An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors


{kind=link}
{kind=link}
Abstract
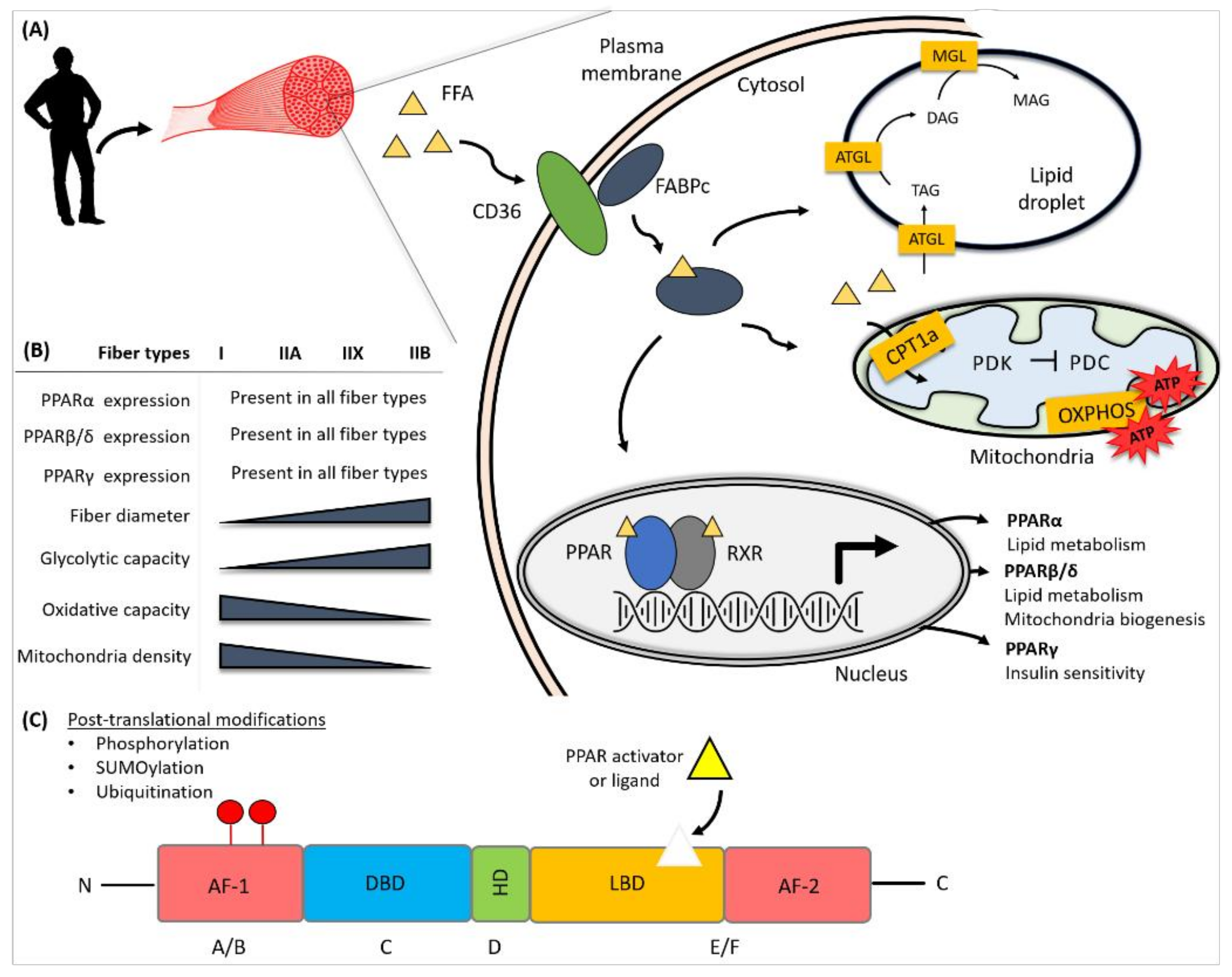
:1. Skeletal Muscle
2. Transcription Regulation by PPARs
3. Nutrient Sensing by PPARs
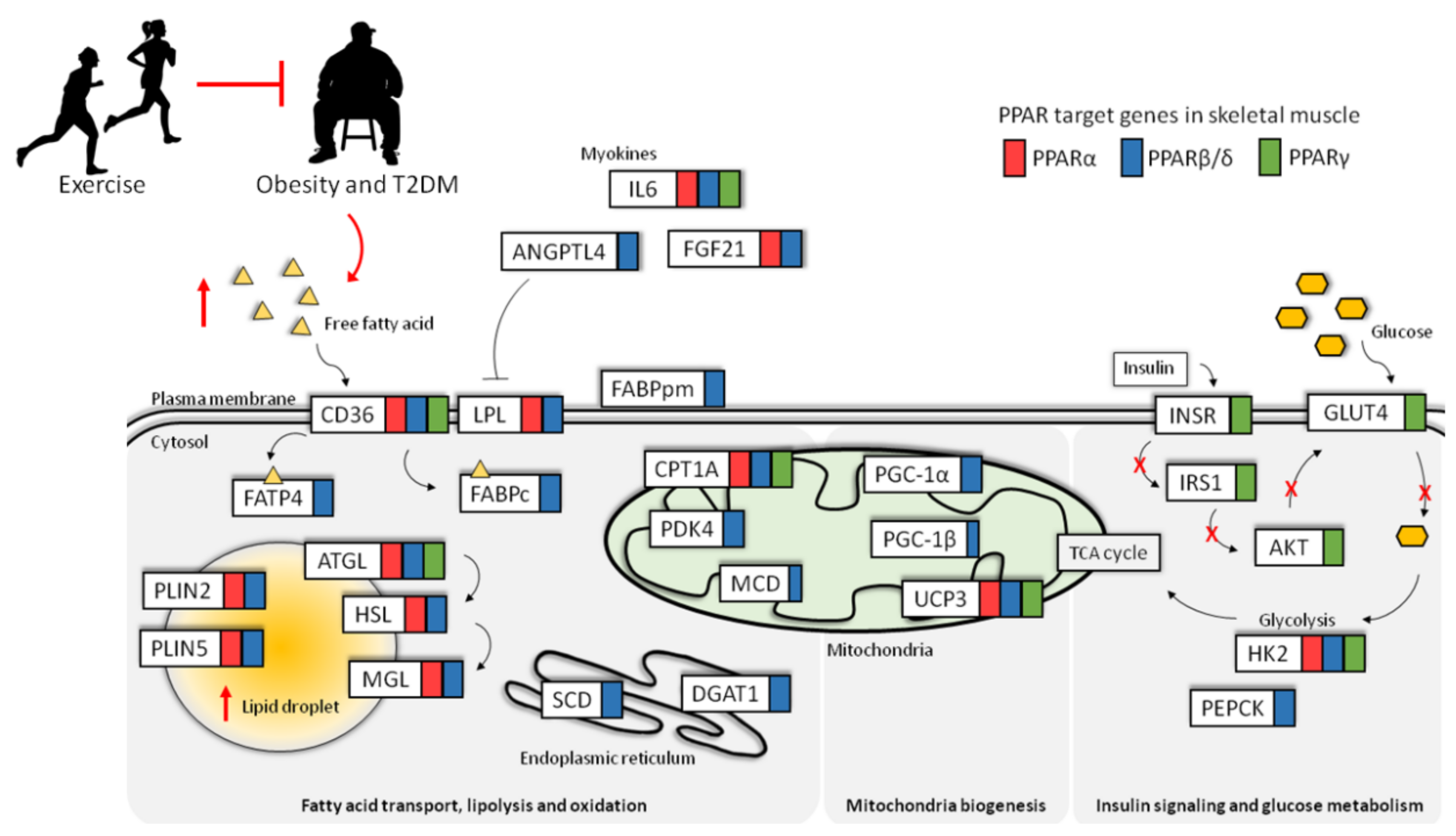
4. Regulation of Lipid Metabolism in Skeletal Muscle by PPARs
4.1. Regulation of Lipid Transport in Skeletal Muscle by PPARs
4.2. Regulation of Muscle Lipolysis by PPARs
4.3. Regulation of Muscle Lipid Storage by PPARs
5. Regulation of Mitochondrial Biogenesis and Function by PPARs
6. Dysregulation of Lipid Metabolism and PPAR during Insulin Resistance and T2DM
6.1. PPARγ Agonists and Insulin Resistance and T2DM Treatment
6.2. PPARα Agonists and Insulin Resistance and T2DM Treatment
6.3. Evidence for PPARβ/δ Agonist Treatment of Insulin Resistance and T2DM
7. Regulation of PPARs during Physical Exercise
8. Regulation of Skeletal Muscle Regeneration by PPARs
8.1. Roles of PPARβ/δ Regulation in Satellite Cells during Muscle Regeneration
8.2. PPAR-Regulated Paracrine Networks between Muscle and Other Cell Types
9. Regulation of PPARs during Aging
Evidence for the Involvement of PPARs during Aging
10. Concluding Remarks and Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| acetyl-CoA | acetyl-coenzyme A |
| AF1 | activation function 1 |
| AF2 | activation function 2 |
| AKT/PKB | protein kinase B |
| AMPK | AMP-activated protein kinase |
| ANGPTL4 | angiopoietin-like 4 |
| ATGL | adipose triglyceride lipase |
| BMI | body mass index |
| CD36 | cluster of differentiation 36/SR-B2 |
| CPT1 | carnitine palmitoyltransferase I |
| CRY1 | Cryptochrome 1 |
| DBD | DNA binding domain |
| DGAT1 | diacylglycerol acyltransferase 1 |
| ER | endoplasmic reticulum |
| FABP3 | fatty acid binding protein 3 |
| FATP | fatty acid transport protein |
| FATPc | cytoplasmic FABP |
| FATPpm | plasma membrane FABP |
| GLUT4 | glucose transporter 4 |
| HSL | hormone-sensitive lipase |
| IGFBP3 | insulin-like growth factor-binding protein 3 |
| IL6 | interleukin-6 |
| IMCL | intramyocellular lipid |
| IRS1 | insulin receptor substrate 1 |
| LBD | ligand binding domain |
| LD | lipid droplets |
| LPL | lipoprotein lipase |
| MAG | monoacylglycerol |
| MAPK | mitogen-activated protein kinase |
| MGL | monoacylglycerol lipase |
| NRF | nuclear respiratory factor |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase kinase |
| PGC-1 | PPARγ-coactivator 1 |
| PI3K | phosphatidylinositol-3 kinase |
| PKC | protein kinase C |
| PLIN | perilipin |
| PP2A | protein phosphatase 2A |
| PPAR | peroxisome proliferator-activated receptor |
| PPRE | peroxisome proliferator response element |
| RXR | retinoid X receptors |
| SCD | stearoyl-CoA desaturase |
| T2DM | type 2 diabetes mellitus |
| TA | tibialis anterior |
| TCA | tricarboxylic acid |
| TFAM | mitochondrial transcription factor A |
| TFB1M | mitochondrial transcription factors B1 |
| TFB2M | mitochondrial transcription factors B2 |
| TZD | Thiazolidinediones |
References
- McLeod, M.; Breen, L.; Hamilton, D.L.; Philp, A. Live strong and prosper: The importance of skeletal muscle strength for healthy ageing. Biogerontology 2016, 17, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, X.; Chen, Y.; Nie, Y.; Huang, H.; Chen, H.; Mo, D. Not all the number of skeletal muscle fibers is determined prenatally. BMC Dev. Biol. 2015, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.H.; Pedersen, A.J.; Birk, J.B.; Kristensen, D.E.; Vind, B.F.; Baba, O.; Nohr, J.; Hojlund, K.; Wojtaszewski, J.F. Human muscle fiber type-specific insulin signaling: Impact of obesity and type 2 diabetes. Diabetes 2015, 64, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Dube, J.J.; Coen, P.M.; DiStefano, G.; Chacon, A.C.; Helbling, N.L.; Desimone, M.E.; Stafanovic-Racic, M.; Hames, K.C.; Despines, A.A.; Toledo, F.G.; et al. Effects of acute lipid overload on skeletal muscle insulin resistance, metabolic flexibility, and mitochondrial performance. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1117–E1124. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Ehrenborg, E.; Krook, A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol. Rev. 2009, 61, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural peroxisome proliferator-activated receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Vazquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor pparbeta/delta. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspe, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human ppargamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Wahli, W. Peroxisome proliferator-activated receptors: Insight into multiple cellular functions. Mutat. Res. 2000, 448, 121–138. [Google Scholar] [CrossRef]
