iTRAQ-Based Proteomics Analyses of Sterile/Fertile Anthers from a Thermo-Sensitive Cytoplasmic Male-Sterile Wheat with Aegilops kotschyi Cytoplasm

Abstract
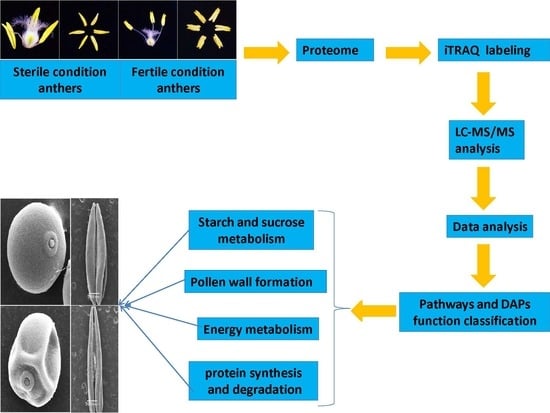
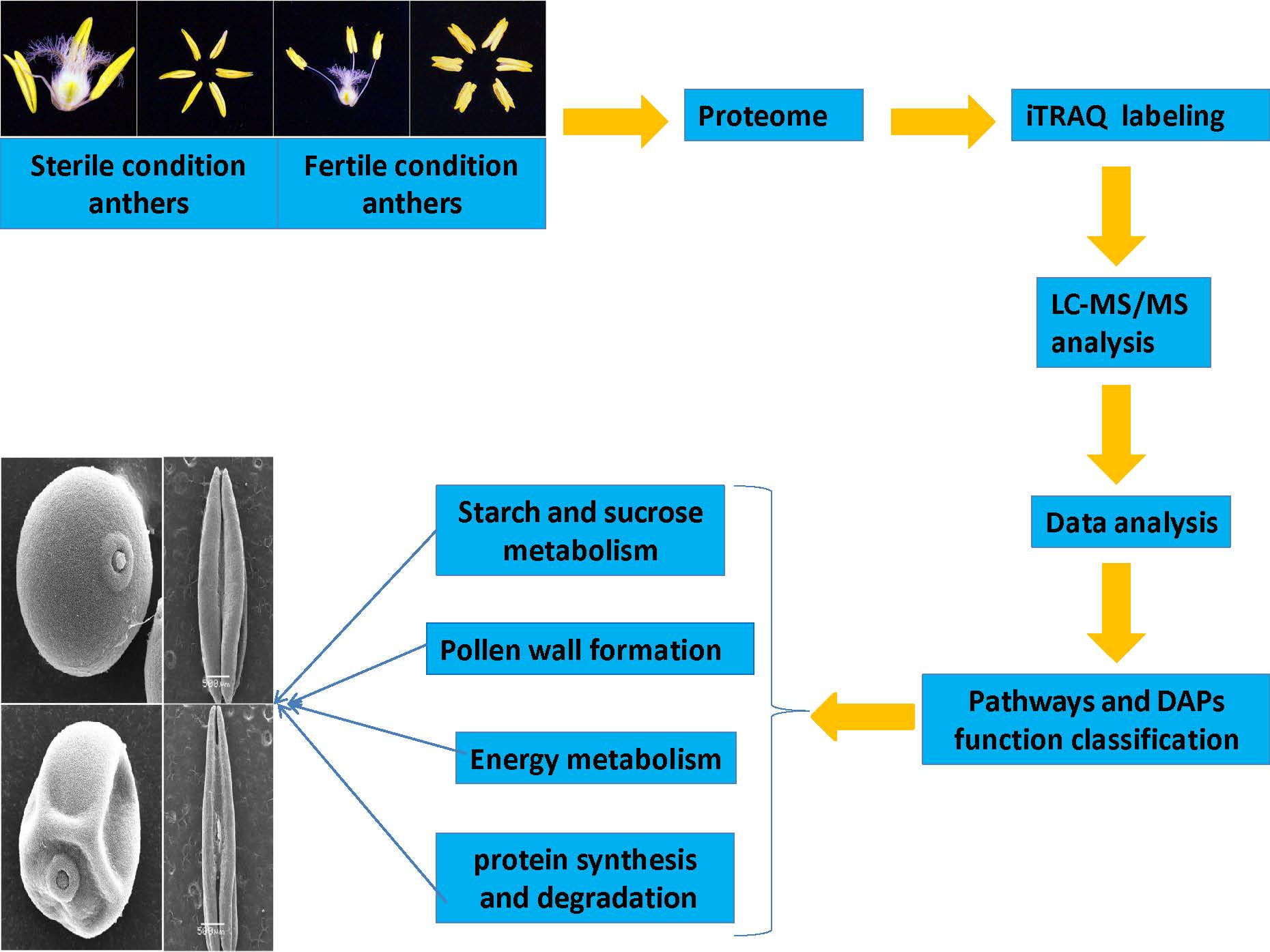
:
1. Introduction
2. Results
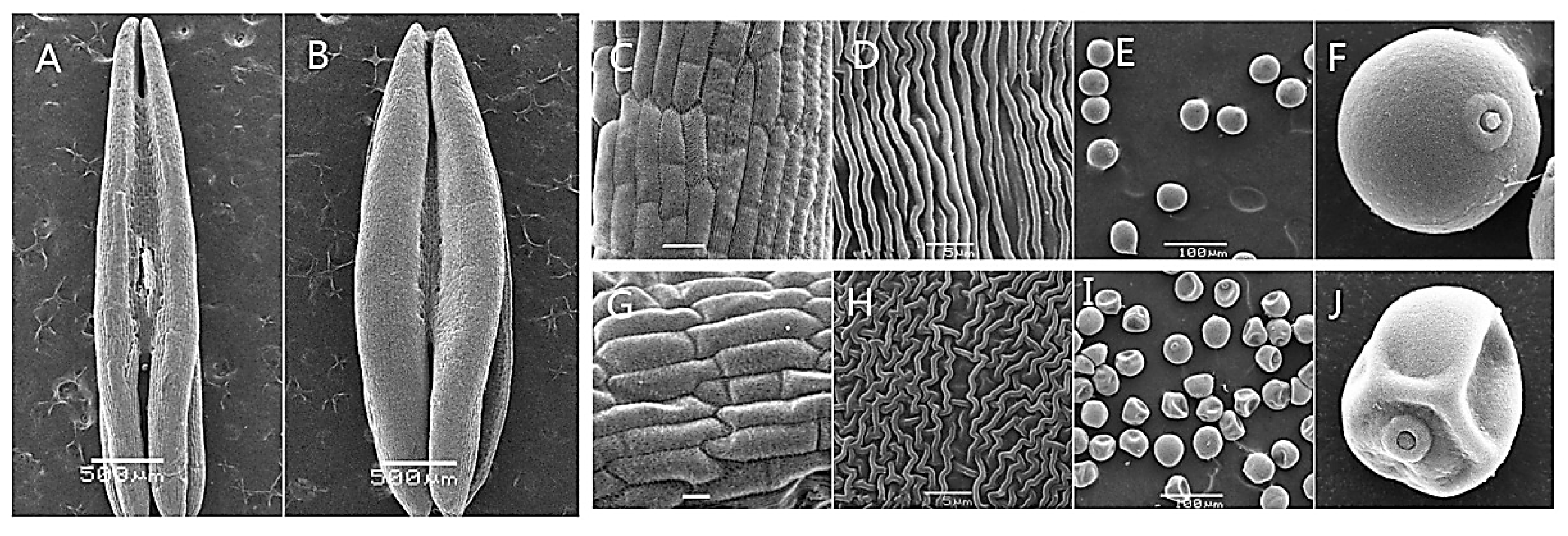
2.1. Anther Development and Phenotypic Characterization
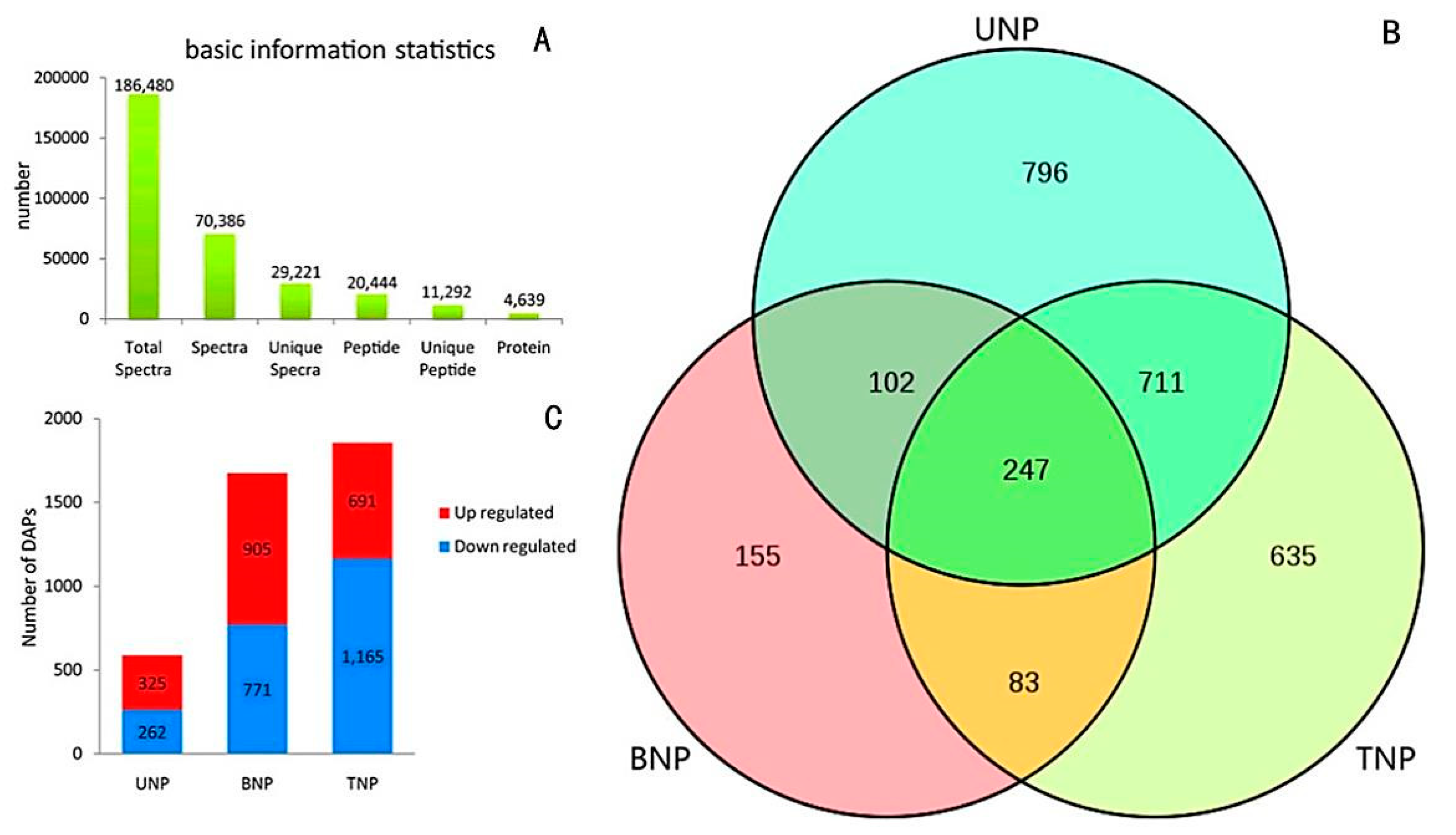
2.2. Assessment of Sequencing Results and Workflow Followed to Obtain Anther Proteomes
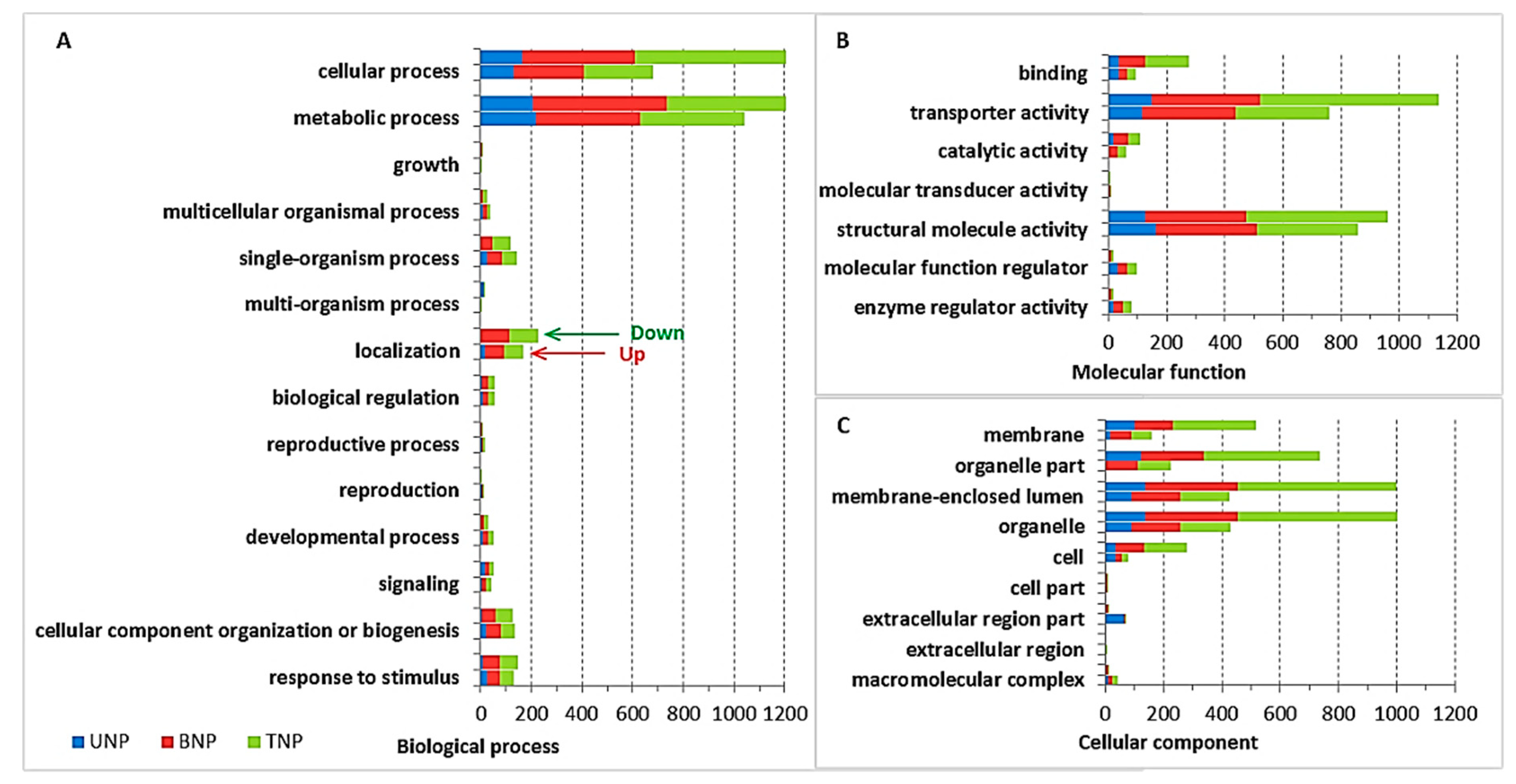
2.3. Gene Ontology (GO) Enrichment Analyses of the DAPs
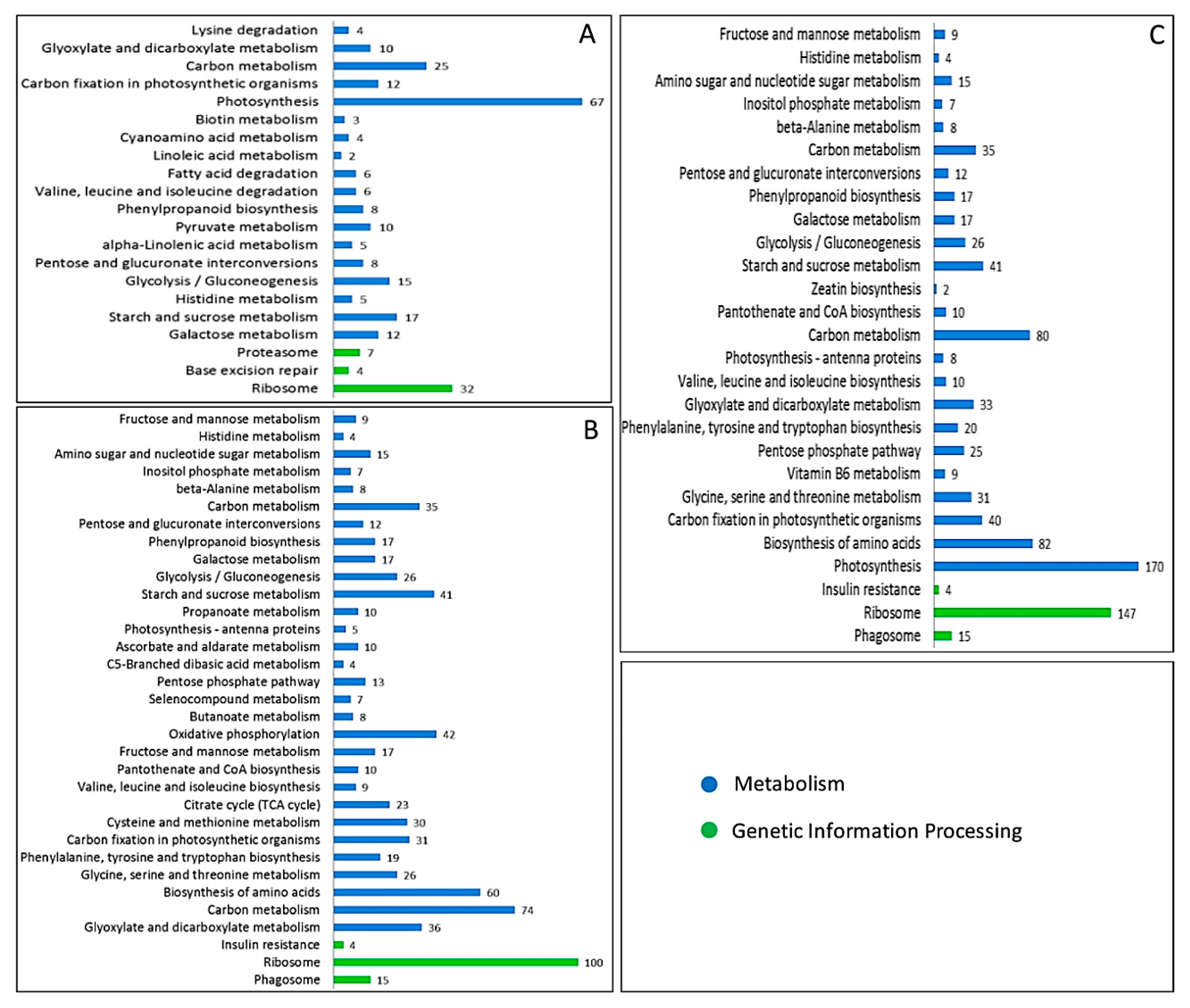
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DAPs
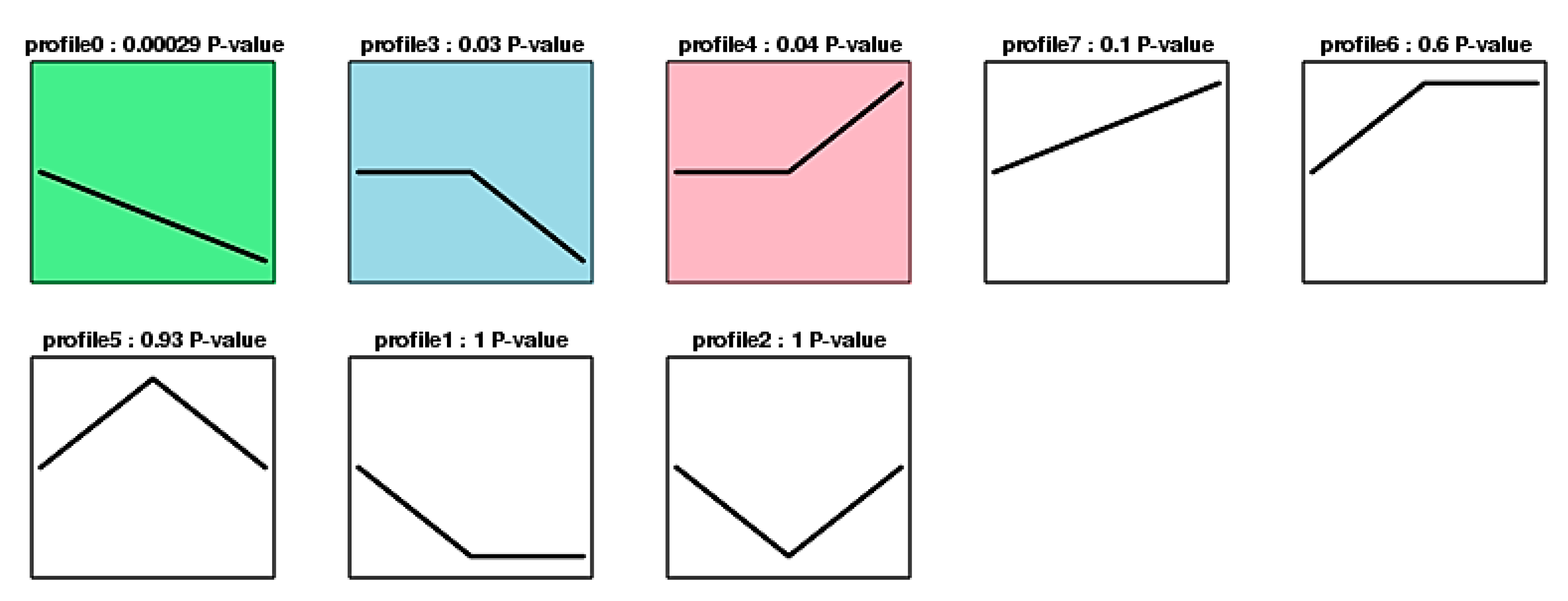
2.5. Analysis of Trends in DAPs
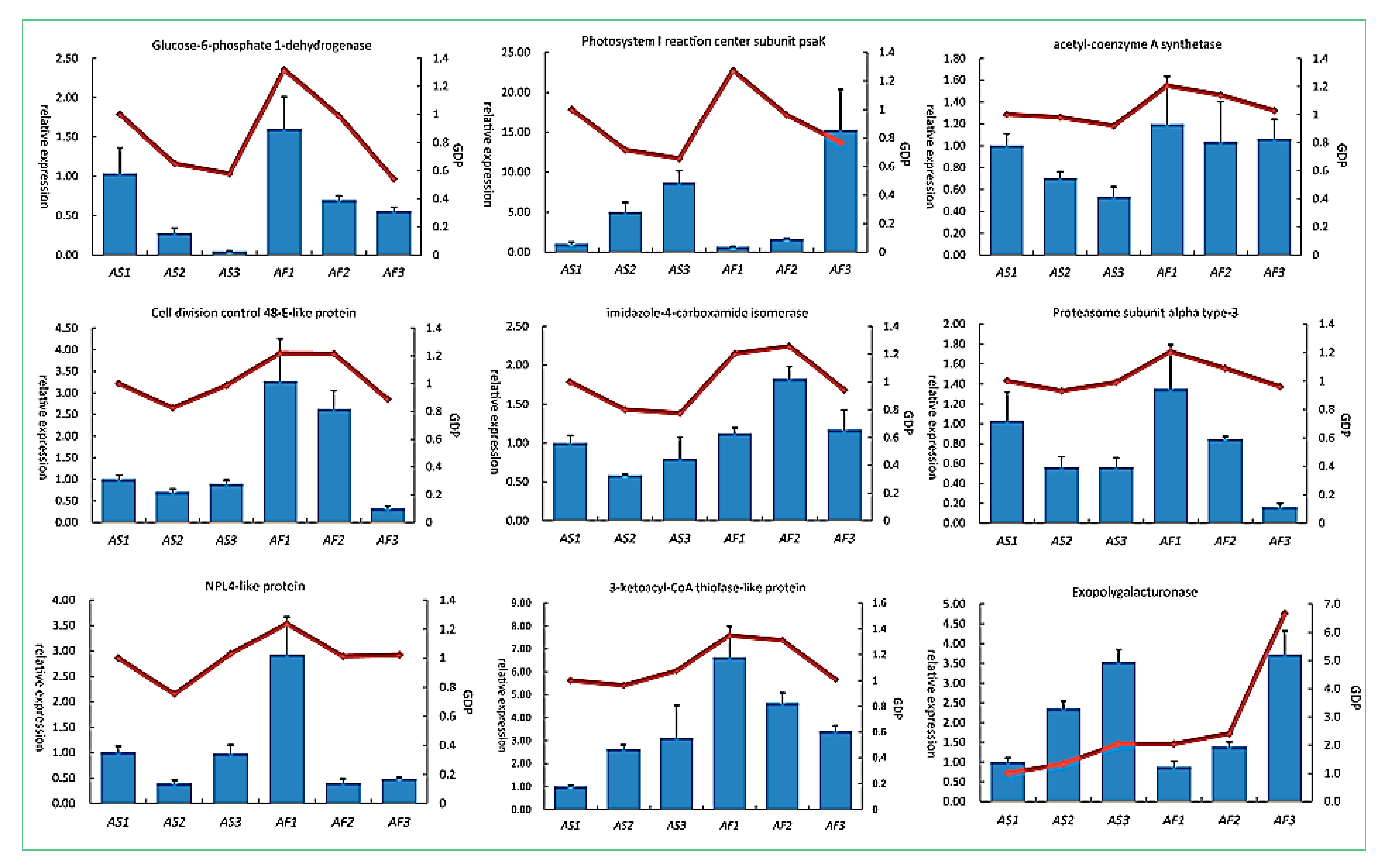
2.6. Relationships between DAPs and Their Corresponding Transcripts
3. Discussion
3.1. Starch and Sucrose Metabolism during Anther Development
3.2. DAPs Related to Pollen Wall Formation
3.3. DAPs Related to Disrupted Energy Metabolism during Anther Development
3.4. DAPs Related to Protein Synthesis and Degradation
4. Materials and Methods
4.1. Plant Materials, Plant Growth, and Anther Collection
4.2. Anther Development and Phenotypic Characterization
4.3. Sample Preparation, Protein Extraction and iTRAQ Labeling
4.4. LC-MS/MS Measurements and Data Analysis
4.5. Functional Category and Clustering Analyses of DAPs
4.6. RNA Extraction and qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Edgerton, M.D. Increasing crop productivity to meet global needs for feed, food, and fuel. Plant Physiol. 2009, 149, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hallauer, A.R. Heterosis: What have we learned? What have we done? Where are we headed? In The Genetics and Exploitation of Heterosis in Crops; Coors, J.G., Pandey, S., Eds.; American Society of Agronomy, Crop Science Society of America, Soil Science Society of America: Madison, WI, USA, 1999; Volume 21, pp. 483–492. [Google Scholar]
- Longin, C.F.; Muehleisen, J.; Maurer, H.P.; Zhang, H.; Gowda, M.; Reif, J.C. Hybrid breeding in autogamous cereals. Theor. Appl. Genet. 2012, 125, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Atienza, S.G.; Martín, A.C. Fertility of CMS wheat is restored by two Rf loci located on a recombined acrocentric chromosome. J. Exp. Bot. 2014, 65, 6667–6677. [Google Scholar] [CrossRef] [PubMed]
