Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease

 ,
,  ,
,
Abstract
:1. Introduction
2. Connexins: From Gene to Protein
2.1. Gene Structure and Splicing
2.2. Transcription Factors and Epigenetics
2.3. RNA Stability and MicroRNAs
2.4. Translational Regulation
2.4.1. Internal Ribosome Entry Site (IRES)
2.4.2. Alternative Translation of Truncated Connexin Forms
3. Post-Translational Regulation of Connexins
3.1. Phosphorylation
3.2. S-Nitrosylation
3.3. Other Post-Translational Modifications: SUMOylation, Ubiquitination, and Acetylation
4. Connexin Trafficking
4.1. Control of Oligomerization
4.2. Connexin Quality Control
4.3. Connexin Cytoplasmic Domains and the Cytoskeleton
4.4. Regulation of Gap Junction Plaque Morphology
5. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Laird, D.W. Next-generation connexin and pannexin cell biology. Trends Cell Biol. 2016, 26, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and disease. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Simek, J.; Laird, D.W. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015, 360, 701–721. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.L.; Phillips, A.R.; Duft, B.J.; Kim, Y.; Green, C.R. Translating connexin biology into therapeutics. Semin. Cell Dev. Biol. 2016, 50, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Rhett, J.M.; Ghatnekar, G.S. Cardiac to cancer: Connecting connexins to clinical opportunity. FEBS Lett. 2014, 588, 1349–1364. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Huang, C.H. Lasagna-search: An integrated web tool for transcription factor binding site search and visualization. BioTechniques 2013, 54, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Dupays, L.; Mazurais, D.; Rücker-Martin, C.; Calmels, T.; Bernot, D.; Cronier, L.; Malassiné, A.; Gros, D.; Théveniau-Ruissy, M. Genomic organization and alternative transcripts of the human connexin40 gene. Gene 2003, 305, 79–90. [Google Scholar] [CrossRef]
- Essenfelder, G.M.; Larderet, G.; Waksman, G.; Lamartine, J. Gene structure and promoter analysis of the human gjb6 gene encoding connexin30. Gene 2005, 350, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Theis, M.; Hallas, G.; Brambach, S.; Dahl, E.; Kidder, G.; Willecke, K. A new alternatively spliced transcript of the mouse connexin32 gene is expressed in embryonic stem cells, oocytes, and liver. Exp. Cell Res. 2001, 266, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, I.; Anderson, C.; Werner, R.; Oltra, E. Redefining the structure of the mouse connexin43 gene: Selective promoter usage and alternative splicing mechanisms yield transcripts with different translational efficiencies. Nucleic Acids Res. 2004, 32, 4550–4562. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Zundel, M.A.; Werner, R. Variable promoter usage and alternative splicing in five mouse connexin genes. Genomics 2005, 85, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Cicirata, F.; Parenti, R.; Spinella, F.; Giglio, S.; Tuorto, F.; Zuffardi, O.; Gulisano, M. Genomic organization and chromosomal localization of the mouse connexin36 (mCx36) gene. Gene 2000, 251, 123–130. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Euwens, C.; Willecke, K.; Söhl, G. The novel mouse connexin39 gene is expressed in developing striated muscle fibers. J. Cell Sci. 2004, 117, 5381–5392. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Janssen-Bienhold, U.; Sohl, G.; Schubert, T.; Bussow, H.; Ott, T.; Weiler, R.; Willecke, K. Functional expression of connexin57 in horizontal cells of the mouse retina. Eur. J. Neurosci. 2004, 19, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Joussen, A.; Kociok, N.; Willecke, K. Expression of connexin genes in the human retina. BMC Ophthalmol. 2010, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta Biomembr. 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, I.M.; Bone, L.; Wang, S.; Ionasescu, V.; Werner, R. The human connexin32 gene is transcribed from two tissue-specific promoters. Biosci. Rep. 1996, 16, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.M.; Polke, J.; Manji, H.; Blake, J.; Reiniger, L.; Sweeney, M.; Houlden, H.; Brandner, S.; Reilly, M.M. A novel mutation in the nerve-specific 5’ UTR of the GJB1 gene causes X-linked charcot-marie-tooth disease. J. Peripher. Nerv. Syst. JPNS 2011, 16, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, R.; Burton-Jones, S.; Antoniadi, T.; Rogers, M.; Jaunmuktane, Z.; Brandner, S.; Kiely, N.; Manuel, R.; Willis, T. Deletion of p2 promoter of GJB1 gene a cause of charcot-marie-tooth disease. Neuromuscul. Disord. NMD 2017, 27, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Al-Yahyaee, S.A.; Al-Kindi, M.; Jonghe, P.D.; Al-Asmi, A.; Al-Futaisi, A.; Vriendt, E.D.; Deconinck, T.; Chand, P. Pelizaeus-merzbacher-like disease in a family with variable phenotype and a novel splicing GJC2 mutation. J. Child Neurol. 2013, 28, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Gandia, M.; Del Castillo, F.J.; Rodriguez-Alvarez, F.J.; Garrido, G.; Villamar, M.; Calderon, M.; Moreno-Pelayo, M.A.; Moreno, F.; del Castillo, I. A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PLoS ONE 2013, 8, e73566. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M.; Bier, A.; Carystinos, G.D.; Alaoui-Jamali, M.A.; Batist, G. Connexin43 pseudogene is expressed in tumor cells and inhibits growth. Oncogene 2004, 23, 4763–4770. [Google Scholar] [CrossRef] [PubMed]
- Bier, A.; Oviedo-Landaverde, I.; Zhao, J.; Mamane, Y.; Kandouz, M.; Batist, G. Connexin43 pseudogene in breast cancer cells offers a novel therapeutic target. Mol. Cancer Ther. 2009, 8, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.M.; Yang, J.J.; Shieh, J.C.; Lin, M.L.; Li, S.Y. Novel mutations in the connexin43 (GJA1) and GJA1 pseudogene may contribute to nonsyndromic hearing loss. Hum. Genet. 2010, 127, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Rackauskas, M.; Neverauskas, V.; Skeberdis, V.A. Diversity and properties of connexin gap junction channels (review). Medicina (Kaunas Lithuania) 2010, 46, 1–12. [Google Scholar] [PubMed]
- Di, W.L.; Rugg, E.L.; Leigh, I.M.; Kelsell, D.P. Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. J. Investig. Dermatol. 2001, 117, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.A.; Tattersall, D.; O’Toole, E.A.; Kelsell, D.P. Connexins in epidermal homeostasis and skin disease. Biochim. Biophys. Acta 2012, 1818, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Faniku, C.; Wright, C.S.; Martin, P.E. Connexins and pannexins in the integumentary system: The skin and appendages. Cell. Mol. Life Sci. CMLS 2015, 72, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E.; Sellitto, C.; Milstone, L.M.; White, T.W. Connexin channels in congenital skin disorders. Semin. Cell Dev. Biol. 2016, 50, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.J.; Kiang, D.T. Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim. Biophys. Acta 1998, 1443, 169–181. [Google Scholar] [CrossRef]
- Bai, S.; Spray, D.C.; Burk, R.D. Identification of proximal and distal regulatory elements of the rat connexin32 gene. BBA Gene Struct. Expr. 1993, 1216, 197–204. [Google Scholar] [CrossRef]
- Field, J.M.L.; Tate, L.A.; Chipman, J.K.; Minchin, S.D. Identification of functional regulatory regions of the connexin32 gene promoter. Biochim. Biophys. Acta Gene Struct. Expr. 2003, 1628, 22–29. [Google Scholar] [CrossRef]
- Seul, K.H.; Tadros, P.N.; Beyer, E.C. Mouse connexin40: Gene structure and promoter analysis. Genomics 1997, 46, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Bierhuizen, M.F.A.; Van Amersfoorth, S.C.M.; Groenewegen, W.A.; Vliex, S.; Jongsma, H.J. Characterization of the rat connexin40 promoter: Two Sp1/Sp3 binding sites contribute to transcriptional activation. Cardiovasc. Res. 2000, 46, 511–522. [Google Scholar] [CrossRef]
- Echetebu, C.O.; Ali, M.; Izban, M.G.; MacKay, L.; Garfield, R.E. Localization of regulatory protein binding sites in the proximal region of human myometrial connexin 43 gene. Mol. Hum. Reprod. 1999, 5, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, B.E.J.; van Amersfoorth, S.C.M.; Opthof, T.; Jongsma, H.J.; Bierhuizen, M.F.A. Sp1 and Sp3 activate the rat connexin40 proximal promoter. Biochem. Biophys. Res. Commun. 2002, 292, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Linhares, V.L.F.; Almeida, N.A.S.; Menezes, D.C.; Elliott, D.A.; Lai, D.; Beyer, E.C.; Campos De Carvalho, A.C.; Costa, M.W. Transcriptional regulation of the murine connexin40 promoter by cardiac factors nkx2-5, gata4 and tbx5. Cardiovasc. Res. 2004, 64, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Geimonen, E.; Boylston, E.; Royek, A.; Andersen, J. Elevated connexin-43 expression in term human myometrium correlates with elevated C-jun expression and is independent of myometrial estrogen receptors. J. Clin. Endocrinol. Metab. 1998, 83, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cobo, M.; Stewart, D.; Drujan, D.; De Maio, A. Promoter activity of the rat connexin 43 gene in nrk cells. J. Cell. Biochem. 2001, 81, 514–522. [Google Scholar] [CrossRef]
