Sumbria, R.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Neuroprotection with a brain-penetrating biologic tumor necrosis factor inhibitor. J. Pharmacol. Exp. Ther. 2011, 339, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1beta and tumor necrosis factor-alpha: Reliable targets for protective therapies in Parkinson’s disease? Front. Cell. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Cerami, A. The value of failure: The discovery of TNF and its natural inhibitor erythropoietin. J. Intern. Med. 2011, 269, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Anzueto, A.; Gutierrez, G.; Tessler, S.; San Pedro, G.; Wunderink, R.; Dal Nogare, A.; Nasraway, S.; Berman, S.; Cooney, R.; et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. Norasept II study group. Lancet 1998, 351, 929–933. [Google Scholar] [CrossRef]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1992, 89, 9784–9788. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Katsikis, P.; Brennan, F.M.; Walker, J.; Bijl, H.; Ghrayeb, J.; et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993, 36, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.A.; Eisen, D.B. Treatment of hidradenitis suppurativa with biologic medications. J. Am. Acad. Dermatol. 2015, 73, S82–S88. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.H.; Racewicz, A.; Brzezicki, J.; Yatsyshyn, R.; Arteaga, E.T.; Baranauskaite, A.; Abud-Mendoza, C.; Navarra, S.; Kadinov, V.; Sariego, I.G.; et al. A phase III randomized study to evaluate the efficacy and safety of CT-p13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the planetra study. Arthritis Res. Ther. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Von Richter, O.; Skerjanec, A.; Afonso, M.; Sanguino Heinrich, S.; Poetzl, J.; Woehling, H.; Velinova, M.; Koch, A.; Kollins, D.; Macke, L.; et al. Gp2015, a proposed etanercept biosimilar: Pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br. J. Clin. Pharmacol. 2017, 83, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Yoo, D.H.; Prodanovic, N.; Jaworski, J.; Miranda, P.; Ramiterre, E.; Lanzon, A.; Baranauskaite, A.; Wiland, P.; Abud-Mendoza, C.; Oparanov, B.; et al. Efficacy and safety of CT-p13 (biosimilar infliximab) in patients with rheumatoid arthritis: Comparison between switching from reference infliximab to CT-p13 and continuing CT-p13 in the planetra extension study. Ann. Rheum. Dis. 2017, 76, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.K.; Shih, D.Q.; Chen, G.C. Insights on the use of biosimilars in the treatment of inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, S.; Mrowietz, U.; Augustin, M.; Ralph von, K.; Enk, A.; Stromer, K.; Schon, M.P.; Radtke, M.A. Biosimilars in dermatology—Theory becomes reality. J. Ger. Soc. Dermatol. JDDG 2018, 16, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Marotte, H.; Cimaz, R. Etanercept—TNF receptor and IgG1 Fc fusion protein: Is it different from other TNF blockers? Expert Opin. Biol. Ther. 2014, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Thaci, D.; Gerdes, S.; Arenberger, P.; Pulka, G.; Kingo, K.; Weglowska, J.; Hattebuhr, N.; Poetzl, J.; Woehling, H.; et al. The EGALITY study: A confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. The originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br. J. Dermatol. 2017, 176, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Khaliq-Kareemi, M.; Lawrance, I.C.; Thomsen, O.O.; Hanauer, S.B.; McColm, J.; Bloomfield, R.; Sandborn, W.J. Maintenance therapy with certolizumab pegol for Crohn’s disease. N. Engl. J. Med. 2007, 357, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Hutas, G. Golimumab as the first monthly subcutaneous fully human anti-TNF-alpha antibody in the treatment of inflammatory arthropathies. Immunotherapy 2010, 2, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.S.; Hartman, D.; Lemos, B.R.; Erlich, E.C.; Spence, S.; Kennedy, S.; Ptak, T.; Pruitt, R.; Vermeire, S.; Fox, B.S. AVX-470, an orally delivered anti-tumour necrosis factor antibody for treatment of active ulcerative colitis: Results of a first-in-human trial. J. Crohn’s Colitis 2016, 10, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Hartman, D.S.; Tracey, D.E.; Lemos, B.R.; Erlich, E.C.; Burton, R.E.; Keane, D.M.; Patel, R.; Kim, S.; Bhol, K.C.; Harris, M.S.; et al. Effects of AVX-470, an oral, locally acting anti-tumour necrosis factor antibody, on tissue biomarkers in patients with active ulcerative colitis. J. Crohn’s Colitis 2016, 10, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Lichtenstein, G.; Radford-Smith, G.; Patel, J.; Innes, A. CDP571, a humanized monoclonal antibody to tumour necrosis factor-alpha, for steroid-dependent Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Aliment. Pharmacol. Ther. 2006, 23, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Nayiager, S.; Louw, I.; Rojkovich, B.; Fu, C.; Udata, C. A multiple ascending dose/proof of concept study of ATN-103 (ozoralizumab) in rheumatoid arthritis subjects on a background of methotrexate. Arthritis Rheum. 2011, 63, 2630. [Google Scholar]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent Llama single-domain antibody fragments against tumor necrosis factor have picomolar potencies due to intramolecular interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.K., 3rd. PEGylated recombinant human soluble tumour necrosis factor receptor type I (r-Hu-sTNF-RI): Novel high affinity TNF receptor designed for chronic inflammatory diseases. Ann. Rheum. Dis. 1999, 58 (Suppl. 1), I73–I81. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.W.; Feige, U.; Bendele, A.M.; Martin, S.W.; Edwards, C.K., 3rd. Treatment of rheumatoid arthritis with PEGylated recombinant human soluble tumour necrosis factor receptor type I: A clinical update. Ann. Rheum. Dis. 2000, 59 (Suppl. 1), i41–i43. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, G.; Gu, C.; Chen, B.; Chen, A.; Li, H.; Gao, B.; Liang, C.; Wu, J.; Yang, T.; et al. T0001, a variant of TNFR2-Fc fusion protein, exhibits improved Fc effector functions through increased binding to membrane-bound TNF-alpha. PLoS ONE 2017, 12, e0177891. [Google Scholar]
- Rice, T.W.; Wheeler, A.P.; Morris, P.E.; Paz, H.L.; Russell, J.A.; Edens, T.R.; Bernard, G.R. Safety and efficacy of affinity-purified, anti-tumor necrosis factor-alpha, ovine fab for injection (CytoFab) in severe sepsis. Crit. Care Med. 2006, 34, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Furst, D.E.; Fleischmann, R.; Kopp, E.; Schiff, M.; Edwards, C.K.; Solinger, A.; Macri, M.; Grp, S. A phase 2 dose-finding study of pegylated recombinant methionyl human soluble tumor necrosis factor type I in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 2303–2310. [Google Scholar] [PubMed]
- Durez, P.; Vandepapeliere, P.; Miranda, P.; Toncheva, A.; Berman, A.; Kehler, T.; Mociran, E.; Fautrel, B.; Mariette, X.; Dhellin, O.; et al. Therapeutic vaccination with TNF-kinoid in TNF antagonist-resistant rheumatoid arthritis: A phase II randomized, controlled clinical trial. PLoS ONE 2014, 9, e113465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandepapeliere, P.; Malen, F.; Rogler, G.; Van der Bijl, A.; Kruger, F.C.; Kruger, D.W.; Grouard-Vogel, G.; Dhellin, O.; Fanget, B.; Michetti, P.F. Safety, immunogenicity and clinical phase I-II results of TNFalpha-kinoid immunotherapeutic in Crohn’s disease patients. Gastroenterology 2011, 140, S-123. [Google Scholar] [CrossRef]
- Spohn, G.; Guler, R.; Johansen, P.; Keller, I.; Jacobs, M.; Beck, M.; Rohner, F.; Bauer, M.; Dietmeier, K.; Kundig, T.M.; et al. A virus-like particle-based vaccine selectively targeting soluble TNF-alpha protects from arthritis without inducing reactivation of latent tuberculosis. J. Immunol. 2007, 178, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, R.K.; Boado, R.J.; Pardridge, W.M. Brain protection from stroke with intravenous tnfalpha decoy receptor-trojan horse fusion protein. J. Cereb. Blood Flow Metab. 2012, 32, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Olesen, C.M.; Coskun, M.; Peyrin-Biroulet, L.; Nielsen, O.H. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol. Ther. 2016, 159, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.D.; Wildenberg, M.E.; van den Brink, G.R. Mechanism of action of anti-TNF therapy in inflammatory bowel disease. J. Crohn’s Colitis 2016, 10, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Scallon, B.; Cai, A.; Solowski, N.; Rosenberg, A.; Song, X.Y.; Shealy, D.; Wagner, C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J. Pharmacol. Exp. Ther. 2002, 301, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm. Bowel Dis. 2007, 13, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Bloemendaal, F.M.; Levin, A.D.; Wildenberg, M.E.; Koelink, P.J.; McRae, B.L.; Salfeld, J.; Lum, J.; van der Neut Kolfschoten, M.; Claassens, J.W.; Visser, R.; et al. Anti-tumor necrosis factor with a glyco-engineered Fc-region has increased efficacy in mice with colitis. Gastroenterology 2017, 153, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Ehrenstein, M.R. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J. Exp. Med. 2016, 213, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Mauri, C.; Ehrenstein, M.R. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J. Exp. Med. 2007, 204, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, L.J.; Zochling, J.; Boonen, A.; Singh, J.A.; Veras, M.M.; Tanjong Ghogomu, E.; Benkhalti Jandu, M.; Tugwell, P.; Wells, G.A. TNF-alpha inhibitors for ankylosing spondylitis. Cochrane Database Syst. Rev. 2015, 3, CD005468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Jobanputra, P.; Barton, P.; Jowett, S.; Bryan, S.; Clark, W.; Fry-Smith, A.; Burls, A. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol. Assess. 2006, 10, 1–229. [Google Scholar] [CrossRef]