- Song, X.Y.; Zhang, L.L.; Zeng, J.L.; Qian, H.H.; Li, H.B.; He, B.R. Development of thermo-sensitive cytoplasmic male sterile (TCMS) lines of wheat characterized by complete male sterility at lower-temperatures and partially restored fertility at higher-temperatures. Euphytica 2013, 192, 393–399. [Google Scholar] [CrossRef]
- Song, X.Y.; Fang, P.; Ma, L.J.; He, B.R. A comparison of wheat CMS lines of Ae. kotschyi cytoplasm of No 1B/1R type and 1B/1R type. J. Northwest A&F Univ. 2002, 30, 1–4. [Google Scholar]
- Molloy, M.P.; Herbert, B.R.; Walsh, B.J.; Tyler, M.I.; Traini, M.; Sanchez, J.C.; Hochstrasser, D.F.; Williams, K.L.; Gooley, A.A. Extraction of membrane proteins by differential solubilization for separation using two-dimensional gel electrophoresis. Electrophoresis 1998, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Hao, P.; Cao, M.; Guo, G.; Lv, D.; Subburaj, S.; Li, X.; Yan, X.; Xiao, J.; Ma, W.; et al. iTRAQ-based quantitative proteomic analysis reveals new metabolic pathways of wheat seedling growth under hydrogen peroxide stress. Proteomics 2013, 13, 3046–3058. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tian, H.; Wang, S.; Shao, J.; Zheng, Y.; Zhang, H.; Guo, L.; Ding, Y. Pollen developmental defects in ZD-CMS rice line explored by cytological, molecular and proteomic approaches. J. Proteom. 2014, 108, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, C.; Wei, H.; Song, M.; Meng, Y.; Ma, J.; Fan, S.; Yu, S. iTRAQ-facilitated proteomic profiling of anthers from a photosensitive male sterile mutant and wild-type cotton (Gossypium hirsutum L.). J. Proteom. 2015, 126, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.B.; Fang, Y.N.; Pan, Z.Y.; Sun, L.; Deng, X.X.; Grosser, J.W.; Guo, W.W. iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile cybrid pummel. J. Proteome Res. 2014, 13, 2998–3015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lan, Y.; Wen, C.; Zhao, H.; Wang, J.; Wang, Y. Transcriptome Sequencing Analyses between the Cytoplasmic Male Sterile Line and Its Maintainer Line in Welsh Onion (Allium fistulosum L.). J. Mol. Sci. 2016, 17, 1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.J.; Seo, M.; Jang, Y.J.; Cho, S.; Lee, G.P. Transcriptome profiling of differentially expressed genes in floral buds and flowers of male sterile and fertile lines in watermelon. BMC Genom. 2015, 9, 914. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, I.S.; Sawhney, V.K. Proteome analysis of the normal and Ogura (ogu) CMS anthers of Brassica napus to identify proteins associated with male sterility. Botany 2010, 88, 217–230. [Google Scholar] [CrossRef]