- Teunissen, B.E.J.; Jansen, A.T.; Van Amersfoorth, S.C.M.; O’Brien, T.X.; Jongsma, H.J.; Bierhuizen, M.F.A. Analysis of the rat connexin 43 proximal promoter in neonatal cardiomyocytes. Gene 2003, 322, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Vine, A.L.; Leung, Y.M.; Bertram, J.S. Transcriptional regulation of connexin 43 expression by retinoids and carotenoids: Similarities and differences. Mol. Carcinog. 2005, 43, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Shao, Q.; Yang, X.J.; Luh, S.P.; Kandouz, M.; Batist, G.; Laird, D.W.; Alaoui-Jamali, M.A. A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 2006, 66, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Villares, G.J.; Dobroff, A.S.; Wang, H.; Zigler, M.; Melnikova, V.O.; Huang, L.; Bar-Eli, M. Overexpression of protease-activated receptor-1 contributes to melanoma metastasis via regulation of connexin 43. Cancer Res. 2009, 69, 6730–6737. [Google Scholar] [CrossRef] [PubMed]
- Geimonen, E.; Jiang, W.; Ali, M.; Fishman, G.I.; Garfield, R.E.; Andersen, J. Activation of protein kinase c in human uterine smooth muscle induces connexin-43 gene transcription through an AP-1 site in the promoter sequence. J. Biol. Chem. 1996, 271, 23667–23674. [Google Scholar] [CrossRef] [PubMed]
- Ghouili, F.; Martin, L.J. Cooperative regulation of GJA1 expression by members of the AP-1 family cjun and cfos in TM3 leydig and TM4 sertoli cells. Gene 2017, 635, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, D.; Lecanda, F.; Shin, C.S.; Stains, J.; Civitelli, R. Sequence and structure of the mouse connexin45 gene. Biosci. Rep. 2001, 21, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Van der Heyden, M.A.; Rook, M.B.; Hermans, M.M.; Rijksen, G.; Boonstra, J.; Defize, L.H.; Destree, O.H. Identification of connexin43 as a functional target for Wnt signalling. J. Cell Sci. 1998, 111 Pt 12, 1741–1749. [Google Scholar] [PubMed]
- Ai, Z.; Fischer, A.; Spray, D.C.; Brown, A.M.; Fishman, G.I. Wnt-1 regulation of connexin43 in cardiac myocytes. J. Clin. Investig. 2000, 105, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, B.E.; Bierhuizen, M.F. Transcriptional control of myocardial connexins. Cardiovasc. Res. 2004, 62, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Koffler, L.D.; Fernstrom, M.J.; Akiyama, T.E.; Gonzalez, F.J.; Ruch, R.J. Positive regulation of connexin32 transcription by hepatocyte nuclear factor-1α. Arch. Biochem. Biophys. 2002, 407, 160–167. [Google Scholar] [CrossRef]
- Rukstalis, J.M.; Kowalik, A.; Zhu, L.; Lidington, D.; Pin, C.L.; Konieczny, S.F. Exocrine specific expression of connexin32 is dependent on the basic helix-loop-helix transcription factor mist1. J. Cell Sci. 2003, 116, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Bondurand, N.; Girard, M.; Pingault, V.; Lemort, N.; Dubourg, O.; Goossens, M. Human connexin 32, a gap junction protein altered in the X-linked form of charcot-marie-tooth disease, is directly regulated by the transcription factor SOX10. Hum. Mol. Genet. 2001, 10, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Petrocelli, T.; Lye, S.J. Regulation of transcripts encoding the myometrial gap junction protein, connexin-43, by estrogen and progesterone. Endocrinology 1993, 133, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Recouvreux, M.S.; Grasso, E.N.; Echeverria, P.C.; Rocha-Viegas, L.; Castilla, L.H.; Schere-Levy, C.; Tocci, J.M.; Kordon, E.C.; Rubinstein, N. RUNX1 and FOXP3 interplay regulates expression of breast cancer related genes. Oncotarget 2016, 7, 6552–6565. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. Regulation of connexin signaling by the epigenetic machinery. Biochim. Biophys. Acta 2015, 1859, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huhn, D.; Knosel, T.; Pacyna-Gengelbach, M.; Deutschmann, N.; Petersen, I. Downregulation of connexin 26 in human lung cancer is related to promoter methylation. Int. J. Cancer 2005, 113, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.W.; Bianco, T.; Dobrovic, A. Variable promoter region cpg island methylation of the putative tumor suppressor gene connexin 26 in breast cancer. Carcinogenesis 2002, 23, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Hirai, A.; Yano, T.; Nishikawa, K.; Suzuki, K.; Asano, R.; Satoh, H.; Hagiwara, K.; Yamasaki, H. Down-regulation of connexin 32 gene expression through DNA methylation in a human renal cell carcinoma cell. Am. J. Nephrol. 2003, 23, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Cheng, Y.W.; Chou, M.C.; Sen-Lin, T.; Lai, W.W.; Ho, W.L.; Lee, H. The correlation between aberrant connexin 43 mRNA expression induced by promoter methylation and nodal micrometastasis in non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 4200–4204. [Google Scholar] [PubMed]
- Wang, Y.; Huang, L.H.; Xu, C.X.; Xiao, J.; Zhou, L.; Cao, D.; Liu, X.M.; Qi, Y. Connexin 32 and 43 promoter methylation in helicobacter pylori-associated gastric tumorigenesis. World J. Gastroenterol. WJG 2014, 20, 11770–11779. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.C.; Wang, H.; Zhang, G.Y.; Xia, B. Downregulation of connexin 43 in nasopharyngeal carcinoma cells is related to promoter methylation. Oral Oncol. 2007, 43, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Sirnes, S.; Honne, H.; Ahmed, D.; Danielsen, S.A.; Rognum, T.O.; Meling, G.I.; Leithe, E.; Rivedal, E.; Lothe, R.A.; Lind, G.E. DNA methylation analyses of the connexin gene family reveal silencing of GJC1 (connexin45) by promoter hypermethylation in colorectal cancer. Epigenetics 2011, 6, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Lin, X.; Andrews, L.; Patel, D.; Lampe, P.D.; Veenstra, R.D. Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front. Pharmacol. 2013, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Han, H.B.; Zhang, Z.Q. Suppression of lung cancer cell invasion and metastasis by connexin43 involves the secretion of follistatin-like 1 mediated via histone acetylation. Int. J. Biochem. Cell Biol. 2011, 43, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Thiel, G. Cell type-specific regulation of RE-1 silencing transcription factor (REST) target genes. Eur. J. Neurosci. 2005, 22, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossi, S.; Musso, E.; Macchi, E.; Mai, A.; et al. Nepsilon-lysine acetylation determines dissociation from gap junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Berni, R.; Rosati, J.; Straino, S.; Vitale, S.; Spallotta, F.; Baruffi, S.; Bocchi, L.; Delucchi, F.; Rossi, S.; et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces cardiac arrhythmias in dystrophic mice. Cardiovasc. Res. 2010, 87, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Forster, T.; Rausch, V.; Zhang, Y.; Isayev, O.; Heilmann, K.; Schoensiegel, F.; Liu, L.; Nessling, M.; Richter, K.; Labsch, S.; et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated CX43-mediated gap junctional intercellular communication. Oncotarget 2014, 5, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Salat-Canela, C.; Munoz, M.J.; Sese, M.; Ramon y Cajal, S.; Aasen, T. Post-transcriptional regulation of connexins. Biochem. Soc. Trans. 2015, 43, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Zhou, Y.; Du, J.; Fan, S.; Pan, B.; Wang, Y.; Fan, L.; Jiang, J. Identification of miR-200a as a novel suppressor of connexin 43 in breast cancer cells. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, J.H.; Song, B.; Xiong, E.Q.; Chen, Z.W.; Zhou, Z.S.; Su, Y.P. Suppression of CX43 expression by miR-20a in the progression of human prostate cancer. Cancer Biol. Ther. 2012, 13, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, C.; Zhang, A.; Wang, K.; Jia, Z.; Wang, G.; Han, L.; Kang, C.; Pu, P. MiR-221/222 is the regulator of CX43 expression in human glioblastoma cells. Oncol. Rep. 2012, 27, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Catoe, H.; Werner, R. MiR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef] [PubMed]
- Hak, K.K.; Yong, S.L.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, X.J.; Wang, H.J.; Li, W.C.; Chen, L.; Ma, D.; Huang, G.Y. Expression of CX43-related microRNAs in patients with tetralogy of fallot. World J. Pediatr. WJP 2014, 10, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Sugino, Y.; Long, X.; Slivano, O.J.; Nishikawa, N.; Yoshimura, N.; Miano, J.M. Myocardin and microRNA-1 modulate bladder activity through connexin 43 expression during post-natal development. J. Cell. Physiol. 2013, 228, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Donahue, H.J.; Qu, R.W.; Genetos, D.C. Joint diseases: From connexins to gap junctions. Nat. Rev. Rheumatol. 2017, 14, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Gindin, Y.; Jiang, Y.; Francis, P.; Walker, R.L.; Abaan, O.D.; Zhu, Y.J.; Meltzer, P.S. MiR-23a impairs bone differentiation in osteosarcoma via down-regulation of GJA1. Front. Genet. 2015, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Zhang, J.F.; Xu, J.; Xu, L.L.; Wu, T.Y.; Wang, B.; Pan, X.H.; Li, G. MicroRNA-144-3p inhibits bone formation in distraction osteogenesis through targeting connexin 43. Oncotarget 2017, 8, 89913–89922. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.M.; Pacheco-Costa, R.; Atkinson, E.G.; Brun, L.R.; Gortazar, A.R.; Harris, J.; Hiasa, M.; Bolarinwa, S.A.; Yoneda, T.; Ivan, M.; et al. Disruption of the CX43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 2017, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Pacheco-Costa, R.; Davis, H.M. MicroRNAs and connexins in bone: Interaction and mechanisms of delivery. Curr. Mol. Biol. Rep. 2017, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ale-Agha, N.; Galban, S.; Sobieroy, C.; Abdelmohsen, K.; Gorospe, M.; Sies, H.; Klotz, L.O. Hur regulates gap junctional intercellular communication by controlling β-catenin levels and adherens junction integrity. Hepatology 2009, 50, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Chun, K.S.; Lee, J.S.; Kang, K.S.; Surh, Y.J.; Lee, H.J. Inhibition of cyclooxygenase-2 expression and restoration of gap junction intercellular communication in h-ras-transformed rat liver epithelial cells by caffeic acid phenethyl ester. Ann. N. Y. Acad. Sci. 2004, 1030, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Ul-Hussain, M.; Dermietzel, R.; Zoidl, G. Connexins and cap-independent translation: Role of internal ribosome entry sites. Brain Res. 2012, 1487, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Werner, R. IRES elements in connexin genes: A hypothesis explaining the need for connexins to be regulated at the translational level. IUBMB Life 2000, 50, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Hudder, A.; Werner, R. Connexin43 mRNA contains a functional internal ribosome entry site. FEBS Lett. 1999, 464, 118–122. [Google Scholar] [CrossRef]