- Feagan, B.G.; McDonald, J.W.; Panaccione, R.; Enns, R.A.; Bernstein, C.N.; Ponich, T.P.; Bourdages, R.; Macintosh, D.G.; Dallaire, C.; Cohen, A.; et al. Methotrexate in combination with infliximab is no more effective than infliximab alone in patients with Crohn’s disease. Gastroenterology 2014, 146, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.; Tsynman, D.N.; Kim, A.; Arora, J.; Pietras, T.; Messing, S.; St Hilaire, L.; Yoon, S.; Decross, A.; Shah, A.; et al. Sustained improvement in health-related quality of life measures in patients with inflammatory bowel disease receiving prolonged anti-tumor necrosis factor therapy. J. Dig. Dis. 2014, 15, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Shah, N.; Deshpande, G.; Tang, D.H.; Eisenberg, D.F. Comparing biologic cost per treated patient across indications among adult us managed care patients: A retrospective cohort study. Drugs Real World Outcomes 2016, 3, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.S.; Hart, A.; De Cruz, P. Systematic review: Predicting and optimising response to anti-TNF therapy in Crohn’s disease—Algorithm for practical management. Aliment. Pharmacol. Ther. 2016, 43, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Lemann, M. Review article: Remission rates achievable by current therapies for inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 33, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Seidman, E. Predicting durable response or resistance to antitumor necrosis factor therapy in inflammatory bowel disease. Ther. Adv. Gastroenterol. 2016, 9, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Mullikin, J.C.; Facio, F.M.; Turner, C.; Cherukuri, P.F.; Blakesley, R.W.; Bouffard, G.G.; Chines, P.S.; Cruz, P.; Hansen, N.F.; et al. The ClinSeq project: Piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009, 19, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Verweij, C.L. Predicting the future of anti-tumor necrosis factor therapy. Arthritis Res. Ther. 2009, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deora, A.; Hegde, S.; Lee, J.; Choi, C.H.; Chang, Q.; Lee, C.; Eaton, L.; Tang, H.; Wang, D.; Lee, D.; et al. Transmembrane tnf-dependent uptake of anti-TNF antibodies. mAbs 2017, 9, 680–695. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Chowers, Y. Review article: Loss of response to anti-TNF treatments in Crohn’s disease. Aliment. Pharmacol. Ther. 2011, 33, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Noman, M.; Van Assche, G.; Baert, F.; D’Haens, G.; Rutgeerts, P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn’s disease. Gut 2007, 56, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Steenholdt, C.; Svenson, M.; Bendtzen, K.; Thomsen, O.O.; Brynskov, J.; Ainsworth, M.A. Acute and delayed hypersensitivity reactions to infliximab and adalimumab in a patient with Crohn’s disease. J. Crohn’s Colitis 2012, 6, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Vande Casteele, N.; Gils, A.; Singh, S.; Ohrmund, L.; Hauenstein, S.; Rutgeerts, P.; Vermeire, S. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am. J. Gastroenterol. 2013, 108, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Pouillon, L.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Noman, M.; Sabino, J.; Vande Casteele, N.; Gils, A.; Vermeire, S. Mucosal healing and long-term outcomes of patients with inflammatory bowel diseases receiving clinic-based vs. trough concentration-based dosing of infliximab. Clin. Gastroenterol. Hepatol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Casteele, N.V.; Van Stappen, T.; Van Assche, G.; Ferrante, M.; Vermeire, S.; Gils, A. Patients with high anti-drug antibody titers require a higher cumulative infliximab dose to achieve target drug concentrations: A post-hoc analysis of the taxit trial. Am. J. Gastroenterol. 2016, 111, S319. [Google Scholar]
- Vande Casteele, N.; Compernolle, G.; Ballet, V.; Van Assche, G.; Gils, A.; Vermeire, S.; Rutgeerts, P.J. Results on the optimisation phase of the prospective controlled trough level adapted infliximab treatment (taxit) trial. Gastroenterology 2012, 142, S211–S212. [Google Scholar] [CrossRef]
- Nunez Martinez, O.; Ripoll Noiseux, C.; Carneros Martin, J.A.; Gonzalez Lara, V.; Gregorio Maranon, H.G. Reactivation tuberculosis in a patient with anti-TNF-alpha treatment. Am. J. Gastroenterol. 2001, 96, 1665–1666. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, F.; Chen, J.W.; Wang, J. Risk of tuberculosis infection in anti-TNF-alpha biological therapy: From bench to bedside. J. Microbiol. Immunol. Infect. 2014, 47, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gasalla, M.; Fernandez-Baca, V.; Juan-Mas, A.; Payeras-Cifre, A.; Cifuentes-Luna, C.; Taberner-Ferrer, R.; Riera-Oliver, J.; Ros-Villamajo, I.; Navarro-Fernandez, V.; Morey Torrandell, C.; et al. Use of Quantiferon-TB-Gold in tube(®) test for detecting latent tuberculosis in patients considered as candidates for anti-TNF therapy in routine clinical practice. Enferm. Infecc. Microbiol. Clin. 2013, 31, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Fellermann, K. Adverse events of tumor necrosis factor inhibitors. Dig. Dis. 2013, 31, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Mak, T.W.; Ohashi, P.S. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845. [Google Scholar] [PubMed]
- Pereira, R.; Lago, P.; Faria, R.; Torres, T. Safety of anti-TNF therapies in immune-mediated inflammatory diseases: Focus on infections and malignancy. Drug Dev. Res. 2015, 76, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L. Inflammatory bowel disease therapies and cancer risk: Where are we and where are we going? Gut 2012, 61, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Beyaert, R.; Beaugerie, L.; Van Assche, G.; Brochez, L.; Renauld, J.C.; Viguier, M.; Cocquyt, V.; Jerusalem, G.; Machiels, J.P.; Prenen, H.; et al. Cancer risk in immune-mediated inflammatory diseases (IMID). Mol. Cancer 2013, 12, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltsonoudis, E.; Voulgari, P.V.; Konitsiotis, S.; Drosos, A.A. Demyelination and other neurological adverse events after anti-TNF therapy. Autoimmun. Rev. 2014, 13, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kemanetzoglou, E.; Andreadou, E. Cns demyelination with TNF-alpha blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Genovese, M.C.; Moreland, L.W. Demyelinating and neurologic events reported in association with tumor necrosis factor alpha antagonism: By what mechanisms could tumor necrosis factor alpha antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum. 2001, 44, 1977–1983. [Google Scholar] [CrossRef]
- Wang, F.; Lin, X.; Zhao, Q.; Li, J. Adverse symptoms with anti-TNF-alpha therapy in inflammatory bowel disease: Systematic review and duration-response meta-analysis. Eur. J. Clin. Pharmacol. 2015, 71, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Druce, K.L.; Bhattacharya, Y.; Jones, G.T.; Macfarlane, G.J.; Basu, N. Most patients who reach disease remission following anti-TNF therapy continue to report fatigue: Results from the British society for rheumatology biologics register for rheumatoid arthritis. Rheumatology 2016, 55, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Perez-De-Lis, M.; Retamozo, S.; Flores-Chavez, A.; Kostov, B.; Perez-Alvarez, R.; Brito-Zeron, P.; Ramos-Casals, M. Autoimmune diseases induced by biological agents. A review of 12,731 cases (biogeas registry). Expert Opin. Drug Saf. 2017, 16, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Tillack, C.; Ehmann, L.M.; Friedrich, M.; Laubender, R.P.; Papay, P.; Vogelsang, H.; Stallhofer, J.; Beigel, F.; Bedynek, A.; Wetzke, M.; et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-gamma-expressing Th1 cells and IL-17a/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut 2014, 63, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.L.; Fraunfelder, F.W.; Rosenbaum, J.T. Do tumor necrosis factor inhibitors cause uveitis? A registry-based study. Arthritis Rheum. 2007, 56, 3248–3252. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Houvenagel, E.; Goeb, V.; Fouache, D.; Martin, A.; Le Dantec, P.; Dernis, E.; Wendling, D.; Ansemant, T.; Berthelot, J.M.; et al. Development of inflammatory bowel disease during anti-TNF-alpha therapy for inflammatory rheumatic disease: A nationwide series. Jt. Bone Spine 2012, 79, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Prati, C. Paradoxical effects of anti-TNF-alpha agents in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.L.; Gadola, S.; Edwards, C.J. Anti-TNF-induced lupus. Rheumatology 2009, 48, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Steiner, G.; Graninger, W.B.; Hofler, E.; Steiner, C.W.; Smolen, J.S. Effects of short-term infliximab therapy on autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. Dual inhibition of TNFR1 and IFNAR1 in imiquimod-induced psoriasiform skin inflammation in mice. J. Immunol. 2015, 194, 5094–5102. [Google Scholar] [CrossRef] [PubMed]
- Joyau, C.; Veyrac, G.; Dixneuf, V.; Jolliet, P. Anti-tumour necrosis factor alpha therapy and increased risk of de novo psoriasis: Is it really a paradoxical side effect? Clin. Exp. Rheumatol. 2012, 30, 700–706. [Google Scholar] [PubMed]
