Knocking down Insulin Receptor in Pancreatic Beta Cell lines with Lentiviral-Small Hairpin RNA Reduces Glucose-Stimulated Insulin Secretion via Decreasing the Gene Expression of Insulin, GLUT2 and Pdx1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
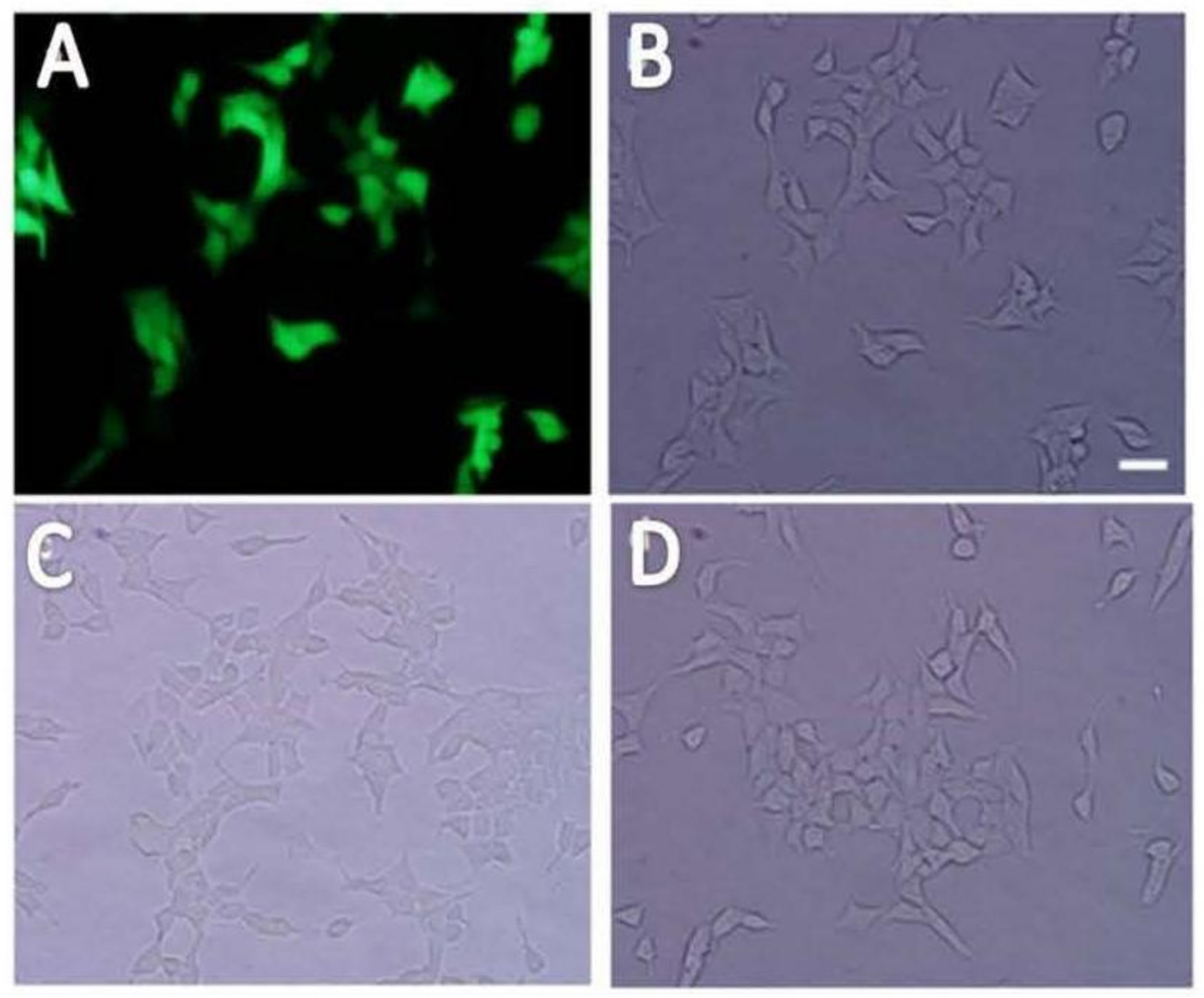
2.1. Cloning shRNAs into pLL3.7 Plasmid and Lentiviral Production
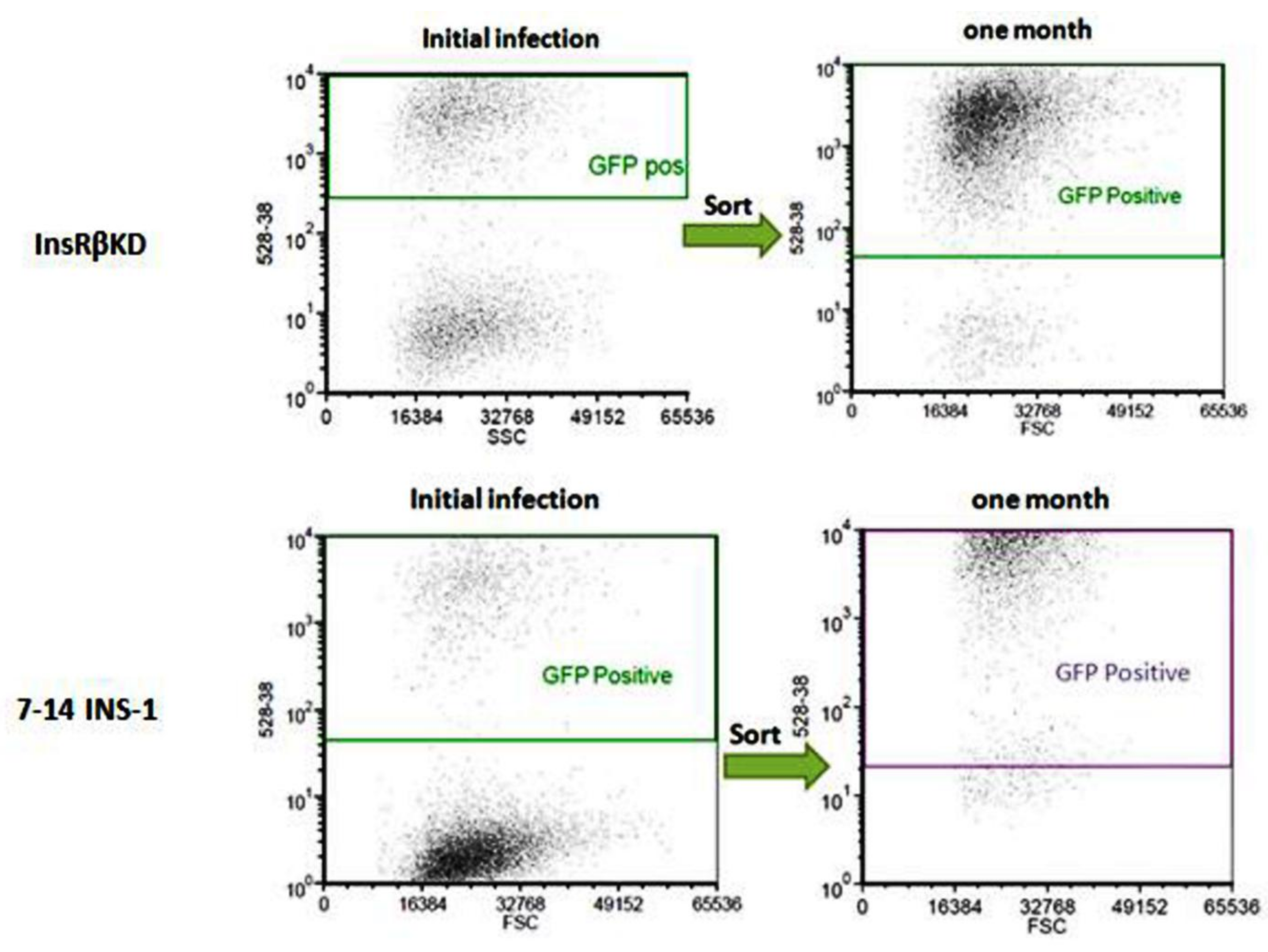
2.2. Establishment of Stable Insulin Receptor Knocking down Cell Line
