Complete Chloroplast Genome of Cercis chuniana (Fabaceae) with Structural and Genetic Comparison to Six Species in Caesalpinioideae


Abstract
:
1. Introduction
2. Results and Discussion
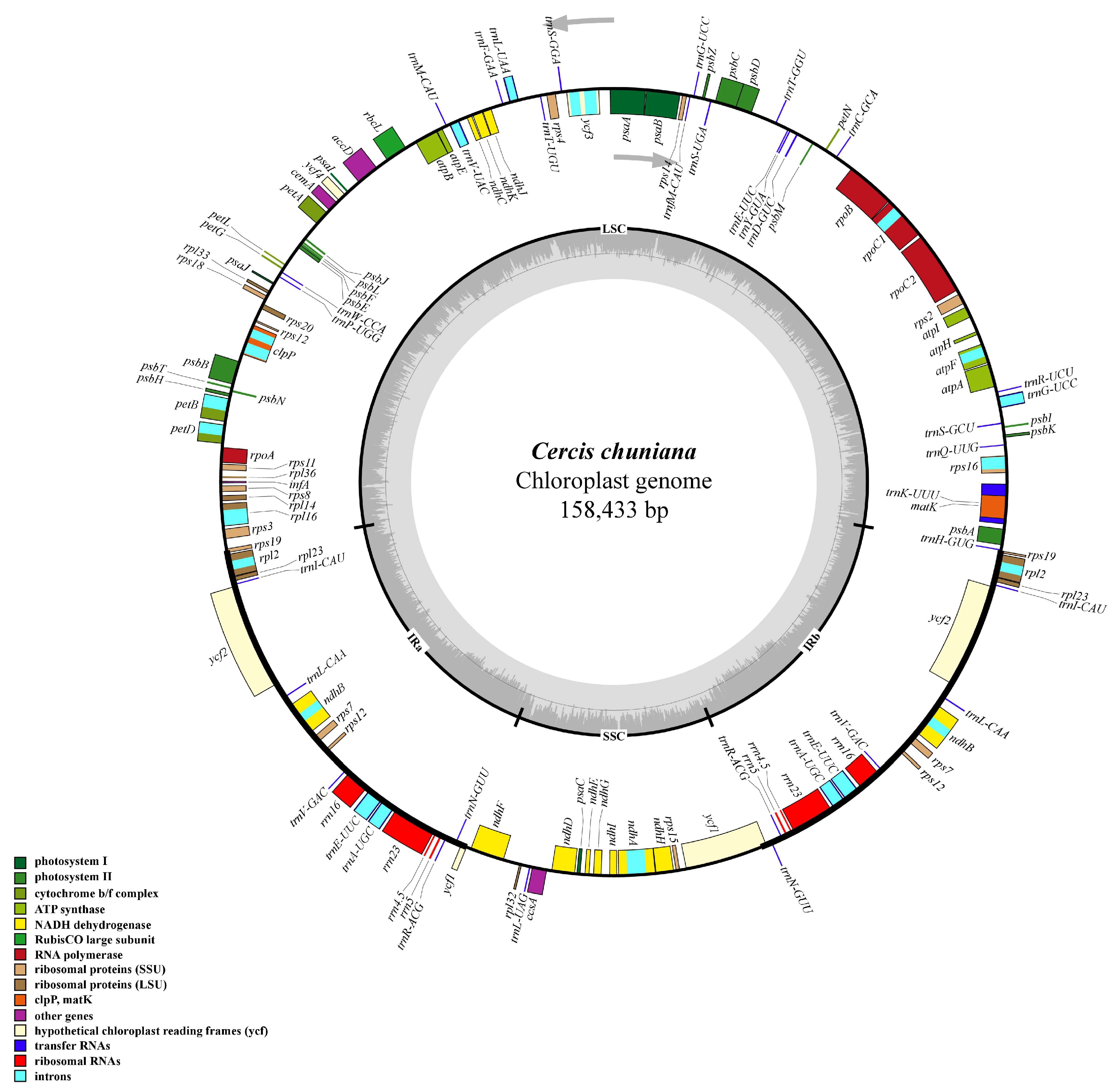
2.1. Genome Organization and Features of C. chuniana
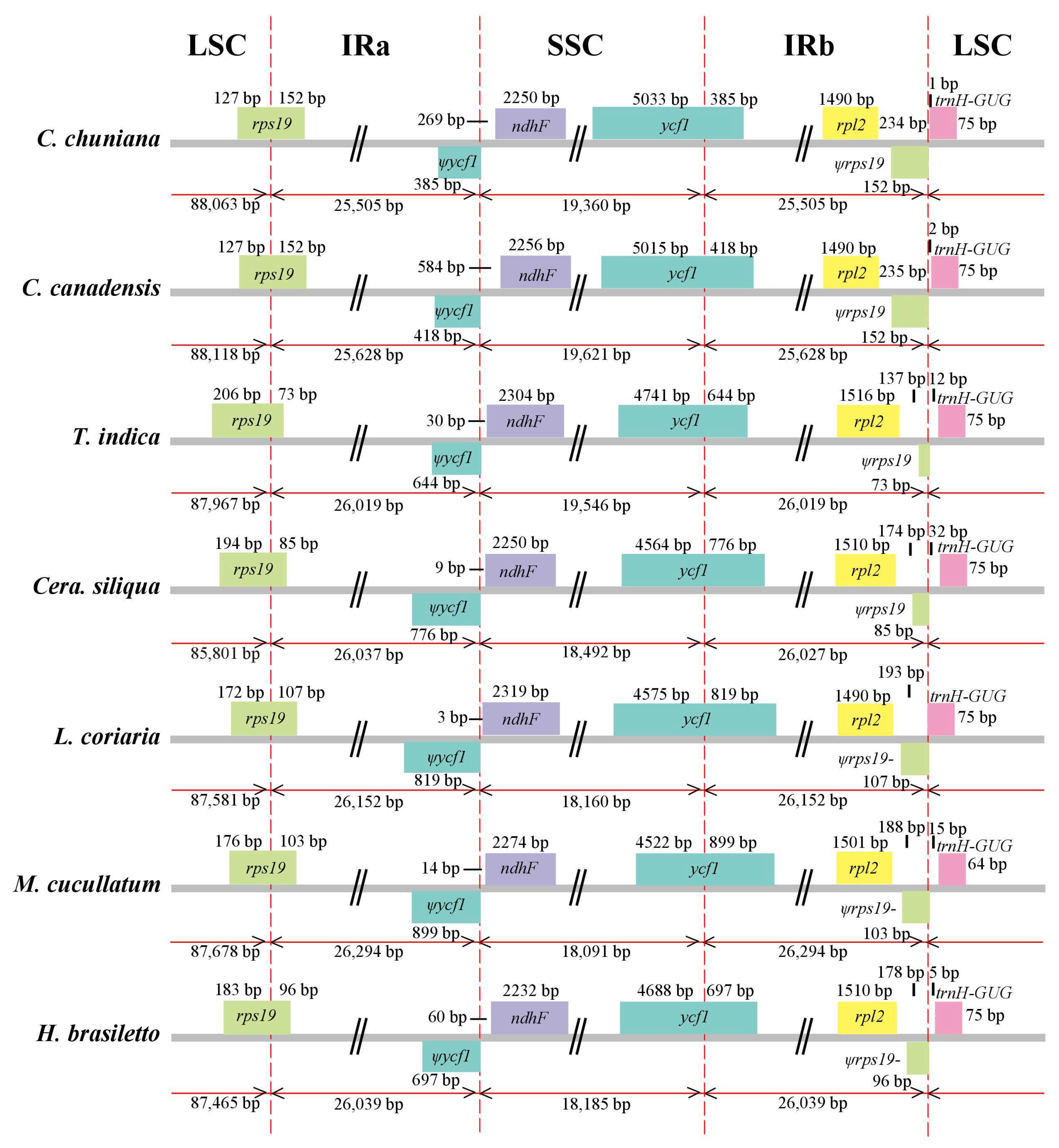
2.2. Comparative Analysis of Genomic Structure
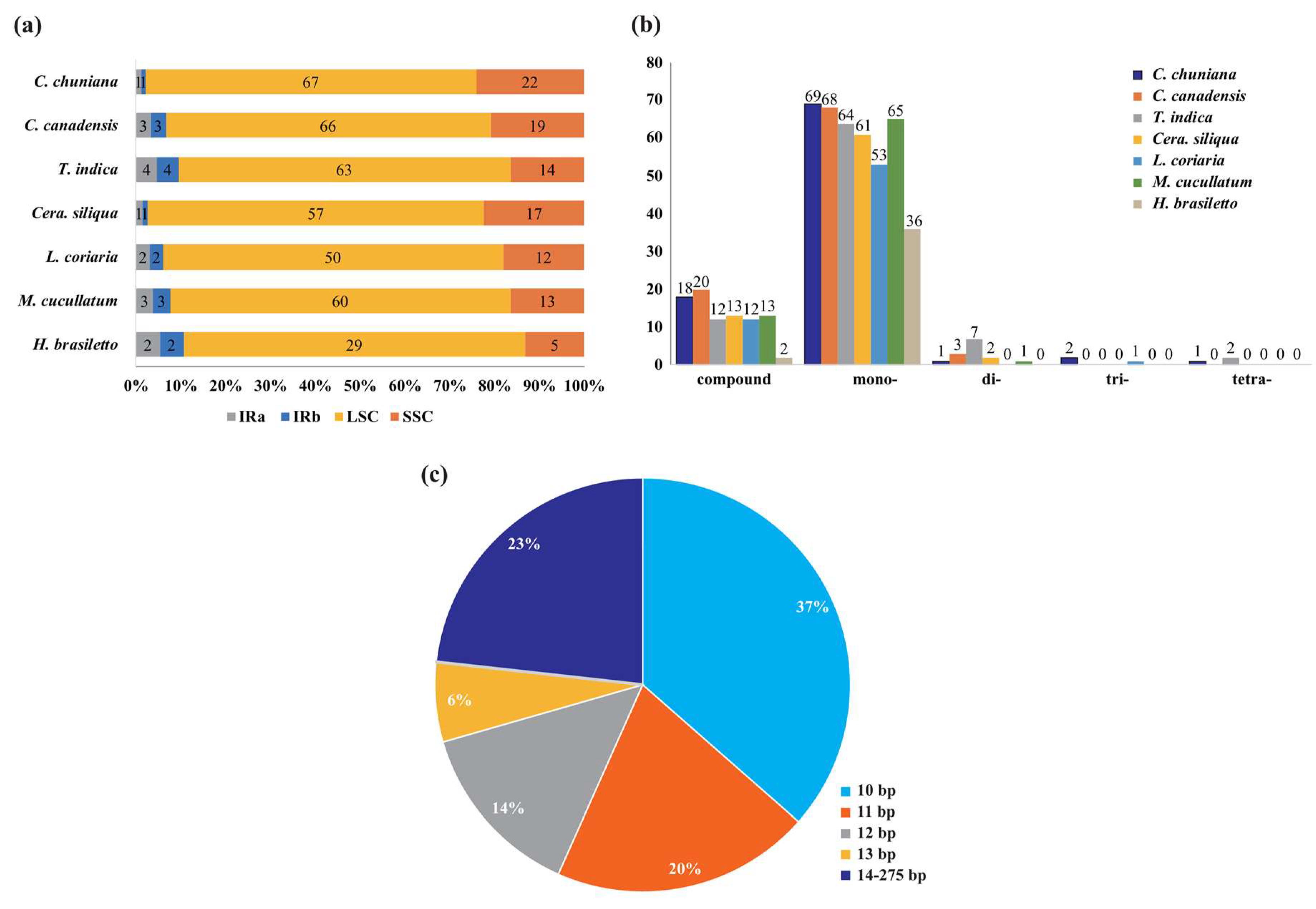
2.3. Characterization of Simple Sequence Repeats
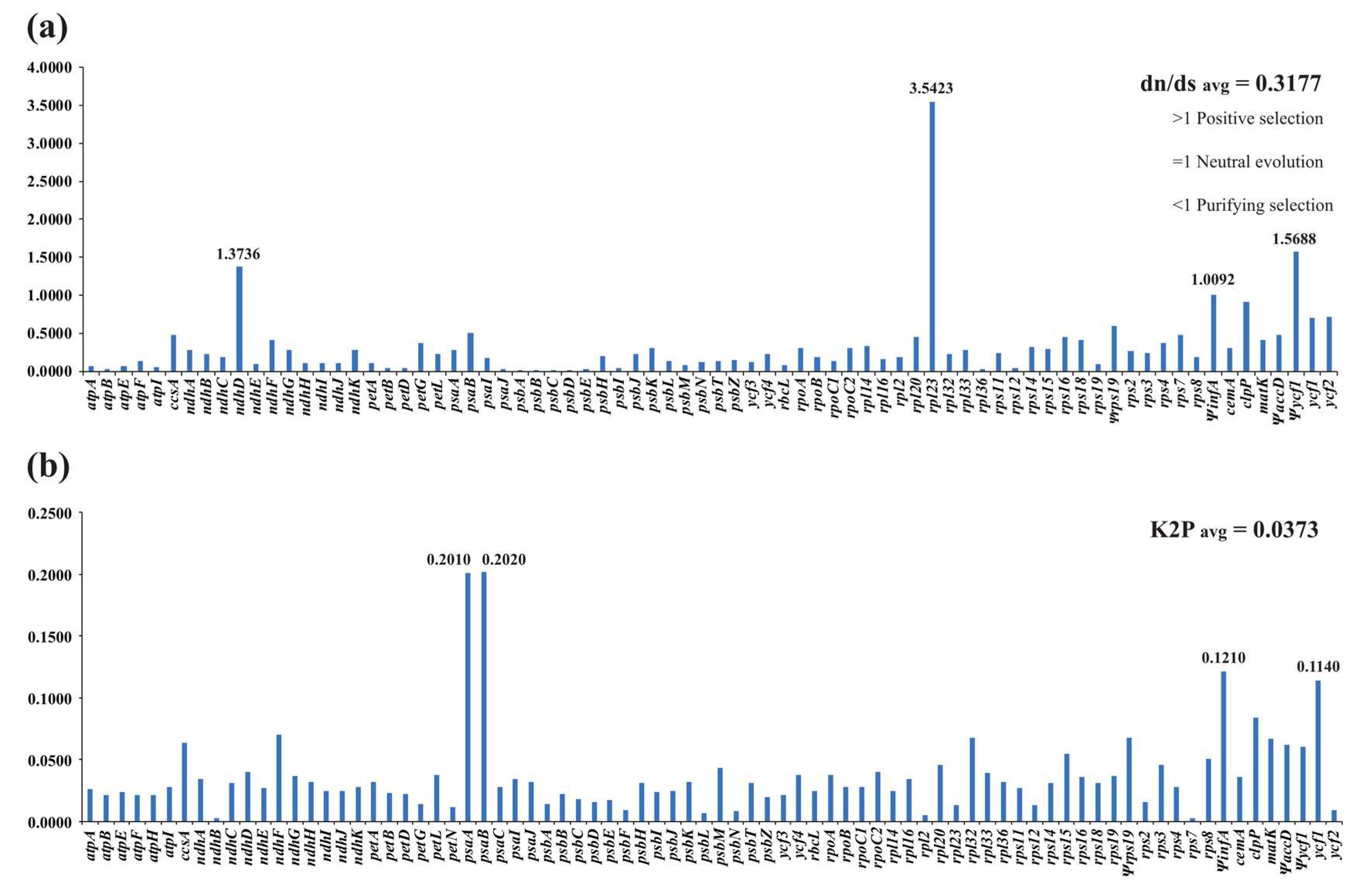
2.4. Sequence Divergence and Nucleotide Diversity
2.5. dn/ds Ratio and Kimura 2-Parameter (K2P) Genetic Distance
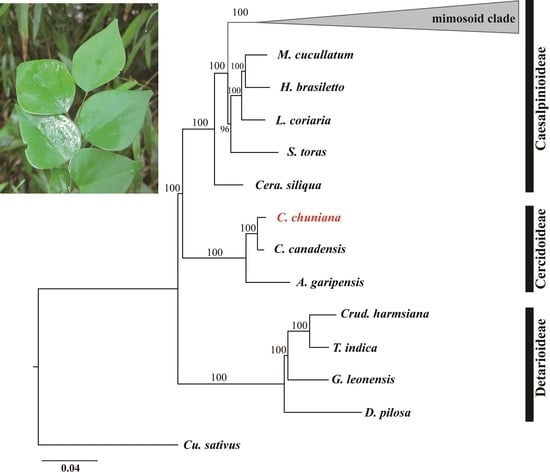
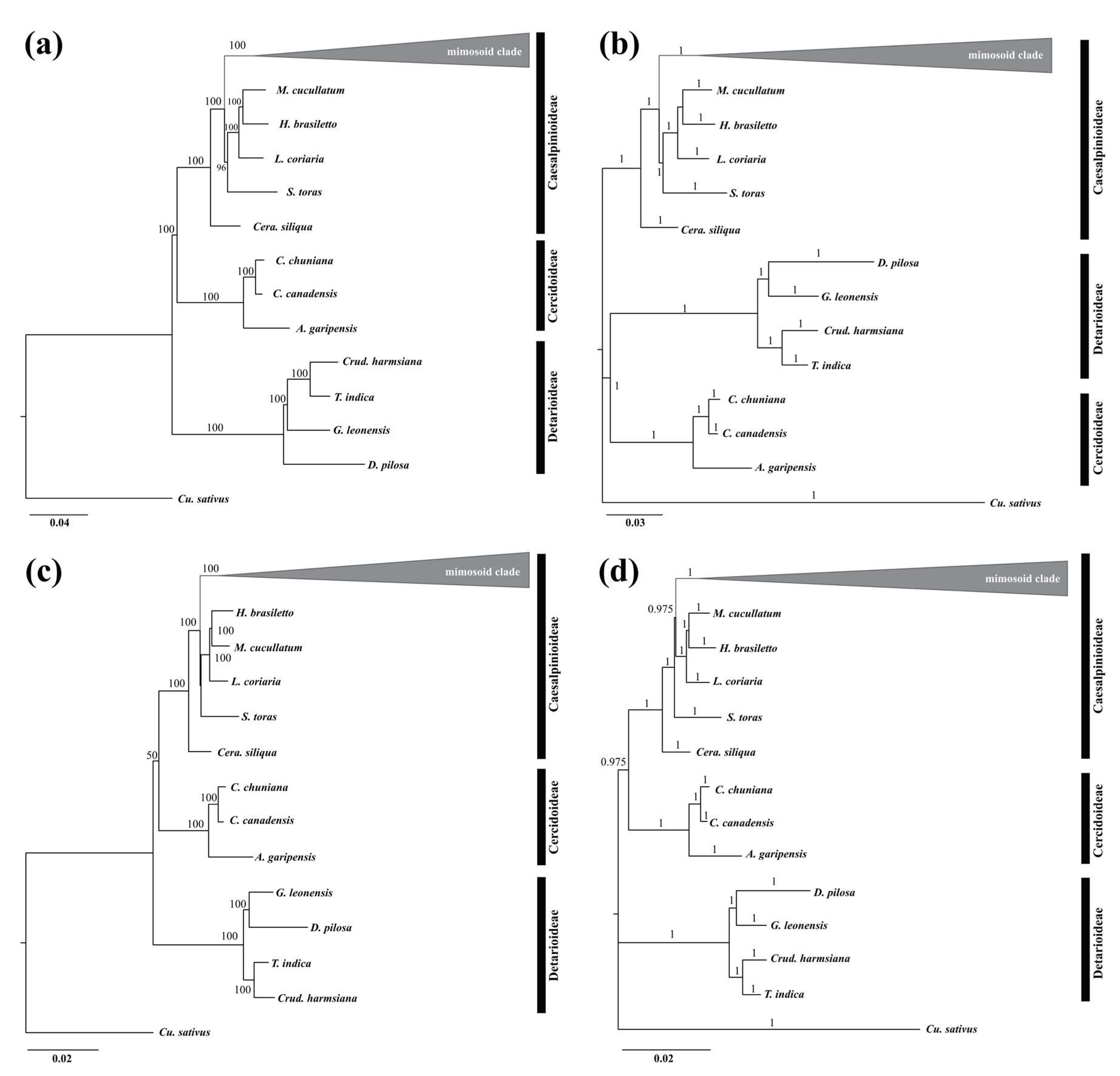
2.6. Phylogenetic Analyses
3. Materials and Methods
3.1. Ethics Statement
3.2. Plant Samples
3.3. DNA Extraction and PCR Amplification
3.4. Chloroplast Genome Sequencing, Assembly and Annotation
3.5. Genome Comparison
3.6. Simple Sequence Repeats Analysis
3.7. Sequence Divergence, dn/ds Ratio and K2P Genetic Distance
3.8. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
| Cp | Chloroplast |
| ML | Maximum likelihood |
| BI | Bayesian inference |
| A | Adenine |
| T | Thymine |
| G | Guanine |
| C | Cytosine |
References
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K. Emerging roles of the chloroplast outer envelope membrane. Trends Plant Sci. 2011, 16, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Ueda, M.; Kadowaki, K.; Tsutsumi, N. Different status of the gene for ribosomal protein S16 in the chloroplast genome during evolution of the genus Arabidopsis and closely related species. Genes Genet. Syst. 2010, 85, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Ni, D.; Wang, Y.; Shao, J.; Wang, X.; Yang, D.; Wang, J.; Chen, H.; Liu, C. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 2016, 6, 21669. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, Z.; Sablok, G.; Daskalova, E.; Zahmanova, G.; Apostolova, E.; Yahubyan, G.; Baev, V. Chloroplast genome analysis of resurrection Tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.; Yang, L.; Takuno, S.; Eguiarte, L.E. The patterns and causes of variation in plant nucleotide substitution rates. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 245–266. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Shen, Y.; Kang, F.; Wang, J. Characterization of the complete chloroplast genomes of the endangered shrub species Prunus mongolica and Prunus pedunculata (Rosales: Rosaceae). Conserv. Genet. Resour. 2018, 1–4. [Google Scholar] [CrossRef]
- Wang, H.; Park, S.; Lee, A.; Jang, S.; Im, D.; Jun, T.; Lee, J.; Chung, J.; Ham, T.; Kwon, S. Next-generation sequencing yields the complete chloroplast genome of C. goeringii acc. smg222 and phylogenetic analysis. Mitochondrial DNA Part B 2018, 3, 215–216. [Google Scholar] [CrossRef]
- Lewis, G.P.; Schrire, B.D.; Mackinder, B.A.; Lock, J.M. (Eds.) Legumes of the World; Royal Botanic Gardens, Kew: Richmond, UK, 2005. [Google Scholar]
- Käss, E.; Wink, M. Molecular evolution of the Leguminosae: Phylogeny of the three subfamilies based on rbcL-sequences. Biochem. Syst. Ecol. 1996, 24, 365–378. [Google Scholar] [CrossRef]
- Doyle, J.; Ballenger, J.; Dickson, E.; Kajita, T.; Ohashi, H. A phylogeny of the chloroplast gene rbcL in the Leguminosae: Taxonomic correlations and insights into the evolution of nodulation. Am. J. Bot. 1997, 84, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Chappill, J.A.; Bailey, C.D.; Kajita, T. Towards a comprehensive phylogeny of legumes: Evidence from rbcL sequences and non-molecular data. In Advances in Legume Systematics; Herendeen, P.S., Bruneau, A., Eds.; Royal Botanic Gardens, Kew: Richmond, UK, 2000; pp. 1–20. [Google Scholar]