- Kodama, S.; Davis, M.; Faustman, D.L. The therapeutic potential of tumor necrosis factor for autoimmune disease: A mechanistically based hypothesis. Cell. Mol. Life Sci. CMLS 2005, 62, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Puimege, L.; Vandenbroucke, R.E.; Van Hauwermeiren, F.; Haustraete, J.; Devoogdt, N.; Hulpiau, P.; Leroux-Roels, G.; Laukens, D.; Meuleman, P.; et al. Generation and characterization of small single domain antibodies inhibiting human tumor necrosis factor receptor 1. J. Biol. Chem. 2015, 290, 4022–4037. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, K.; de Haard, H.; Beirnaert, E.; Dreier, T.; Lauwereys, M.; Huyck, L.; Van Huysse, J.; Demetter, P.; Steidler, L.; Remaut, E.; et al. Orally administered L. lactis secreting an anti-TNF nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol. 2010, 3, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ubah, O.C.; Steven, J.; Kovaleva, M.; Ferguson, L.; Barelle, C.; Porter, A.J.R.; Barelle, C.J. Novel, anti-hTNF-alpha variable new antigen receptor formats with enhanced neutralizing potency and multifunctionality, generated for therapeutic development. Front. Immunol. 2017, 8, 1780. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, Z.; Wu, F.; Tan, J.; Shen, Y.; Li, E.; Dai, J.; Shen, R.; Li, G.; Wu, J.; et al. A variant of TNFR2-fc fusion protein exhibits improved efficacy in treating experimental rheumatoid arthritis. PLoS Comput. Biol. 2010, 6, e1000669. [Google Scholar] [CrossRef] [PubMed]
- Papp, K. Clinical development of onercept, a tumor necrosis factor binding protein, in psoriasis. Curr. Med. Res. Opin. 2010, 26, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; Sandborn, W.J.; Fedorak, R.N.; Rachmilewitz, D.; Tarabar, D.; Gibson, P.; Haagen Nielsen, O.; Wild, G.; Schreiber, S.; Pena Rossi, C.; et al. Onercept for moderate-to-severe Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2006, 4, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hettinghouse, A.; Liu, C. The role of progranulin in arthritis. Ann. N. Y. Acad. Sci. 2016, 1383, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.M.Z.; Mundra, J.J.; Jian, J.L.; Tian, Q.Y.; Gonzalez-Gugel, E.; Richbourgh, B.; Liu, C.J. Progranulin inhibition of TNF alpha. Immunol. Cell Biol. 2014, 92, 299–300. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Alleyne, C.H., Jr.; Dhandapani, K.M. TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci. Lett. 2013, 542, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Sumbria, R.K.; Zhou, Q.H.; Hui, E.K.; Lu, J.Z.; Boado, R.J.; Pardridge, W.M. Pharmacokinetics and brain uptake of an IgG-TNF decoy receptor fusion protein following intravenous, intraperitoneal, and subcutaneous administration in mice. Mol. Pharm. 2013, 10, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gong, H.; Zhu, H.; Ji, Q.; Su, P.; Liu, P.; Cao, S.; Yao, J.; Jiang, L.; Han, M.; et al. A novel small-molecule tumor necrosis factor alpha inhibitor attenuates inflammation in a hepatitis mouse model. J. Biol. Chem. 2014, 289, 12457–12466. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mrna degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of beta-secretase in a mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e55091. [Google Scholar]
- Alkam, T.; Nitta, A.; Mizoguchi, H.; Saito, K.; Seshima, M.; Itoh, A.; Yamada, K.; Nabeshima, T. Restraining tumor necrosis factor-alpha by thalidomide prevents the amyloid beta-induced impairment of recognition memory in mice. Behav. Brain Res. 2008, 189, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. Biofactors 2013, 39, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Semmler, J.; Gebert, U.; Eisenhut, T.; Moeller, J.; Schonharting, M.M.; Allera, A.; Endres, S. Xanthine derivatives: Comparison between suppression of tumour necrosis factor-alpha production and inhibition of camp phosphodiesterase activity. Immunology 1993, 78, 520–525. [Google Scholar] [PubMed]
- Brustolim, D.; Ribeiro-dos-Santos, R.; Kast, R.E.; Altschuler, E.L.; Soares, M.B. A new chapter opens in anti-inflammatory treatments: The antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int. Immunopharmacol. 2006, 6, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Becnel, J.; Zerfaoui, M.; Rohatgi, R.; Boulares, A.H.; Nichols, C.D. Serotonin 5-hydroxytryptamine(2A) receptor activation suppresses tumor necrosis factor-alpha-induced inflammation with extraordinary potency. J. Pharmacol. Exp. Ther. 2008, 327, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Siegel, R.M. Wishing away inflammation? New links between serotonin and TNF signaling. Mol. Interve. 2009, 9, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L. TNF, TNF inducers, and TNFR2 agonists: A new path to type 1 diabetes treatment. Diabetes/Metab. Res. Rev. 2018, 34, e2941. [Google Scholar] [CrossRef] [PubMed]
- Cleynen, I.; Vermeire, S. Paradoxical inflammation induced by anti-TNF agents in patients with IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF receptor one-specific antibody (ATROSAB): Receptor binding and in vitro bioactivity. PLoS ONE 2013, 8, e72156. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Munkel, S.; Neumeyer, J.; Muller, D.; Branschadel, M.; Scheurich, P.; Pfizenmaier, K. A humanized tumor necrosis factor receptor 1 (TNFR1)-specific antagonistic antibody for selective inhibition of tumor necrosis factor (TNF) action. J. Immunother. 2008, 31, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Yoshioka, Y.; Abe, Y.; Ohkawa, A.; Nomura, T.; Minowa, K.; Mukai, Y.; Nakagawa, S.; Taniai, M.; Ohta, T.; et al. The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 2009, 30, 6638–6647. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, M.; Isoda, K.; Kamada, H.; Kobayashi, T.; Tsunoda, S.; Tsutsumi, Y.; Niida, T.; Kujiraoka, T.; Ishigami, N.; Ishihara, M.; et al. Novel TNF-alpha receptor 1 antagonist treatment attenuates arterial inflammation and intimal hyperplasia in mice. J. Atheroscler. Thromb. 2012, 19, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.M.; Davies, M.; Mistry, P.; Green, P.; Giddins, G.; Feldmann, M.; Stoop, A.A.; Brennan, F.M. Selective blockade of tumor necrosis factor receptor I inhibits proinflammatory cytokine and chemokine production in human rheumatoid arthritis synovial membrane cell cultures. Arthritis Rheum. 2013, 65, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Bertok, S.; Wilson, M.R.; Morley, P.J.; de Wildt, R.; Bayliffe, A.; Takata, M. Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax 2012, 67, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Wakabayashi, K.; Bertok, S.; Oakley, C.M.; Patel, B.V.; O’Dea, K.P.; Cordy, J.C.; Morley, P.J.; Bayliffe, A.I.; Takata, M. Inhibition of TNF receptor p55 by a domain antibody attenuates the initial phase of acid-induced lung injury in mice. Front. Immunol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.C.; Wurthner, J.U.; Morley, P.J.; Birchler, M.A.; Lambert, J.; Albayaty, M.; Serone, A.P.; Wilson, R.; Chen, Y.; Forrest, R.M.; et al. Autoantibodies to variable heavy (VH) chain ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-alpha receptor 1. J. Clin. Immunol. 2013, 33, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.C.; Morley, P.J.; Wright, T.J.; Birchler, M.A.; Lewis, A.P.; Emmins, R.; Chen, Y.Z.; Powley, W.M.; Bareille, P.J.; Wilson, R.; et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-alpha receptor 1. Clin. Exp. Immunol. 2015, 182, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Chou, F.C.; Chen, S.J.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting pre-ligand assembly domain of TNFR1 ameliorates autoimmune diseases—an unrevealed role in downregulation of Th17 cells. J. Autoimmun. 2011, 37, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.M.; Zheng, L.; Chan, F.K.; Lenardo, M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat. Med. 2005, 11, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discov. 2017, 22, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, R.; Hu, H.; Hu, Y.; Chen, X. Modulation of regulatory T cell activity by TNF receptor type II-targeting pharmacological agents. Front. Immunol. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Polz, J.; Remke, A.; Weber, S.; Schmidt, D.; Weber-Steffens, D.; Pietryga-Krieger, A.; Muller, N.; Ritter, U.; Mostbock, S.; Mannel, D.N. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun. Inflamm. Dis. 2014, 2, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Zhang, H.; Wu, M.; Wang, Y. Myeloid-derived suppressor cells protect mouse models from autoimmune arthritis via controlling inflammatory response. Inflammation 2014, 37, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Falter, L.; Weber, S.; Muller, N.; Molitor, K.; Zeller, D.; Weber-Steffens, D.; Hehlgans, T.; Wajant, H.; Mostbock, S.; et al. Chronic inflammation increases the sensitivity of mouse Treg for TNFR2 costimulation. Front. Immunol. 2017, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, H.; Stueber, D.; Banner, D.; Mackay, F.; Lesslauer, W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J. Biol. Chem. 1993, 268, 26350–26357. [Google Scholar] [PubMed]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef] [PubMed]