- Karp, N.A.; Huber, W.; Sadowski, P.G.; Charles, P.D.; Hseter, S.V.; Lilley, K.S. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell Proteom. 2010, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.Y.; Zhou, J.W.; Chen, G.X.; Bian, Y.W.; Lv, D.G.; Li, X.H.; Wang, Z.M.; Yan, Y.M. iTRAQ-based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genom. 2014, 15, 1029. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.L.; Cassin, A.; Bacic, A. Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front. Plant Sci. 2011, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.Y.; Liu, Z.H.; Zhang, L.L.; Hu, G.; Song, X.Y. Cytological characterization of a thermo-sensitive cytoplasmic male-sterile wheat line having K-type cytoplasm of Aegilops kotschyi. Breed Sci. 2016, 66, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Qin, Y.; Deng, Y.; Kong, F.; Wang, Z.; Shen, G.; Wang, J.; Duan, B.; Li, R. Expression profiles of a cytoplasmic male sterile line of Gossypium harknessii and its fertility restorer and maintainer lines revealed by RNA-Seq. Plant Physiol. Biochem. 2017, 116, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kafri, M.; Metzl-Raz, E.; Jona, G.; Barkai, N. The cost of protein production. Cell Rep. 2016, 14, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Liu, W.; Zhang, G.S.; Ye, J.X. Mitochondrial Proteomic Analysis of Cytoplasmic Male Sterility Line and Its Maintainer in Wheat (Triticum aestivum L.). Agric. Sci. China 2010, 6, 771–782. [Google Scholar] [CrossRef]
- Zhang, L.P.; Zhao, C.P.; Shan, F.H.; Zhang, F.T.; Ye, Z.J. The mixed genetic analysis of photoperiod-temperature sensitive male sterility of BS210 in wheat. Acta Agron. Sin. 2007, 33, 1553–1557. [Google Scholar]
- Keeling, P.L.; Wood, J.R.; Tyson, R.H.; Bridges, I.G. Starch biosynthesis in developing wheat grain: Evidence against the direct involvement of triose phosphates in the metabolic pathway. Plant Physiol. 1988, 87, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.; Godt, D.E.; Guivarc'h, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci. USA 2001, 98, 6522–6527. [Google Scholar] [CrossRef] [PubMed]