- Hudder, A.; Werner, R. Analysis of a charcot-marie-tooth disease mutation reveals an essential internal ribosome entry site element in the connexin-32 gene. J. Biol. Chem. 2000, 275, 34586–34591. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, H.; Fanjul, M.; Pradayrol, L.; Susini, C.; Pyronnet, S. Restoration of functional gap junctions through internal ribosome entry site-dependent synthesis of endogenous connexins in density-inhibited cancer cells. Mol. Cell. Biol. 2005, 25, 4034–4045. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Lozano, G.; Fernandez-Chamorro, J.; Francisco-Velilla, R.; Galan, A.; Diaz, R. RNA-binding proteins impacting on internal initiation of translation. Int. J. Mol. Sci. 2013, 14, 21705–21726. [Google Scholar] [CrossRef] [PubMed]
- Faye, M.D.; Holcik, M. The role of IRES trans-acting factors in carcinogenesis. Biochim. Biophys. Acta 2015, 1849, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Mazumder, B.; Merrick, W.C. A new framework for understanding IRES-mediated translation. Gene 2012, 502, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.R. So you want to know if your message has an IRES? Wiley Interdiscip. Rev. RNA 2012, 3, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Overbaugh, J. Evidence that an IRES within the Notch2 coding region can direct expression of a nuclear form of the protein. Mol. Cell 2000, 6, 939–945. [Google Scholar] [CrossRef]
- Ul-Hussain, M.; Zoidl, G.; Klooster, J.; Kamermans, M.; Dermietzel, R. IRES-mediated translation of the carboxy-terminal domain of the horizontal cell specific connexin CX55.5 in vivo and in vitro. BMC Mol. Biol. 2008, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Ul-Hussain, M.; Dermietzel, R.; Zoidl, G. Characterization of the internal IRES element of the zebrafish connexin55.5 reveals functional implication of the polypyrimidine tract binding protein. BMC Mol. Biol. 2008, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Mukherjee, R.; Coombs, W.; Burrer, C.; de Mora, I.A.; Delmar, M.; Taffet, S.M. Evidence for the presence of a free C-terminal fragment of cx43 in cultured cells. Cell Commun. Adhes. 2007, 14, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Shaw, R.M. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013, 5, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Salat-Canela, C.; Sese, M.; Peula, C.; Ramon y Cajal, S.; Aasen, T. Internal translation of the connexin 43 transcript. Cell Commun. Signal. 2014, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Ul-Hussain, M.; Olk, S.; Schoenebeck, B.; Wasielewski, B.; Meier, C.; Prochnow, N.; May, C.; Galozzi, S.; Marcus, K.; Zoidl, G.; et al. Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of CX43 and is responsive to hypoxic conditions. J. Biol. Chem. 2014, 289, 20979–20990. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k arranges actin to guide CX43 delivery to cardiac intercalated discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, S.S.; Xiao, S.; Basheer, W.A.; Baum, R.; Epifantseva, I.; Hong, T.; Shaw, R.M. Cx43 isoform GJA1-20k promotes microtubule dependent mitochondrial transport. Front. Physiol. 2017, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, R.; Rashid, R.; Ismail, R.; Niaz, S.; Chowdri, N.A.; Hussain, M.U. The carboxy-terminal domain of connexin 43 (CT-CX43) modulates the expression of p53 by altering miR-125b expression in low-grade human breast cancers. Cell. Oncol. 2015, 38, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Foote, C.I.; Zhou, L.; Zhu, X.; Nicholson, B.J. The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J. Cell Biol. 1998, 140, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.R.; Billaud, M.; Lohman, A.W.; Taddeo, E.P.; Isakson, B.E. Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 2012, 245, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. 1), 11. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim. Biophys. Acta 2005, 1711, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Martinez, A.D.; Branes, M.C.; Gonzalez, H.E. Regulation of gap junctions by protein phosphorylation. Braz. J. Med. Biol. Res. 1998, 31, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Stultz, C.M.; Levin, A.D.; Edelman, E.R. Phosphorylation-induced conformational changes in a mitogen-activated protein kinase substrate. Implications for tyrosine hydroxylase activation. J. Biol. Chem. 2002, 277, 47653–47661. [Google Scholar] [CrossRef] [PubMed]
- Diestel, S.; Eckert, R.; Hülser, D.; Traub, O. Exchange of serine residues 263 and 266 reduces the function of mouse gap junction protein connexin31 and exhibits a dominant-negative effect on the wild-type protein in hela cells. Exp. Cell Res. 2004, 294, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Chang, M.; Wang, S.; Liu, Z.; Zhu, W.; Wang, Y.; Yan, F.; Li, J.; Zhang, B.; Dou, G.; et al. Connexin 32-mediated cell-cell communication is essential for hepatic differentiation from human embryonic stem cells. Sci. Rep. 2016, 6, 37388. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Ghosh, S.; Das, S. Self-regulation of rat liver gap junction by phosphorylation. Biochim. Biophys. Acta 2002, 1564, 500–504. [Google Scholar] [CrossRef]
- Ghosh, P. Self-phosphorylation modulates the gating of rat liver gap junction channels: A nonstationary noise analysis. Biophys. Chem. 2007, 127, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.L.; Pontifex, T.K.; Li, H.; Solan, J.L.; Lampe, P.D.; Sorgen, P.L.; Burt, J.M. Regulation of cx37 channel and growth-suppressive properties by phosphorylation. J. Cell Sci. 2017, 130, 3308–3321. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Kanter, E.M.; Huang, R.Y.; Maxeiner, S.; Frank, M.; Zhang, Y.; Schuessler, R.B.; Smith, T.W.; Townsend, R.R.; Rohrs, H.W.; et al. Residual Cx45 and its relationship to Cx43 in murine ventricular myocardium. Channels (Austin) 2011, 5, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.R.; Ross, J.; Rizzo, M.J.; Straub, A.C.; Lampe, P.D.; Leitinger, N.; Isakson, B.E. Oxidized phospholipid species promote in vivo differential Cx43 phosphorylation and vascular smooth muscle cell proliferation. Am. J. Pathol. 2009, 175, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.R.; Kroncke, B.M.; Straub, A.C.; Best, A.K.; Dunn, C.A.; Mitchell, L.A.; Peskova, Y.; Nakamoto, R.K.; Koval, M.; Lo, C.W.; et al. Mapk phosphorylation of connexin 43 promotes binding of cyclin e and smooth muscle cell proliferation. Circ. Res. 2012, 111, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.M.; Akpovi, C.D.; Chen, L.; Kumar, N.M.; Vitale, M.L. Complementary expression and phosphorylation of Cx46 and Cx50 during development and following gene deletion in mouse and in normal and orchitic mink testes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R255–R276. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.J.; Zeilinger, C.; Bintig, W.; Kolb, H.A.; Ngezahayo, A. Phosphorylation in the C-terminus of the rat connexin46 (rCx46) and regulation of the conducting activity of the formed connexons. J. Bioenergy Biomembr. 2008, 40, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ek Vitorin, J.F.; Weintraub, S.T.; Gu, S.; Shi, Q.; Burt, J.M.; Jiang, J.X. Phosphorylation of connexin 50 by protein kinase a enhances gap junction and hemichannel function. J. Biol. Chem. 2011, 286, 16914–16928. [Google Scholar] [CrossRef] [PubMed]
- May, D.; Tress, O.; Seifert, G.; Willecke, K. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of connexin43 protein in astrocytes. J. Neurosci. 2013, 33, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Traub, O.; Look, J.; Dermietzel, R.; Brummer, F.; Hulser, D.; Willecke, K. Comparative characterization of the 21-kD and 26-kD gap junction proteins in murine liver and cultured hepatocytes. J. Cell Biol. 1989, 108, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Elvira, M.; Díez, J.A.; Wang, K.K.; Villalobo, A. Phosphorylation of connexin-32 by protein kinase C prevents its proteolysis by mu-calpain and m-calpain. J. Biol. Chem. 1993, 268, 14294–14300. [Google Scholar] [PubMed]