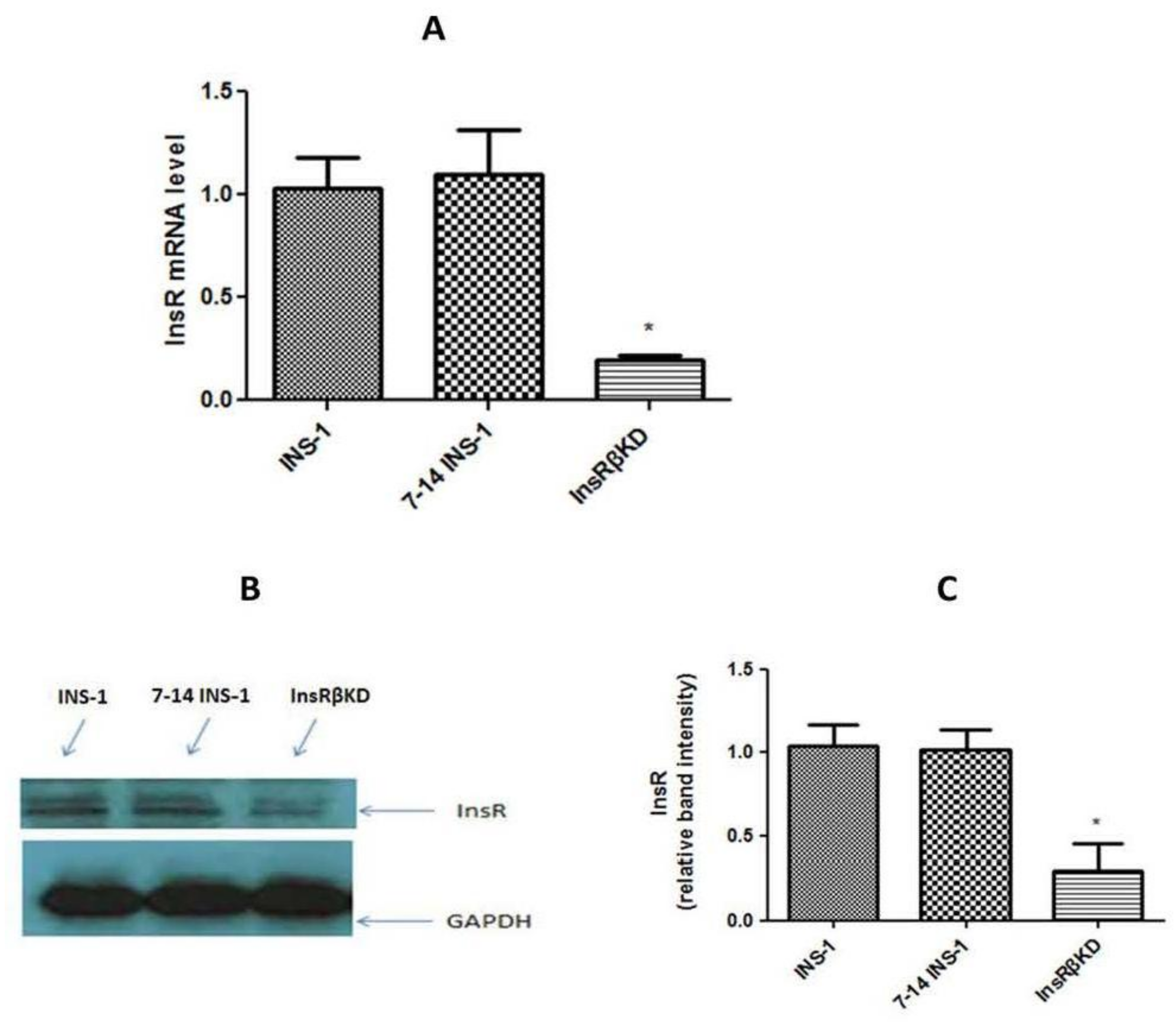
2.3. Confirmation of Gene Silence
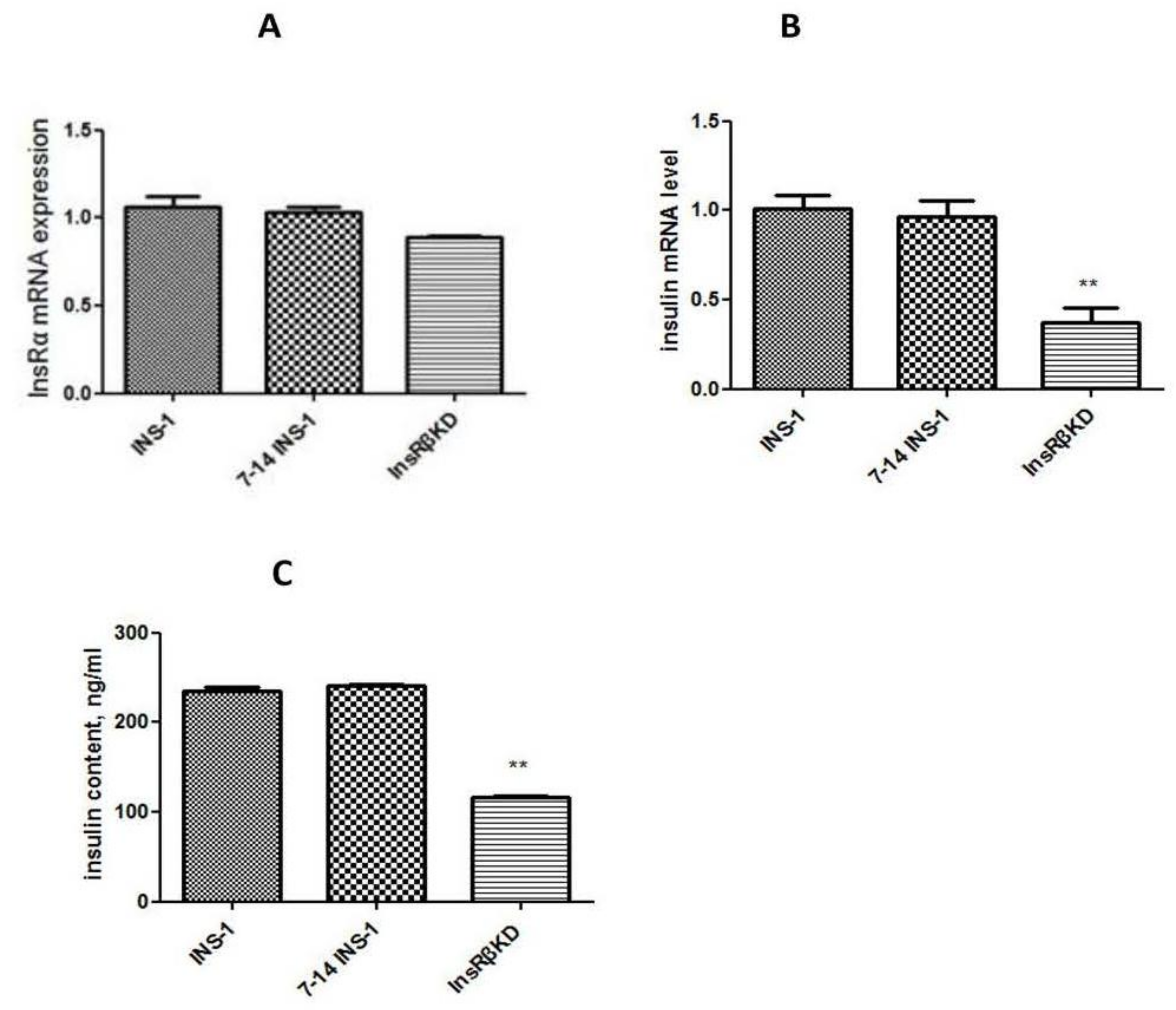
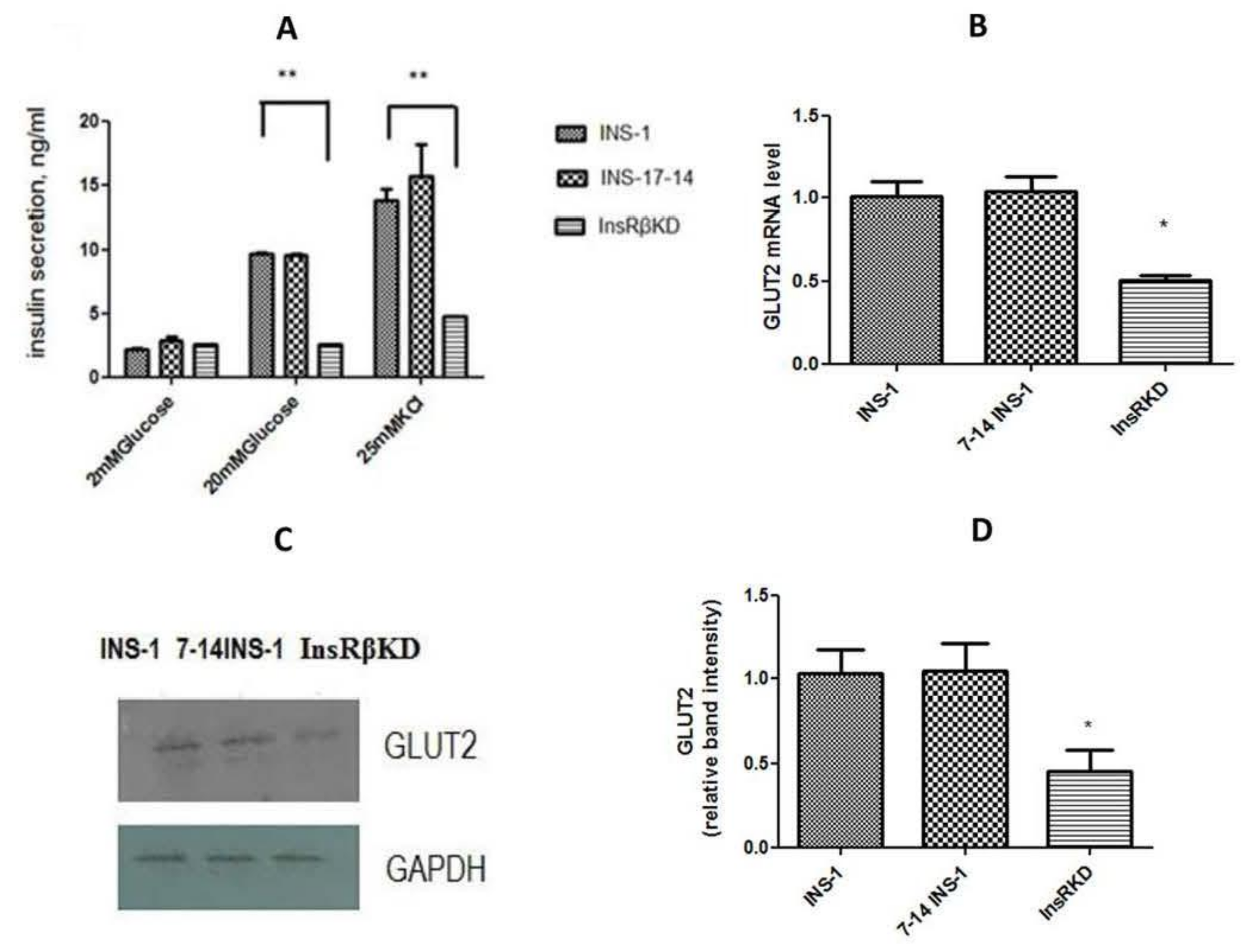
2.4. Reduced Insulin Expression and GSIS in InsRβKD Cells
2.5. Reduced Glucose Influx through GLUT2 and Pdx1 Expression in InsRβKD Cells
3. Discussion
4. Materials and Methods
4.1. Small Hairpin RNA (shRNA) Design, Cloning and Sequencing
- -InsRβ-1: 5′TGATGACAGCAGAAATTGCCTTCAAGAGAGGCAATTTCTGCTGTCATCTTTTTTC;
- -InsRβ-2: 5′TGAATGACGAGAGACATCTATTCAAGAGATAGATGTCTCTCGTCATTCTTTTTTC;
- -InsRβ-3: 5′TTCACTGCTTCTTCCGACATTTCAAGAGAATGTCGGAAGAAGCAGTGATTTTTTC;
- -InsRβ-4: 5′TGTTCGAGGACATGGAGAATTTCAAGAGAATTCTCCATGTCCTCGAACTTTTTTC.