- Lamontain, V.; Schmid, T.; Weber-Steffens, D.; Zeller, D.; Jenei-Lanzl, Z.; Wajant, H.; Straub, R.H.; Mannel, D.N. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Torrey, H.; Butterworth, J.; Zheng, H.; Faustman, D.L. Treg activation defect in type 1 diabetes: Correction with TNFR2 agonism. Clin. Transl. Immunol. 2016, 5, e56. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Herrera, P.L.; Harlan, D.M.; Vassalli, P. A mouse CD8 T cell-mediated acute autoimmune diabetes independent of the perforin and fas cytotoxic pathways: Possible role of membrane TNF. Proc. Natl. Acad. Sci. USA 2000, 97, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, J.M.; Barthold, D.; Brinton, R.D.; Joyce, G. Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol. 2017, 74, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; Sabbagh, M.; Connor, D.; Soares, H.; Lopez, J.; Stankovic, G.; Johnson-Traver, S.; Ziolkowski, C.; Browne, P. Statin therapy in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 2006, 185, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Nijholt, I.M.; Ostroveanu, A.; Ten Bosch, Q.; Luiten, P.G.; Eisel, U.L. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J. Alzheimer’s Dis. JAD 2008, 13, 111–122. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; He, J.; Shirota, H.; Trivett, A.L.; Yang; Klinman, D.M.; Oppenheim, J.J.; Chen, X. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci. Signal. 2018, 11, eaan0790. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L.; Wang, L.; Okubo, Y.; Burger, D.; Ban, L.; Man, G.; Zheng, H.; Schoenfeld, D.; Pompei, R.; Avruch, J.; et al. Proof-of-concept, randomized, controlled clinical trial of Bacillus-Calmette-Guerin for treatment of long-term type 1 diabetes. PLoS ONE 2012, 7, e41756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolillo, A.; Buzzi, M.G.; Giugni, E.; Sabatini, U.; Bastianello, S.; Pozzilli, C.; Salvetti, M.; Ristori, G. The effect of bacille calmette-guerin on the evolution of new enhancing lesions to hypointense T1 lesions in relapsing remitting MS. J. Neurol. 2003, 250, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Ristori, G.; Romano, S.; Cannoni, S.; Visconti, A.; Tinelli, E.; Mendozzi, L.; Cecconi, P.; Lanzillo, R.; Quarantelli, M.; Buttinelli, C.; et al. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology 2014, 82, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Secher, T.; Ezhevsky, S.A.; Janot, L.; Steed, P.M.; O’Brien, C.; Eivazi, A.; Kung, J.; Nguyen, D.H.; Doberstein, S.K.; et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J. Immunol. 2007, 179, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.L.; Di Polo, A. Neuroinflammation in glaucoma: Soluble tumor necrosis factor alpha and the connection with excitotoxic damage. Neural Regener. Res. 2016, 11, 424–426. [Google Scholar]
- Bedrosian, T.A.; Weil, Z.M.; Nelson, R.J. Chronic dim light at night provokes reversible depression-like phenotype: Possible role for TNF. Mol. Psychiatry 2013, 18, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef] [PubMed]
- Winsauer, C.; Kruglov, A.A.; Chashchina, A.A.; Drutskaya, M.S.; Nedospasov, S.A. Cellular sources of pathogenic and protective TNF and experimental strategies based on utilization of TNF humanized mice. Cytokine Growth Factor Rev. 2014, 25, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, A.V.; Grivennikov, S.I.; Kruglov, A.A.; Shebzukhov, Y.V.; Koroleva, E.P.; Piao, Y.; Cui, C.Y.; Kuprash, D.V.; Nedospasov, S.A. Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood 2010, 116, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Corazza, N.; Eichenberger, S.; Eugster, H.P.; Mueller, C. Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG)2(−/−) mice upon transfer of CD4(+)CD45RB(hi) T cells. J. Exp. Med. 1999, 190, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Boulougouris, G.; Manoloukos, M.; Armaka, M.; Apostolaki, M.; Pizarro, T.; Kotlyarov, A.; Forster, I.; Flavell, R.; Gaestel, M.; et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J. Exp. Med. 2002, 196, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Drutskaya, M.S.; Efimov, G.A.; Kruglov, A.A.; Nedospasov, S.A. Can we design a better anti-cytokine therapy? J. Leukoc. Biol. 2017, 102, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Efimov, G.A.; Kruglov, A.A.; Khlopchatnikova, Z.V.; Rozov, F.N.; Mokhonov, V.V.; Rose-John, S.; Scheller, J.; Gordon, S.; Stacey, M.; Drutskaya, M.S.; et al. Cell-type-restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Nosenko, M.A.; Atretkhany, K.N.; Mokhonov, V.V.; Efimov, G.A.; Kruglov, A.A.; Tillib, S.V.; Drutskaya, M.S.; Nedospasov, S.A. VHH-based bispecific antibodies targeting cytokine production. Front. Immunol. 2017, 8, 1073. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Therapy: Cell-type-specific approach to TNF inhibition. Nat. Rev. Rheumatol. 2016, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Roulis, M.; Armaka, M.; Manoloukos, M.; Apostolaki, M.; Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 5396–5401. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Ospelt, C.; Pasparakis, M.; Kollias, G. The p55TNFR-IKK2-Ripk3 axis orchestrates arthritis by regulating death and inflammatory pathways in synovial fibroblasts. Nat. Commun. 2018, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Garcin, G.; Paul, F.; Staufenbiel, M.; Bordat, Y.; Van der Heyden, J.; Wilmes, S.; Cartron, G.; Apparailly, F.; De Koker, S.; Piehler, J.; et al. High efficiency cell-specific targeting of cytokine activity. Nat. Commun. 2014, 5, 3016. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Tumanov, A.V.; Liepinsh, D.J.; Kruglov, A.A.; Marakusha, B.I.; Shakhov, A.N.; Murakami, T.; Drutskaya, L.N.; Forster, I.; Clausen, B.E.; et al. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: Protective and deleterious effects. Immunity 2005, 22, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.; Romanelli, M.; Chiricozzi, A. A revolutionary therapeutic approach for psoriasis: Bispecific biological agents. Expert Opin. Investig. Drugs 2016, 25, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Khatri, A.; Othman, A.A. Population pharmacokinetics of the TNF-alpha and IL-17A dual-variable domain antibody ABT-122 in healthy volunteers and subjects with psoriatic or rheumatoid arthritis: Analysis of phase 1 and 2 clinical trials. J. Clin. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Wagner, F.; Kivitz, A.J.; Mansikka, H.T.; Khan, N.; Othman, A.A.; Khatri, A.; Hong, F.; Jiang, P.; Ruzek, M.; et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. 2017, 69, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Li, R.; Chang, C. Bispecific Antibodies That Neutralize Both TNF-Alpha and IL-6: Novel Therapeutic Agent for Autoimmune Disease. U.S. Patent No. 9,416,197, 16 August 2016. [Google Scholar]
- Beidler, C.B.; Millican, R.L.; Na, S.; Seo, N. Anti-TNF/Anti-IL-23 Bispecific Antibodies. U.S. Patent No. 9,718,884, 1 August 2017. [Google Scholar]
- Kim, Y.; Yi, H.; Jung, H.; Rim, Y.A.; Park, N.; Kim, J.; Jung, S.M.; Park, S.H.; Park, Y.W.; Ju, J.H. A dual target-directed agent against interleukin-6 receptor and tumor necrosis factor alpha ameliorates experimental arthritis. Sci. Rep. 2016, 6, 20150. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Cohen, S.; Moreland, L.; Lium, D.; Robbins, S.; Newmark, R.; Bekker, P. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004, 50, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-kappab inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [PubMed]
- Kanakaraj, P.; Puffer, B.A.; Yao, X.T.; Kankanala, S.; Boyd, E.; Shah, R.R.; Wang, G.; Patel, D.; Krishnamurthy, R.; Kaithamana, S.; et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. mAbs 2012, 4, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.; Faurholm, B.; Dell’Accio, F.; Manzo, A.; Seed, M.; Eltawil, N.; Marrelli, A.; Gould, D.; Subang, C.; Al-Kashi, A.; et al. Human single-chain variable fragment that specifically targets arthritic cartilage. Arthritis Rheum. 2010, 62, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Huang, Y.; Liu, T.T.; Guo, R.; Wang, B.; Wang, X.L.; Chen, L.H.; Zhou, Y.; Ji, R.R.; Liu, T. TNF-alpha/TNFR1 signaling is required for the full expression of acute and chronic itch in mice via peripheral and central mechanisms. Neurosci. Bull. 2018, 34, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Lau, W.B.; Jiao, L.-Y.Y.; Liu, B.; Yuan, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.-L.L.; et al. Tumor necrosis factor-α and lymphotoxin-α mediate myocardial ischemic injury via TNF receptor 1, but are cardioprotective when activating TNF receptor 2. PLoS ONE 2013, 8, e60227. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.L.; Wang, M.; Crisostomo, P.R.; Abarbanell, A.M.; Herrmann, J.L.; Weil, B.R.; Meldrum, D.R. TNF receptor 2, not TNF receptor 1, enhances mesenchymal stem cell-mediated cardiac protection following acute ischemia. Shock 2010, 33, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Heusch, G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and yang in myocardial infarction? Circulation 2009, 119, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, F.; Yun, H.; Chen, L.; Muntner, P.; Levitan, E.B.; Safford, M.M.; Kent, S.T.; Osterman, M.T.; Lewis, J.D.; et al. Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Di Genaro, M.S.; Cargnelutti, D.E.; Elicabe, J.R.; Lacoste, M.G.; Valdez, S.; Gomez, N.; de Guzman, A.M. Role of TNFRp55 in Yersinia enterocolitica O:3-induced arthritis: Triggering bacterial antigens and articular immune response. Rheumatology 2007, 46, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, H.; Holtmann, H.; Brakebusch, C.; Avni, Y.S.; Sarov, I.; Nophar, Y.; Hadas, E.; Leitner, O.; Wallach, D. Antibodies to a soluble form of a tumor necrosis factor (TNF) receptor have TNF-like activity. J. Biol. Chem. 1990, 265, 14497–14504. [Google Scholar] [PubMed]
- Grell, M.; Scheurich, P.; Meager, A.; Pfizenmaier, K. TR60 and TR80 tumor necrosis factor (TNF)-receptors can independently mediate cytolysis. Lymphokine Cytokine Res. 1993, 12, 143–148. [Google Scholar] [PubMed]
- Hoos, A.; Anderson, J.; Boutin, M.; Dewulf, L.; Geissler, J.; Johnston, G.; Joos, A.; Metcalf, M.; Regnante, J.; Sargeant, I.; et al. Partnering with patients in the development and lifecycle of medicines: A call for action. Ther. Innov. Regul. Sci. 2015, 49, 929–939. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cytokine kinetics in sepsis. Tumor necrosis factor (TNF) and interleukin (IL-)1 are the first cytokines to be released in sepsis and promote the secretion of IL-6. Together, these cytokines are the orchestrators during the pro-inflammatory phase in sepsis. After some time, compensation mechanisms arise to dampen the pro-inflammatory response such as IL-10, IL-1 receptor antagonist (IL-1Ra) and soluble TNF receptor (sTNFR). Figure adapted from [63].