- Locke, D.; Koreen, I.V.; Harris, A.L. Isoelectric points and post-translational modifications of connexin26 and connexin32. FASEB J. 2006, 20, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Locke, D.; Bian, S.; Li, H.; Harris, A.L. Post-translational modifications of connexin26 revealed by mass spectrometry. Biochem. J. 2009, 424, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Beyer, E.C.; Kurata, W.E.; Lau, A.F.; Lampe, P.D. The gap-junction protein connexin 56 is phosphorylated in the intracellular loop and the carboxy-terminal region. Eur. J. Biochem. 1997, 244, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Winbow, V.M.; Patel, L.S.; Burr, G.S.; Mitchell, C.K.; O’Brien, J. Protein kinase a mediates regulation of gap junctions containing connexin35 through a complex pathway. Brain Res. Mol. Brain Res. 2005, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.; Isakson, B.; Locke, D. Biological and biophysical properties of vascular connexin channels. Int. Rev. Cell Mol. Biol. 2009, 278, 69–118. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Gouw, J.W.; Naus, C.C.; Foster, L.J. Connexin multi-site phosphorylation: Mass spectrometry-based proteomics fills the gap. Biochim. Biophys. Acta 2013, 1828, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Duffy, H.S.; Spray, D.C.; Delmar, M. Ph-dependent dimerization of the carboxyl terminal domain of cx43. Biophys. J. 2004, 87, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Duffy, H.S.; Sahoo, P.; Coombs, W.; Delmar, M.; Spray, D.C. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J. Biol. Chem. 2004, 279, 54695–54701. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, D.; Kieken, F.; Kellezi, A.; Sorgen, P.L. Structural changes in the carboxyl terminus of the gap junction protein connexin 40 caused by the interaction with c-Src and zonula occludens-1. Cell Commun. Adhes. 2008, 15, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta 2018, 1860, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grosely, R.; Kopanic, J.L.; Nabors, S.; Kieken, F.; Spagnol, G.; Al-Mugotir, M.; Zach, S.; Sorgen, P.L. Effects of phosphorylation on the structure and backbone dynamics of the intrinsically disordered connexin43 C-terminal domain. J. Biol. Chem. 2013, 288, 24857–24870. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Duffy, H.S.; Cahill, S.M.; Coombs, W.; Spray, D.C.; Delmar, M.; Girvin, M.E. Sequence-specific resonance assignment of the carboxyl terminal domain of connexin43. J. Biomol. NMR 2002, 23, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Banks, E.A.; Yu, X.S.; Gu, S.; Lauer, J.; Fields, G.B.; Jiang, J.X. Amino acid residue val362 plays a critical role in maintaining the structure of C terminus of connexin 50 and in lens epithelial-fiber differentiation. J. Biol. Chem. 2010, 285, 18415–18422. [Google Scholar] [CrossRef] [PubMed]
- Kopanic, J.L.; Sorgen, P.L. Chemical shift assignments of the connexin45 carboxyl terminal domain: Monomer and dimer conformations. Biomol. NMR Assign. 2013, 7, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Kopanic, J.L.; Al-mugotir, M.H.; Kieken, F.; Zach, S.; Trease, A.J.; Sorgen, P.L. Characterization of the connexin45 carboxyl-terminal domain structure and interactions with molecular partners. Biophys. J. 2014, 106, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.W.; Berthoud, V.M.; Kurutz, J.; Minogue, P.J.; Greenspan, M.; Hanck, D.A.; Beyer, E.C. The n terminus of connexin37 contains an α-helix that is required for channel function. J. Biol. Chem. 2009, 284, 20418–20427. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, D.; Spagnol, G.; Chenavas, S.; Kieken, F.; Vitrac, H.; Brownell, S.; Kellezi, A.; Forge, V.; Sorgen, P.L. Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl-terminal and cytoplasmic loop domains. J. Biol. Chem. 2009, 284, 34257–34271. [Google Scholar] [CrossRef] [PubMed]
- Grosely, R.; Kieken, F.; Sorgen, P.L. 1h, 13c, and 15n backbone resonance assignments of the connexin43 carboxyl terminal domain attached to the 4th transmembrane domain in detergent micelles. Biomol. NMR Assign. 2013, 7, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Solan, J.L.; Gaietta, G.M.; Ngan, L.; Lee, G.J.; Mackey, M.R.; Lampe, P.D. The C-terminus of connexin43 adopts different conformations in the golgi and gap junction as detected with structure-specific antibodies. Biochem. J. 2007, 408, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Grosely, R.; Kieken, F.; Sorgen, P.L. Optimizing the solution conditions to solve the structure of the connexin43 carboxyl terminus attached to the 4(th) transmembrane domain in detergent micelles. Cell Commun. Adhes. 2010, 17, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Hirst-Jensen, B.J.; Sahoo, P.; Kieken, F.; Delmar, M.; Sorgen, P.L. Characterization of the PH-dependent interaction between the gap junction protein connexin43 carboxyl terminus and cytoplasmic loop domains. J. Biol. Chem. 2007, 282, 5801–5813. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Sorgen, P.L.; Girvin, M.E.; O’Donnell, P.; Coombs, W.; Taffet, S.M.; Delmar, M.; Spray, D.C. Ph-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J. Biol. Chem. 2002, 277, 36706–36714. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; Burt, J.M. Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim. Biophys. Acta 2013, 1828, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.P. Connexin phosphorylation as a regulatory event linked to channel gating. Biochim. Biophys. Acta 2005, 1711, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Nairn, A.C.; Czernik, A.J.; Spray, D.C.; Hertzberg, E.L.; Greengard, P.; Bennett, M.V. Phosphorylation of connexin 32, a hepatocyte gap-junction protein, by cAMP-dependent protein kinase, protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Eur. J. Biochem. 1990, 192, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Saheki, S.; Shimazu, T.; Takeuchi, N. Phosphorylation of the 27-kDa gap junction protein by protein kinase c in vitro and in rat hepatocytes. J. Biochem. 1989, 106, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Diez, J.A.; Elvira, M.; Villalobo, A. The epidermal growth factor receptor tyrosine kinase phosphorylates connexin32. Mol. Cell. Biochem. 1998, 187, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kothmann, W.W.; Li, X.; Burr, G.S.; O’Brien, J. Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis. Neurosci. 2007, 24, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chuang, A.Z.; O’Brien, J. Photoreceptor coupling is controlled by connexin 35 phosphorylation in zebrafish retina. J. Neurosci. 2009, 29, 15178–15186. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.S.; Mitchell, C.K.; Dubinsky, W.P.; O’Brien, J. Regulation of gap junction coupling through the neuronal connexin Cx35 by nitric oxide and cGMP. Cell Commun. Adhes. 2006, 13, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chuang, A.Z.; O’Brien, J. Regulation of photoreceptor gap junction phosphorylation by adenosine in zebrafish retina. Vis. Neurosci. 2014, 31, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Kjenseth, A.; Fykerud, T.A.; Sirnes, S.; Bruun, J.; Yohannes, Z.; Kolberg, M.; Omori, Y.; Rivedal, E.; Leithe, E. The gap junction channel protein connexin 43 is covalently modified and regulated by sumoylation. J. Biol. Chem. 2012, 287, 15851–15861. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Laing, J.G.; Kanter, E.M.; Berthoud, V.M.; Bao, M.; Rohrs, H.W.; Townsend, R.R.; Yamada, K.A. Identification of CaMKII phosphorylation sites in connexin43 by high-resolution mass spectrometry. J. Proteome Res. 2011, 10, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Lin, R.; Warn-Cramer, B.J.; Lau, A.F.; Burt, J.M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 2003, 284, C511–C520. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Warn-Cramer, B.J.; Kurata, W.E.; Lau, A.F. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J. Cell Biol. 2001, 154, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Hengeveld, T.; Postma, F.R.; Moolenaar, W.H. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J. Biol. Chem. 2001, 276, 8544–8549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kasperek, E.M.; Nicholson, B.J. Dissection of the molecular basis of pp60(v-Src) induced gating of connexin 43 gap junction channels. J. Cell Biol. 1999, 144, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Swenson, K.I.; Piwnica-Worms, H.; McNamee, H.; Paul, D.L. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-Src-induced inhibition of communication. Cell Regul. 1990, 1, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Kurata, W.E.; Warn-Cramer, B.J.; Lau, A.F. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 1998, 111 Pt 6, 833–841. [Google Scholar] [PubMed]
- Kanemitsu, M.Y.; Jiang, W.; Eckhart, W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998, 9, 13–21. [Google Scholar] [PubMed]
- Sirnes, S.; Kjenseth, A.; Leithe, E.; Rivedal, E. Interplay between pkc and the map kinase pathway in connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Dang, X.; Ping, P.; Fandrich, R.R.; Nickel, B.E.; Jin, Y.; Cattini, P.A.; Kardami, E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004, 117, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Srisakuldee, W.; Jeyaraman, M.M.; Nickel, B.E.; Tanguy, S.; Jiang, Z.S.; Kardami, E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 2009, 83, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Billaud, M.; Johnstone, S.R.; Best, A.K.; Yemen, S.; Dwyer, S.T.; Looft-Wilson, R.; Lysiak, J.J.; Gaston, B.; Palmer, L.; et al. Compartmentalized connexin 43 S-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.J.; Chen, Q.; Chen, L.J.; Shu, Y.; Bu, L.L.; Shao, X.Y.; Zhang, P.; Jiao, F.J.; Shi, J.; Tian, B. Phosphorylation of connexin 43 by cdk5 modulates neuronal migration during embryonic brain development. Mol. Neurobiol. 2016, 53, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Procida, K.; Jorgensen, L.; Schmitt, N.; Delmar, M.; Taffet, S.M.; Holstein-Rathlou, N.H.; Nielsen, M.S.; Braunstein, T.H. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm 2009, 6, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Decker, K.F.; Kanter, E.; Mohler, P.J.; Boyden, P.A.; Schuessler, R.B.; Yamada, K.A.; Rudy, Y. Role of activated CaMKII in abnormal calcium homeostasis and i(na) remodeling after myocardial infarction: Insights from mathematical modeling. J. Mol. Cell. Cardiol. 2008, 45, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.D.; Lampe, P.D. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 2002, 277, 44962–44968. [Google Scholar] [CrossRef] [PubMed]