- Shalev, A.; Siegrist-Kaiser, C.A.; Yen, P.M.; Wahli, W.; Burger, A.G.; Chin, W.W.; Meier, C.A. The peroxisome proliferator-activated receptor alpha is a phosphoprotein: Regulation by insulin. Endocrinology 1996, 137, 4499–4502. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, C.; Barbier, O.; Fruchart, J.C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptor alpha (PPARα) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J. Biol. Chem. 2002, 277, 37254–37259. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Catapano, C.V. Block of nuclear receptor ubiquitination. A mechanism of ligand-dependent control of peroxisome proliferator-activated receptor delta activity. J. Biol. Chem. 2007, 282, 11776–11785. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Willis, M.S. The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and sumoylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.D.; Kriebs, A.; Vaughan, M.; Duglan, D.; Fan, W.; Henriksson, E.; Huber, A.L.; Papp, S.J.; Nguyen, M.; Afetian, M.; et al. Cry1/2 selectively repress PPARδ and limit exercise capacity. Cell Metab. 2017, 26, 243–255.e6. [Google Scholar] [CrossRef] [PubMed]
- Abbott, B.D. Review of the expression of peroxisome proliferator-activated receptors alpha (PPARα), beta (PPARβ), and gamma (PPARγ) in rodent and human development. Reprod. Toxicol. 2009, 27, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Lazar, M.A. The many faces of ppargamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.G.M.; Desvergne, B. Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl. Recept. Signal. 2015, 13, e001. [Google Scholar]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Ragni, M.; Silvestri, E.; Moreno, M.; Schiavo, L.; Lombardi, A.; Farina, P.; Feola, A.; Goglia, F.; Lanni, A. Combined cdna array/RT-PCR analysis of gene expression profile in rat gastrocnemius muscle: Relation to its adaptive function in energy metabolism during fasting. FASEB J. 2004, 18, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) α knock-out mice. Evidence for compensatory regulation by ppar delta. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Peeters, A.; Baes, M. Role of PPARα in hepatic carbohydrate metabolism. PPAR Res. 2010, 2010, 572405. [Google Scholar] [CrossRef] [PubMed]
- Kido, Y.; Nakae, J.; Accili, D. Clinical review 125: The insulin receptor and its cellular targets. J. Clin. Endocrinol. Metab. 2001, 86, 972–979. [Google Scholar] [PubMed]
- Foley, K.; Boguslavsky, S.; Klip, A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 2011, 50, 3048–3061. [Google Scholar] [CrossRef] [PubMed]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Hue, L.; Taegtmeyer, H. The randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J. Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R661–R668. [Google Scholar] [CrossRef] [PubMed]
- Spriet, L.L.; Tunstall, R.J.; Watt, M.J.; Mehan, K.A.; Hargreaves, M.; Cameron-Smith, D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. (1985) 2004, 96, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.; Saramaki, A.; Malinen, M.; Rieck, M.; Vaisanen, S.; Huotari, A.; Herzig, K.H.; Muller, R.; Carlberg, C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor β/δ. J. Mol. Biol. 2007, 372, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Nahle, Z.; Hsieh, M.; Pietka, T.; Coburn, C.T.; Grimaldi, P.A.; Zhang, M.Q.; Das, D.; Abumrad, N.A. CD36-dependent regulation of muscle foxo1 and PDK4 in the PPAR β/δ -mediated adaptation to metabolic stress. J. Biol. Chem. 2008, 283, 14317–14326. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Arner, P.; Yki-Jarvinen, H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem. 2006, 42, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Cha, B.S.; Park, K.S.; Carter, L.; Mudaliar, S.R.; Henry, R.R. Free fatty acid metabolism in human skeletal muscle is regulated by PPARγ and RXR agonists. Ann. N. Y. Acad. Sci. 2002, 967, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. Ppars and errs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator-activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Zhang, C.-L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. Pgc1alpha expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Harmon, C.M.; Luce, P.; Beth, A.H.; Abumrad, N.A. Labeling of adipocyte membranes by sulfo-n-succinimidyl derivatives of long-chain fatty acids: Inhibition of fatty acid transport. J. Membr. Biol. 1991, 121, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. From fat to fat (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Hoy, A.J. Lipid metabolism in skeletal muscle: Generation of adaptive and maladaptive intracellular signals for cellular function. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1315–E1328. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, J.G.; Alkhateeb, H.; Benton, C.R.; Lally, J.; Nickerson, J.; Han, X.X.; Wilson, M.H.; Jain, S.S.; Snook, L.A.; Glatz, J.F.; et al. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J. Biol. Chem. 2009, 284, 16522–16530. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P.; Lally, J.; Nickerson, J.G.; Alkhateeb, H.; Snook, L.A.; Heigenhauser, G.J.; Calles-Escandon, J.; Glatz, J.F.; Luiken, J.J.; Spriet, L.L.; et al. Fatty acid binding protein facilitates sarcolemmal fatty acid transport but not mitochondrial oxidation in rat and human skeletal muscle. J. Physiol. 2007, 582, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Thumser, A.E. Tissue-specific functions in the fatty acid-binding protein family. J. Biol. Chem. 2010, 285, 32679–32683. [Google Scholar] [CrossRef] [PubMed]