- Wojciechowski, M.F.; Lavin, M.; Sanderson, M.J. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am. J. Bot. 2004, 91, 1846–1862. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [PubMed]
- The Legume phylogeny Working Group. Legume phylogeny and classification in the 21st century: Progress, prospects and lessons for other species-rich clades. Taxon 2013, 62, 217–248. [Google Scholar]
- The Legume Phylogeny Working Group. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 2017, 66, 44–77. [Google Scholar]
- Li, H. Taxonomy and distribution of the genus Cercis in China. Bull. Torrey Bot. Club 1944, 71, 419–425. [Google Scholar] [CrossRef]
- Robertson, K.R. Cercis: The redbuds. Arnoldia 1976, 36, 37–49. [Google Scholar]
- Hao, G.; Zhang, D.; Guo, L.; Zhang, M.; Deng, Y.; Wen, X. A phylogenetic and biogeographic study of Cercis (Leguminosae). Acta Bot. Sin. 2001, 43, 1275–1278. [Google Scholar]
- Davis, C.C.; Fritsch, P.W.; Li, J.; Donoghue, M.J. Phylogeny and biogeography of Cercis (Fabaceae): Evidence from nuclear ribosomal ITS and chloroplast ndhF sequence data. Syst. Bot. 2002, 27, 289–302. [Google Scholar]
- Fritsch, P.W.; Cruz, B.C. Phylogeny of Cercis based on DNA sequences of nuclear ITS and four plastid regions: Implications for transatlantic historical biogeography. Mol. Phylogenet. Evol. 2012, 62, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Dezhao, C.; Dianxiang, Z.; Larsen, S.S.; Vincent, M.A. Cercis. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden: St. Louis, MO, USA, 2010; Volume 10, pp. 5–6. [Google Scholar]
- Metcalf, F.P. Eight new species of Leguminosae from Southeastern China. Lingnan Sci. J. 1940, 19, 549–563. [Google Scholar]
- Coşkun, F.; Parks, C.R. A molecular phylogenetic study of red buds (Cercis L., Fabaceae) based on ITS nrDNA sequences. Pak. J. Bot. 2009, 41, 1577–1586. [Google Scholar]
