Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
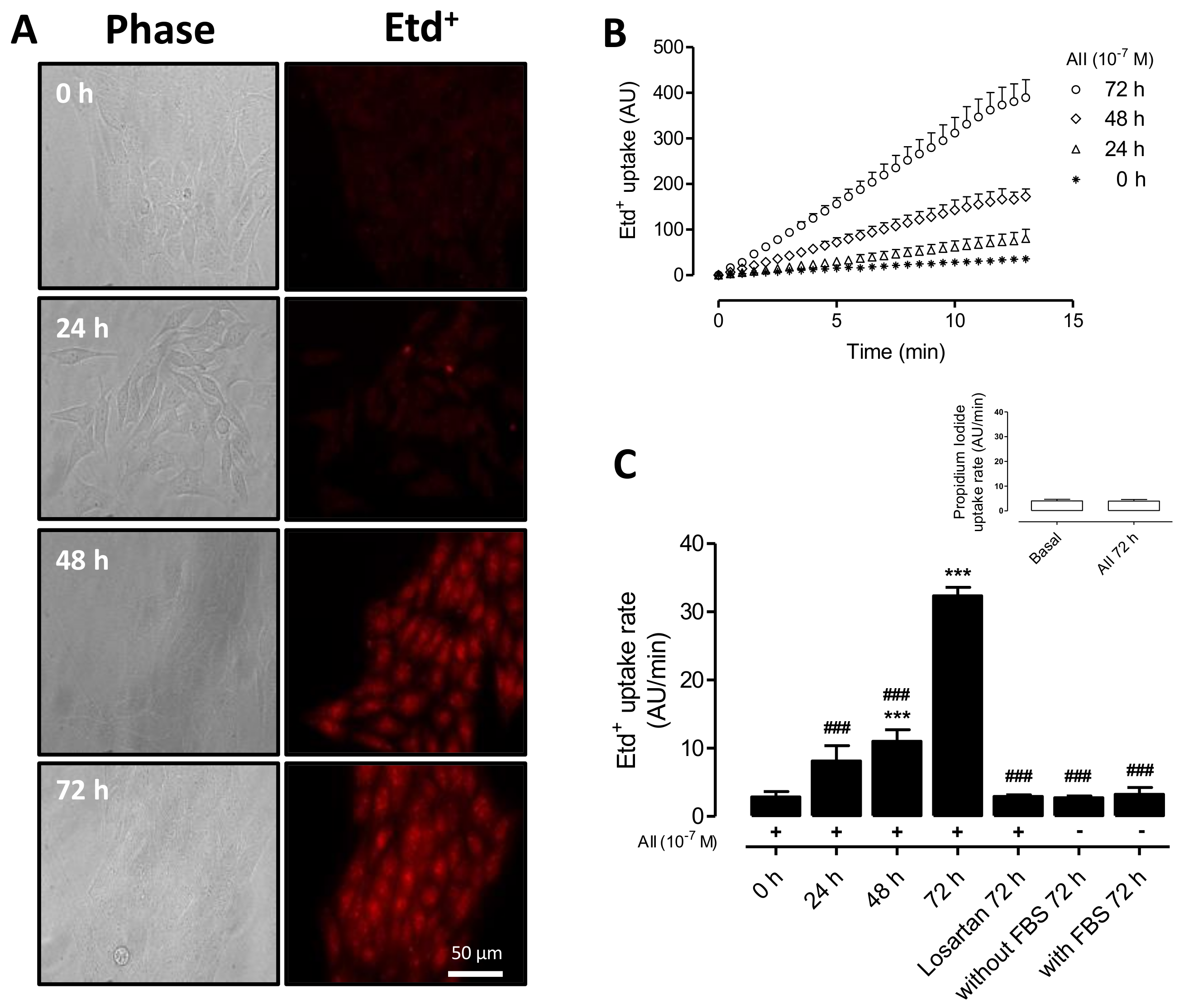
2.1. AngII Increases Cell Membrane Permeability of MES-13 Cells
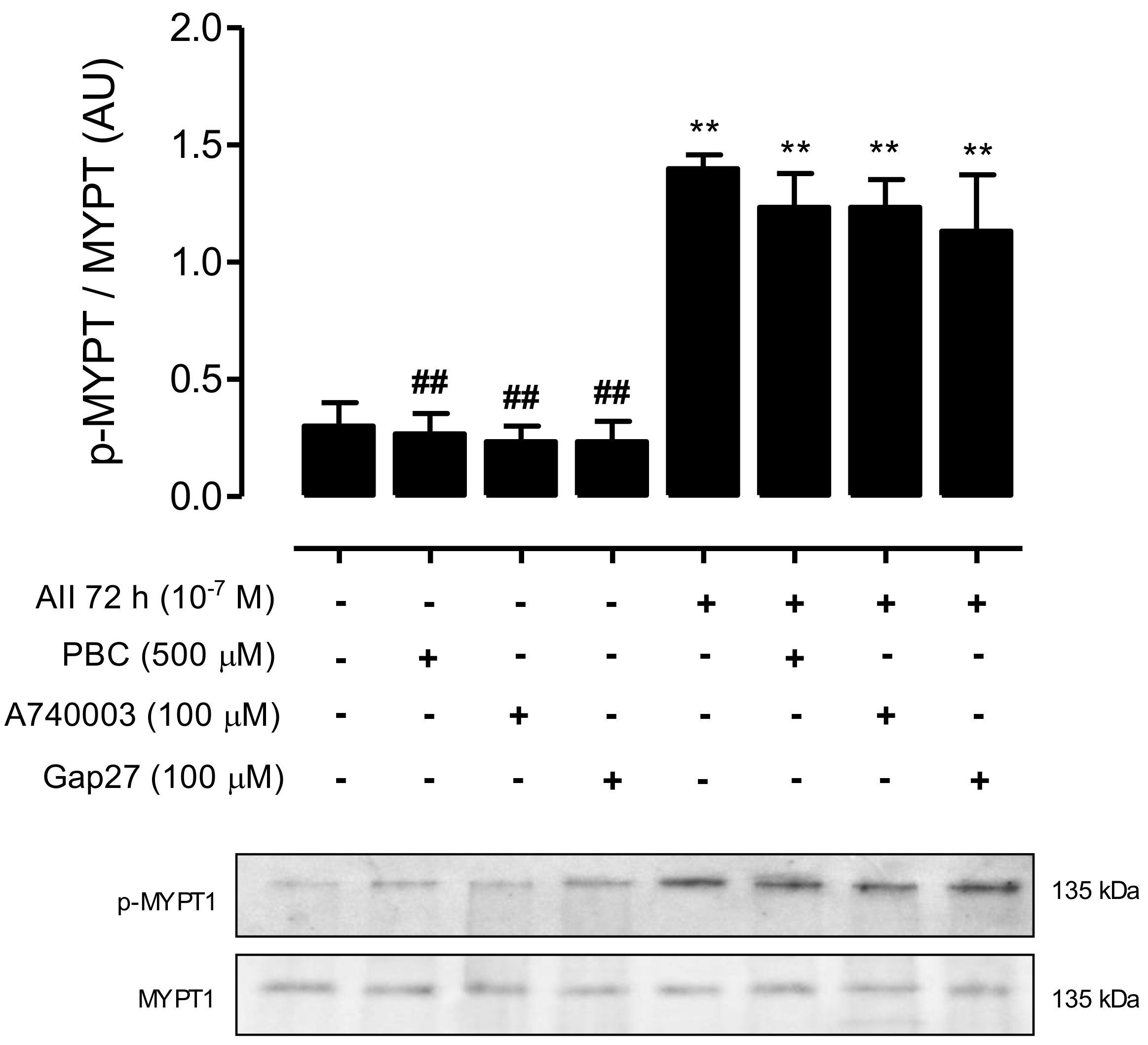
2.2. AngII Promotes Phosphorylation of MYPT and Increases the Amount of Cx43, Panx1 and P2X7R in Mesangial Cells
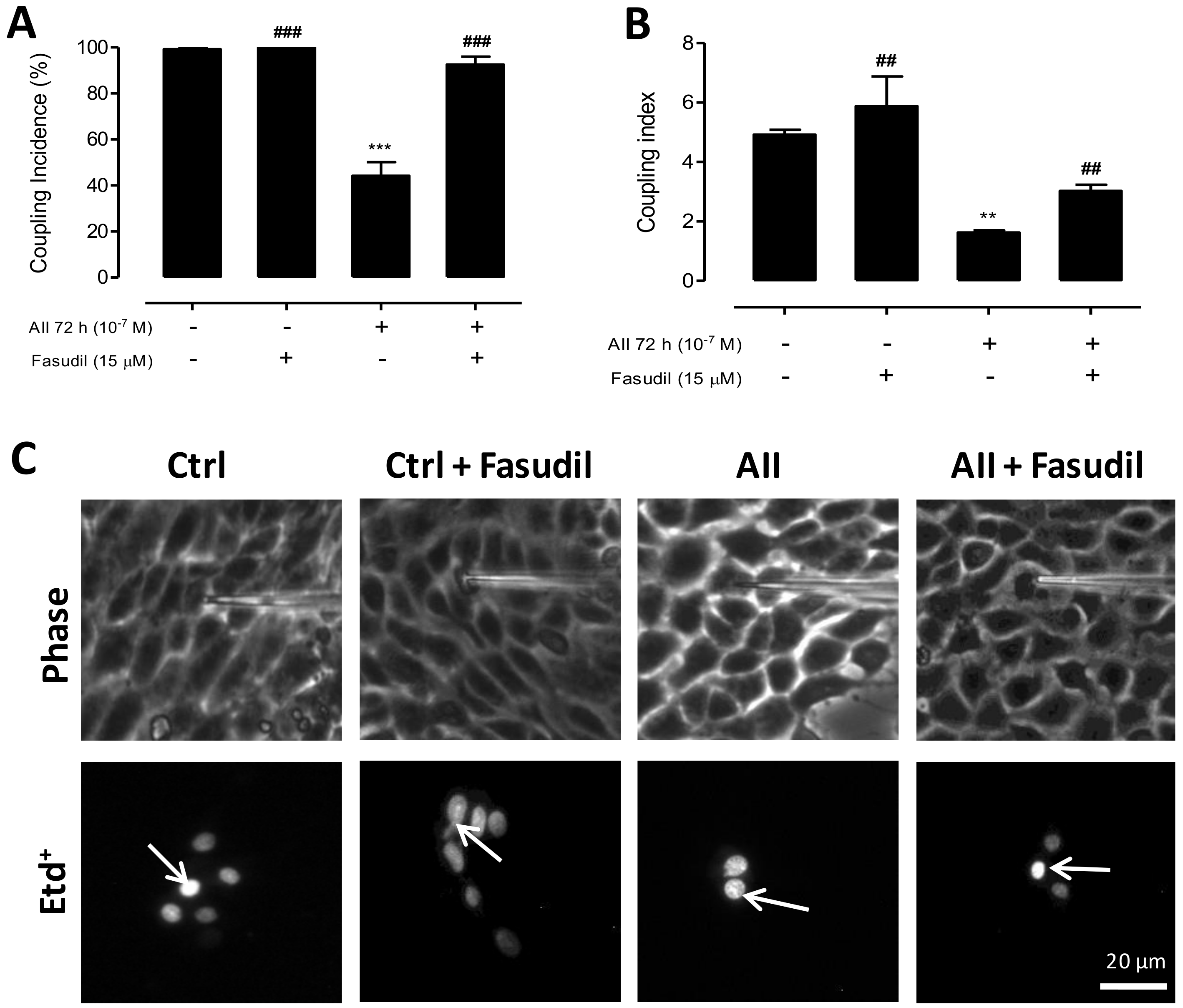
2.3. AngII-Induced Cell Membrane Permeability is Prevented by the Inhibition of a RhoA/ROCK-Dependent Pathway, and Is Mediated by the Activation of Cx43 HCs, Panx1 Chs and P2X7Rs
2.4. AngII Reduces Intercellular Communication Mediated by GJs in MES-13 Cells
2.5. Inhibition of RhoA/ROCK Reduces the Amount of Phosphorylated MYPT and Cx43, but Does Not Impact the Amount of Panx1 or P2X7 Receptors in AngII-Treated MES-13 Cells
2.6. Inhibition of Panx1 Chs, Cx43 HCs, or P2X7Rs Does Not Affect AngII-Induced RhoA/ROCK Activation in MES-13 Cells
2.7. Inhibition of RhoA/ROCK, Cx HCs or P2X7Rs, but Not Panx1 Chs, Prevents Increases in Lipid Peroxidation and Inflammatory Responses Induced by AngII in MES-13 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Dye Uptake
4.4. Dye Coupling
4.5. Western Blot Assays
4.6. Thiobarbituric Acid Reactive Substances (TBARS) Measurement
4.7. Enzyme-Linked Immunosorbent Assay
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cx43 | Connexin 43 |
| Panx1 | Pannexin 1 |
| P2X7R | P2X7 Receptor |
| OS | Oxidative stress |
| TBARS | Thiobarbituric reactive species |
| AngII | Angiotensin II |
| ROCK | Rho kinase |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Cx GJs | Connexin gap junctions |
| Cx HCs | Connexin hemichannels |
| Panx Chs | Pannexin channels |
| FBS | Fetal bovine serum |
| CBX | Carbenoxolone |
| PBC | Probenecid |
| MDA | Malondialdehyde |
| Etd+ | Ethidium |
References
- Ozawa, Y.; Kobori, H.; Suzaki, Y.; Navar, L.G. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am. J. Physiol. Ren. Physiol. 2007, 292, F330–F339. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Noble, N.A. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 1998, 31 Pt 2, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Rúperez, M.; Sánchez-Lopez, E.; Blanco-Colio, L.M.; Esteban, V.; Rodriguez-Vita, J.; Plaza, J.J.; Egido, J.; Ruíz-Ortega, M. The Rho-kinase pathway regulates angiotensin II-induced renal damage. Kidney Int. Suppl. 2005, 99, S39–S45. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Rolli-Derkinderen, M.; Loufrani, L.; Bourge, A.; Henrion, D.; Sabourin, L.; Loirand, G.; Pacaud, P. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ. Res. 2008, 102, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wu, D.; Gao, B.; Ingram, A.J.; Zhang, B.; Chorneyko, K.; McKenzie, R.; Krepinsky, J.C. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes 2008, 57, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Kolavennu, V.; Zeng, L.; Peng, H.; Wang, Y.; Danesh, F.R. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 2008, 57, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, F.; Huang, X.R.; Liu, F.; Chen, H.; Chung, A.C.; Shi, J.; Wei, L.; Lan, H.Y.; Fu, P. Amelioration of albuminuria in ROCK1 knockout mice with streptozotocin-induced diabetic kidney disease. Am. J. Nephrol. 2011, 34, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Loirand, G. Rho Kinases in Health and Disease: From Basic Science to Translational Research. Pharmacol. Rev. 2015, 67, 1074–1095. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, T.; Oda, T.; Yamamoto, K.; Higashi, K.; Watanabe, A.; Takechi, H.; Uchida, T.; Oshima, N.; Sakurai, Y.; Miura, S.; et al. Protective effects of Rho kinase inhibitor fasudil on rats with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F1325–F1334. [Google Scholar] [CrossRef] [PubMed]
- Toubas, J.; Beck, S.; Pageaud, A.L.; Huby, A.C.; Mael-Ainin, M.; Dussaule, J.C.; Chatziantoniou, C.; Chadjichristos, C.E. Alteration of connexin expression is an early signal for chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F24–F32. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.C.; Berthoud, V.M.; Brañes, M.C.; Martínez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Paul, D.L.; Goodenough, D.A. Connexin43: A protein from rat heart homologous to a gap junction protein from liver. J. Cell Biol. 1987, 105 Pt 1, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Hernández, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.; Giaume, C.; Sáez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Boassa, D.; Dermietzel, R.; Duffy, H.S.; Laird, D.W.; MacVicar, B.; Naus, C.C.; Penuela, S.; Scemes, E.; Spray, D.C.; et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011, 5, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Volonte, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2X7 receptors: Channels, pores and more. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Hanner, F.; Lam, L.; Nguyen, M.T.; Yu, A.; Peti-Peterdi, J. Intrarenal localization of the plasma membrane ATP channel pannexin1. Am. J. Physiol. Ren. Physiol. 2012, 303, F1454–F1459. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Renal connexins and blood pressure. Biochim. Biophys. Acta 2012, 1818, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Menzies, R.I.; Howarth, A.R.; Unwin, R.J.; Tam, F.W.; Mullins, J.J.; Bailey, M.A. Inhibition of the purinergic P2X7 receptor improves renal perfusion in angiotensin-II-infused rats. Kidney Int. 2015, 88, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, J.A.; Krattinger, N.; Martin, D.; Pedrazzini, T.; Capponi, A.; Doring, B.; Plum, A.; Charollais, A.; Willecke, K.; Meda, P. Connexin43-dependent mechanism modulates renin secretion and hypertension. J. Clin. Investig. 2006, 116, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Hillis, G.S.; Duthie, L.A.; Mlynski, R.; McKay, N.G.; Mistry, S.; MacLeod, A.M.; Simpson, J.G.; Haites, N.E. The expression of connexin 43 in human kidney and cultured renal cells. Nephron 1997, 75, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Vonend, O.; Turner, C.M.; Chan, C.M.; Loesch, A.; Dell’Anna, G.C.; Srai, K.S.; Burnstock, G.; Unwin, R.J. Glomerular expression of the ATP-sensitive P2X receptor in diabetic and hypertensive rat models. Kidney Int. 2004, 66, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Vergara, L.; Bao, X.; Cooper, M.; Bello-Reuss, E.; Reuss, L. Gap-junctional hemichannels are activated by ATP depletion in human renal proximal tubule cells. J. Membr. Biol. 2003, 196, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Palacios-Prado, N.; Orellana, J.A.; Sáez, J.C. Currently used methods for identification and characterization of hemichannels. Cell Commun. Adhes. 2008, 15, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.J.; Onstead-Haas, L.; Lee, T.; Torfah, M.; Mooradian, A.D. Angiotensin II receptor one (AT1) mediates dextrose induced endoplasmic reticulum stress and superoxide production in human coronary artery endothelial cells. Int. J. Cardiol. 2016, 220, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Sáez, P.J.; Shoji, K.F.; Schalper, K.A.; Palacios-Prado, N.; Velarde, V.; Giaume, C.; Bennett, M.V.; Sáez, J.C. Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox Signal. 2009, 11, 369–399. [Google Scholar] [CrossRef] [PubMed]
- Shoji, K.F.; Sáez, P.J.; Harcha, P.A.; Aguila, H.L.; Sáez, J.C. Pannexin1 channels act downstream of P2X 7 receptors in ATP-induced murine T-cell death. Channels (Austin) 2014, 8, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Hanner, F.; Sorensen, C.M.; Holstein-Rathlou, N.H.; Peti-Peterdi, J. Connexins and the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1143–R11455. [Google Scholar] [CrossRef] [PubMed]
