The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Neurovascular Coupling
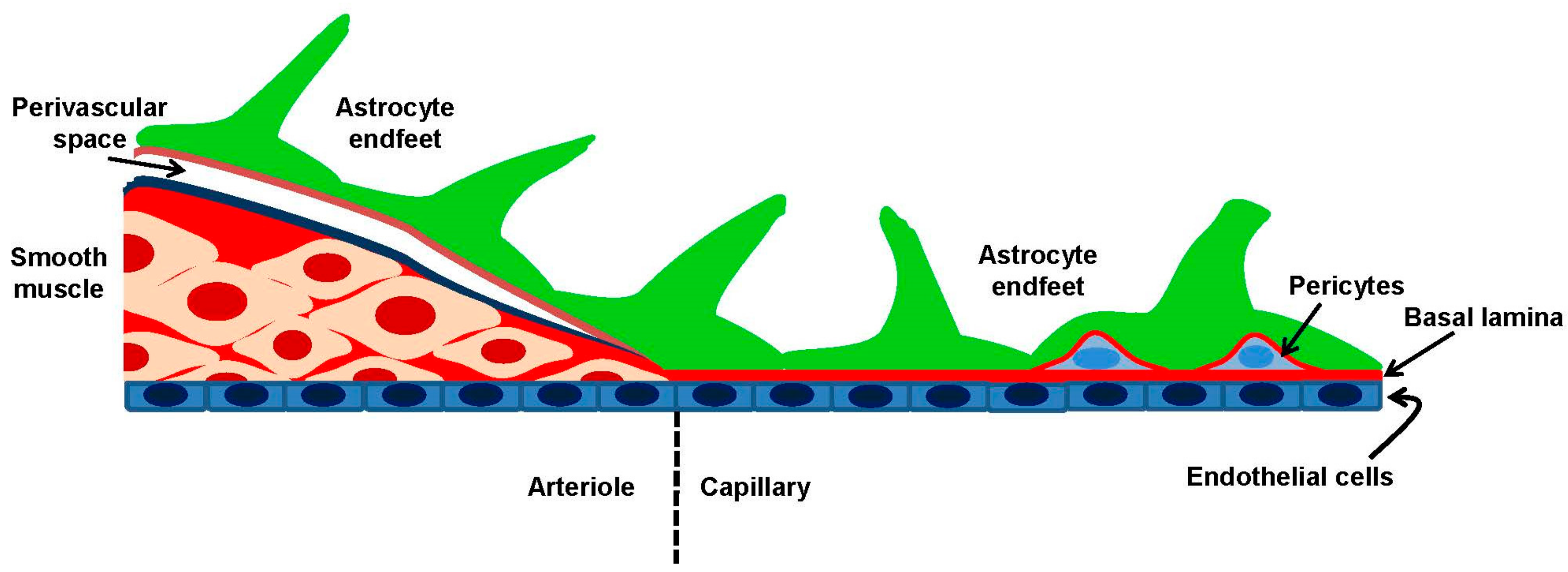
2.1. Cerebral Circulation and the Neurovascular Unit
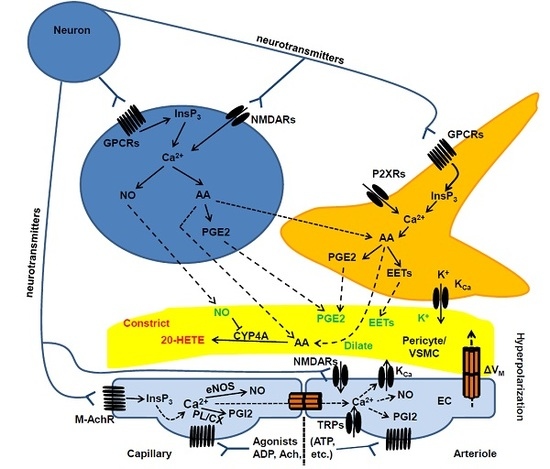
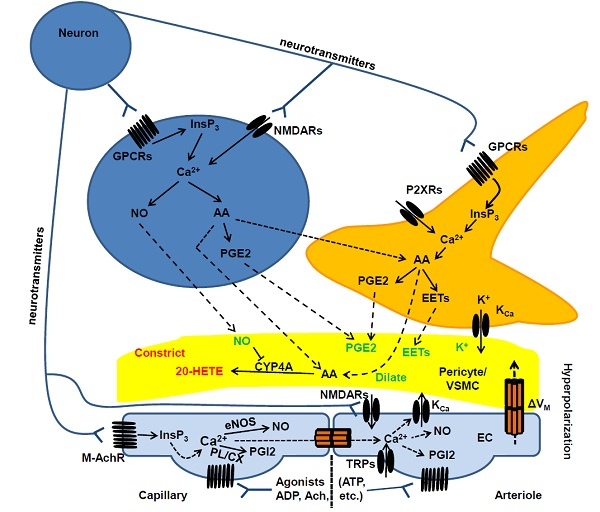
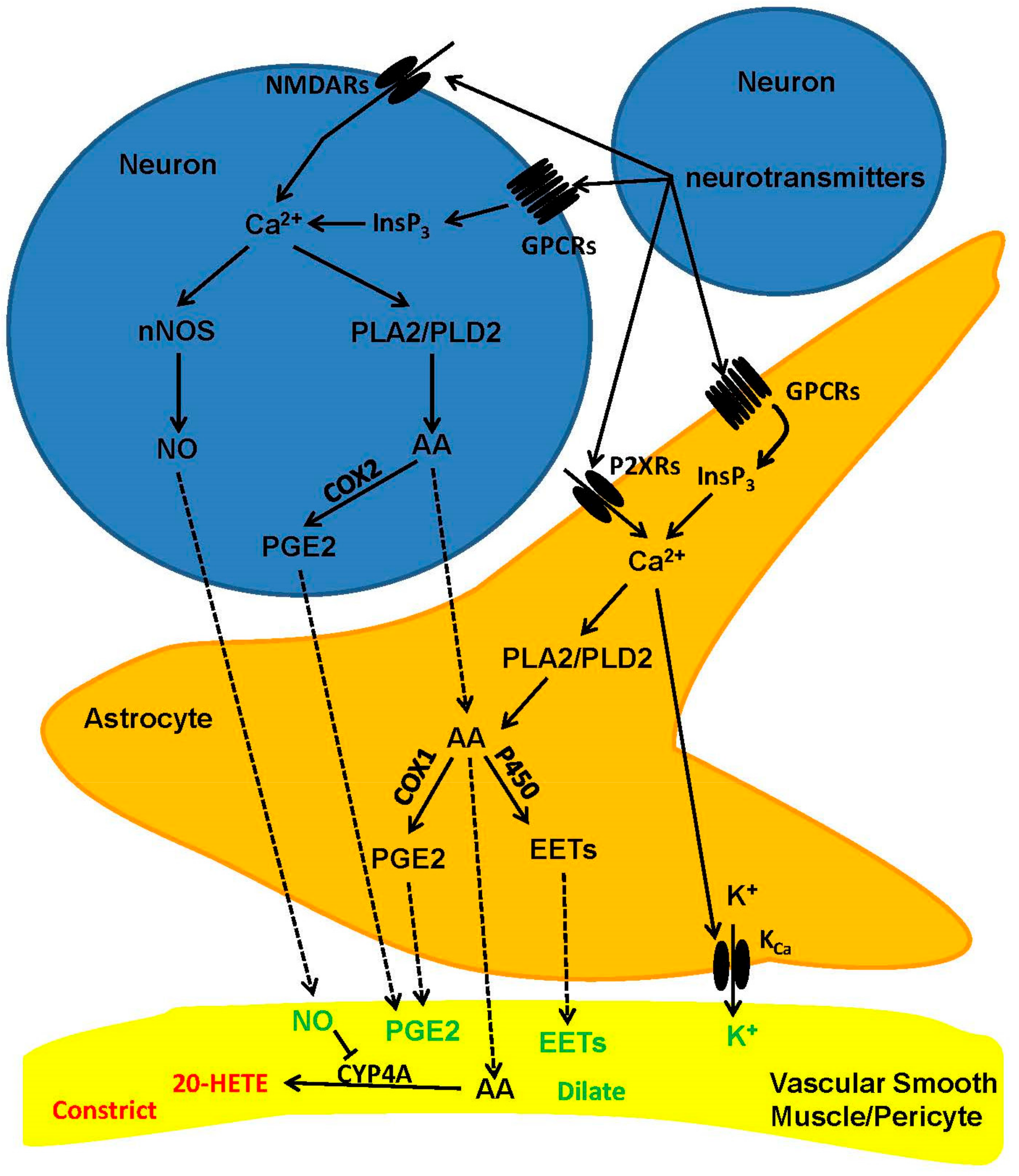
2.2. Cellular and Biochemical Pathways of Neurovascular Coupling: The Role of Neurons and Astrocytes in Arterioles and Capillaries
2.3. Is There a Role for Endothelial Ca2+ Signaling in Neurovascular Coupling?
3. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling
3.1. Endothelial Ca2+ Signaling in Brief
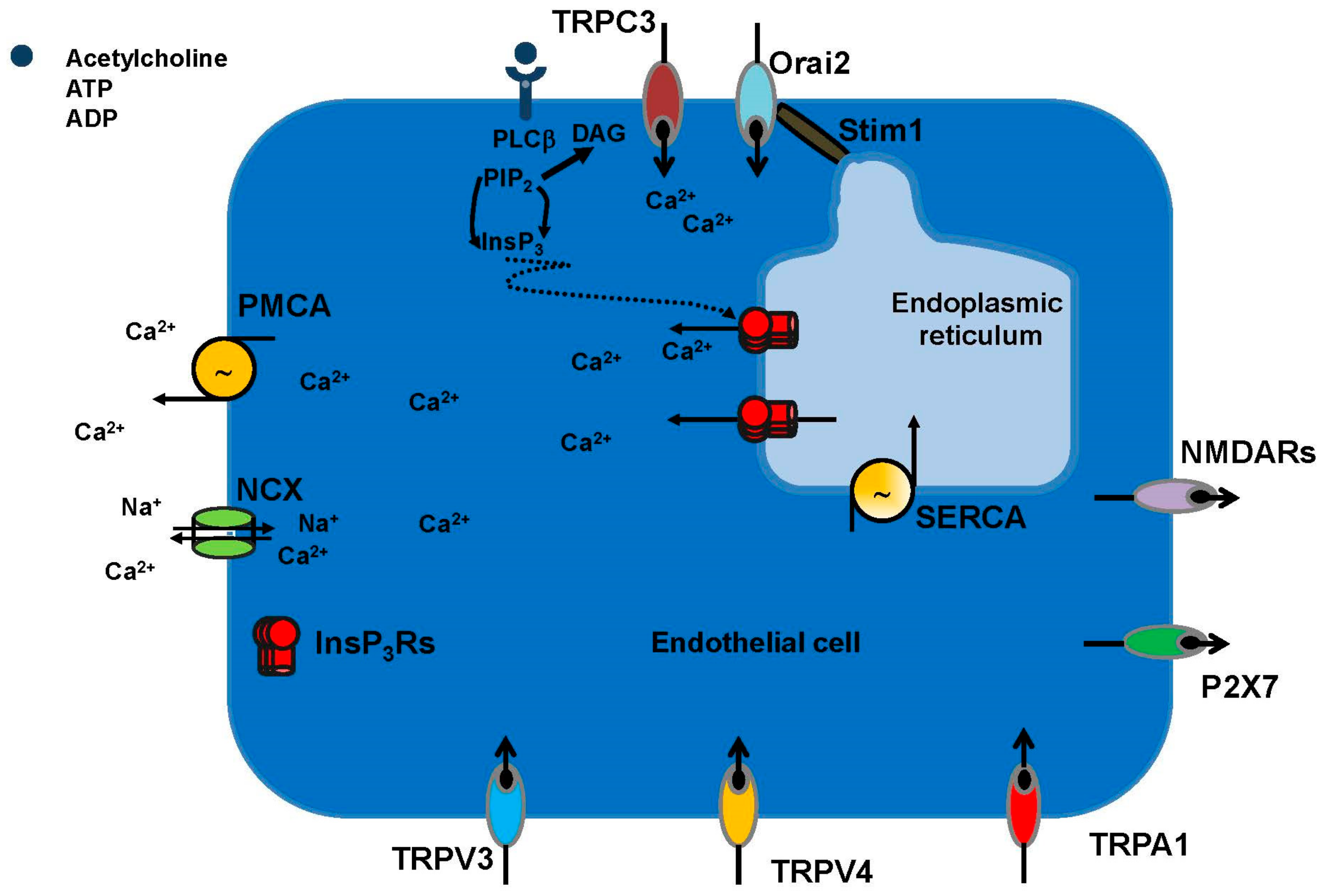
3.1.1. The Endothelial Ca2+ Toolkit in Brain Microvascular Endothelial Cells: Endogenous Ca2+ Release
3.1.2. The Endothelial Ca2+ Toolkit in Brain Microvascular Endothelial Cells: Endogenous Ca2+ Release
3.2. Endothelial NMDA Receptors Trigger Glutamate-Induced Nitric Oxide-Mediated Vasodilation
3.3. Intracellular Ca2+ Signals Drive Acetylcholine-Induced Nitric Oxide Release from Brain Microvascular Endothelial Cells
3.4. Endothelial Ca2+ Signals Could Mediate ATP-Induced Vasodilation
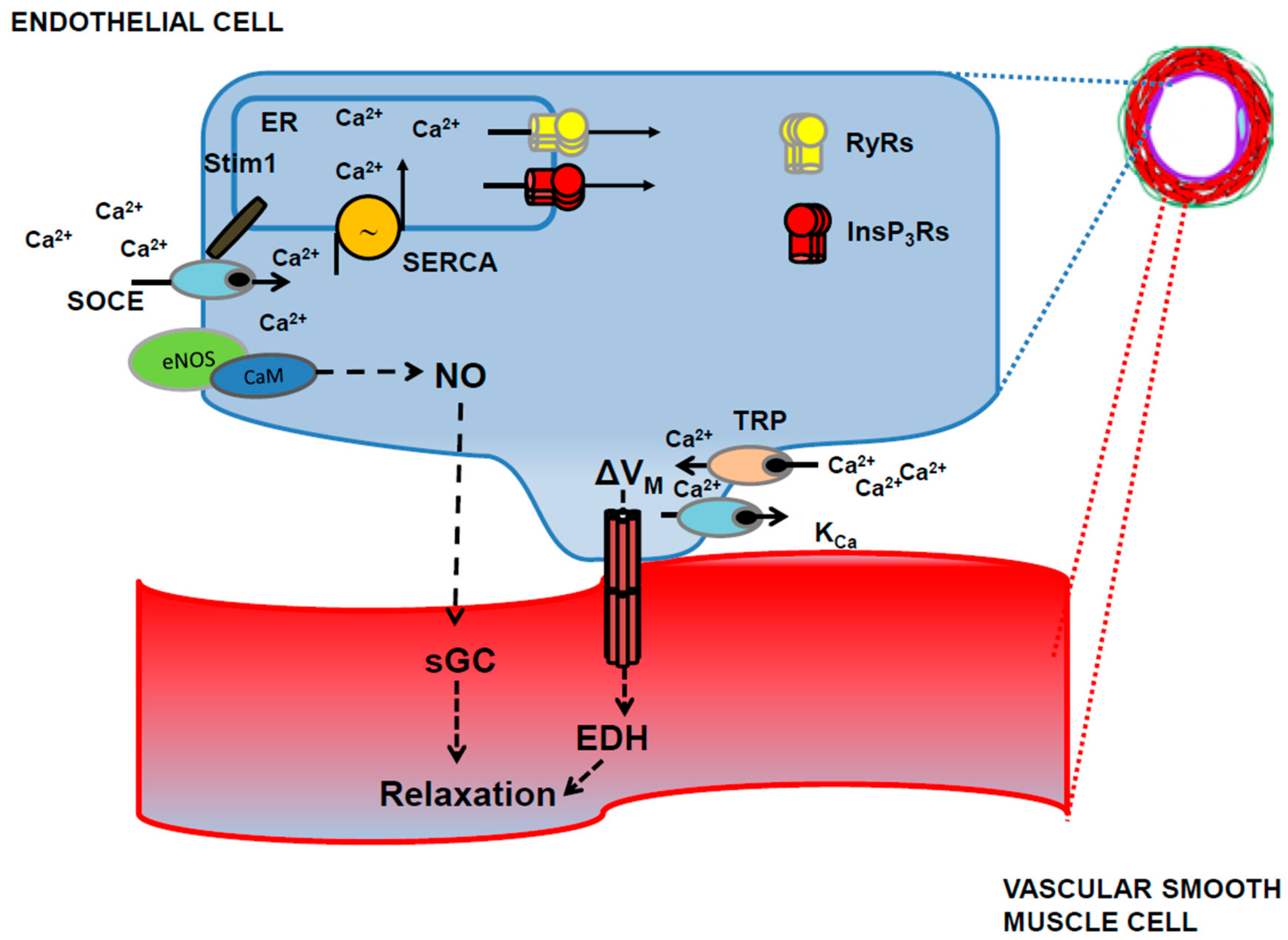
3.5. TRP Channels Trigger Endothelial-Dependent Hyperpolarization in Cerebrovascular Endothelial Cells
3.5.1. TRPV4
3.5.2. TRPA1, TRPC3 and TRPV3
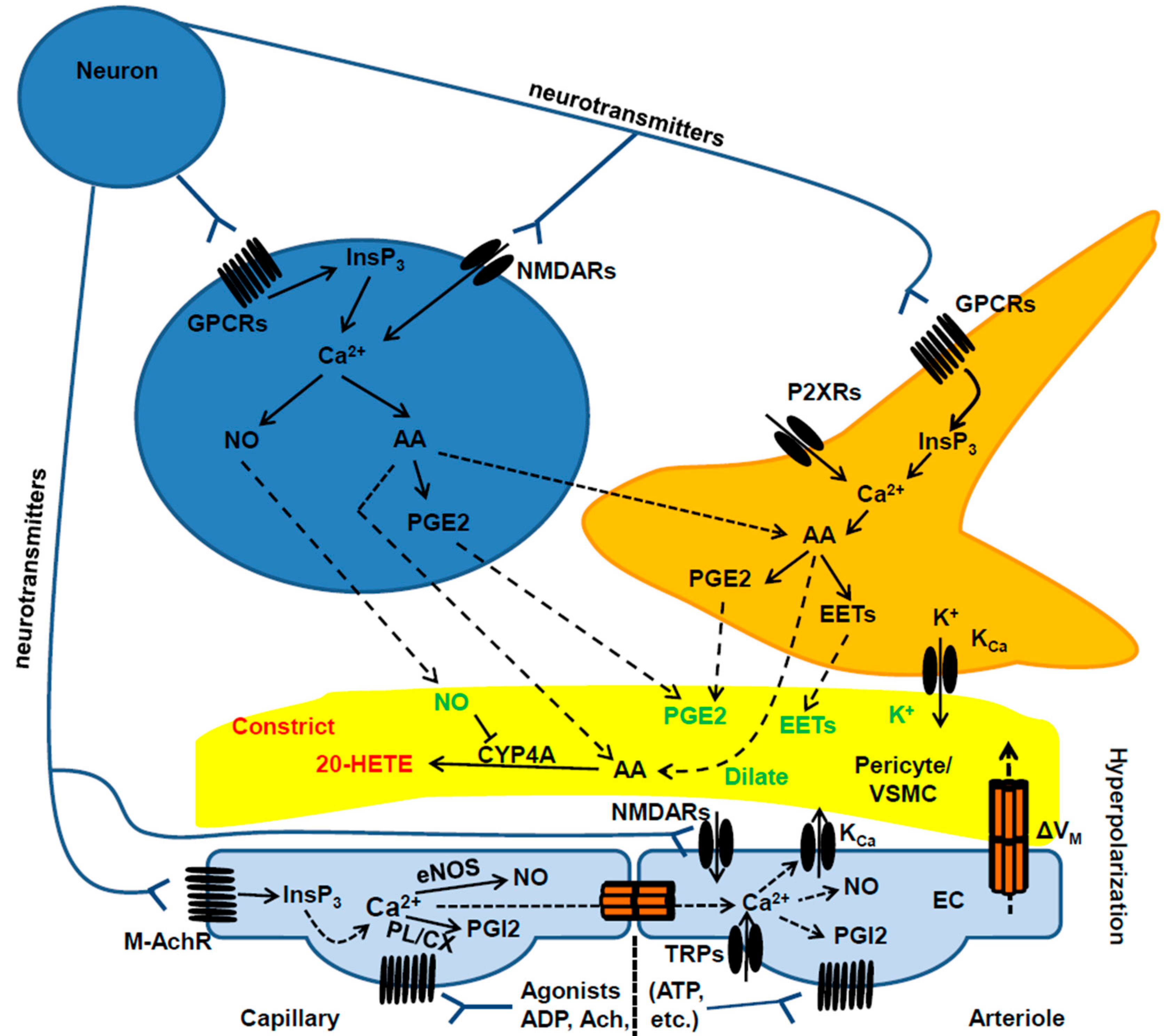
4. How to Integrate Endothelial Ca2+ Signals in the Current Models of Neurovascular Coupling and Propagated Vasodilation
5. Conclusions
Conflicts of Interest
Abbreviations
| 20-HETE | 20-hydroxyeicosatetraenoic acid |
| AA | arachidonic acid |
| AMPA | a-amino-3-hydroxy-5-methyl-4-isoxazol epropionic acid |
| BKCa | large-conductance Ca2+-activated K+ channels |
| BOLD | blood oxygenation level dependent |
| CaM | Calmodulin |
| CBF | cerebral blood flow |
| cGMP | cyclic guanosine-3′,5′-monophosphate |
| CICR | Ca2+-induced Ca2+ release |
| CYP4A | cytochrome P450 4A |
| DAG | Diacylglycerol |
| EDH | endothelial-dependent hyperpolarization |
| EETs | epoxyeicosatrienoic acids |
| EL | Endolysosomal |
| eNOS | neuronal NO synthase |
| ER | endoplasmic reticulum |
| GPCRs | G-protein-coupled receptors |
| IKCa | intermediate-conductance Ca2+-activated K+ channels |
| InsP3 | inositol-1,4,5-trisphosphate |
| InsP3Rs | InsP3 receptors |
| KCa | Ca2+-activated K+ channels |
| Kir | inward rectifying K+ channels |
| M-AchRs | muscarinic acetylcholine receptors |
| MCU | mitochondrial Ca2+ uniporter |
| MEGJs | myo-endothelial gap-junctions |
| mGluRs | metabotropic glutamate receptors |
| NA | neuronal activity |
| NAADP | nicotinic acid adenine dinucleotide phosphate |
| NMDA | N-methyl-d-aspartate |
| NMDARs | NMDA receptors |
| NO | nitric oxide |
| nNOS | neuronal NO synthase |
| NOX2 | NADPH oxidase isoform 2 |
| NVC | neurovascular coupling |
| NVU | neurovascular unit |