- Koonen, D.P.; Glatz, J.F.; Bonen, A.; Luiken, J.J. Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim. Biophys. Acta 2005, 1736, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Binas, B.; Danneberg, H.; van der Vusse, G.J.; Glatz, J.F. Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ. Res. 1999, 85, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Escriba, P.V.; Gonzalez-Ros, J.M.; Goni, F.M.; Kinnunen, P.K.; Vigh, L.; Sanchez-Magraner, L.; Fernandez, A.M.; Busquets, X.; Horvath, I.; Barcelo-Coblijn, G. Membranes: A meeting point for lipids, proteins and therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, W.J.; van der Luit, A.H.; Veldman, R.J.; Verheij, M.; Borst, J. Ceramide: Second messenger or modulator of membrane structure and dynamics? Biochem. J. 2003, 369, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.H.; Goswami, D.; Griffin, P.R.; Noy, N.; Ortlund, E.A. Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J. Biol. Chem. 2014, 289, 14941–14954. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Borrmann, C.M.; Borchers, T.; Spener, F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors α—and γ-mediated gene expression via liver fatty acid binding protein: A signaling path to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Ayers, S.D.; Nedrow, K.L.; Gillilan, R.E.; Noy, N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARγ by FABP4. Biochemistry 2007, 46, 6744–6752. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Shaw, N.S.; Vinckenbosch, N.; Liu, P.; Yasmin, R.; Desvergne, B.; Wahli, W.; Noy, N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 2002, 22, 5114–5127. [Google Scholar] [CrossRef] [PubMed]
- Bezaire, V.; Langin, D. Regulation of adipose tissue lipolysis revisited. Proc. Nutr. Soc. 2009, 68, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Alsted, T.J.; Ploug, T.; Prats, C.; Serup, A.K.; Hoeg, L.; Schjerling, P.; Holm, C.; Zimmermann, R.; Fledelius, C.; Galbo, H.; et al. Contraction-induced lipolysis is not impaired by inhibition of hormone-sensitive lipase in skeletal muscle. J. Physiol. 2013, 591, 5141–5155. [Google Scholar] [CrossRef] [PubMed]
- Gronke, S.; Mildner, A.; Fellert, S.; Tennagels, N.; Petry, S.; Muller, G.; Jackle, H.; Kuhnlein, R.P. Brummer lipase is an evolutionary conserved fat storage regulator in drosophila. Cell Metab. 2005, 1, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.; Ghosh, M.; Kumar, S.; Chakrabarti, P. PPARα-ATGL pathway improves muscle mitochondrial metabolism: Implication in aging. FASEB J. 2016, 30, 3822–3834. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via ppar-alpha and pgc-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Huijsman, E.; van de Par, C.; Economou, C.; van der Poel, C.; Lynch, G.S.; Schoiswohl, G.; Haemmerle, G.; Zechner, R.; Watt, M.J. Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E505–E513. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Mason, R.M.; Martin, S.D.; McGee, S.L.; Bruce, C.R.; Watt, M.J. ATGL-mediated triglyceride turnover and the regulation of mitochondrial capacity in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E960–E970. [Google Scholar] [CrossRef] [PubMed]
- Jocken, J.W.; Smit, E.; Goossens, G.H.; Essers, Y.P.; van Baak, M.A.; Mensink, M.; Saris, W.H.; Blaak, E.E. Adipose triglyceride lipase (ATGL) expression in human skeletal muscle is type I (oxidative) fiber specific. Histochem. Cell Biol. 2008, 129, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Yang, H.; Wang, S.P.; Soni, K.G.; Brunel-Guitton, C.; Mitchell, G.A. Inborn errors of cytoplasmic triglyceride metabolism. J. Inherit. Metab. Dis. 2015, 38, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, T.; Havekes, B.; Bilet, L.; Hoeks, J.; Sparks, L.; Bosma, M.; Paglialunga, S.; Jorgensen, J.; Janssen, M.C.; Schaart, G.; et al. Effects of bezafibrate treatment in a patient and a carrier with mutations in the PNPLA2 gene, causing neutral lipid storage disease with myopathy. Circ. Res. 2013, 112, e51–e54. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.J.; Samjoo, I.A.; Devries, M.C.; Stevic, I.; Robertshaw, H.A.; Tarnopolsky, M.A. Perilipin family (PLIN) proteins in human skeletal muscle: The effect of sex, obesity, and endurance training. Appl. Physiol. Nutr. Metab. 2012, 37, 724–735. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, J.; Smit, E.; Snepvangers, F.J.; de Wit, N.W.; Mohren, R.; Hulshof, M.F.; Mariman, E.C. Adipophilin protein expression in muscle—A possible protective role against insulin resistance. FEBS J. 2010, 277, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.Z.; Nikolic, N.; Bakke, S.S.; Boekschoten, M.V.; Kersten, S.; Kase, E.T.; Rustan, A.C.; Thoresen, G.H. PPARδ activation in human myotubes increases mitochondrial fatty acid oxidative capacity and reduces glucose utilization by a switch in substrate preference. Arch. Physiol. Biochem. 2014, 120, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARδ agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Bindesboll, C.; Berg, O.; Arntsen, B.; Nebb, H.I.; Dalen, K.T. Fatty acids regulate perilipin5 in muscle by activating ppardelta. J. Lipid Res. 2013, 54, 1949–1963. [Google Scholar] [CrossRef] [PubMed]