- Paulson, A.F.; Lampe, P.D.; Meyer, R.A.; TenBroek, E.; Atkinson, M.M.; Walseth, T.F.; Johnson, R.G. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J. Cell Sci. 2000, 113 Pt 17, 3037–3049. [Google Scholar] [PubMed]
- Darrow, B.J.; Fast, V.G.; Kleber, A.G.; Beyer, E.C.; Saffitz, J.E. Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl camp-treated cultured cardiac myocytes. Circ. Res. 1996, 79, 174–183. [Google Scholar] [CrossRef] [PubMed]
- TenBroek, E.M.; Lampe, P.D.; Solan, J.L.; Reynhout, J.K.; Johnson, R.G. Ser364 of connexin43 and the upregulation of gap junction assembly by camp. J. Cell Biol. 2001, 155, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, L.N.; Stahlhut, M.; Mohammed, S.; Larsen, B.D.; Nielsen, M.S.; Holstein-Rathlou, N.H.; Andersen, S.; Jensen, O.N.; Hennan, J.K.; Kjolbye, A.L. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J. Mol. Cell. Cardiol. 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Martinez, A.M.; Fletcher, W.H. The connexin43 gap junction protein is phosphorylated by protein kinase a and protein kinase c: In vivo and in vitro studies. Mol. Cell. Biochem. 2002, 238, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Yogo, K.; Ogawa, T.; Akiyama, M.; Ishida, N.; Takeya, T. Identification and functional analysis of novel phosphorylation sites in Cx43 in rat primary granulosa cells. FEBS Lett. 2002, 531, 132–136. [Google Scholar] [CrossRef]
- Zou, J.; Yue, X.Y.; Zheng, S.C.; Zhang, G.; Chang, H.; Liao, Y.C.; Zhang, Y.; Xue, M.Q.; Qi, Z. Cholesterol modulates function of connexin 43 gap junction channel via PKC pathway in H9c2 cells. Biochim. Biophys. Acta 2014, 1838, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; King, T.J.; Heyman, N.S.; Lampe, P.D.; Burt, J.M. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 2006, 98, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Hu, S.; Wei, Y. Cardiomyopathy-associated gene 1-sensitive PKC-dependent connexin 43 expression and phosphorylation in left ventricular noncompaction cardiomyopathy. Cell. Physiol. Biochem. 2017, 44, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Altenberg, G.A.; Reuss, L. Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C-mediated phosphorylation. Am. J. Physiol. Cell Physiol. 2004, 286, C647–C654. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.K.; Cheng, H.H.; Wang, S.D.; Yeih, D.F.; Wang, S.M. Pkcvarepsilon mediates serine phosphorylation of connexin43 induced by lysophosphatidylcholine in neonatal rat cardiomyocytes. Toxicology 2013, 314, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Lampe, P.D. Injury-triggered akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Wallick, C.J.; Martyn, K.D.; Lau, A.F.; Jin, C.; Warn-Cramer, B.J. Akt phosphorylates connexin43 on ser373, a “mode-1” binding site for 14-3-3. Cell Commun. Adhes. 2007, 14, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Westphale, E.M.; Grigoryeva, A.; Beyer, E.C. Pkc isoenzymes in the chicken lens and TPA-induced effects on intercellular communication. Investig. Ophthalmol. Vis. Sci. 2000, 41, 850–858. [Google Scholar]
- Isakson, B.E. Localized expression of an Ins(1,4,5)P3 receptor at the myoendothelial junction selectively regulates heterocellular Ca2+ communication. J. Cell Sci. 2008, 121, 3664–3673. [Google Scholar] [CrossRef] [PubMed]
- Isakson, B.E.; Ramos, S.I.; Duling, B.R. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ. Res. 2007, 100, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Johnstone, S.R.; Heberlein, K.R.; Rizzo, M.J.; Best, A.K.; Boitano, S.; Isakson, B.E. Site-specific connexin phosphorylation is associated with reduced heterocellular communication between smooth muscle and endothelium. J. Vasc. Res. 2010, 47, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Revel, J.P.; Karnovsky, M.J. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J. Cell Biol. 1967, 33, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at s365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of connexin43 phosphorylated at s325, s328 and s330 in normoxic and ischemic heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Jabr, R.I.; Hatch, F.S.; Salvage, S.C.; Orlowski, A.; Lampe, P.D.; Fry, C.H. Regulation of gap junction conductance by calcineurin through Cx43 phosphorylation: Implications for action potential conduction. Pflugers Arch. 2016, 468, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Gres, P.; Skyschally, A.; Duschin, A.; Belosjorow, S.; Konietzka, I.; Heusch, G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J. 2003, 17, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Gourdie, R.G. The perinexus: A new feature of Cx43 gap junction organization. Heart Rhythm 2012, 9, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, J.A.; O’Quinn, M.P.; Barker, R.J.; Harris, B.S.; Jourdan, J.; Gourdie, R.G. ZO-1 determines adherens and gap junction localization at intercalated disks. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H583–H594. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Barker, R.J.; Hunter, A.W.; Zhang, Y.; Jourdan, J.; Gourdie, R.G. Quantitative analysis of ZO-1 colocalization with Cx43 gap junction plaques in cultures of rat neonatal cardiomyocytes. Microsc. Microanal. 2005, 11, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Görge, P.M.; Görbe, A.; Ferdinandy, P.; Lampe, P.D.; Leybaert, L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 2015, 153, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Soder, B.L.; Propst, J.T.; Brooks, T.M.; Goodwin, R.L.; Friedman, H.I.; Yost, M.J.; Gourdie, R.G. The connexin43 carboxyl-terminal peptide act1 modulates the biological response to silicone implants. Plast. Reconstr. Surg. 2009, 123, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Su, G.Y.; Wang, J.; Xu, Z.X.; Qiao, X.J.; Zhong, J.Q.; Zhang, Y. Effects of rotigaptide (ZP123) on connexin43 remodeling in canine ventricular fibrillation. Mol. Med. Rep. 2015, 12, 5746–5752. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, M.; Petersen, J.S.; Hennan, J.K.; Ramirez, M.T. The antiarrhythmic peptide rotigaptide (ZP123) increases connexin 43 protein expression in neonatal rat ventricular cardiomyocytes. Cell Commun. Adhes. 2006, 13, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Larsen, B.D.; Petersen, J.S.; Mohr, F.W. Effects of the new antiarrhythmic peptide ZP123 on epicardial activation and repolarization pattern. Cell Commun. Adhes. 2003, 10, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Kjolbye, A.L.; Nielsen, M.S.; Petersen, J.S.; Harlow, K.W.; Holstein-Rathlou, N.H.; Martins, J.B. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J. Cardiovasc. Electrophysiol. 2003, 14, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Kjølbye, A.L.; Haugan, K.; Hennan, J.K.; Petersen, J.S. Pharmacological modulation of gap junction function with the novel compound rotigaptide: A promising new principle for prevention of arrhythmias. Basic Clin. Pharmacol. Toxicol. 2007, 101, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Walter, B.; Schultz Hansen, R.; Heusch, G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Pidkovka, N.A.; Sarmento, O.F.; Yoshida, T.; Gan, Q.; Adiguzel, E.; Bendeck, M.P.; Berliner, J.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce type viii collagen expression and vascular smooth muscle cell migration. Circ. Res. 2009, 104, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.S.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, N. Oxidized phospholipids as triggers of inflammation in atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Berliner, J.A.; Subbanagounder, G.G.; Bhunia, A.K.; Koh, S. Identification of a biologically active component in minimally oxidized low density lipoprotein (MM-LDL) responsible for aortic smooth muscle cell proliferation. Glycoconj. J. 2004, 20, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Nelson, T.K.; Simon, A.M.; Burt, J.M. A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 2011, 124, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Ek-Vitorín, J.F.; Burt, J.M. Extracellular loop cysteine mutant of Cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 2012, 245, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Ek-Vitorin, J.F.; Burt, J.M. Structural determinants and proliferative consequences of connexin 37 hemichannel function in insulinoma cells. J. Biol. Chem. 2014, 289, 30379–30386. [Google Scholar] [CrossRef] [PubMed]
- Traub, O.; Hertlein, B.; Kasper, M.; Eckert, R.; Krisciukaitis, A.; Hülser, D.; Willecke, K. Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human hela cells. Eur. J. Cell Biol. 1998, 77, 313–322. [Google Scholar] [CrossRef]
- Morel, S.; Burnier, L.; Roatti, A.; Chassot, A.; Roth, I.; Sutter, E.; Galan, K.; Pfenniger, A.; Chanson, M.; Kwak, B.R. Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis 2010, 31, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.M.; Seul, K.H.; Berthoud, V.M.; Lau, A.F.; Sagar, G.D.; Beyer, E.C. Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell. Biol. Res. Commun. 2000, 3, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Vanhamme, L.; Rolin, S.; Szpirer, C. Inhibition of gap-junctional intercellular communication between epithelial cells transformed by the activated h-ras-1 oncogene. Exp. Cell Res. 1989, 180, 297–301. [Google Scholar] [CrossRef]