| O2 | Oxygen |
| P450 | cytochrome P450 |
| PGE2 | prostaglandin E2 |
| PIP2 | phosphatidylinositol-4,5-bisphosphate |
| PLA2 | phospholipase A2 |
| PLD2 | phospholipase D2 |
| RyRs | ryanodine receptors |
| SERCA | sarco-endoplasmic reticulum Ca2+-ATPase |
| SKCa | small-conductance Ca2+-activated K+ channels |
| SOCE | store-operated Ca2+ entry |
| Stim | stromal interaction molecule |
| TPCs | two-pore channels |
| TRPs | TRP channels |
| TRPA1 | ankyrin TRP 1 |
| TRPC | canonical TRP |
| TRPV4 | vanilloid TRP 4 |
| VDACs | voltage-dependent anion channels |
| VSMC | vascular smooth muscle cells |
References
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Toth, P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J. Vasc. Res. 2012, 49, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Hillman, E.M. Coupling mechanism and significance of the BOLD signal: A status report. Annu. Rev. Neurosci. 2014, 37, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Iadecola, C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002, 25, 621–625. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Rancillac, A.; Rossier, J.; Guille, M.; Tong, X.K.; Geoffroy, H.; Amatore, C.; Arbault, S.; Hamel, E.; Cauli, B. Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the Cerebellum. J. Neurosci. 2006, 26, 6997–7006. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Rosenegger, D.G.; Gordon, G.R. A slow or modulatory role of astrocytes in neurovascular coupling. Microcirculation 2015, 22, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, M.H.; Davis, M.J.; Secher, N.H.; van Lieshout, J.J.; Arce-Esquivel, A.A.; Simmons, G.H.; Bender, S.B.; Padilla, J.; Bache, R.J.; Merkus, D.; et al. Peripheral circulation. Compr. Physiol. 2012, 2, 321–447. [Google Scholar] [PubMed]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Pla, A.F.; Moccia, F.; Tanzi, F.; Munaron, L. Old and new gasotransmitters in the cardiovascular system: Focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 2011, 12, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Tang, E.H. Endothelium-dependent contractions: When a good guy turns bad! J. Physiol. 2008, 586, 5295–5304. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen sulfide and endothelial dysfunction: Relationship with nitric oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J. Calcium and electrical signaling in arterial endothelial tubes: New insights into cellular physiology and cardiovascular function. Microcirculation 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiologica 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Hill-Eubanks, D.C.; Nelson, M.T. Ion channel networks in the control of cerebral blood flow. J. Cereb. Blood Flow Metab. 2016, 36, 492–512. [Google Scholar] [CrossRef] [PubMed]