- Tetlow, I.J.; Wait, R.; Lu, Z.; Akkasaeng, R.; Bowsher, C.G.; Esposito, S.; Kosar-Hashemi, B.; Morell, M.K.; Emes, M.J. Protein phosphorylation in amyloplasts regulates starch branching enzyme activity and protein-protein interactions. Plant Cell 2004, 16, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, X.D. Tapetum: Regulation and role in sporopollenin biosynthesis in Arabidopsis. Plant Mol. Biol. 2013, 83, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.T.; Wan, Z.Y.; Li, S.; Zhang, Y. Spatiotemporal Production of Reactive Oxygen Species by NADPH Oxidase Is Critical for Tapetal Programmed Cell Death and Pollen Development in Arabidopsis. Plant Cell 2014, 26, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, G.C.; Xue, Y.L. Physiological role and regulation of phenylpropanoid metabolism in plant. Plant Physiol. Commun. 1988, 3, 9–16. [Google Scholar]
- Ye, J.L.; Duan, Y.; Hu, G.; Geng, X.X.; Zhang, G.M.; Yan, P.J.; Liu, Z.H.; Zhang, L.L.; Song, X.Y. Identification of candidate genes and biosynthesis pathways related to fertility conversion by wheat KTM3315A transcriptome profiling. Front. Plant Sci. 2017, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Tsuchiya, T.; Kishitani, S.; Tanaka, Y.; Toriyama, K. Partial male sterility in transgenic tobacco carrying antisense and sense PAL cDNA under the control of a tapetum-specific promoter. Plant Cell Physiol. 1996, 37, 215–222. [Google Scholar] [CrossRef]
- Chang, F.; Wang, Y.; Wang, S.; Ma, H. Molecular control of microsporogenesis in Arabidopsis. Curr. Opin. Plant Biol. 2011, 14, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Annu. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Liu, S.N.; Wei, L.; Hu, Y.J.; Yu, J.H.; Zhao, J.; Ding, Y. The calcium distribution in the anther of cytoplasmic male sterile line of Yunnan purple rice during anther development. J. Wuhan Bot. Res. 2005, 23, 101–106. [Google Scholar]
- Zhang, M.Y.; Liang, J.Y.; Huang, Y.W.; Liu, H.X. Comparative of respiratory pathway of CMS and its maintainer rice (Oryza sativa L.). J. Plant Physiol. 1998, 14, 55–58. [Google Scholar]
- Zhang, M.Y.; Xu, S.X. Identification of a rice cDNA encoding the acyl CoA binding protein (ACBP). Acta Phytophysiol. Sin. 1999, 25, 327–331. [Google Scholar]