- Minnaard, R.; Schrauwen, P.; Schaart, G.; Jorgensen, J.A.; Lenaers, E.; Mensink, M.; Hesselink, M.K. Adipocyte differentiation-related protein and OXPAT in rat and human skeletal muscle: Involvement in lipid accumulation and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2009, 94, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.S.; Sherlock, M.; Stewart, P.M.; Wagenmakers, A.J. Adipophilin distribution and colocalization with lipid droplets in skeletal muscle. Histochem. Cell Biol. 2009, 131, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.A.; Skinner, J.R.; Shew, T.M.; Pietka, T.A.; Abumrad, N.A.; Wolins, N.E. Perilipin 5-driven lipid droplet accumulation in skeletal muscle stimulates the expression of fibroblast growth factor 21. Diabetes 2015, 64, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C.; Nebb, H.I. LSDP5 is a pat protein specifically expressed in fatty acid oxidizing tissues. Biochim. Biophys. Acta 2007, 1771, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C.; Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Kern, P.A.; et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, S.; Nguyen-Tran, V.; Bare, O.; Huang, X.; Spiegelman, B.; Wu, Z. PPARδ agonism activates fatty acid oxidation via PGC-1α but does not increase mitochondrial gene expression and function. J. Biol. Chem. 2009, 284, 18624–18633. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Pineda-Torra, I.; Iglesias, R.; Staels, B.; Villarroya, F.; Giralt, M. Ppardelta, but not PPARα, activates PGC-1alpha gene transcription in muscle. Biochem. Biophys. Res. Commun. 2007, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Hancock, C.R.; Han, D.H.; Chen, M.; Terada, S.; Yasuda, T.; Wright, D.C.; Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 7815–7820. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1 α expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Lebrasseur, N.; Morris, C.; Smith, E.; Yang, W.; Ma, Y.; Chin, S.; Spiegelman, B.M. The transcriptional coactivator PGC-1β drives the formation of oxidative type IIx fibers in skeletal muscle. Cell Metab. 2007, 5, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.H. PPARβ is essential for maintaining normal levels of PGC-1alpha and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef] [PubMed]
- Gemmink, A.; Goodpaster, B.H.; Schrauwen, P.; Hesselink, M.K.C. Intramyocellular lipid droplets and insulin sensitivity, the human perspective. Biochim. Biophys. Acta 2017, 1862, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Zhang, X.; Yi, Z.; Cichello, S. Skeletal intramyocellular lipid metabolism and insulin resistance. Biophys. Rep. 2015, 1, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1 h NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Szczepaniak, L.S.; Bentley, B.; Esser, V.; Myhill, J.; McGarry, J.D. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes 2001, 50, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Machann, J.; Rett, K.; Brechtel, K.; Volk, A.; Renn, W.; Maerker, E.; Matthaei, S.; Schick, F.; Claussen, C.D.; et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 1999, 48, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 2001, 50, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated pkb pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55α regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ promotes running endurance by preserving glucose. Cell Metab. 2017, 25, 1186–1193.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Chen, N.; Shi, X.; Tsang, B.; Yu, Y.H. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Investig. 2007, 117, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Dallas-Yang, Q.; Li, Z.; Szalkowski, D.; Liu, F.; Shen, X.; Wu, M.; Zhou, G.; Doebber, T.; Berger, J.; et al. Potentiation of insulin signaling in tissues of zucker obese rats after acute and long-term treatment with PPARγ agonists. Diabetes 2002, 51, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Ciaraldi, T.P.; Kong, A.; Kim, D.; Chu, N.; Mohideen, P.; Mudaliar, S.; Henry, R.R.; Kahn, B.B. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases P110β protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 2002, 51, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; He, H.; Mandarino, L.J.; DeFronzo, R.A. Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes 2003, 52, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Wiley, S.E.; Rogers, G.W.; Andreyev, A.Y.; Petrosyan, S.; Loviscach, M.; Wall, E.A.; Yadava, N.; Heuck, A.P.; Ferrick, D.A.; et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA 2013, 110, 5422–5427. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Vigueira, P.A.; Chambers, K.T.; Hall, A.M.; Mitra, M.S.; Qi, N.; McDonald, W.G.; Colca, J.R.; Kletzien, R.F.; Finck, B.N. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor γ-sparing thiazolidinedione. J. Biol. Chem. 2012, 287, 23537–23548. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Ciaraldi, T.P.; Abrams-Carter, L.; Mudaliar, S.; Nikoulina, S.E.; Henry, R.R. PPAR-γ gene expression is elevated in skeletal muscle of obese and type II diabetic subjects. Diabetes 1997, 46, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Marin-Juez, R.; Diaz, M.; Morata, J.; Planas, J.V. Mechanisms regulating GLUT4 transcription in skeletal muscle cells are highly conserved across vertebrates. PLoS ONE 2013, 8, e80628. [Google Scholar] [CrossRef] [PubMed]