- Cauli, B.; Hamel, E. Revisiting the role of neurons in neurovascular coupling. Front. Neuroenerg. 2010, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Cauli, B.; Tong, X.K.; Rancillac, A.; Serluca, N.; Lambolez, B.; Rossier, J.; Hamel, E. Cortical GABA interneurons in neurovascular coupling: Relays for subcortical vasoactive pathways. J. Neurosci. 2004, 24, 8940–8949. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Toussay, X.; Kocharyan, A.; Fernandes, P.; Neupane, S.; Levesque, M.; Plaisier, F.; Shmuel, A.; Cauli, B.; Hamel, E. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J. Neurosci. 2011, 31, 9836–9847. [Google Scholar] [CrossRef] [PubMed]
- Lecrux, C.; Hamel, E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. pii: 20150350. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J. Appl. Physiol. (1985) 2006, 100, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Attwell, D. Control of brain energy supply by astrocytes. Curr. Opin. Neurobiol. 2017, 47, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, T.; Alarcon-Martinez, L. Cerebral microvascular pericytes and neurogliovascular signaling in health and disease. Brain Res. 2015, 1623, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M. Reading vascular changes in brain imaging: Is dendritic calcium the key? Nat. Rev. Neurosci. 2005, 6, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.F.; Ledo, A.; Barbosa, R.M.; Laranjinha, J. Neurovascular-neuroenergetic coupling axis in the brain: Master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic. Biol. Med. 2017, 108, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Chen, G.; Ebner, T.J.; Iadecola, C. Nitric oxide is the predominant mediator of cerebellar hyperemia during somatosensory activation in rats. Am. J. Physiol. 1999, 277 (Pt 2), R1760–R1770. [Google Scholar] [CrossRef] [PubMed]
- Lovick, T.A.; Brown, L.A.; Key, B.J. Neurovascular relationships in hippocampal slices: Physiological and anatomical studies of mechanisms underlying flow-metabolism coupling in intraparenchymal microvessels. Neuroscience 1999, 92, 47–60. [Google Scholar] [CrossRef]
- Duchemin, S.; Boily, M.; Sadekova, N.; Girouard, H. The complex contribution of NOS interneurons in the physiology of cerebrovascular regulation. Front. Neural Circ. 2012, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Morrison, H.W.; Iddings, J.A.; Du, W.; Kim, K.J. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience 2016, 323, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lou, N.; Xu, Q.; Tian, G.F.; Peng, W.G.; Han, X.; Kang, J.; Takano, T.; Nedergaard, M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006, 9, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A. Binaural blood flow control by astrocytes: Listening to synapses and the vasculature. J. Physiol. 2017, 595, 1885–1902. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Iddings, J.A.; Stern, J.E.; Blanco, V.M.; Croom, D.; Kirov, S.A.; Filosa, J.A. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J. Neurosci. 2015, 35, 8245–8257. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Ramiro Diaz, J.; Iddings, J.A.; Filosa, J.A. Vasculo-Neuronal Coupling: Retrograde Vascular Communication to Brain Neurons. J. Neurosci. 2016, 36, 12624–12639. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Arcuino, G.; Takano, T.; Liu, Q.S.; Nedergaard, M. Signaling at the gliovascular interface. J. Neurosci. 2003, 23, 9254–9262. [Google Scholar] [PubMed]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerg. 2010, 2. pii: 5. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, E. The organization of plasticity in the cerebellar cortex: From synapses to control. Prog. Brain Res. 2014, 210, 31–58. [Google Scholar] [PubMed]
- Berra-Romani, R.; Avelino-Cruz, J.E.; Raqeeb, A.; Della Corte, A.; Cinelli, M.; Montagnani, S.; Guerra, G.; Moccia, F.; Tanzi, F. Ca(2)(+)-dependent nitric oxide release in the injured endothelium of excised rat aorta: A promising mechanism applying in vascular prosthetic devices in aging patients. BMC Surg. 2013, 13 (Suppl. 2), S40. [Google Scholar] [CrossRef] [PubMed]
- Andresen, J.; Shafi, N.I.; Bryan, R.M., Jr. Endothelial influences on cerebrovascular tone. J. Appl. Physiol. (1985) 2006, 100, 318–327. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Johnson, T.D.; Childres, W.F.; Bryan, R.M., Jr. Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am. J. Physiol. 1997, 273 (Pt 2), H1472–H1477. [Google Scholar] [CrossRef] [PubMed]
- Elhusseiny, A.; Cohen, Z.; Olivier, A.; Stanimirovic, D.B.; Hamel, E. Functional acetylcholine muscarinic receptor subtypes in human brain microcirculation: Identification and cellular localization. J. Cereb. Blood Flow Metab. 1999, 19, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Elhusseiny, A.; Hamel, E. Muscarinic--but not nicotinic--acetylcholine receptors mediate a nitric oxide-dependent dilation in brain cortical arterioles: A possible role for the M5 receptor subtype. J. Cereb. Blood Flow Metab. 2000, 20, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, J.L.; Sanders, S.A.; Stobart, M.J.; Lu, L.; Knox, J.D.; Anderson, H.D.; Anderson, C.M. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow Metab. 2012, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Lu, L.; Anderson, H.D.; Mori, H.; Anderson, C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.S., Jr.; Reynolds, J.J.; Bearden, S.E. Metabotropic glutamate receptor 5 mediates phosphorylation of vascular endothelial cadherin and nuclear localization of beta-catenin in response to homocysteine. Vascul. Pharmacol. 2012, 56, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.M.; Guo, X.M.; Chen, B.; Lei, Q.; Pan, Y.J.; Yang, Q. The Noncompetitive AMPAR Antagonist Perampanel Abrogates Brain Endothelial Cell Permeability in Response to Ischemia: Involvement of Claudin-5. Cell. Mol. Neurobiol. 2016, 36, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Park, K.A.; Montalto, M.C.; Alapati, S.; Buras, J.A.; Stahl, G.L.; Colgan, S.P. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J. Biol. Chem. 2002, 277, 14801–14811. [Google Scholar] [CrossRef] [PubMed]