- Coşkun, F.; Parks, C.R. A molecular phylogeny of Cercis L. (Fabaceae) using the chloroplast trnL-F DNA sequences. Pak. J. Bot. 2009, 41, 1587–1592. [Google Scholar]
- Kong, W.Q.; Yang, J.H. The complete chloroplast genome sequence of Morus cathayana and Morus multicaulis, and comparative analysis within genus Morus L. PeerJ 2017, 5, e3037. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Castillo-Ramírez, S.; González, V.; Bustos, P.; Fernández-Vázquez, J.L.; Santamaría, R.I.; Arellano, J.; Cevallos, M.A.; Dávila, G. Rapid evolutionary change of common bean (Phaseolus vulgaris L.) plastome, and the genomic diversification of legume chloroplasts. BMC Genom. 2007, 8, 228. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.V.; Boykin, L.M.; Howell, K.A.; Nevill, P.G.; Small, I. Correction: The complete sequence of the Acacia ligulata chloroplast genome reveals a highly divergent clpP1 gene. PLoS ONE 2015, 10, e138367. [Google Scholar] [CrossRef] [PubMed]
- Kaila, T.; Chaduvla, P.K.; Saxena, S.; Bahadur, K.; Gahukar, S.J.; Chaudhury, A.; Sharma, T.R.; Singh, N.K.; Gaikwad, K. Chloroplast genome sequence of Pigeonpea (Cajanus cajan (L.) Millspaugh) and Cajanus scarabaeoides (L.) Thouars: Genome organization and comparison with other legumes. Front. Plant Sci. 2016, 7, 1847. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qu, X.; Chen, S.; Li, D.; Yi, T. Plastomes of Mimosoideae: Structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet. Genomes 2017, 13, 41. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Yi, T.; Wang, Y. The complete chloroplast genomes of Adenolobus garipensis and Cercis glabra (Cercidoideae, Fabaceae). Conserv. Genet. Resour. 2017, 9, 635–638. [Google Scholar] [CrossRef]