- Azarnia, R.; Reddy, S.; Kmiecik, T.E.; Shalloway, D.; Loewenstein, W.R. The cellular SRC gene product regulates junctional cell-to-cell communication. Science 1988, 239, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, A.; Jaraiz-Rodriguez, M.; Dominguez-Prieto, M.; Herrero-Gonzalez, S.; Medina, J.M.; Tabernero, A. Connexin43 recruits PTEN and Csk to inhibit c-Src activity in glioma cells and astrocytes. Oncotarget 2016, 7, 49819–49833. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Mitra, S.; Katoch, P.; Kelsey, L.S.; Johnson, K.R.; Mehta, P.P. Phosphorylation on ser-279 and ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 2013, 24, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Brissette, J.L.; Kumar, N.M.; Gilula, N.B.; Dotto, G.P. The tumor promoter 12-O-tetradecanoylphorbol-13-acetate and the ras oncogene modulate expression and phosphorylation of gap junction proteins. Mol. Cell. Biol. 1991, 11, 5364–5371. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Grupen, C.G.; Murray, A.W. Phorbol ester induces phosphorylation and down-regulation of connexin 43 in wb cells. Biochim. Biophys. Acta 1991, 1094, 243–245. [Google Scholar] [CrossRef]
- Asamoto, M.; Oyamada, M.; el Aoumari, A.; Gros, D.; Yamasaki, H. Molecular mechanisms of TPA-mediated inhibition of gap-junctional intercellular communication: Evidence for action on the assembly or function but not the expression of connexin 43 in rat liver epithelial cells. Mol. Carcinog. 1991, 4, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Ruch, R.J.; Trosko, J.E.; Madhukar, B.V. Inhibition of connexin43 gap junctional intercellular communication by tpa requires erk activation. J. Cell. Biochem. 2001, 83, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Y.; Jiang, Q.H.; Hong, T.; Zhang, Z.Y.; Yang, R.J.; Huang, J.Q.; Hu, K.; Peng, Y.P. Altered expression of connexin43 and phosphorylation connexin43 in glioma tumors. Int. J. Clin. Exp. Pathol. 2015, 8, 4296–4306. [Google Scholar] [PubMed]
- Wu, J.F.; Ji, J.; Dong, S.Y.; Li, B.B.; Yu, M.L.; Wu, D.D.; Tao, L.; Tong, X.H. Gefitinib enhances oxaliplatin-induced apoptosis mediated by Src and PKC-modulated gap junction function. Oncol. Rep. 2016, 36, 3251–3258. [Google Scholar] [CrossRef] [PubMed]
- Peterson-Roth, E.; Brdlik, C.M.; Glazer, P.M. Src-induced cisplatin resistance mediated by cell-to-cell communication. Cancer Res. 2009, 69, 3619–3624. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Tan, T.; Chan, C.; Laxton, V.; Chan, Y.W.; Liu, T.; Wong, W.T.; Tse, G. The role of connexins in wound healing and repair: Novel therapeutic approaches. Front. Physiol. 2016, 7, 596. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.L.; Thrasivoulou, C.; Phillips, A.R. Connexins in wound healing; perspectives in diabetic patients. Biochim. Biophys. Acta 2012, 1818, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, B.; Vinken, M.; Silva, T.C.; Araújo, C.M.M.; Aloia, T.P.A.; Chaible, L.M.; Mori, C.M.C.; Dagli, M.L.Z. Connexin 43 deficiency accelerates skin wound healing and extracellular matrix remodeling in mice. J. Dermatol. Sci. 2015, 79, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Ryu, J.M.; Suh, H.N.; Park, S.H.; Oh, Y.M.; Lee, S.H.; Han, H.J. Camp promotes cell migration through cell junctional complex dynamics and actin cytoskeleton remodeling: Implications in skin wound healing. Stem Cells Dev. 2015, 24, 2513–2524. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.P.; Yamamoto, M.; Rose, B. Transcription of the gene for the gap junctional protein connexin43 and expression of functional cell-to-cell channels are regulated by camp. Mol. Biol. Cell 1992, 3, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.S.; Dunn, C.A.; Carter, W.G.; Usui, M.L.; Olerud, J.E.; Lampe, P.D. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 2004, 167, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Pollok, S.; Pfeiffer, A.C.; Lobmann, R.; Wright, C.S.; Moll, I.; Martin, P.E.; Brandner, J.M. Connexin 43 mimetic peptide gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J. Cell. Mol. Med. 2011, 15, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Kinase programs spatiotemporally regulate gap junction assembly and disassembly: Effects on wound repair. Semin. Cell Dev. Biol. 2016, 50, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Lincoln, J.; Cook, J.E.; Becker, D.L. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes 2007, 56, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Houdek, P.; Hüsing, B.; Kaiser, C.; Moll, I. Connexins 26, 30, and 43: Differences among spontaneous, chronic, and accelerated human wound healing. J. Investig. Dermatol. 2004, 122, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A multicenter randomized controlled trial evaluating a Cx43-mimetic peptide in cutaneous scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; García, I.E.; Pinto, B.I.; Pupo, A.; Báez, D.; Stehberg, J.; Del Rio, R.; González, C. Extracellular cysteine in connexins: Role as redox sensors. Front. Physiol. 2016, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.E.; Sánchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Sáez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Sáez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channels. Neuropharmacology 2013, 75, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Füller, M.; Pohl, U.; Kameritsch, P. No, via its target Cx37, modulates calcium signal propagation selectively at myoendothelial gap junctions. Cell Commun. Signal. CCS 2014, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Mannell, H.; Blodow, S.; Schneider, H.; Schubert, K.M.; Qiu, J.; Schmidt, A.; Imhof, A.; Beck, H.; Tanase, L.I.; et al. No augments endothelial reactivity by reducing myoendothelial calcium signal spreading: A novel role for Cx37 (connexin 37) and the protein tyrosine phosphatase SHP-2. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The sumo pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Paul, D.L.; Goodenough, D.A. Posttranslational phosphorylation of lens fiber connexin46: A slow occurrence. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3558–3565. [Google Scholar]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Fallon, R.F.; Goodenough, D.A. Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 1981, 90, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.P.; Baena, V.; Terasaki, M. Localization of phosphorylated connexin 43 using serial section immunogold electron microscopy. J. Cell Sci. 2017, 130, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Boassa, D.; Solan, J.L.; Papas, A.; Thornton, P.; Lampe, P.D.; Sosinsky, G.E. Trafficking and recycling of the connexin43 gap junction protein during mitosis. Traffic 2010, 11, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Piehl, M.; Lehmann, C.; Gumpert, A.; Denizot, J.P.; Segretain, D.; Falk, M.M. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol. Biol. Cell 2007, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.W.; Gourdie, R.G. The second pdz domain of zonula occludens-1 is dispensable for targeting to connexin 43 gap junctions. Cell Commun. Adhes. 2008, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial f-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Sirnes, S.; Fykerud, T.; Kjenseth, A.; Rivedal, E. Endocytosis and post-endocytic sorting of connexins. Biochim. Biophys. Acta 2012, 1818, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.M.; Fong, J.T.; Kells, R.M.; O’Laughlin, M.C.; Kowal, T.J.; Thevenin, A.F. Degradation of endocytosed gap junctions by autophagosomal and endo-/lysosomal pathways: A perspective. J. Membr. Biol. 2012, 245, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Rivedal, E. Ubiquitination of gap junction proteins. J. Membr. Biol. 2007, 217, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Townley-Tilson, W.H.; Kang, E.Y.; Homeister, J.W.; Patterson, C. Sent to destroy: The ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ. Res. 2010, 106, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26s proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Girao, H.; Yuste, A.; Patel, B.; Marques, C.; Spray, D.C.; Pereira, P.; Cuervo, A.M. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol. Cell 2012, 23, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.T.; Kells, R.M.; Gumpert, A.M.; Marzillier, J.Y.; Davidson, M.W.; Falk, M.M. Internalized gap junctions are degraded by autophagy. Autophagy 2012, 8, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M. Autophagy: A pathway that contributes to connexin degradation. J. Cell Sci. 2011, 124, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Yuste, A.; Patel, B.; Stout, R.F., Jr.; Spray, D.C.; Cuervo, A.M. Connexins modulate autophagosome biogenesis. Nat. Cell Biol. 2014, 16, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, G.G.; Shah, M.H.; Halperin, V.L.; Cooke, C.A.; Akar, F.G.; Yen, T.E.; Kass, D.A.; Machamer, C.E.; Van Eyk, J.E.; Tomaselli, G.F. Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ. Res. 2010, 106, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Rivedal, E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J. Biol. Chem. 2004, 279, 50089–50096. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Ruan, Z.; Yang, X.; Chu, K.; Wu, H.; Li, Y.; Huang, Y. Connexin 31.1 degradation requires the clathrin-mediated autophagy in NSCLC cell h1299. J. Cell. Mol. Med. 2015, 19, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Catarino, S.; Ramalho, J.S.; Marques, C.; Pereira, P.; Girao, H. Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem. J. 2011, 437, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Girao, H.; Pereira, P. The proteasome regulates the interaction between Cx43 and ZO-1. J. Cell. Biochem. 2007, 102, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of AKT, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Totland, M.Z.; Bergsland, C.H.; Fykerud, T.A.; Knudsen, L.M.; Rasmussen, N.L.; Eide, P.W.; Yohannes, Z.; Sorensen, V.; Brech, A.; Lothe, R.A.; et al. The E3 ubiquitin ligase nedd4 induces endocytosis and lysosomal sorting of connexin 43 to promote loss of gap junctions. J. Cell Sci. 2017, 130, 2867–2882. [Google Scholar] [CrossRef] [PubMed]