- Warner, A.; Clements, D.K.; Parikh, S.; Evans, W.H.; DeHaan, R.L. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 1995, 488 Pt 3, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverria, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Dahl, G.; Qiu, F.; Wang, J. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology 2013, 75, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaracharya, A.; Dao-Ung, P.; Jalilian, I.; Spildrejorde, M.; Skarratt, K.K.; Fuller, S.J.; Sluyter, R.; Stokes, L. Probenecid blocks human P2X7 receptor-induced dye uptake via a pannexin-1 independent mechanism. PLoS ONE 2014, 9, e93058. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Diaz, E.; Schalper, K.A.; Vargas, A.A.; Bennett, M.V.; Sáez, J.C. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem. Biophys. Res. Commun. 2011, 409, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Liu, M.; Inoue, K.; Ohno, Y. Potent inhibition by trivalent cations of ATP-gated channels. Eur. J. Pharmacol. 1997, 325, 237–243. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine prevents renal alterations in diabetic rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wei, C.; Chen, X.; Wang, J.; Cheng, H.; Zhang, X.; Hong, Q.; Shi, S.; Fu, B.; Wei, R. Essential role of Ca2+ release channels in angiotensin II-induced Ca2+ oscillations and mesangial cell contraction. Kidney Int. 2006, 70, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Ji, Z. AngII-induced glomerular mesangial cell proliferation inhibited by losartan via changes in intracellular calcium ion concentration. Clin. Exp. Med. 2014, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney fibrosis in hypertensive rats: Role of oxidative stress. Am. J. Nephrol. 2008, 28, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.E.; Sáez, J.C.; Reuss, L.; Altenberg, G.A. Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 2012, 287, 40826–40834. [Google Scholar] [CrossRef] [PubMed]
- Van Kats, J.P.; de Lannoy, L.M.; Jan Danser, A.H.; van Meegen, J.R.; Verdouw, P.D.; Schalekamp, M.A. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension 1997, 30 Pt 1, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Reuveny, M.; Heller, H.; Bengal, E. RhoA controls myoblast survival by inducing the phosphatidylinositol 3-kinase-Akt signaling pathway. FEBS Lett. 2004, 569, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Puebla, C.; Cisterna, B.A.; Salas, D.P.; Delgado-López, F.; Lampe, P.D.; Sáez, J.C. Linoleic acid permeabilizes gastric epithelial cells by increasing connexin 43 levels in the cell membrane via a GPR40- and Akt-dependent mechanism. Biochim. Biophys. Acta 2016, 1861, 439–448. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, E.; Wang, N.; Decrock, E.; De Bock, M.; Vinken, M.; Van Moorhem, M.; Lai, C.; Culot, M.; Rogiers, V.; Cecchelli, R.; et al. Ca2+ regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 2009, 46, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Piskuric, N.A.; Vollmer, C.; Nurse, C.A. P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: potential role in amplifying the neurotransmitter ATP. J. Physiol. 2012, 590, 4335–4350. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Sáez, J.C. Connexin hemichannels explain the ionic imbalance and lead to atrophy in denervated skeletal muscles. Biochim. Biophys. Acta 2016, 1862, 2168–2176. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, X.; Cai, G.Y.; Lv, Y.; Zhuo, L.; Gao, J.J.; Cui, S.Y.; Feng, Z.; Fu, B.; Chen, X.M. High glucose-induced hypertrophy of mesangial cells is reversed by connexin43 overexpression via PTEN/Akt/mTOR signaling. Nephrol. Dial. Transplant. 2012, 27, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Wakino, S.; Kanda, T.; Homma, K.; Sugano, N.; Saruta, T. Molecular mechanisms and therapeutic strategies of chronic renal injury: role of rho-kinase in the development of renal injury. J. Pharmacol. Sci. 2006, 100, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.C.; Stone, C.; Tkach, L.; SundarRaj, N. Rho and Rho-kinase (ROCK) signaling in adherens and gap junction assembly in corneal epithelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 978–986. [Google Scholar]
- Langevin, H.M.; Fujita, T.; Bouffard, N.A.; Takano, T.; Koptiuch, C.; Badger, G.J.; Nedergaard, M. Fibroblast cytoskeletal remodeling induced by tissue stretch involves ATP signaling. J. Cell. Physiol. 2013, 228, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Sáez, P.J.; Cortés-Campos, C.; Elizondo, R.J.; Shoji, K.F.; Contreras-Duarte, S.; Figueroa, V.; Velarde, V.; Jiang, J.X.; Nualart, F.; et al. Glucose increases intracellular free Ca2+ in tanycytes via ATP released through connexin 43 hemichannels. Glia 2012, 60, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Saffitz, J.E.; Laing, J.G.; Yamada, K.A. Connexin expression and turnover: Implications for cardiac excitability. Circ. Res. 2000, 86, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Sáez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channels. Neuropharmacology 2013, 75, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Satou, R.; Shao, W.; Prieto, M.C.; Urushihara, M.; Kobori, H.; Navar, L.G. ROCK/NF-kappaB axis-dependent augmentation of angiotensinogen by angiotensin II in primary-cultured preglomerular vascular smooth muscle cells. Am. J. Physiol. Ren. Physiol. 2014, 306, F608–F618. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Nishikimi, T.; Akimoto, K.; Ishimura, K.; Ono, H.; Matsuoka, H. Long-term administration of rho-kinase inhibitor ameliorates renal damage in malignant hypertensive rats. Hypertension 2006, 47, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Sáez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Cogliati, B.; Pereira, I.V.A.; da Silva, T.C.; Crespo Yanguas, S.; Maes, M.; Govoni, V.M.; Lima, A.; Felisbino, D.A.; Decrock, E.; et al. Inhibition of connexin hemichannels alleviates non-alcoholic steatohepatitis in mice. Sci. Rep. 2017, 7, 8268. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Green, C.R.; Kho, D.T.; Zhang, J.; Graham, E.S.; Acosta, M.L.; Rupenthal, I.D. The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta 2018, 1862, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Chaudhari, S.; Li, W. Canonical Transient Receptor Potential 6 Channel: A New Target of Reactive Oxygen Species in Renal Physiology and Pathology. Antioxid. Redox Signal. 2016, 25, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.; Frank, M.; Willecke, K.; Sáez, J.C. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.E.; Sánchez, H.A.; Eugenín, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Sáez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Ramanathan, L.; Das, N.P.; Li, Q.T. Studies on lipid oxidation in fish phospholipid liposomes. Biol. Trace Elem. Res. 1994, 40, 59–70. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, G.I.; Fernández, P.; Velarde, V.; Sáez, J.C. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. Int. J. Mol. Sci. 2018, 19, 957. https://doi.org/10.3390/ijms19040957
Gómez GI, Fernández P, Velarde V, Sáez JC. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. International Journal of Molecular Sciences. 2018; 19(4):957. https://doi.org/10.3390/ijms19040957
Chicago/Turabian StyleGómez, Gonzalo I., Paola Fernández, Victoria Velarde, and Juan C. Sáez. 2018. "Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels" International Journal of Molecular Sciences 19, no. 4: 957. https://doi.org/10.3390/ijms19040957