- Gillard, S.E.; Tzaferis, J.; Tsui, H.C.; Kingston, A.E. Expression of metabotropic glutamate receptors in rat meningeal and brain microvasculature and choroid plexus. J. Comp. Neurol. 2003, 461, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Suzuki, N. Control of brain capillary blood flow. J. Cereb. Blood Flow Metab. 2012, 32, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.S. Integration and Modulation of Intercellular Signaling Underlying Blood Flow Control. J. Vasc. Res. 2015, 52, 136–157. [Google Scholar] [CrossRef] [PubMed]
- Tallini, Y.N.; Brekke, J.F.; Shui, B.; Doran, R.; Hwang, S.M.; Nakai, J.; Salama, G.; Segal, S.S.; Kotlikoff, M.I. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: Measurements in Cx40BAC GCaMP2 transgenic mice. Circ. Res. 2007, 101, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Wolfle, S.E.; Chaston, D.J.; Goto, K.; Sandow, S.L.; Edwards, F.R.; Hill, C.E. Non-linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J. Physiol. 2011, 589 (Pt 10), 2607–2623. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. STIM1 and ORAI1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1alpha Promotes Endothelial Colony-Forming Cell Migration Through the Ca(2+)-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [PubMed]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca2+ signalling in endothelial progenitor cells: A novel means to improve cell-based therapy and impair tumour vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5018. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Shipsky, M.M.; Yan, G.; Abood, M.E.; Brailoiu, G.C. Mechanisms of modulation of brain microvascular endothelial cells function by thrombin. Brain Res. 2017, 1657, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Domotor, E.; Abbott, N.J.; Adam-Vizi, V. Na+-Ca2+ exchange and its implications for calcium homeostasis in primary cultured rat brain microvascular endothelial cells. J. Physiol. 1999, 515 (Pt 1), 147–155. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Scharbrodt, W.; Abdallah, Y.; Kasseckert, S.A.; Gligorievski, D.; Piper, H.M.; Boker, D.K.; Deinsberger, W.; Oertel, M.F. Cytosolic Ca2+ oscillations in human cerebrovascular endothelial cells after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2009, 29, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Earley, S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J. Cardiovasc. Pharmacol. 