- Spagnol, G.; Kieken, F.; Kopanic, J.L.; Li, H.; Zach, S.; Stauch, K.L.; Grosely, R.; Sorgen, P.L. Structural studies of the NEDD4 WW domains and their selectivity for the connexin43 (Cx43) carboxyl terminus. J. Biol. Chem. 2016, 291, 7637–7650. [Google Scholar] [CrossRef] [PubMed]
- Girao, H.; Catarino, S.; Pereira, P. Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp. Cell Res. 2009, 315, 3587–3597. [Google Scholar] [CrossRef] [PubMed]
- Auth, T.; Schluter, S.; Urschel, S.; Kussmann, P.; Sonntag, S.; Hoher, T.; Kreuzberg, M.M.; Dobrowolski, R.; Willecke, K. The TSG101 protein binds to connexins and is involved in connexin degradation. Exp. Cell Res. 2009, 315, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Kristensen, A.R.; Foster, L.J.; Naus, C.C. Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J. Proteome Res. 2012, 11, 6134–6146. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Harris, B.S.; Mentrup, H.L.; Abreha, M.; Thames, E.L.; Lea, J.B.; Swing, D.A.; Copeland, N.G.; Jenkins, N.A.; Price, R.L.; et al. Cardiomyocyte-specific overexpression of the ubiquitin ligase WWP1 contributes to reduction in connexin 43 and arrhythmogenesis. J. Mol. Cell. Cardiol. 2015, 88, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fykerud, T.A.; Kjenseth, A.; Schink, K.O.; Sirnes, S.; Bruun, J.; Omori, Y.; Brech, A.; Rivedal, E.; Leithe, E. Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J. Cell Sci. 2012, 125, 3966–3976. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.L.; Lai, S.Y.; Lai, W.A.; Lee, M.T.; Liao, C.F.; Ke, F.C.; Hwang, J.J. CRTC2 and NEDD4 ligase involvement in FSH and TGFβ1 upregulation of connexin43 gap junction. J. Mol. Endocrinol. 2015, 55, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Gurtner, A.; Rosati, J.; Illi, B.; Ragone, G.; Piaggio, G.; Moggio, M.; Lamperti, C.; D’Angelo, G.; Clementi, E.; et al. Nitric oxide deficiency determines global chromatin changes in duchenne muscular dystrophy. FASEB J. 2009, 23, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Azzimato, V.; Colussi, C.; Florio, M.C.; Binda, A.; Panariti, A.; Qanud, K.; Suffredini, S.; Gennaccaro, L.; Miragoli, M.; et al. Acetylation mediates Cx43 reduction caused by electrical stimulation. J. Mol. Cell. Cardiol. 2015, 87, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Carette, D.; Gilleron, J.; Denizot, J.P.; Grant, K.; Pointis, G.; Segretain, D. New cellular mechanisms of gap junction degradation and recycling. Biol. Cell Auspices Eur. Cell Biol. Org. 2015, 107, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Koval, M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006, 16, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Smith, T.D.; Sarma, J.D.; Ritzenthaler, J.D.; Maza, J.; Kaplan, B.E.; Cunningham, L.A.; Suaud, L.; Hubbard, M.J.; Rubenstein, R.C.; et al. ERP29 restricts connexin43 oligomerization in the endoplasmic reticulum. Mol. Biol. Cell 2009, 20, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Maza, J.; Das Sarma, J.; Koval, M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 21115–21121. [Google Scholar] [CrossRef] [PubMed]
- Jara, O.; Acuna, R.; Garcia, I.E.; Maripillan, J.; Figueroa, V.; Saez, J.C.; Araya-Secchi, R.; Lagos, C.F.; Perez-Acle, T.; Berthoud, V.M.; et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell 2012, 23, 3299–3311. [Google Scholar] [CrossRef] [PubMed]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Koval, M.; Harley, J.E.; Hick, E.; Steinberg, T.H. Connexin46 is retained as monomers in a trans-golgi compartment of osteoblastic cells. J. Cell Biol. 1997, 137, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.D.; Mohankumar, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M.; Koval, M. Cytoplasmic amino acids within the membrane interface region influence connexin oligomerization. J. Membr. Biol. 2012, 245, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Lagree, V.; Brunschwig, K.; Lopez, P.; Gilula, N.B.; Richard, G.; Falk, M.M. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J. Cell Sci. 2003, 116, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.A.; Stauffer, B.; Moriarty, H.K.; Kim, A.H.; McCarty, N.A.; Koval, M. Junctional abnormalities in human airway epithelial cells expressing F508DEL CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L475–L487. [Google Scholar] [CrossRef] [PubMed]
- Suaud, L.; Miller, K.; Alvey, L.; Yan, W.; Robay, A.; Kebler, C.; Kreindler, J.L.; Guttentag, S.; Hubbard, M.J.; Rubenstein, R.C. Erp29 regulates deltaf508 and wild-type cystic fibrosis transmembrane conductance regulator (CFTR) trafficking to the plasma membrane in cystic fibrosis (CF) and non-CF epithelial cells. J. Biol. Chem. 2011, 286, 21239–21253. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, J.; Kaplan, B.E.; Willemsen, D.; Koval, M. Identification of Rab20 as a potential regulator of connexin43 trafficking. Cell Commun. Adhes. 2008, 15, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Appelskog, I.B.; Ammerpohl, O.; Ekstrom, T.J.; Almqvist, P.M. Histone deacetylase inhibitor 4-phenylbutyrate modulates glial fibrillary acidic protein and connexin 43 expression, and enhances gap-junction communication, in human glioblastoma cells. Eur. J Cancer 2004, 40, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Fukushima, M.; Maitani, Y. Non-viral delivery of the connexin 43 gene with histone deacetylase inhibitor to human nasopharyngeal tumor cells enhances gene expression and inhibits in vivo tumor growth. Int. J. Oncol. 2007, 30, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Akhtar, M.; Asklund, T.; Juliusson, B.; Almqvist, P.M.; Ekstrom, T.J. Hdac inhibition amplifies gap junction communication in neural progenitors: Potential for cell-mediated enzyme prodrug therapy. Exp. Cell Res. 2007, 313, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Minogue, P.J.; Guo, J.; Williamson, E.K.; Xu, X.; Ebihara, L.; Beyer, E.C. Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur. J. Cell Biol. 2003, 82, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; Gordon, C.; Bergamaschi, R.; Wang, H.Z.; Cohen, I.S.; Valiunas, V.; Brink, P.R. The effects of the histone deacetylase inhibitor 4-phenylbutyrate on gap junction conductance and permeability. Front. Pharmacol. 2013, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Minogue, P.J.; Lambert, P.A.; Snabb, J.I.; Beyer, E.C. The cataract-linked mutant connexin50D47A causes endoplasmic reticulum stress in mouse lenses. J. Biol. Chem. 2016, 291, 17569–17578. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.; Gaietta, G.M.; Deerinck, T.J.; Crum, J.; Sosinsky, G.E.; Beyer, E.C.; Berthoud, V.M. The cytoplasmic accumulations of the cataract-associated mutant, connexin50P88S, are long-lived and form in the endoplasmic reticulum. Exp. Eye Res. 2009, 88, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Alapure, B.V.; Stull, J.K.; Firtina, Z.; Duncan, M.K. The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp. Eye Res. 2012, 102, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, D.; Scott, C.A.; Gray, C.; Zicha, D.; Kelsell, D.P. Ekv mutant connexin 31 associated cell death is mediated by er stress. Hum. Mol. Genet. 2009, 18, 4734–4745. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Ma, H.; Xiong, H.; Pan, Q.; Huang, L.; Wang, D.; Zhang, Z. Trafficking abnormality and ER stress underlie functional deficiency of hearing impairment-associated connexin-31 mutants. Protein Cell 2010, 1, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, D. Friend or foe: Endoplasmic reticulum protein 29 (ERP29) in epithelial cancer. FEBS Open Biol. 2015, 5, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Spagnol, G.; Pontifex, T.K.; Burt, J.M.; Sorgen, P.L. Chemical shift assignments of the connexin37 carboxyl terminal domain. Biomol. NMR Assign. 2017, 11, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, B.; Schadzek, P.; Hemmerling, F.; Schaarschmidt, F.; Heisterkamp, A.; Ngezahayo, A. The role of the C-terminus in functional expression and internalization of rat connexin46 (rcx46). J. Bioenergy Biomembr. 2013, 45, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Laing, J.G.; Koval, M.; Steinberg, T.H. Association with ZO-1 correlates with plasma membrane partitioning in truncated connexin45 mutants. J. Membr. Biol. 2005, 207, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Trease, A.J.; Capuccino, J.M.V.; Contreras, J.; Harris, A.L.; Sorgen, P.L. Intramolecular signaling in a cardiac connexin: Role of cytoplasmic domain dimerization. J. Mol. Cell. Cardiol. 2017, 111, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Calero, G.; Kanemitsu, M.; Taffet, S.M.; Lau, A.F.; Delmar, M. A 17MER peptide interferes with acidification-induced uncoupling of connexin43. Circ. Res. 1998, 82, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulos, K.; Alvarado, J.L.; Mastroianni, M.; Ek-Vitorin, J.F.; Taffet, S.M.; Delmar, M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 1999, 84, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Das, S.; Molina, S.A.; Madgwick, D.; Katz, M.R.; Jena, S.; Bossmann, L.K.; Pal, D.; Takemoto, D.J. Investigation of the reciprocal relationship between the expression of two gap junction connexin proteins, connexin46 and connexin43. J. Biol. Chem. 2011, 286, 24519–24533. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.; Shaw, R. The “tail” of connexin43: An unexpected journey from alternative translation to trafficking. Biochim. Biophys. Acta 2016, 1863, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [PubMed]