- Sabar, M.; Gagliardi, D.; Balk, J.; Leaver, C.J. ORFB is a subunit of F1F (O)-ATP synthase: Insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003, 4, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Giegé, P.; Heazlewood, J.L.; Roessner-Tunali, U.; Millar, A.H.; Fernie, A.R.; Leaver, C.J.; Sweetlove, L.J. Enzymes of glycolysis are functionally associated with the mitochondrion in Arabidopsis cells. Plant Cell 2003, 15, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Ogino, A.; Kasai, M.; Sano, Y.; Kanazawa, A. Fertility restoration by Ifr1 in rice with BT-type cytoplasmic male sterility is associated with a reduced level, but not processing, of atp6-orf79 co-transcribed RNA. Plant Cell Rep. 2010, 29, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F.; Karpen, J.W.; Critchfield, J.M.; Strye, L. Stereochemical course of the reaction catalyzed by the cyclic GMP phosphodiesterase from retinal rod outer segments. J. Biol. Chem. 1988, 5, 14080–14085. [Google Scholar]
- Wilkinson, B.; Xiao, R.; Gilbert, H.F. A structural disulfide of yeast protein-disulfide isomerase destabilizes the active site disulfide of the N-terminal thioredoxin domain. J. Biol. Chem. 2005, 280, 11483–11487. [Google Scholar] [CrossRef] [PubMed]
- D’Aloisio, E.; Paolacci, A.R.; Dhanapal, A.P.; Tanzarella, O.A.; Porceddu, E.; Ciaffi, M. The protein disulfide isomerase gene family in bread wheat (T. aestivum L.). BMC Plant Biol. 2010, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Yue, S.J.; Xu, X.; Liu, J.; Xu, Q.; Wang, X.; Han, L.; Yu, D. Proteome analysis of the wild and YX-1 male sterile mutant anthers of Wolfberry (Lycium barbarum L.). PLoS ONE 2012, 7, e41861. [Google Scholar] [CrossRef] [PubMed]
- Kurepa, J.; Toh-E, A.; Smalle, J.A. 26S proteasome regulatory particle mutants have increased oxidative stress tolerance. Plant J. 2008, 53, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Kurepa, J.; Wang, S.; Li, Y.; Zaitlin, D.; Pierce, A.J.; Smalle, J.A. Loss of 26S proteasome function leads to increased cell size and decreased cell number in Arabidopsis shoot organs. Plant Physiol. 2009, 150, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Gallois, J.L.; Guyon-Debast, A.; Lecureuil, A.; Vezon, D.; Carpentier, V.; Bonhomme, S.; Guerche, P. The Arabidopsis proteasome RPT5 subunits are essential for gametophyte development and show accession-Dependent redundancy. Plant Cell 2009, 21, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Matsui, K.; Ishiguro, S.; Kato, T.; Tabata, S.; Kobayashi, M.; Seki, M.; Shinozaki, K.; Okada, K. Arabidopsis RPT2a encoding the 26S proteasome subunit is required for various aspects of root meristem maintenance, and regulates gametogenesis redundantly with its homolog, RPT2b. Plant Cell Physiol. 