2011, 57, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Manoonkitiwongsa, P.S.; Whitter, E.F.; Wareesangtip, W.; McMillan, P.J.; Nava, P.B.; Schultz, R.L. Calcium-dependent ATPase unlike ecto-ATPase is located primarily on the luminal surface of brain endothelial cells. Histochem. J. 2000, 32, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Signorelli, S.; Castelli, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002, 364 (Pt 1), 235–244. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Knipe, B.; Gorantla, S.; Zheng, J.; Persidsky, Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J. Neurochem. 2007, 100, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Hertle, D.N.; Yeckel, M.F. Distribution of inositol-1,4,5-trisphosphate receptor isotypes and ryanodine receptor isotypes during maturation of the rat hippocampus. Neuroscience 2007, 150, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Czupalla, C.J.; Liebner, S.; Devraj, K. In vitro models of the blood-brain barrier. Methods Mol. Biol. 2014, 1135, 415–437. [Google Scholar] [PubMed]
- Pedriali, G.; Rimessi, A.; Sbano, L.; Giorgi, C.; Wieckowski, M.R.; Previati, M.; Pinton, P. Regulation of Endoplasmic Reticulum-Mitochondria Ca2+ Transfer and Its Importance for Anti-Cancer Therapies. Front. Oncol. 2017, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e7. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Remodelling of the Ca2+ Toolkit in Tumor Endothelium as a Crucial Responsible for the Resistance to Anticancer Therapies. Curr. Signal Transduct. Ther. 2017, 12. [Google Scholar] [CrossRef]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [PubMed]
- Malli, R.; Frieden, M.; Hunkova, M.; Trenker, M.; Graier, W.F. Ca2+ refilling of the endoplasmic reticulum is largely preserved albeit reduced Ca2+ entry in endothelial cells. Cell Calcium 2007, 41, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Malli, R.; Frieden, M.; Osibow, K.; Zoratti, C.; Mayer, M.; Demaurex, N.; Graier, W.F. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J. Biol. Chem. 2003, 278, 44769–44779. [Google Scholar] [CrossRef] [PubMed]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar] [PubMed]
- Gerencser, A.A.; Adam-Vizi, V. Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration. Cell Calcium 2001, 30, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Moreira, P.I.; Oliveira, C.R.; Cardoso, S.M.; Pinton, P.; Pereira, C.F. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol. 2015, 51, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Ko, Y.; Mata, W.; Saquib, M.; Eldridge, J.; Cohen-Gadol, A.; Leaver, H.A.; Wang, S.; Rizzo, M.T. Arachidonic acid induces brain endothelial cell apoptosis via p38-MAPK and intracellular calcium signaling. Microvasc. Res. 2015, 98, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Katakam, P.V.; Wappler, E.A.; Katz, P.S.; Rutkai, I.; Institoris, A.; Domoki, F.; Gaspar, T.; Grovenburg, S.M.; Snipes, J.A.; Busija, D.W. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca(2)(+)-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca(2)(+) signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Lim, D.; Poletto, V.; Guerra, G.; Tanzi, F.; Rosti, V.; Moccia, F. Acidic Ca2+ stores interact with the endoplasmic reticulum to shape intracellular Ca2+ signals in human endothelial progenitor cells. Vascul. Pharmacol. 2015, 75, 70–71. [Google Scholar] [CrossRef]
- Di Nezza, F.; Zuccolo, E.; Poletto, V.; Rosti, V.; De Luca, A.; Moccia, F.; Guerra, G.; Ambrosone, L. Liposomes as a Putative Tool to Investigate NAADP Signaling in Vasculogenesis. J. Cell. Biochem. 2017, 118, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca(2+) Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. ORAI1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca(2)+ entry both by store-dependent, STIM1/ORAI1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]
- Regan, E.R.; Aird, W.C. Dynamical systems approach to endothelial heterogeneity. Circ. Res. 2012, 111, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Adams, D.J.; Taglietti, V.; Tanzi, F. Basal nonselective cation permeability in rat cardiac microvascular endothelial cells. Microvasc. Res. 2002, 64, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca(2+) Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bruns, A.F.; Hou, B.; Rode, B.; Webster, P.J.; Bailey, M.A.; Appleby, H.L.; Moss, N.K.; Ritchie, J.E.; Yuldasheva, N.Y.; et al. ORAI3 Surface Accumulation and Calcium Entry Evoked by Vascular Endothelial Growth Factor. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca(2+) Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19. pii: E217. [Google Scholar] [CrossRef] [PubMed]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Wu, S.; Chen, H.; Alexeyev, M.; St Croix, C.M.; Pitt, B.R.; Uhlig, S.; Stevens, T. ORAI1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ. Res. 2012, 110, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Cioffi, D.L.; Alexeyev, M.; Rich, T.C.; Stevens, T. Sodium entry through endothelial store-operated calcium entry channels: Regulation by ORAI1. Am. J. Physiol. Cell Physiol. 2015, 308, C277–C288. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Barry, C.; Stevens, T. Store-operated calcium entry channels in pulmonary endothelium: The emerging story of TRPCS and Orai1. Adv. Exp. Med. Biol. 2010, 661, 137–154. [Google Scholar] [PubMed]