Figure 1.
Cytokine kinetics in sepsis. Tumor necrosis factor (TNF) and interleukin (IL-)1 are the first cytokines to be released in sepsis and promote the secretion of IL-6. Together, these cytokines are the orchestrators during the pro-inflammatory phase in sepsis. After some time, compensation mechanisms arise to dampen the pro-inflammatory response such as IL-10, IL-1 receptor antagonist (IL-1Ra) and soluble TNF receptor (sTNFR). Figure adapted from [63].

Figure 2.
Multiple roles for tumor necrosis factor (TNF) receptor (TNFR) signaling in multiple sclerosis (MS) pathology. In the central nervous system (CNS), TNF is primarily expressed by astrocytes, microglia and neurons and can stimulate its own release via TNFR1. A detrimental role for TNFR1 has been described in the pathology of MS. TNFR1 signaling triggers oligodendrocyte (OLG) death and contributes to primary demyelination via macrophages. Conversely, TNFR2 has protective effects in the CNS, as the interaction with tmTNF on astrocytes stimulates remyelination and neuronal TNFR2 protects against excitotoxicity. In the periphery, TNFR2 induces the development of effector T cells, but in the CNS, microglial-expressed TNFR2 is protective. TNF, possibly via TNFR2, mediates regression of activated myelin-specific T cells. Furthermore, TNFR2 signaling also facilitates the expansion of regulatory T cells (Tregs) and improves their suppressive capacities against effector T cells. Normal arrows indicate the action of a mediator or the processes that are induced; bold arrows represent mediators produced by a specific cellular subset. T bars represent the inhibition of the indicated pathway.
Figure 2.
Multiple roles for tumor necrosis factor (TNF) receptor (TNFR) signaling in multiple sclerosis (MS) pathology. In the central nervous system (CNS), TNF is primarily expressed by astrocytes, microglia and neurons and can stimulate its own release via TNFR1. A detrimental role for TNFR1 has been described in the pathology of MS. TNFR1 signaling triggers oligodendrocyte (OLG) death and contributes to primary demyelination via macrophages. Conversely, TNFR2 has protective effects in the CNS, as the interaction with tmTNF on astrocytes stimulates remyelination and neuronal TNFR2 protects against excitotoxicity. In the periphery, TNFR2 induces the development of effector T cells, but in the CNS, microglial-expressed TNFR2 is protective. TNF, possibly via TNFR2, mediates regression of activated myelin-specific T cells. Furthermore, TNFR2 signaling also facilitates the expansion of regulatory T cells (Tregs) and improves their suppressive capacities against effector T cells. Normal arrows indicate the action of a mediator or the processes that are induced; bold arrows represent mediators produced by a specific cellular subset. T bars represent the inhibition of the indicated pathway.

Figure 3.
Structure (A); and mechanisms of action (B) of anti-TNF biologics. All anti-TNF biologics neutralize membrane-bound (tmTNF) and soluble TNF (sTNF) but, in addition to that, some inhibitors also induce outside-to-inside signaling via tmTNF and their Fc-regions mediate antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). ADCC: antibody-dependent cellular cytotoxicity; CDC: complement-dependent cytotoxicity, Fab: fragment antigen binding; IgG: immunoglobulin G; IL: interleukin; NK: natural killer; ROS: reactive oxygen species tmTNF: transmembrane TNF.
Figure 3.
Structure (A); and mechanisms of action (B) of anti-TNF biologics. All anti-TNF biologics neutralize membrane-bound (tmTNF) and soluble TNF (sTNF) but, in addition to that, some inhibitors also induce outside-to-inside signaling via tmTNF and their Fc-regions mediate antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). ADCC: antibody-dependent cellular cytotoxicity; CDC: complement-dependent cytotoxicity, Fab: fragment antigen binding; IgG: immunoglobulin G; IL: interleukin; NK: natural killer; ROS: reactive oxygen species tmTNF: transmembrane TNF.
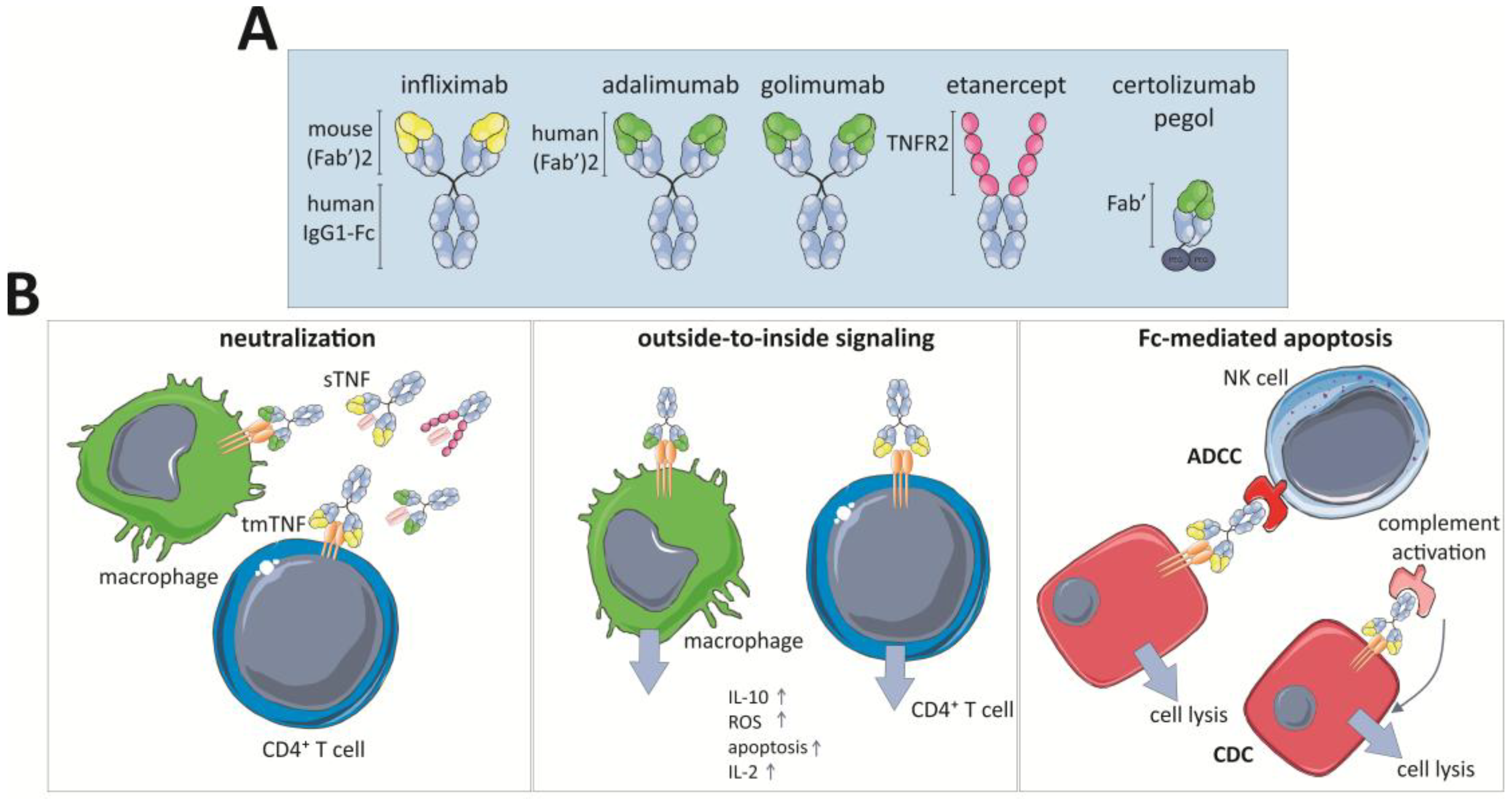
Figure 4.