- Hammarstedt, A.; Smith, U. Thiazolidinediones (PPARγ ligands) increase IRS-1, UCP-2 and C/EBPα expression, but not transdifferentiation, in l6 muscle cells. Diabetologia 2003, 46, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.H.; Mathews, S.T.; Camp, H.S.; Ding, L.; Leff, T. Selective activation of PPARγ in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E28–E37. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific PPARG deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.W.; Chen, L.; Fisher, S.J.; Szanto, I.; Ristow, M.; Jozsi, A.C.; Hirshman, M.F.; Rosen, E.D.; Goodyear, L.J.; Gonzalez, F.J.; et al. Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Investig. 2003, 112, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Muller-Wieland, D.; Pfeiffer, A.; Krone, W.; Kahn, C.R. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N. Engl. J. Med. 1998, 339, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A PRO12ALA substitution in PPARγ 2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.C.; Nemesh, J.; Lane, C.R.; Schaffner, S.F.; Bolk, S.; Brewer, C.; et al. The common PPARγ PRO12ALA polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000, 26, 76–80. [Google Scholar] [PubMed]
- Jones, P.H. Chapter 26—fibrates A2—Ballantyne, christie m. In Clinical Lipidology; W.B. Saunders: Philadelphia, PA, USA, 2009; pp. 315–325. [Google Scholar]
- Koh, K.K.; Han, S.H.; Quon, M.J.; Yeal Ahn, J.; Shin, E.K. Beneficial effects of fenofibrate to improve endothelial dysfunction and raise adiponectin levels in patients with primary hypertriglyceridemia. Diabetes Care 2005, 28, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Saitoh, Y.; Mizuta, M.; Shiiya, T.; Noma, K.; Mashiba, S.; Kojima, S.; Nakazato, M. Fenofibrate ameliorates insulin resistance, hypertension and novel oxidative stress markers in patients with metabolic syndrome. Obes. Res. Clin. Pract. 2011, 5, e267–e360. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Huypens, P.; Neschen, S.; Irmler, M.; Rozman, J.; Rathkolb, B.; Neff, F.; Prehn, C.; Dubois, G.; Baumann, M.; et al. Bezafibrate improves insulin sensitivity and metabolic flexibility in STZ-induced diabetic mice. Diabetes 2016, 65, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. Mbx-8025, a novel peroxisome proliferator receptor-δ agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.P.; Fisher, J.S. Skeletal muscle insulin resistance: Roles of fatty acid metabolism and exercise. Phys. Ther. 2008, 88, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.P.; Fluckey, J.D.; Lambert, B.S.; Greene, E.S.; Riechman, S.E.; Crouse, S.F. Regulators of blood lipids and lipoproteins? PPARδ and AMPK, induced by exercise, are correlated with lipids and lipoproteins in overweight/obese men and women. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1212–E1221. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.B.; Yoshioka, J. Regulating PPARδ signaling as a potential therapeutic strategy for skeletal muscle disorders in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H967–H969. [Google Scholar] [CrossRef] [PubMed]
- Lagu, B.; Kluge, A.F.; Fredenburg, R.A.; Tozzo, E.; Senaiar, R.S.; Jaleel, M.; Panigrahi, S.K.; Tiwari, N.K.; Krishnamurthy, N.R.; Takahashi, T.; et al. Novel highly selective peroxisome proliferator-activated receptor delta (PPARδ) modulators with pharmacokinetic properties suitable for once-daily oral dosing. Bioorg. Med. Chem. Lett. 2017, 27, 5230–5234. [Google Scholar] [CrossRef] [PubMed]
- Lagu, B.; Kluge, A.F.; Goddeeris, M.M.; Tozzo, E.; Fredenburg, R.A.; Chellur, S.; Senaiar, R.S.; Jaleel, M.; Babu, D.R.K.; Tiwari, N.K.; et al. Highly selective peroxisome proliferator-activated receptor delta (PPARδ) modulator demonstrates improved safety profile compared to GW501516. Bioorg. Med. Chem. Lett. 2018, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Feilchenfeldt, J.; Schreiber, S.; Praz, M.; Crettenand, A.; Gobelet, C.; Meier, C.A.; Bell, D.R.; Kralli, A.; Giacobino, J.P.; et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-γ coactivator-1 and peroxisome proliferator-activated receptor-α in skeletal muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.K.; Ahlsen, M.; Norrbom, J.; Jansson, E.; Hjeltnes, N.; Gustafsson, T.; Krook, A. Human skeletal muscle fibre type variations correlate with PPARα, PPARδ and PGC-1α MRNA. Acta Physiol. (Oxf.) 2006, 188, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Tuvblad, C.; Forero, D.A. Sports genetics: The PPARA gene and athletes’ high ability in endurance sports. A systematic review and meta-analysis. Biol. Sport 2016, 33, 3–6. [Google Scholar] [PubMed]