2011, 52, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Minami, A.; Marshall, R.S.; Book, A.J.; Farmer, L.M.; Walker, J.M.; VierstraM, R.D. The RPT2 subunit of the 26S proteasome directs complex assembly, histone dynamics, and gametophyte and sporophyte development in Arabidopsis. Plant Cell 2011, 23, 4298–4317. [Google Scholar] [CrossRef] [PubMed]
- Book, A.J.; Smalle, J.; Lee, K.H.; Yang, P.; Walker, J.M.; Casper, S.; Holmes, J.H.; Russo, L.A.; Buzzinotti, Z.W.; Jenik, P.D.; et al. The RPN5 subunit of the 26s proteasome is essential for gametogenesis, sporophyte development, and complex assembly in Arabidopsis. Plant Cell 2009, 21, 460–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Li, C.Z.; Huang, H.Q.; Wang, Z.Y.; Peng, Y.K. Different proteins in mitochondrial proteome of T-type maize cytoplasmic male-sterile line and its maintainer line. Fen Zi Xi Bao Sheng Wu Xue Bao 2007, 40, 410–418. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 22DDCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Description | BLASTp Result | AS vs. AF a | |||
|---|---|---|---|---|---|---|
| E-Value | Score | UNP | BNP | TNP | ||
| Traes_7DS_529BAB150.1 | Sucrose synthase 1 | 6.00 × 10−8 | 57 | 1.33 | 1.49 | 0.82 |
| Traes_6BL_FBF9DA7CE.1 | S-adenosylmethionine synthase 1 | 1.00 × 10−96 | 296 | 0.73 | 0.59 | 0.38 |
| Traes_5DS_69B96465C.1 | Calcium-dependent protein kinase | 0 | 539 | 1.46 | 1.25 | 0.73 |
| Traes_7AS_EBD5F1F54.1 | Flowering locus T | 6.00 × 10−6 | 46.2 | 1.96 | 2.17 | 3.06 |
| Traes_7BS_581AA844D.1 | Flowering locus T | 3.00 × 10−6 | 46.6 | 1.96 | 2.17 | 3.06 |
| Traes_1DS_486DB7BFE.1 | NADP-dependent malic enzyme | 0 | 555 | 1.21 | 1.39 | 1.76 |
| Traes_2AL_7A5A988CA.1 | Acetyl-coenzyme A synthetase | 0 | 736 | 1.20 | 1.16 | 1.12 |
| Traes_2DL_06E6A2543.1 | Glucose-6-phosphate 1-dehydrogenase | 7.00 × 10−178 | 520 | 1.32 | 1.52 | 0.94 |
| Traes_4AS_91D4C5213.1 | Fructose-1,6-bisphosphatase | 2.00 × 10−93 | 288 | 0.73 | 0.72 | 0.64 |
| GeneID | Description | BLASTp Result | AS vs. AF | |||
|---|---|---|---|---|---|---|
| E-Value | Score | UNP | BNP | TNP | ||
| Traes_4DS_29F298799.1 | Glyco_hydro_1 domain-containing protein | 1.00 × 10−152 | 449 | −1.41 | −3.52 | −0.57 |
| Traes_3AL_9A3B8E4D9.1 | Alpha-glucan phosphorylase, H isozyme | 8.00 × 10−104 | 329 | 2.20 | 2.10 | 1.57 |