Beneficial and side effects of anti-TNF medication. In addition to the well-known beneficial effects in several autoimmune diseases, anti-TNF medication is associated with many side effects.
Figure 4.
Beneficial and side effects of anti-TNF medication. In addition to the well-known beneficial effects in several autoimmune diseases, anti-TNF medication is associated with many side effects.


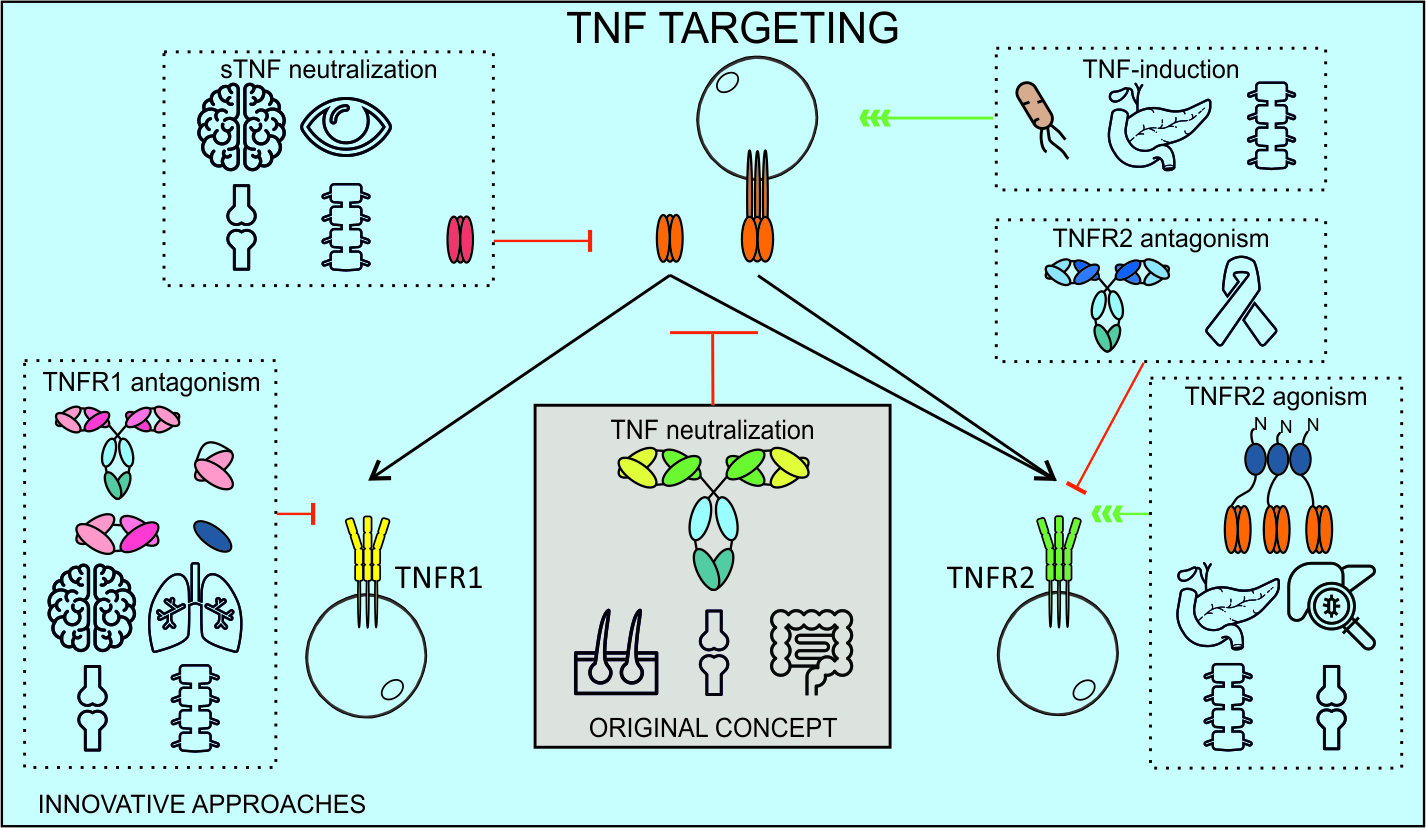{kind=link}
{kind=link}
{kind=link}
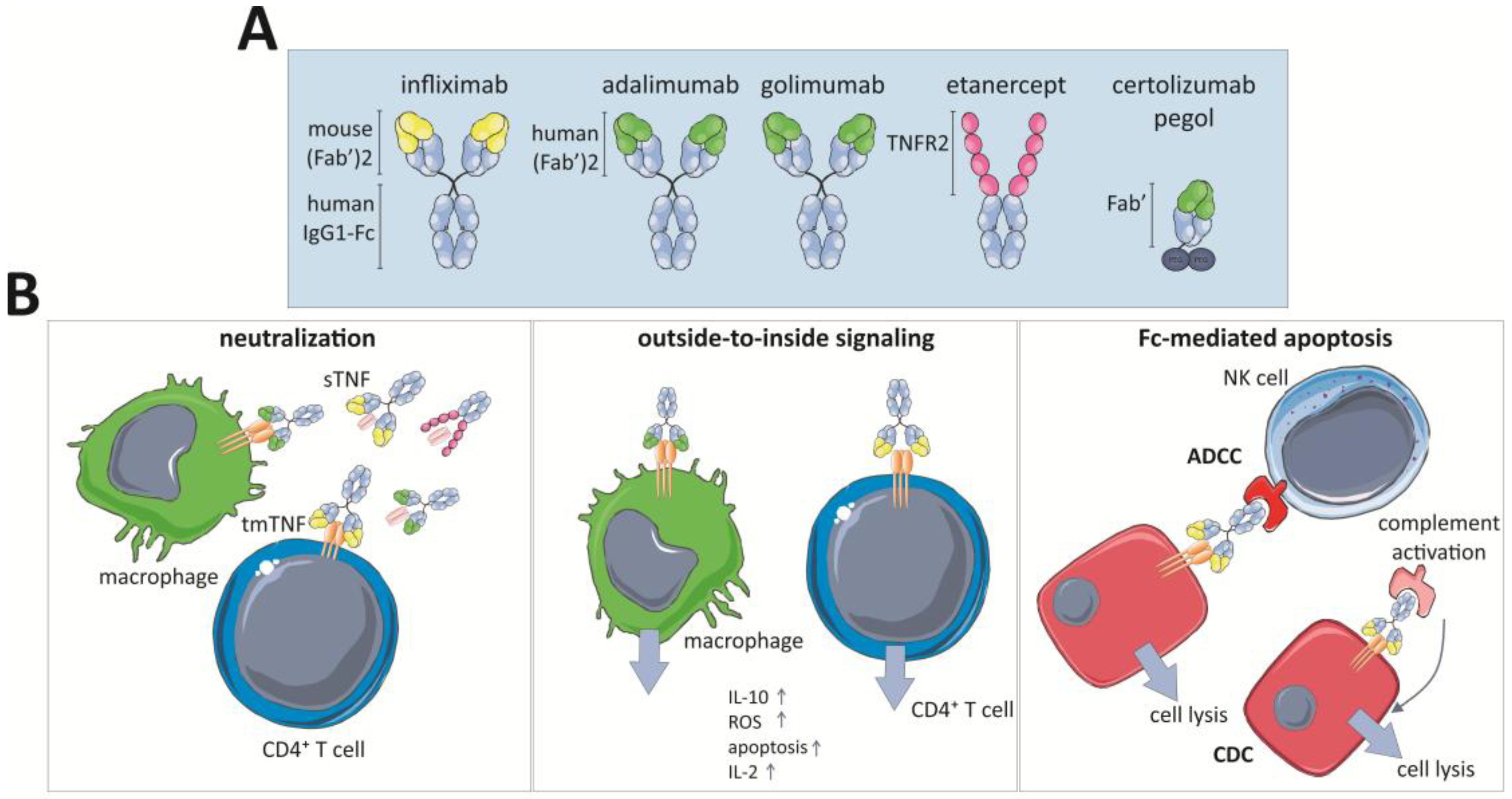{kind=link}
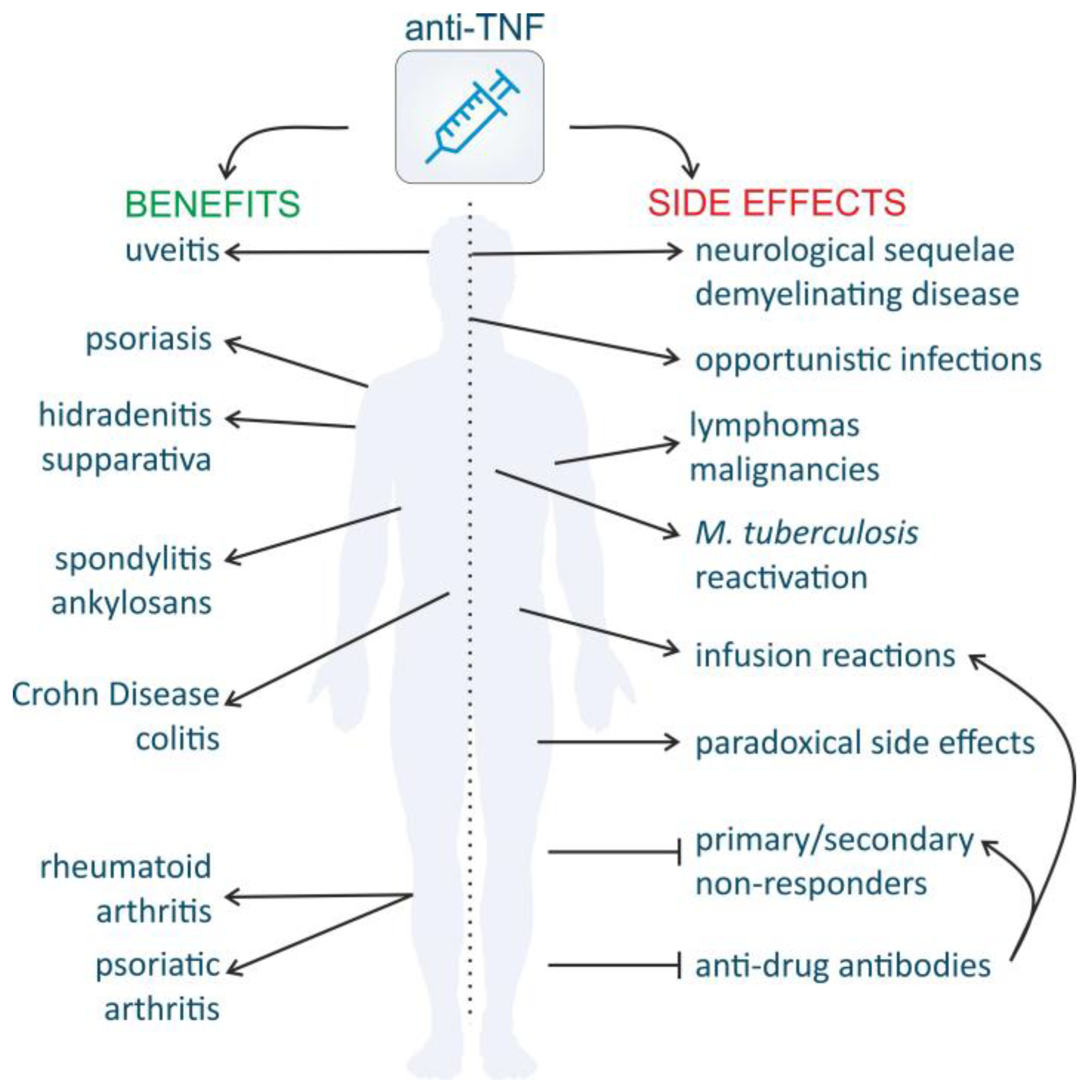{kind=link}
Table 1.