- Ahmetov, I.I.; Mozhayskaya, I.A.; Flavell, D.M.; Astratenkova, I.V.; Komkova, A.I.; Lyubaeva, E.V.; Tarakin, P.P.; Shenkman, B.S.; Vdovina, A.B.; Netreba, A.I.; et al. PPARα gene variation and physical performance in russian athletes. Eur. J. Appl. Physiol. 2006, 97, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska-Karlowska, A.; Sawczuk, M.; Cieszczyk, P.; Zarebska, A.; Sawczyn, S. Association between the PRO12ALA polymorphism of the peroxisome proliferator-activated receptor gamma gene and strength athlete status. PLoS ONE 2013, 8, e67172. [Google Scholar] [CrossRef] [PubMed]
- Huard, J.; Li, Y.; Fu, F.H. Muscle injuries and repair: Current trends in research. J. Bone Jt. Surg. Am. 2002, 84-A, 822–832. [Google Scholar] [CrossRef]
- Jarvinen, T.A.; Jarvinen, T.L.; Kaariainen, M.; Kalimo, H.; Jarvinen, M. Muscle injuries: Biology and treatment. Am. J. Sports Med. 2005, 33, 745–764. [Google Scholar] [CrossRef] [PubMed]
- Delos, D.; Maak, T.G.; Rodeo, S.A. Muscle injuries in athletes: Enhancing recovery through scientific understanding and novel therapies. Sports Health 2013, 5, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Jude, E.B.; Eleftheriadou, I.; Tentolouris, N. Peripheral arterial disease in diabetes—A review. Diabet. Med. 2010, 27, 4–14. [Google Scholar] [CrossRef] [PubMed]
- St Pierre, B.A.; Tidball, J.G. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J. Appl. Physiol. (1985) 1994, 77, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Chazaud, B.; Sonnet, C.; Lafuste, P.; Bassez, G.; Rimaniol, A.C.; Poron, F.; Authier, F.J.; Dreyfus, P.A.; Gherardi, R.K. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 2003, 163, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N.; Bassel-Duby, R. Skeletal muscle remodeling. Curr. Opin. Rheumatol. 2007, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Hoier, B.; Hellsten, Y. Exercise-induced capillary growth in human skeletal muscle and the dynamics of VEGF. Microcirculation 2014, 21, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.C.; Copp, S.W.; Ferguson, S.K.; Musch, T.I. Skeletal muscle capillary function: Contemporary observations and novel hypotheses. Exp. Physiol. 2013, 98, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. All muscle satellite cells are equal, but are some more equal than others? J. Cell Sci. 2008, 121, 2975–2982. [Google Scholar] [CrossRef] [PubMed]
- Angione, A.R.; Jiang, C.; Pan, D.; Wang, Y.X.; Kuang, S. PPARδ regulates satellite cell proliferation and skeletal muscle regeneration. Skelet Muscle 2011, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, P.; Manickam, R.; Ge, X.; Bonala, S.; McFarlane, C.; Sharma, M.; Wahli, W.; Kambadur, R. Inactivation of PPARβ/δ adversely affects satellite cells and reduces postnatal myogenesis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E122–E131. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Arauzo-Bravo, M.J.; Kovacs, W.J.; Karalay, O.; Suter, U.; Machado, R.A.; et al. Metabolic control of adult neural stem cell activity by FASN-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Chakkalakal, J.V.; Boudreault, L.; Belanger, G.; Hebert, R.L.; Renaud, J.M.; Jasmin, B.J. Pharmacological activation of PPARβ/δ stimulates utrophin a expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum. Mol. Genet. 2009, 18, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Mounier, R.; Patsalos, A.; Gogolak, P.; Peloquin, M.; Horvath, A.; Pap, A.; Daniel, B.; Nagy, G.; Pintye, E.; et al. Macrophage PPARγ, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity 2016, 45, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Latroche, C.; Weiss-Gayet, M.; Muller, L.; Gitiaux, C.; Leblanc, P.; Liot, S.; Ben-Larbi, S.; Abou-Khalil, R.; Verger, N.; Bardot, P.; et al. Coupling between myogenesis and angiogenesis during skeletal muscle regeneration is stimulated by restorative macrophages. Stem Cell Rep. 2017, 9, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Latroche, C.; Gitiaux, C.; Chretien, F.; Desguerre, I.; Mounier, R.; Chazaud, B. Skeletal muscle microvasculature: A highly dynamic lifeline. Physiology (Bethesda) 2015, 30, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Han, J.K.; Kim, H.L.; Jeon, K.H.; Choi, Y.E.; Lee, H.S.; Kwon, Y.W.; Jang, J.J.; Cho, H.J.; Kang, H.J.; Oh, B.H.; et al. Peroxisome proliferator-activated receptor-delta activates endothelial progenitor cells to induce angio-myogenesis through matrix metallo-proteinase-9-mediated insulin-like growth factor-1 paracrine networks. Eur. Heart J. 2013, 34, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Nwadozi, E. Regulation of skeletal muscle capillary growth in exercise and disease. Appl. Physiol. Nutr. Metab. 2015, 40, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Hwa, V.; Oh, Y.; Rosenfeld, R.G. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sweeney, G. Adiponectin action in skeletal muscle. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 2005, 48, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; McAinch, A.J.; Macaulay, S.L.; Castelli, L.A.; O’Brien P, E.; Dixon, J.B.; Cameron-Smith, D.; Kemp, B.E.; Steinberg, G.R. Impaired activation of AMP-kinase and fatty acid oxidation by globular adiponectin in cultured human skeletal muscle of obese type 2 diabetics. J. Clin. Endocrinol. Metab. 2005, 90, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Kinney, B.; Yoo, H.S.; Lee, B.; Schaack, J.; Shao, J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes 2012, 61, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Vavvas, D.; Apazidis, A.; Saha, A.K.; Gamble, J.; Patel, A.; Kemp, B.E.; Witters, L.A.; Ruderman, N.B. Contraction-induced changes in acetyl-CoA carboxylase and 5′-amp-activated kinase in skeletal muscle. J. Biol. Chem. 1997, 272, 13255–13261. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.K.; Schwarsin, A.J.; Roduit, R.; Masse, F.; Kaushik, V.; Tornheim, K.; Prentki, M.; Ruderman, N.B. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside. J. Biol. Chem. 2000, 275, 24279–24283. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K.; Nagaretani, H.; Matsuda, M.; Komuro, R.; Ouchi, N.; et al. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chewchuk, S.; Lavigne, C.; Brule, S.; Pilon, G.; Houde, V.; Xu, A.; Marette, A.; Sweeney, G. Functional significance of skeletal muscle adiponectin production, changes in animal models of obesity and diabetes, and regulation by rosiglitazone treatment. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E657–E664. [Google Scholar] [CrossRef] [PubMed]
- Erol, A. The functions of PPARs in aging and longevity. PPAR Res. 2007, 2007, 39654. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.; Engelhardt, M. Strength and muscle mass loss with aging process. Age and strength loss. Muscles Ligaments Tendons J. 2013, 3, 346–350. [Google Scholar] [PubMed]
- Miljkovic, N.; Lim, J.Y.; Miljkovic, I.; Frontera, W.R. Aging of skeletal muscle fibers. Ann. Rehabil. Med. 2015, 39, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Karlsson, J. Isometric and dynamic endurance as a function of age and skeletal muscle characteristics. Acta Physiol. Scand. 1978, 104, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.C.; Taylor, J.L. Age-related changes in motor cortical properties and voluntary activation of skeletal muscle. Curr. Aging Sci. 2011, 4, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, L.B.; Koopman, R.; Schaart, G.; Meijer, K.; Savelberg, H.H.; van Loon, L.J. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E151–E157. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Suh, D.; Krivickas, L.S.; Hughes, V.A.; Goldstein, R.; Roubenoff, R. Skeletal muscle fiber quality in older men and women. Am. J. Physiol. Cell Physiol. 2000, 279, C611–C618. [Google Scholar] [CrossRef] [PubMed]
- Atherton, H.J.; Gulston, M.K.; Bailey, N.J.; Cheng, K.K.; Zhang, W.; Clarke, K.; Griffin, J.L. Metabolomics of the interaction between PPAR-α and age in the PPAR-α-null mouse. Mol. Syst. Biol. 2009, 5, 259. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Rousseau, A.S.; Wagner, N.; Gaudel, C.; Murdaca, J.; Jehl-Pietri, C.; Sibille, B.; Grimaldi, P.A.; Lopez, P. Peroxisome proliferator-activated receptor beta activation promotes myonuclear accretion in skeletal muscle of adult and aged mice. Pflugers Arch. 2009, 458, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Zhang, X.J.; Wang, Z.J.; Zhang, C. Effect of aging on the expression of peroxisome proliferator-activated receptor γ and the possible relation to insulin resistance. Gerontology 2006, 52, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.J.; Tchernof, A. Lipid metabolism in the elderly. Eur. J. Clin. Nutr. 2000, 54 (Suppl. 3), 121S–S125. [Google Scholar] [CrossRef]
- Johannsen, D.L.; Conley, K.E.; Bajpeyi, S.; Punyanitya, M.; Gallagher, D.; Zhang, Z.; Covington, J.; Smith, S.R.; Ravussin, E. Ectopic lipid accumulation and reduced glucose tolerance in elderly adults are accompanied by altered skeletal muscle mitochondrial activity. J. Clin. Endocrinol. Metab. 2012, 97, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Gaudel, C.; Schwartz, C.; Giordano, C.; Abumrad, N.A.; Grimaldi, P.A. Pharmacological activation of PPARβ promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E297–E304. [Google Scholar] [CrossRef] [PubMed]
- Hodel, C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol. Lett. 2002, 128, 159–168. [Google Scholar] [CrossRef]
- Burri, L.; Thoresen, G.H.; Berge, R.K. The role of pparalpha activation in liver and muscle. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Faiola, B.; Falls, J.G.; Peterson, R.A.; Bordelon, N.R.; Brodie, T.A.; Cummings, C.A.; Romach, E.H.; Miller, R.T. PPARα, more than PPARδ, mediates the hepatic and skeletal muscle alterations induced by the PPAR agonist GW0742. Toxicol. Sci. 2008, 105, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Guillet, C.; Boirie, Y. Insulin resistance: A contributing factor to age-related muscle mass loss? Diabetes Metab. 2005, 31, 5S20–5S26. [Google Scholar] [CrossRef]