4.2. Cell Lines and Maintenance
4.3. Production of Lentiviruses and Transduction of INS-1 Cells
4.4. Establishment of InsRβKD Cells
4.5. Quantitative PCR (qPCR)
4.6. Western Blotting Analysis
4.7. Insulin Assay
4.8. Glucose Uptake Assay
4.9. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Fact Sheet No. 312; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Grundy, S.M. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J. Am. Coll. Cardiol. 2012, 59, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Gastaldelli, A.; Iozzo, P. Pathophysiology of prediabetes. Med. Clin. N. Am. 2011, 95, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Montminy, M.; Koo, S.H. Diabetes: Outfoxing insulin resistance? Nature 2004, 432, 958–959. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar] [PubMed]
- Kulkarni, R.N.; Bruning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef]
- Ohsugi, M.; Cras-Meneur, C.; Zhou, Y.; Bernal-Mizrachi, E.; Johnson, J.D.; Luciani, D.S.; Polonsky, K.S.; Permutt, M.A. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J. Biol. Chem. 2005, 280, 4992–5003. [Google Scholar] [CrossRef] [PubMed]
- Beith, J.L.; Alejandro, E.U.; Johnson, J.D. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology 2008, 149, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Gould, G.W.; Holman, G.D. The glucose transporter family: Structure, function and tissue-specific expression. Biochem. J. 1993, 295 Pt 2, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Thorens, B. Glucose transporters in the regulation of intestinal, renal, and liver glucose fluxes. Am. J. Physiol. 1996, 270, G541–G553. [Google Scholar] [CrossRef] [PubMed]
- Colville, C.A.; Seatter, M.J.; Jess, T.J.; Gould, G.W.; Thomas, H.M. Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: Substrate specificities and effects of transport inhibitors. Biochem. J. 1993, 290 Pt 3, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Wu, Y.J.; Leahy, J.L.; Weir, G.C. The loss of GLUT2 expression by glucose-unresponsive beta cells of db/db mice is reversible and is induced by the diabetic environment. J. Clin. Investig. 1992, 90, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Guillam, M.T.; Beermann, F.; Burcelin, R.; Jaquet, M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J. Biol. Chem. 2000, 275, 23751–23758. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Waeber, G.; Thompson, N.; Nicod, P.; Bonny, C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol. Endocrinol. 1996, 10, 1327–1334. [Google Scholar] [PubMed]
- Wang, H.; Maechler, P.; Ritz-Laser, B.; Hagenfeldt, K.A.; Ishihara, H.; Philippe, J.; Wollheim, C.B. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J. Biol. Chem. 2001, 276, 25279–25286. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y.; Fujitani, S.; Boyer, D.F.; Gannon, M.; Kawaguchi, Y.; Ray, M.; Shiota, M.; Stein, R.W.; Magnuson, M.A.; Wright, C.V. Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 2006, 20, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Payne, E.; Sun, S.; Burgess, M.; McMillan, N.A. Inhibition of cervical cancer cell growth in vitro and in vivo with dual shRNAs. Cancer Gene Ther. 2011, 18, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, R.J.; Kruithof, E.K. Short-term cytotoxic effects and long-term instability of RNAi delivered using lentiviral vectors. BMC Mol. Biol. 2004, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.S.; Jan, M.S.; Chou, C.K.; Chen, P.H.; Ke, N.J. Is green fluorescent protein toxic to the living cells? Biochem. Biophys. Res. Commun. 1999, 260, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M. A hard way to the nucleus. Mol. Med. 2004, 10, 1–5. [Google Scholar] [PubMed]