- Choi, I.; Choi, B. The distinct plastid genome structure of Maackia fauriei (Fabaceae: Papilionoideae) and its systematic implications for genistoids and tribe Sophoreae. PLoS ONE 2017, 12, e173766. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Li, X.; Qian, J.; Wang, L.; Ma, L.; Tian, X.; Wang, Y. The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 2016, 21, 1029. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S. Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PLoS ONE 2017, 12, e183197. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jansen, R.K.; Park, S.J. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Li, P.; Qiu, Y. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Liu, W.; Yao, G.; Gong, W. A comparison of chloroplast genome sequences in Aconitum (Ranunculaceae): A traditional herbal medicinal genus. PeerJ 2017, 5, e4018. [Google Scholar] [CrossRef] [PubMed]
- Piovani, P.; Leonardi, S.; Piotti, A.; Menozzi, P. Conservation genetics of small relic populations of silver fir (Abies alba Mill.) in the northern Apennines. Plant Biosyst. 2010, 144, 683–691. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.; Chen, G.; Wang, C.; Su, Y. Invasive chloroplast population genetics of Mikania micrantha in China: No local adaptation and negative correlation between diversity and geographic distance. Front. Plant Sci. 2016, 7, 1426. [Google Scholar] [CrossRef] [PubMed]
- Der, J.P.; Thomson, J.A.; Stratford, J.K.; Wolf, P.G. Global chloroplast phylogeny and biogeography of bracken (Pteridium; Dennstaedtiaceae). Am. J. Bot. 2009, 96, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Rauwolf, U.; Meurer, J.; Herrmann, R.G. The role of plastids in plant speciation. Mol. Ecol. 2011, 20, 671–691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.; Yang, T. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Myszczyński, K.; Bączkiewicz, A.; Buczkowska, K.; Ślipiko, M.; Szczecińska, M.; Sawicki, J. The extraordinary variation of the organellar genomes of the Aneura pinguis revealed advanced cryptic speciation of the early land plants. Sci. Rep. 2017, 7, 9804. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wicke, S.; Wang, H.; Jin, J.; Chen, S.; Zhang, S.; Li, D.; Yi, T. Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Front. Plant Sci. 2018, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Zhang, T.; Zeng, C.X.; Yang, J.B.; Li, H.T.; Li, D.Z. Fifteen novel universal primer pairs for sequencing whole chloroplast genomes and a primer pair for nuclear ribosomal DNAs. J. Syst. Evol. 2016, 54, 219–227. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2014, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.N.; Ruhlman, T.A.; Sabir, J.S.M.; Hajrah, N.H.; Alharbi, N.S.; Al Malki, A.L.; Bailey, C.D.; Jansen, R.K. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 2015, 53, 458–468. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, H. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete chloroplast genome of Tanaecium tetragonolobum: The first Bignoniaceae plastome. PLoS ONE 2015, 10, e129930. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for General Users and for Biologist Programmers; Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 365–386. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evol. Bioinform. 2015, 11, S21501. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, D.; Michalak, I. raxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | C. chuniana | C. canadensis | T. indica | Cera. siliqua | L. coriaria | M. cucullatum | H. brasiletto |
|---|---|---|---|---|---|---|---|
| GenBank Accession No. | MF741770 | KF856619 | KJ468103 | KJ468096 | KJ468095 | KU569489 | KJ468097 |