- Morley, J.E. Hormones and the aging process. J. Am. Geriatr. Soc. 2003, 51, S333–S337. [Google Scholar] [CrossRef]
- Houmard, J.A.; Weidner, M.D.; Dolan, P.L.; Leggett-Frazier, N.; Gavigan, K.E.; Hickey, M.S.; Tyndall, G.L.; Zheng, D.; Alshami, A.; Dohm, G.L. Skeletal muscle GLUT4 protein concentration and aging in humans. Diabetes 1995, 44, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Lai, Y.M.; Ryan, K.K. Ppargamma and stress: Implications for aging. Exp. Gerontol. 2013, 48, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Investigators, D.T.; Gerstein, H.C.; Yusuf, S.; Bosch, J.; Pogue, J.; Sheridan, P.; Dinccag, N.; Hanefeld, M.; Hoogwerf, B.; Laakso, M.; et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: A randomised controlled trial. Lancet 2006, 368, 1096–1105. [Google Scholar]
- Nolan, J.J.; Ludvik, B.; Beerdsen, P.; Joyce, M.; Olefsky, J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 1994, 331, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Sanguino, E.; Roglans, N.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Different response of senescent female Sprague-Dawley rats to gemfibrozil and rosiglitazone administration. Exp. Gerontol. 2005, 40, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-γ deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Miles, P.D.; Barak, Y.; Evans, R.M.; Olefsky, J.M. Effect of heterozygous PPARγ deficiency and TZD treatment on insulin resistance associated with age and high-fat feeding. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E618–E626. [Google Scholar] [CrossRef] [PubMed]
- Stephens, F.B.; Tsintzas, K. Metabolic and molecular changes associated with the increased skeletal muscle insulin action 24–48 h after exercise in young and old humans. Biochem. Soc. Trans. 2018, 46, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Post, S.M.; Duez, H.; Gervois, P.P.; Staels, B.; Kuipers, F.; Princen, H.M. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-α-mediated downregulation of cholesterol 7α-hydroxylase and sterol 27-hydroxylase expression. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1840–1845. [Google Scholar] [CrossRef] [PubMed]
- Kostapanos, M.S.; Florentin, M.; Elisaf, M.S. Fenofibrate and the kidney: An overview. Eur J. Clin Investig. 2013, 43, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.M.; Kwok, C.S.; Chen-Turner, C.; Maduakor, C.A.; Singh, S.; Loke, Y.K. Thiazolidinediones and associated risk of bladder cancer: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2014, 78, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Loke, Y.K.; Furberg, C.D. Thiazolidinediones and heart failure: A teleo-analysis. Diabetes Care 2007, 30, 2148–2153. [Google Scholar] [CrossRef] [PubMed]
- Geiger, L.N.; Dunsford, W.S.; Lewis, D.J.; Brennan, C.; Liu, K.C.; Newsholme, S.J. Rat Carcinogenicity Study with gw501516, A Ppar Delta Agonist. Toxicol. Sci. 2009, 108, 895. [Google Scholar]
- Catoire, M.; Alex, S.; Paraskevopulos, N.; Mattijssen, F.; Evers-van Gogh, I.; Schaart, G.; Jeppesen, J.; Kneppers, A.; Mensink, M.; Voshol, P.J.; et al. Fatty acid-inducible anGPTL4 governs lipid metabolic response to exercise. Proc. Natl. Acad. Sci. USA 2014, 111, E1043–E1052. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Lichtenstein, L.; Steenbergen, E.; Mudde, K.; Hendriks, H.F.; Hesselink, M.K.; Schrauwen, P.; Muller, M. Caloric restriction and exercise increase plasma ANGPTl4 levels in humans via elevated free fatty acids. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Weihrauch, M.; Handschin, C. Pharmacological targeting of exercise adaptations in skeletal muscle: Benefits and pitfalls. Biochem. Pharmacol. 2018, 147, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.C.; Feng, D.D.; Chen, C.; Lee, H.G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef] [PubMed]
- Al-Lahham, S.H.; Peppelenbosch, M.P.; Roelofsen, H.; Vonk, R.J.; Venema, K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim. Biophys. Acta 2010, 1801, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Bengmark, S. Gut microbiota, immune development and function. Pharmacol. Res. 2013, 69, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Diao, H.; Xiao, Y.; Li, W.; Yu, B.; He, J.; Yu, J.; Zheng, P.; Mao, X.; Luo, Y.; et al. Gut microbiota can transfer fiber characteristics and lipid metabolic profiles of skeletal muscle from pigs to germ-free mice. Sci. Rep. 2016, 6, 31786. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2018, 19, 1425. https://doi.org/10.3390/ijms19051425
Phua WWT, Wong MXY, Liao Z, Tan NS. An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. International Journal of Molecular Sciences. 2018; 19(5):1425. https://doi.org/10.3390/ijms19051425
Chicago/Turabian StylePhua, Wendy Wen Ting, Melissa Xin Yu Wong, Zehuan Liao, and Nguan Soon Tan. 2018. "An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors" International Journal of Molecular Sciences 19, no. 5: 1425. https://doi.org/10.3390/ijms19051425