| Traes_1AL_A1B2A8EB0.1 | ADP-glucose pyrophosphorylase large subunit | 1.00 × 10−99 | 313 | 2.11 | 1.55 | 5.71 |
| Traes_6BS_0AFE47E4B.1 | UTP–glucose-1-phosphate uridylyltransferase | 2.00 × 10−140 | 414 | −1.07 | −2.03 | −1.08 |
| TRAES3BF066900020CFD_t1 | Hexokinase-6 | 6.00 × 10−75 | 250 | −0.76 | −1.30 | −0.81 |
| GeneID | Description | BLASTp Result | AS vs. AF | |||
|---|---|---|---|---|---|---|
| E-Value | Score | UNP | BNP | TNP | ||
| Traes_6DL_7960654CF.2 | 3-Ketoacyl-CoA thiolase-like protein | 2.00 × 10−52 | 186 | 1.35 | 1.36 | 0.94 |
| Traes_2AL_43CCA70E9.1 | 4-Coumarate-CoA ligase-like protein 9 | 3.00 × 10−87 | 285 | 2.75 | 2.87 | 0.61 |
| Traes_5DL_7D83C2AB2.1 | Calreticulin | 6.00 × 10−39 | 145 | −0.72 | −1.54 | 0.07 |
| Traes_5BL_D71543428.1 | ABC transporter C family member 4-like | 0 | 1107 | 0.83 | 0.57 | 1.15 |
| Traes_2BL_E8D5E0972.1 | 3-Oxoacyl-synthase I, chloroplastic-like | 1.00 × 10−16 | 83.6 | 1.35 | −0.36 | 1.36 |
| Traes_6BS_D30CEBBC9.1 | Calnexin-like protein | 4.00 × 10−78 | 259 | −0.94 | −2.50 | −1.02 |
| GeneID | Description | BLASTp Result | AS vs. AF | |||
|---|---|---|---|---|---|---|
| E-Value | Score | UNP | BNP | TNP | ||
| Traes_1AL_4F3CAE982.2 | Vacuolar proton-ATPase C subunit | 2.00 × 10−81 | 257 | 2.00 | 2.22 | 1.92 |
| Traes_5DS_978062D3E.1 | ATP synthase CF1 epsilon subunit | 1.00 × 10−5 | 44.3 | −1.50 | −1.37 | −1.47 |
| Traes_6DS_27D11E4B8.2 | 1.00 × 10−5 | 44.3 | −1.50 | −1.37 | −1.47 | |
| Traes_5BL_D12151D1D.1 | 6-Phosphofructokinase 5 | 4.00 × 10−11 | 66.6 | 1.35 | 1.70 | 2.44 |
| Traes_5DL_27372150C.2 | 2.00 × 10−11 | 67.8 | 1.35 | 1.70 | 2.44 | |
| Traes_4AS_91D4C5213.1 | Fructose-1,6-bisphosphatase | 2.00 × 10−93 | 288 | −1.73 | −1.78 | −2.43 |
| Traes_4BL_B25B3CE481.1 | 5.00 × 10−93 | 287 | −1.73 | −1.78 | −2.43 | |
| GeneID | Description | BLASTp Result | AS vs. AF | |||
|---|---|---|---|---|---|---|
| E-Value | Score | UNP | BNP | TNP | ||
| Traes_1AL_3F7B8E94D.1 | Imidazole-4-carboxamide isomerase | 4.00 × 10−81 | 423 | 1.20 | 1.57 | 1.22 |
| Traes_2AL_F73AFACB4.1 | UBP1-associated protein 2B-like | 4.00 × 10−22 | 100 | 1.14 | 2.07 | 0.12 |
| Traes_6BL_0043F2EE5.1 | Serine/arginine-rich splicing factor 7 | 2.00 × 10−15 | 73.6 | −1.80 | −0.83 | −0.01 |
| Traes_6AL_913A14974.2 | Serine/arginine-rich splicing factor | 3.00 × 10−16 | 74.7 | −1.80 | −0.83 | −0.01 |
| Traes_7BL_AEF6873D9.1 | Small nuclear ribonucleoprotein E-like | 2.00 × 10−6 | 43.5 | 0.80 | 2.50 | 1.69 |