Evidence for specific TNF(R) targeting in neuronal disease.
| Model | In Vivo/In Vitro | Mechanism | References |
|---|---|---|---|
| Multiple Sclerosis | |||
| TNFR1/TNFR2−/−, TNFR1−/−, TNFR2−/− in EAE | In vivo | TNFR1−/− mice are resistant to disease development | [160,161,171] |
| TNFR2−/− mice display exacerbated disease progression | |||
| TNF−/− and TNFR1−/− mice in EAE | In vivo | TNF protects against EAE chronicity by inducing regression of myelin-reactive T cells, independent of TNFR1 | [96] |
| TNF∆1–12 mice in EAE | In vivo | tmTNF protects against disease development | [162] |
| TNFR2−/− in EAE and bone-marrow transplantations | In vivo | TNFR2 on non-hematopoietic cells is required for Treg functioning | [181] |
| OLG-specific TNFR2 KOs (CNP-cre TNFR2fl/fl) in EAE | In vivo | OLG-TNFR2 drives OPC differentiation | [177] |
| CX3cr1-cre TNFR2fl/fl and LysM-cre TNFR2fl/fl in EAE | In vivo | TNFR2 ablation in microglia leads to early EAE onset | [173] |
| TNFR2 ablation in monocytes results in EAE suppression | |||
| TNFR1−/− that conditionally re-express TNFR1 in astrocytes in EAE (hGFAPcreT2/tnfr1cneo/cneo) | In vivo | Astrocyte TNFR1 mediates learning memory impairment | [172] |
| TNF−/−, TNFR1−/− and TNFR2−/− in CPZ model | In vivo | TNFR2 is critical for OLG proliferation and remyelination | [33] |
| TNFR2−/− in CPZ model | In vivo | Astrocyte TNFR2 mediates OPC proliferation and differentiation via CXCL12 | [179] |
| CNS-overexpressing TNF mice in TNFR1 or TNFR2 KO background | In vivo | TNFR1 induces OLG apoptosis | [157] |
| Neuron or astrocyte-overexpressing tmTNF mice | In vivo | Astrocyte tmTNF but not neuron-specific TNF triggers CNS inflammation and neurodegeneration | [145] |
| TNFR1 inhibition in EAE | In vivo | Disease development is reduced | [169,170] |
| Nanobody-based TNFR1 inhibition in EAE | In vivo | Prophylactic and therapeutic administration prevents or stops disease development | [171] |
| sTNF inhibition in EAE | In vivo | Functional outcome is improved | [165] |
| sTNF inhibition in EAE and in astrocyte-neuron coculture | In vivo | tmTNF is neuroprotective via NF-κB | [164] |
| In vitro | sTNF inhibition protects against glucose deprivation | ||
| sTNF inhibition in CPZ model | In vivo | tmTNF is neuroprotective and is needed to maintain myelin sTNF inhibits remyelination and repair | [167] |
| Astrocytes-OPC co-culture | In vitro | Astrocyte TNFR2 promotes OLG maturation via LIF | [178] |
| Rat microglia and OLG | In vitro | tmTNF kills OLGs more efficiently than sTNF | [146] |
| Alzheimer’s Disease | |||
| 5xFAD/Tg197 mice | In vivo | Peripheral hTNF mediates reduced amyloidosis and more microglial and astrocytic activation, but also synaptic loss | [185] |
| TNFR1-overexpressing primary neurons and TNFR1−/− neurons | In vitro | TNFR1 mediates Aβ-induced neuronal death | [129] |
| APP/PS1 TNFR1−/− and icv AβO injection in TNFR1−/− | In vivo | TNFR1 mediates AD-mediated choroid plexus inflammation and TNFR1+/+ mice are protected against cognitive decline, microgliosis and amyloidosis | [186] |
| APP23 TNFR1−/− | In vivo | TNFR1 signaling enhances BACE1 activity and Aβ production. Absence of TNFR1 leads to less memory deficits, neuronal loss and microglia activation | [131] |
| APP23 TNFR2−/− | In vivo | Exacerbated AD pathology | [187] |
| Nanobody-based TNFR1 inhibition icv AβO injection | In vivo | TNFR1 inhibition prevents against cognitive decline | [186] |
| sTNF inhibition in 3×Tg-AD mice | In vivo | sTNF inhibition reduces APP accumulation in hippocampus and restores synaptic dysfunction | [188,189] |
| TNFR2 inhibition in SH-SY5Y cells | In vitro | Enhances Aβ toxicity | [190] |
| Neuronal TNFR2 knockdown in 3×Tg-AD mice | In vivo | Enhances Aβ and Tau-pathology | [191] |
| Transverse hippocampus slices of WT or TNFR1−/− mice | In vitro | TNF via TNFR1 is a critical mediator of the Aβ-induced inhibition of LTP | [192] |
| Parkinson’s Disease | |||
| Double TNFR−/− mice in MPTP model | In vivo | Mice were protected against neurotoxicity, but hippocampal vulnerability increased | [193,194] |
| TNFR1−/−, TNFR2−/− mice in MPTP model | In vivo | Neither TNFR1 nor TNFR2 KO mice were protected against MPTP neurotoxicity | [195] |
| sTNF neutralization in 6-OHDA model | In vivo | Reduced nigral dopaminergic loss and microglia activation | [196,197,198] |
| Spinal Cord Injury and Traumatic Brain Injury | |||
| sTNF inhibition in SCI | In vivo | Protective | [163] |
| Double TNFR−/− mice subjected to TBI | In vivo | Bigger lesion volume and BBB impairment | [199] |
| TNFR1−/− and TNFR2−/− subjected to controlled cortical impact brain injury | In vivo | TNFR1 exacerbates cognitive functioning, TNFR2 attenuates it | [200] |
| TNFR1−/− and TNFR2−/− subjected to TBI | In vivo | TNFR1 exacerbates neurobehavioral deficits and tissue damage, TNFR2 is protective | [201] |
| Stroke, Ischemia and Oxidative Stress | |||
| Double TNFR−/− mice in stroke model | In vivo | More neuronal damage and less injury-induced microglial activation | [138] |
| TNFR1−/−, TNFR2−/− and double TNFR mice in focal cerebral ischemia/reperfusion | In vivo | TNFR1 limits neuronal damage and prevents hippocampal degeneration | [202] |
| TNFR1−/− and TNFR2−/− in model retinal ischemia | In vivo | TNFR1 augments neuronal death, TNFR2 promotes neuroprotection via PI3K-PKB/Akt pathway | [203] |
| TNFR2 agonist in LUHMES cells treated with H2O2 | In vitro | TNFR2 promotes anti-apoptotic response via PI3K-PKB/Akt pathway | [168] |
| hTNFR2-expressing OLG + TNFR2 agonist treated with H2O2 | In vitro | TNFR2 protects OPC against oxidative stress | [175] |
| Excitotoxicity and Seizures | |||
| TNFR2 agonism or TNFR1 inhibition in NMDA-induced neurodegeneration | In vivo | TNFR1 inhibition/TNFR2 agonism protects cholinergic neurons against cell death and reverts neurodegeneration-associated memory impairment | [174] |
| TNFR1−/− and TNFR2−/− mice on kainate seizures | In vivo | TNFR2 exerts anticonvulsant effects, TNFR1 mediates excitotoxicity | [204] |
| Primary cortical cells treated with glutamate | In vitro | TNFR2 protects neurons against excitotoxic insults via activation NMDA-receptor | [128] |
| Microiontophoretic administration of glutamate to TNFR1−/− or TNFR2−/− mice | In vitro | TNFR2 protects hippocampal neurons against excitotoxicity | [205] |
| Brain Inflammation | |||
| Hippocampal TNFR1−/− and TNFR2−/− neurons | In vitro | TNF vulnerability of TNFR2−/− hippocampal neurons is higher than of TNFR1−/− neurons | [130] |
| Cultured microglia | In vitro | TNFR2 upregulation after inflammatory stimuli and TNFR2-mediated induction of anti-inflammatory pathways | [182] |