- Manjunath, N.; Wu, H.; Subramanya, S.; Shankar, P. Lentiviral delivery of short hairpin RNAs. Adv. Drug Deliv. Rev. 2009, 61, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Persaud, S.J.; Asare-Anane, H.; Jones, P.M. Insulin receptor activation inhibits insulin secretion from human islets of Langerhans. FEBS Lett. 2002, 510, 225–228. [Google Scholar] [CrossRef]
- Kubota, N.; Tobe, K.; Terauchi, Y.; Eto, K.; Yamauchi, T.; Suzuki, R.; Tsubamoto, Y.; Komeda, K.; Nakano, R.; Miki, H.; et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 2000, 49, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Goren, H.J. Insulin constitutively secreted by beta-cells is necessary for glucose-stimulated insulin secretion. Diabetes 2003, 52, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.G.; Gao, Z.Y.; Borge, P.D., Jr.; Jegier, P.A.; Young, R.A.; Wolf, B.A. Insulin regulation of beta-cell function involves a feedback loop on SERCA gene expression, Ca(2+) homeostasis, and insulin expression and secretion. Biochemistry 2000, 39, 14912–14919. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Kulkarni, R.N.; Baldwin, A.C.; Krutzfeldt, J.; Ueki, K.; Stoffel, M.; Kahn, C.R.; Polonsky, K.S. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E41–E49. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D.; Ben-Neriah, Y.; Cerasi, E. Glucose modulates the binding of an islet-specific factor to a conserved sequence within the rat I and the human insulin promoters. Proc. Natl. Acad. Sci. USA 1993, 90, 3865–3869. [Google Scholar] [CrossRef] [PubMed]
- Peers, B.; Leonard, J.; Sharma, S.; Teitelman, G.; Montminy, M.R. Insulin expression in pancreatic islet cells relies on cooperative interactions between the helix loop helix factor E47 and the homeobox factor STF-1. Mol. Endocrinol. 1994, 8, 1798–1806. [Google Scholar] [PubMed]
- Andrali, S.S.; Sampley, M.L.; Vanderford, N.L.; Ozcan, S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem. J. 2008, 415, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Guo, M.; Huang, S.; Stein, R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol. Cell. Biol. 2002, 22, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Glick, E.; Leshkowitz, D.; Walker, M.D. Transcription factor BETA2 acts cooperatively with E2A and PDX1 to activate the insulin gene promoter. J. Biol. Chem. 2000, 275, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Aramata, S.; Han, S.I.; Yasuda, K.; Kataoka, K. Synergistic activation of the insulin gene promoter by the beta-cell enriched transcription factors MafA, Beta2, and Pdx1. Biochim. Biophys. Acta 2005, 1730, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Guo, M.; Matsuoka, T.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V.; Stein, R. The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J. Biol. Chem. 2005, 280, 11887–11894. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.C.; Bogardus, C.; Takeda, J.; Knowler, W.C.; Thompson, D.B. Linkage analysis of acute insulin secretion with GLUT2 and glucokinase in Pima Indians and the identification of a missense mutation in GLUT2. Diabetes 1994, 43, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, A.; Tanizawa, Y.; Matsutani, A.; Kaneko, T.; Kaku, K. Sequence variations of the pancreatic islet/liver glucose transporter (GLUT2) gene in Japanese subjects with noninsulin dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1995, 80, 3131–3135. [Google Scholar] [PubMed]