| Traes_1DL_E9432F054.1 | U6 snRNA-associated Sm-like protein LSm8 | 3.00 × 10−10 | 54.7 | 0.47 | 1.06 | 0.08 |
| Traes_5BL_21270EDFB.1 | U2 small nuclear ribonucleoprotein A′ | 7.00 × 10−6 | 48.1 | −0.50 | −1.09 | −0.83 |
| Traes_1AL_51CED3DBF.1 | Heat shock cognate 70 kDa protein 4 | 0 | 872 | −1.80 | −1.14 | −1.59 |
| Traes_4AS_B978C93FA.1 | Heat shock cognate 70 kDa protein | 0 | 980 | −1.25 | −2.20 | −1.51 |
| Traes_5DS_AC5D29D23.1 | Heat shock protein 83 | 0 | 556 | −1.05 | −2.13 | −2.61 |
| Traes_3AL_75B505500.1 | DnaJ homolog subfamily C member 3 | 8.00 × 10−18 | 85.5 | −0.71 | 1.50 | 0.10 |
| Traes_7AS_8B382E1D0.1 | Proteasome subunit alpha type-7-A | 2.00 × 10−74 | 229 | 1.97 | 1.08 | 1.01 |
| Traes_5AL_2FB884126.2 | Proteasome subunit alpha type-3 | 3.00 × 10−32 | 121 | 1.03 | 0.85 | −0.17 |
| Traes_2DL_C6C4CFB34.1 | 26S proteasome non-ATPase regulatory subunit 7 | 3.00 × 10−115 | 341 | −0.49 | −2.00 | −2.20 |
| Traes_2BL_2B983AD37.1 | 26S proteasome regulatory subunit T3 | 0 | 697 | −0.75 | −1.25 | −0.17 |
| Traes_4AS_6285AE7F6.1 | 26S proteasome regulatory subunit T2 | 4.00 × 10−133 | 395 | −0.55 | −1.66 | −0.08 |
| Traes_1BS_548536A26.2 | Proteasome subunit beta type-7-B | 1.00 × 10−89 | 273 | −0.23 | −1.05 | −1.87 |
| Traes_1BL_E29880146.1 | Cell division control 48-E-like protein | 9.00 × 10−97 | 313 | 1.20 | 1.57 | 1.22 |
| Traes_6AS_E60FDC38F1.2 | NPL4-like protein | 3.00 × 10−11 | 66.6 | 1.24 | 1.34 | 0.99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Ye, J.; Jia, Y.; Zhang, L.; Song, X. iTRAQ-Based Proteomics Analyses of Sterile/Fertile Anthers from a Thermo-Sensitive Cytoplasmic Male-Sterile Wheat with Aegilops kotschyi Cytoplasm. Int. J. Mol. Sci. 2018, 19, 1344. https://doi.org/10.3390/ijms19051344
Zhang G, Ye J, Jia Y, Zhang L, Song X. iTRAQ-Based Proteomics Analyses of Sterile/Fertile Anthers from a Thermo-Sensitive Cytoplasmic Male-Sterile Wheat with Aegilops kotschyi Cytoplasm. International Journal of Molecular Sciences. 2018; 19(5):1344. https://doi.org/10.3390/ijms19051344
Chicago/Turabian StyleZhang, Gaoming, Jiali Ye, Yulin Jia, Lingli Zhang, and Xiyue Song. 2018. "iTRAQ-Based Proteomics Analyses of Sterile/Fertile Anthers from a Thermo-Sensitive Cytoplasmic Male-Sterile Wheat with Aegilops kotschyi Cytoplasm" International Journal of Molecular Sciences 19, no. 5: 1344. https://doi.org/10.3390/ijms19051344