| Neuropathic Pain and Hippocampal Neurogenesis | |||
| Healthy or diseased TNFR1−/− and TNFR2−/− mice | In vivo | TNFR1 is a suppressor of adult neurogenesis, absence of TNFR2 leads to reduced hippocampal neurodegeneration | [132,133,134,135] |
| TNFR1−/− and TNFR2−/− mice subjected to CCI | In vivo | TNFR1 induces a neuropathic-pain induced depression | [136] |
| Double TNFR mice and TNFR1 and TNFR2 inhibitors following CCI | In vivo | TNFR1 inhibition prolongs Wallerian degeneration and TNFR1 regulates macrophage influx | [206,207] |
| TNFR1 mediates thermal hyperalgesia | |||
AβO: oligomerized amyloid beta; AD: Alzheimer’s disease; APP/PS1: amyloid precursor protein/presenilin 1; BACE1: beta-secretase 1; CCI: chronic constriction injury; CNP: 2′,3′-cyclic nucleotide 3′-phosphodiesterase; CPZ: cuprizone; CXCR3: CXC motif chemokine receptor 3; EAE: experimental autoimmune encephalomyelitis; icv: intracerebroventricular; KO: knockout; LIF: leukemia inhibiting factor; LTP: long term potentiation; LUHMES: Lund human mesencephalic; LysM: lysin-motif; NMDA: N-methyl-d-aspartate; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; OLG: oligodendrocytes; OPC: oligodendrocyte precursor cells; PI3K-PKB/Akt: phosphoinositide-3-kinase–protein kinase B/Akt; SCI: spinal cord injury; sTNF: soluble TNF; TBI: traumatic brain injury; Tg: transgenic; tmTNF: transmembrane TNF.
Table 2.
Anti-TNF biologics that are approved, in the pipeline or discontinued.
| Current Approved Anti-TNF Biologics | |||||
| Drug | Biosimilars | Structure | Approved Indication | Administration Route | Ref. |
| Infliximab | CT-P13; SB2 | Chimeric mAb | RA, PA, psoriasis, AS, UC, CD, pediatric RA & CD | IV, every 8 weeks following loading at week 0, 2 and 6 | [253,254,255,256] |
| Etanercept | GP2015; SB4 | Fusion protein: human TNFR2:IgG1-Fc | RA, PA, psoriasis, AS, JIA | SC, every week or every two weeks | [256,257,258] |
| Adalimumab | ABP501; ZRC3197 | Human IgG1 mAb | RA, PA, psoriasis, AS, JIA, CD, hidradenitis suppurativa, uveitis | SC, every two weeks | [256,259] |
| Certolizumab pegol | NA | Humanized PEGylated Fab’ fragment of humanized iGG1 | RA, PA, AS, CD (only in US and Switzerland) | SC, every two weeks | [260] |
| Golimumab | NA | Human IgG1 mAb | RA, PA, AS, ulcerative colitis | SC, monthly | [261] |
| Anti-TNF Biologics in the Pipeline or Discontinued | |||||
| Drug | Biosimilar | Structure | Disease Indication | Clinical Phase/State | Ref. |
| Infliximab | BOW015; PF-06438179 | Chimeric mAb | RA | Phase III/ongoing | [256] |
| Etanercept | CHS-0214; HD203 | Fusion protein: human TNFR2:IgG1-Fc | RA, psoriasis | Phase III/ongoing | [256] |
| Adalimumab | BI695501; CHS-1420; GP-2017; M923; SB5; ZRC-3197; FKB327 | Human IgG1 mAb | RA, psoriasis, AS | Phase I, II and III/ongoing | [256] |
| Golimumab | ONS-3035 | Human IgG1 mAb | RA, PsA, AS, UC | Preclinical | |
| SSS-07 | NA | Humanized mAb | RA | Phase I | NCT02460393 |
| AVX-470; Aveximab-TNF | NA | Polyclonal bovine anti-TNF Ab | UC, NEC | Phase I | [262,263], NCT01759056 |
| CDP571 | NA | Humanized IgG4 anti-human TNF mAb | CD | Phase II/discontinued | [264] |
| Ozoralizumab | NA | Trivalent, bispecific anti-TNF Nanobody | RA, discontinued for AS, CD and PsA | Phase IIa (Japan)/ongoing | [265] |
| VHH#1-3 | NA | Bivalent Nanobody | RA | Preclinical | [266] |
| Onercept | NA | PEGylated dimeric soluble human TNFR1 | CD, psoriasis, PsA, RA, sepsis, endometriosis | Phase II and III/discontinued | [267,268] |
| Lenercept | NA | Fusion protein: soluble TNFR1:IgG1-Fc | Severe sepsis, septic shock, RA and MS | Phase II/discontinued | [53,153] |
| Hitanercept (T0001) | NA | TNFR2-Fc fusion protein | RA | Phase I (China) | [269] |
| HL036 | NA | TNFR1 fragment | Dry eye disease | Phase II | NCT03334539 |
| CytoFab | NA | Polyclonal anti-TNF Fab fragment | Severe sepsis, septic shock | Phase II/discontinued | [270] |
| Pegsunercept | NA | PEGylated sTNFR1 | RA | Phase II/discontinued | [271] |
| TNF-kinoid | NA | Vaccine to induce anti-TNF Ab, recombinant human TNF coupled to carrier protein KLH | CD, RA | Phase II/suspended | [272,273] |
| CYT007-TNFQb | NA | Vaccine to induce anti-TNF Ab, recombinant TNF coupled to virus-like particles of the bacteriophage Qbeta | Psoriasis | Phase I and II/discontinued | [274] |
| TfRMAb-TNFR | NA | Fusion protein: extracellular TNFR2 coupled to mAb against TfR | PD, AD, ischemic stroke | Preclinical | [223,242,275] |
(m)Ab: (monoclonal) antibody; AD: Alzheimer’s disease; AS: Ankylosing spondylitis; CD: Crohn’s disease; Fab: fragment antigen binding; JIA: juvenile idiopathic arthritis; KLH: keyhole limpet hemocyanin; MS: multiple sclerosis; NEC: necrotizing enterocolitis; PEG: polyethylene glycol; PsA: psoriatic arthritis; PD: Parkinson’s disease; RA: rheumatoid arthritis; TfR: Transferrin receptor; TNFR: TNF receptor; UC: ulcerative colitis; VHH: variable domain of heavy-chain only antibodies NA: non-applicable.
Table 3.
Distinct functions of TNF produced by T cells and myeloid cells in several experimental mouse diseases.
Table 3.
Distinct functions of TNF produced by T cells and myeloid cells in several experimental mouse diseases.
| Disease Model | Cellular Source of TNF | Ref. | |
|---|---|---|---|
| Myeloid Cells | T Cells | ||
| T cell transfer colitis model | Pathogenic | Non-redundant | [390] |
| TNF∆ARE intestinal inflammation | Pathogenic | Pathogenic | [392] |
| TNF∆ARE joint inflammation | NA | Non-redundant | [85] |
| L. monocytogenes infection | Protective | Protective | [400] |
| M. tuberculosis infection | Dispensable 1 | Protective | [40] |
| Systemic LPS/D-Gal hepatotoxicity | Pathogenic | Dispensable | [40] |
| Autoimmune arthritis | Pathogenic | Protective | [393] |
| EAE | Pathogenic during early phase | Protective and pathogenic | [147] |
| protective in late phase | |||
| ConA-hepatitis | Pathogenic | Pathogenic | [40] |
| Diabetic nephropathy | Pathogenic | ND | [389] |
1 Mediates immune functions of cells at early stages of infections, but dispensable for protection. ConA: Concanavalin-A; EAE: Experimental autoimmune encephalomyelitis; ND: Not determined.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. https://doi.org/10.3390/ijms19051442
AMA Style
Steeland S, Libert C, Vandenbroucke RE. A New Venue of TNF Targeting. International Journal of Molecular Sciences. 2018; 19(5):1442. https://doi.org/10.3390/ijms19051442
Chicago/Turabian StyleSteeland, Sophie, Claude Libert, and Roosmarijn E. Vandenbroucke. 2018. "A New Venue of TNF Targeting" International Journal of Molecular Sciences 19, no. 5: 1442. https://doi.org/10.3390/ijms19051442
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.