- Tanizawa, Y.; Riggs, A.C.; Chiu, K.C.; Janssen, R.C.; Bell, D.S.; Go, R.P.; Roseman, J.M.; Acton, R.T.; Permutt, M.A. Variability of the pancreatic islet beta cell/liver (GLUT 2) glucose transporter gene in NIDDM patients. Diabetologia 1994, 37, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.M.; Jensen, N.M.; Pildal, J.; Drivsholm, T.; Borch-Johnsen, K.; Urhammer, S.A.; Hansen, T.; Pedersen, O. Studies of genetic variability of the glucose transporter 2 promoter in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2001, 86, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Latek, R.; Hossbach, M.; Tuschl, T.; Lewitter, F. siRNA Selection Server: An automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004, 32, W130–W134. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Ma, J.B.; Yuan, Y.R.; Ye, K.; Pei, Y.; Kuryavyi, V.; Malinina, L.; Meister, G.; Tuschl, T. Structural biology of RNA silencing and its functional implications. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Asfari, M.; Janjic, D.; Meda, P.; Li, G.; Halban, P.A.; Wollheim, C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992, 130, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Skelin, M.; Rupnik, M.; Cencic, A. Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX 2010, 27, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Putral, L.N.; Bywater, M.J.; Gu, W.; Saunders, N.A.; Gabrielli, B.G.; Leggatt, G.R.; McMillan, N.A. RNA interference against human papillomavirus oncogenes in cervical cancer cells results in increased sensitivity to cisplatin. Mol. Pharmacol. 2005, 68, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Cochrane, M.; Leggatt, G.R.; Payne, E.; Choyce, A.; Zhou, F.; Tindle, R.; McMillan, N.A. Both treated and untreated tumors are eliminated by short hairpin RNA-based induction of target-specific immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 8314–8319. [Google Scholar] [CrossRef] [PubMed]
- VanGuilder, H.D.; Vrana, K.E.; Freeman, W.M. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 2008, 44, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nakata, M.; Horimoto, N.; Saito, M.; Matsuoka, H.; Inagaki, N. Measurement of glucose uptake and intracellular calcium concentration in single, living pancreatic beta-cells. J. Biol. Chem. 2000, 275, 22278–22283. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.T.; Cheung, A.T.; Riedel, M.J.; Light, P.E.; Cheeseman, C.I.; Kieffer, T.J. Leptin reduces glucose transport and cellular ATP levels in INS-1 beta-cells. J. Mol. Endocrinol. 2004, 32, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kyriazis, G.A.; Soundarapandian, M.M.; Tyrberg, B. Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2012, 109, E524–E532. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.Y.; Rampal, A.L. Cytochalasin B binding sites and glucose transport carrier in human erythrocyte ghosts. J. Biol. Chem. 1977, 252, 5456–5463. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Gu, W.; Chen, C. Knocking down Insulin Receptor in Pancreatic Beta Cell lines with Lentiviral-Small Hairpin RNA Reduces Glucose-Stimulated Insulin Secretion via Decreasing the Gene Expression of Insulin, GLUT2 and Pdx1. Int. J. Mol. Sci. 2018, 19, 985. https://doi.org/10.3390/ijms19040985
Wang J, Gu W, Chen C. Knocking down Insulin Receptor in Pancreatic Beta Cell lines with Lentiviral-Small Hairpin RNA Reduces Glucose-Stimulated Insulin Secretion via Decreasing the Gene Expression of Insulin, GLUT2 and Pdx1. International Journal of Molecular Sciences. 2018; 19(4):985. https://doi.org/10.3390/ijms19040985
Chicago/Turabian StyleWang, Jie, Wenyi Gu, and Chen Chen. 2018. "Knocking down Insulin Receptor in Pancreatic Beta Cell lines with Lentiviral-Small Hairpin RNA Reduces Glucose-Stimulated Insulin Secretion via Decreasing the Gene Expression of Insulin, GLUT2 and Pdx1" International Journal of Molecular Sciences 19, no. 4: 985. https://doi.org/10.3390/ijms19040985