| Size (bp) | 158,433 | 158,995 | 159,551 | 156,367 | 158,045 | 158,357 | 157,728 |
| LSC length (bp) | 88,063 | 88,118 | 87,967 | 85,801 | 87,581 | 87,663 | 87,465 |
| SSC length (bp) | 19,360 | 19,621 | 19,546 | 18,492 | 18,160 | 18,091 | 18,185 |
| IR length (bp) | 25,505 | 25,628 | 26,019 | 26,037 | 26,152 | 26,294 | 26,039 |
| Number of genes | 114 | 113 | 113 | 112 | 113 | 114 | 113 |
| PCGs | 81 | 79 | 79 | 78 | 80 | 80 | 79 |
| tRNA genes | 29 | 30 | 30 | 30 | 29 | 30 | 30 |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| G + C content (%) | 36.10 | 36.20 | 36.20 | 36.70 | 36.50 | 36.40 | 36.70 |
| Species | IRa | IRb | LSC | LSC |
|---|---|---|---|---|
| Ψycf1 | Ψrps19 | ΨinfA | ΨaccD | |
| C. chuniana | 385 bp * | 152 bp * | 1 indel, 91-bp SV * | 5 indels * |
| C. canadensis | 418 bp * | 152 bp * | 1 indel, 91-bp SV * | 5 indels * |
| T. indica | 644 bp * | 73 bp * | 71-bp SV * | - |
| Cera. siliqua | 776 bp * | 85 bp * | - | 4 indels, 63-bp SV * |
| L. coriaria | 819 bp * | 107 bp * | - | 4 indels * |
| M. cucullatum | 899 bp * | 103 bp * | 4 indels * | 3 indels * |
| H. brasiletto | 697 bp * | 96 bp * | 4 indels * | 4 indels * |
| Species | N | LSC | SSC | IRa | IRb | Compound | Mono- (≥10) | Di- (≥6) | Tri- (≥5) | Tetra- (≥5) |
|---|---|---|---|---|---|---|---|---|---|---|
| C. chuniana | 91 | 67 (73.63) | 22 (24.18) | 1 (1.10) | 1 (1.10) | 18 (19.78) | 69 (75.82) | 1 (1.10) | 2 (2.20) | 1 (1.10) |
| C. canadensis | 91 | 66 (72.53) | 19 (20.88) | 3 (3.30) | 3 (3.30) | 20 (21.98) | 68 (74.73) | 3 (3.30) | 0 (0.00) | 0 (0.00) |
| T. indica | 85 | 63 (74.12) | 14 (16.47) | 4 (4.71) | 4 (4.71) | 12 (14.12) | 64 (75.29) | 7 (8.24) | 0 (0.00) | 2 (2.35) |
| Cera. siliqua | 76 | 57 (75.00) | 17 (22.37) | 1 (1.32) | 1 (1.32) | 13 (17.11) | 61 (80.26) | 2 (2.63) | 0 (0.00) | 0 (0.00) |