- James, C.C.; Zeitz, M.J.; Calhoun, P.J.; Lamouille, S.; Smyth, J.W. Altered translation initiation of GJA1 limits gap junction formation during epithelial-mesenchymal transition. Mol. Biol. Cell 2018, 29, 797–808. [Google Scholar] [CrossRef]
- Smyth, J.W.; Vogan, J.M.; Buch, P.J.; Zhang, S.S.; Fong, T.S.; Hong, T.T.; Shaw, R.M. Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circ. Res. 2012, 110, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef]
- Yamane, Y.; Shiga, H.; Asou, H.; Ito, E. Gap junctional channel inhibition alters actin organization and calcium propagation in rat cultured astrocytes. Neuroscience 2002, 112, 593–603. [Google Scholar] [CrossRef]
- Theiss, C.; Meller, K. Microinjected anti-actin antibodies decrease gap junctional intercellular commmunication in cultured astrocytes. Exp. Cell Res. 2002, 281, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Nowak, R.B.; Gao, J.; Sun, X.; Biswas, S.K.; Lo, W.K.; Mathias, R.T.; Fowler, V.M. Lens ion homeostasis relies on the assembly and/or stability of large connexin 46 gap junction plaques on the broad sides of differentiating fiber cells. Am. J. Physiol. Cell Physiol. 2015, 308, C835–C847. [Google Scholar] [CrossRef] [PubMed]
- Lauf, U.; Giepmans, B.N.; Lopez, P.; Braconnot, S.; Chen, S.C.; Falk, M.M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10446–10451. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Thevenin, A.F.; Margraf, R.A.; Fisher, C.G.; Kells-Andrews, R.M.; Falk, M.M. Phosphorylation regulates connexin43/zo-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 2017, 28, 3595–3608. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Fanning, A.S.; Bridges, A.; Anderson, J.M. Zo-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Van Itallie, C.M.; Anderson, J.M. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol. Biol. Cell 2012, 23, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Waxse, B.J.; Sengupta, P.; Hesketh, G.G.; Lippincott-Schwartz, J.; Buss, F. Myosin vi facilitates connexin 43 gap junction accretion. J. Cell Sci. 2017, 130, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lin, Y.P.; Mitchell, C.K.; Ram, S.; O’Brien, J. Two-color fluorescent analysis of connexin 36 turnover: Relationship to functional plasticity. J. Cell Sci. 2015, 128, 3888–3897. [Google Scholar] [CrossRef] [PubMed]
- Windoffer, R.; Beile, B.; Leibold, A.; Thomas, S.; Wilhelm, U.; Leube, R.E. Visualization of gap junction mobility in living cells. Cell Tissue Res. 2000, 299, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Two-Color Fluorescent Analysis of Connexin 36 Turnover—Relationship to Functional plasticity. Ph.D. Dissertation, University of Texas, Houston, UT, USA, May 2015. GSBS Dissertations and Theses (Open Access). [Google Scholar]
- Stahley, S.N.; Saito, M.; Faundez, V.; Koval, M.; Mattheyses, A.L.; Kowalczyk, A.P. Desmosome assembly and disassembly are membrane raft-dependent. PLoS ONE 2014, 9, e87809. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.M.; Tucker, D.K.; Kottke, M.D.; Saito, M.; Delva, E.; Hanakawa, Y.; Amagai, M.; Kowalczyk, A.P. Desmosome disassembly in response to pemphigus vulgaris igg occurs in distinct phases and can be reversed by expression of exogenous DSG3. J. Investig. Dermatol. 2011, 131, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Schlingmann, B.; Overgaard, C.E.; Molina, S.A.; Lynn, K.S.; Mitchell, L.A.; Dorsainvil White, S.; Mattheyses, A.L.; Guidot, D.M.; Capaldo, C.T.; Koval, M. Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat. Commun. 2016, 7, 12276. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Schlingmann, B.; Stecenko, A.A.; Guidot, D.M.; Koval, M. Nf-kb inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 2015, 3, e982424. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.E.; Schlingmann, B.; Dorsainvil White, S.; Ward, C.; Fan, X.; Swarnakar, S.; Brown, L.A.; Guidot, D.M.; Koval, M. The relative balance of GM-CSF and tgfbeta1 regulates lung epithelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1212–L1223. [Google Scholar] [CrossRef] [PubMed]
- Lafemina, M.J.; Rokkam, D.; Chandrasena, A.; Pan, J.; Bajaj, A.; Johnson, M.; Frank, J.A. Keratinocyte growth factor enhances barrier function without altering claudin expression in primary alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L724–L734. [Google Scholar] [CrossRef] [PubMed]



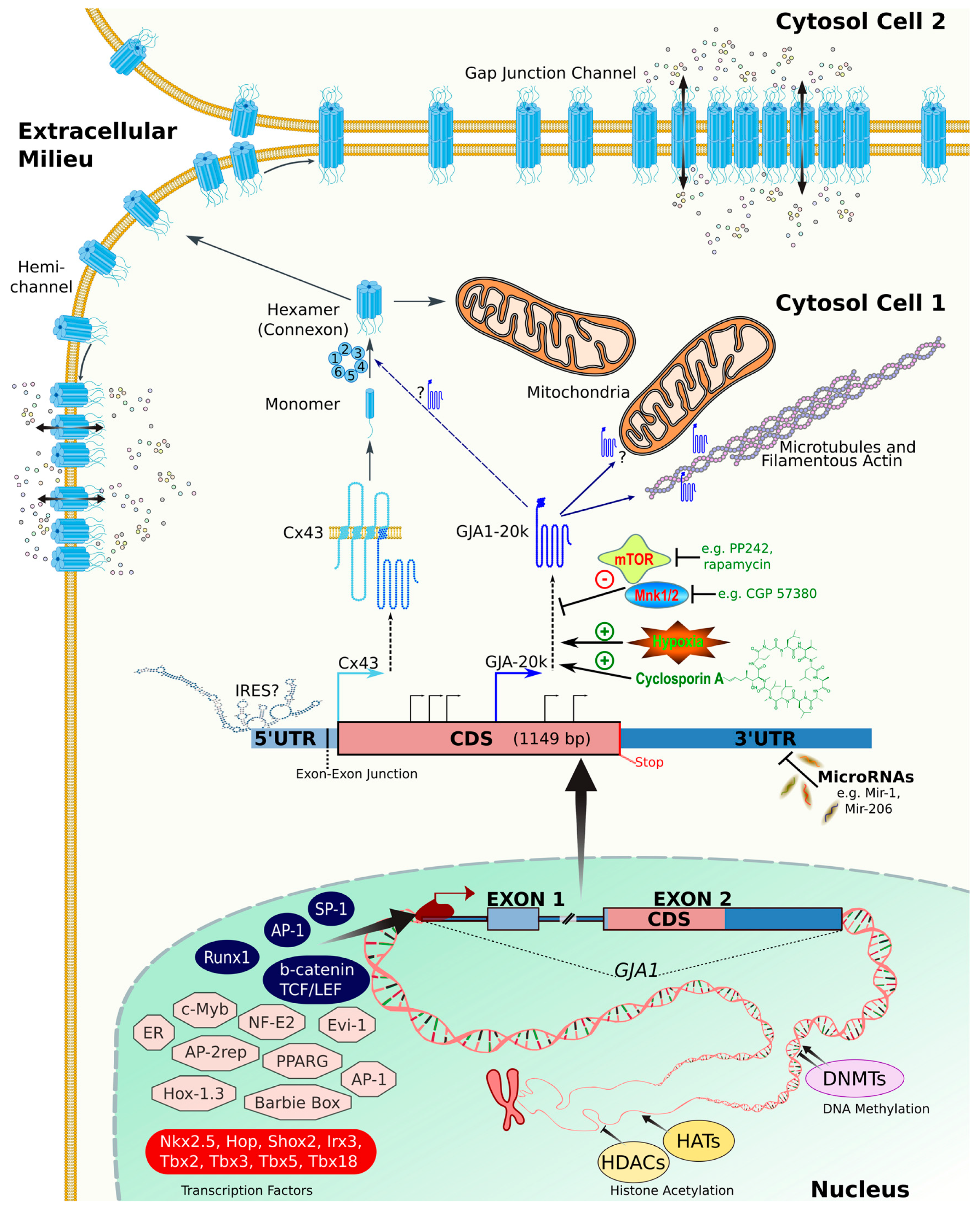{kind=link}
{kind=link}
| Connexin/Residue | PTM | GJIC | Expression | Refs. |
|---|---|---|---|---|
| Cx26 m.s./a: | ||||
| M1/K15/K102/ K103/K105/ K108/K112/K116 | Acetylation | ND | ND | [138,139] |
| N14/N113/ N170/N176 | Hydroxylation | ND | ND | |
| E42/E47/E114 | carboxylation | ND | ND | |
| K61/R75/ K221/K223 | Methylation | ND | ND | |
| T123/T177/S183/ T186/(Y233 or Y235 or Y240) | Phosphorylation | ND | ND | |
| Cx31(m): | ||||
| 263 b | CK1 | No change | No change | [124] |
| 266 b | CK1 | No change | No change | [124] |
| Cx32: | ||||
| S229 | PKC | Increase/Decrease | Increase/Decrease | [165] |
| S233 | PKA/PKC | Increase/Decrease | Increase/Decrease | [120,138,165,166] |
| S240 | ND | ND | ND | [138] |
| Y7/Y243 | EGFR tyrosine kinase | ND | ND | [167] |
| Cx35/Cx36: | ||||
| S110 | PKA/PKG | No change | Decrease | [141,168,169,170] |
| S276/293 | PKA/PKG | No change | Decrease | [141,168,169,170,171] |
| S289 | PKG (NO mediated) | ND | Decrease | [170] |
| Cx37: | ||||
| S275/S285/ S302/S319/S321/ S325/S329 | PKC | Increased | Decrease | [128] |
| Cx43: | ||||
| S5 m.s. | ND | ND | ND | [144] |
| K144 | SUMO | Increase | Increase | [172] |
| K237 | SUMO | Increase | Increase | [172] |
| S244 m.s. | CAMKII | ND | ND | [173] |
| Y247 c | Src | Decrease | Decrease c | [120,146,174,175,176,177,178] |
| S255 m.s. | CAMKII | ND | ND | [173] |
| P34cdc2 | Decrease | Decrease | [179,180] | |
| MAPK | No change/Decrease | No change | [120,131,148,181] | |
| S257 m.s. | PKG/CAMKII | ND | ND | [173] |
| S262 d | P34cdc2 | Decrease | Decrease | [179,180] |
| MAPK | Decrease | Decrease/no change | [120,131,148,182,183] | |
| PKCε a | Decrease | Decrease | [131,181,182] | |
| Y265 c | Src | Decrease | Decrease c | [120,146,174,175,176,177,178] |
| C271 | Nitrosylation | Increase | No change | [184] |
| S279 e | MAPK | Decrease | Decrease/no change | [131,148,174] |
| CDK5 | Decrease | [185] | ||
| S282 e | MAPK | Decrease | Decrease/no change | [131,148,174] |
| CDK5 | Decrease | Decrease | [185] | |
| S296 m.s. | CAMKII | ND | No change | [173,186] |
| S297 m.s. | CAMKII/PKCε | ND | No change | [173,186] |
| S306 m.s. | CAMKII | Decrease | Decrease associated with De-Phosph. | [173,186,187] |
| S314 m.s. | CAMKII | ND | ND | [173] |
| S325 m.s. | CAMKII | ND | ND | [173] |
| CK1 | Increase | Increase | [188] | |
| S328 m.s. | CAMKII | ND | ND | [173] |
| CK1 | Increase | Increase | [188] | |
| S330 m.s. | CAMKII | ND | ND | [173] |
| CK1 | Increase | Increase | [188] | |
| S364 m.s. | CAMKII | ND | ND | [173] |
| PKA | Increase | Increase | [120,189,190,191] | |
| S365 m.s. | CAMKII | ND | ND | [173] |
| PKA | Increase | Increase | ||
| PKC | Decrease | Decrease | [120,192,193,194] | |
| S368 f | PKCα | Increase/Preserved/ | Increase | [192,193,194,195,196] |
| PKCε | Decrease g | Decrease | [120,193,194,195,196,197,198,199] | |
| S369 m.s. | CAMKII | ND | ND | |
| PKA | Increase | No change | [173] | |
| PKC | Increase | Increase | [5,120,192,193,194] | |
| S372 m.s. | CAMKII | ND | ND | [173] |
| PKC | Decrease | Decrease | [5,148,192,193,200,201] | |
| S373 g,m.s. | Akt | Increase | Increase e | [200,201] |
| CAMKII | ND | ND | [173] | |
| PKC | Decrease | Decrease | ||
| PKA | Increase | Increase | [120,148,192,193,194] | |
| Cx45: | ||||
| S326/Y337/ S381/S382/S384/ S385/S387/S393 m.s. | CAMKII | ND | ND | [129] |
| S326/S382/S384/ S387/S393 m.s. | CK1 | ND | ND | [129] |
| Cx46 (Cx56 Chick homologue): | ||||
| S118 | PKCε | ND | Decrease | [140,202] |
| Cx50: | ||||
| S363 | CK1 | Increase | Increase | [120,193] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. https://doi.org/10.3390/ijms19051296
Aasen T, Johnstone S, Vidal-Brime L, Lynn KS, Koval M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. International Journal of Molecular Sciences. 2018; 19(5):1296. https://doi.org/10.3390/ijms19051296
Chicago/Turabian StyleAasen, Trond, Scott Johnstone, Laia Vidal-Brime, K. Sabrina Lynn, and Michael Koval. 2018. "Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease" International Journal of Molecular Sciences 19, no. 5: 1296. https://doi.org/10.3390/ijms19051296