| L. coriaria | 66 | 50 (75.76) | 12 (18.18) | 2 (3.03) | 2 (3.03) | 12 (18.18) | 53 (80.30) | 0 (0.00) | 1 (1.52) | 0 (0.00) |
| M. cucullatum | 79 | 60 (75.95) | 13 (16.46) | 3 (3.80) | 3 (3.80) | 13 (16.46) | 65 (82.28) | 1 (1.27) | 0 (0.00) | 0 (0.00) |
| H. brasiletto | 38 | 29 (76.32) | 5 (13.16) | 2 (5.26) | 2 (5.26) | 2 (5.26) | 36 (94.74) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Average | 75 | 56 (74.76) | 15 (18.81) | 2 (3.22) | 2 (3.22) | 13 (17.14) | 59 (79.24) | 2 (2.67) | 0.43 (0.57) | 0.43 (0.57) |
| Species | C. chuniana | C. canadensis | T. indica | Cera. siliqua | L. coriaria | M. cucullatum | H. brasiletto |
|---|---|---|---|---|---|---|---|
| C. chuniana | - | ||||||
| C. canadensis | 19 | - | |||||
| T. indica | 1 | 0 | - | ||||
| Cera. siliqua | 6 | 4 | 1 | - | |||
| L. coriaria | 4 | 6 | 3 | 7 | - | ||
| M. cucullatum | 6 | 5 | 1 | 1 | 8 | - | |
| H. brasiletto | 4 | 3 | 2 | 6 | 6 | 4 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Kong, H.; Zhou, J.; Fritsch, P.W.; Hao, G.; Gong, W. Complete Chloroplast Genome of Cercis chuniana (Fabaceae) with Structural and Genetic Comparison to Six Species in Caesalpinioideae. Int. J. Mol. Sci. 2018, 19, 1286. https://doi.org/10.3390/ijms19051286
Liu W, Kong H, Zhou J, Fritsch PW, Hao G, Gong W. Complete Chloroplast Genome of Cercis chuniana (Fabaceae) with Structural and Genetic Comparison to Six Species in Caesalpinioideae. International Journal of Molecular Sciences. 2018; 19(5):1286. https://doi.org/10.3390/ijms19051286
Chicago/Turabian StyleLiu, Wanzhen, Hanghui Kong, Juan Zhou, Peter W. Fritsch, Gang Hao, and Wei Gong. 2018. "Complete Chloroplast Genome of Cercis chuniana (Fabaceae) with Structural and Genetic Comparison to Six Species in Caesalpinioideae" International Journal of Molecular Sciences 19, no. 5: 1286. https://doi.org/10.3390/ijms19051286