- Berrout, J.; Jin, M.; O’Neil, R.G. Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood-brain barrier endothelial cells. Brain Res. 2012, 1436, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Rosenbaum, M.A.; Birnbaumer, L.; Graham, L.M. Integration of TRPC6 and NADPH oxidase activation in lysophosphatidylcholine-induced TRPC5 externalization. Am. J. Physiol. Cell Physiol. 2017, 313, C541–C555. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharmacol. 2016, 173, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Malik, A.B. TRP channels and the control of vascular function. Curr. Opin. Pharmacol. 2010, 10, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.O.; Yao, X. TRP channels in vascular endothelial cells. Adv. Exp. Med. Biol. 2011, 704, 759–780. [Google Scholar] [PubMed]
- Ellinsworth, D.C.; Earley, S.; Murphy, T.V.; Sandow, S.L. Endothelial control of vasodilation: Integration of myoendothelial microdomain signalling and modulation by epoxyeicosatrienoic acids. Pflugers Arch. 2014, 466, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H. t.; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Krizbai, I.A.; Deli, M.A.; Pestenacz, A.; Siklos, L.; Szabo, C.A.; Andras, I.; Joo, F. Expression of glutamate receptors on cultured cerebral endothelial cells. J. Neurosci. Res. 1998, 54, 814–819. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, K.; Zhang, X.; Zhang, J.; Xu, B. ATP Induces Disruption of Tight Junction Proteins via IL-1 Beta-Dependent MMP-9 Activation of Human Blood-Brain Barrier In Vitro. Neural Plast. 2016, 2016, 8928530. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, X.; Dai, Z.; Feng, Y.; Li, Q.; Zhang, J.H.; Liu, X.; Chen, Y.; Feng, H. P2X7 Receptor Suppression Preserves Blood-Brain Barrier through Inhibiting RhoA Activation after Experimental Intracerebral Hemorrhage in Rats. Sci. Rep. 2016, 6, 23286. [Google Scholar] [CrossRef] [PubMed]
- De Wit, C.; Wolfle, S.E. EDHF and gap junctions: Important regulators of vascular tone within the microcirculation. Curr. Pharm. Biotechnol. 2007, 8, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Keller, T.C. t.; Begandt, D.; Butcher, J.T.; Biwer, L.; Keller, A.S.; Columbus, L.; Isakson, B.E. Endothelial nitric oxide synthase in the microcirculation. Cell. Mol. Life Sci. 2015, 72, 4561–4575. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 2002, 539 (Pt 1), 77–91. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Functional consequences of activating store-operated CRAC channels. Cell Calcium 2007, 42, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Fagan, K.A.; Li, K.X.; Shaul, P.W.; Cooper, D.M.; Rodman, D.M. Sustained endothelial nitric-oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 2000, 275, 17979–17985. [Google Scholar] [CrossRef] [PubMed]
- Kimura, C.; Oike, M.; Ohnaka, K.; Nose, Y.; Ito, Y. Constitutive nitric oxide production in bovine aortic and brain microvascular endothelial cells: A comparative study. J. Physiol. 2004, 554 (Pt 3), 721–730. [Google Scholar] [CrossRef] [PubMed]
- Boittin, F.X.; Alonso, F.; Le Gal, L.; Allagnat, F.; Beny, J.L.; Haefliger, J.A. Connexins and M3 muscarinic receptors contribute to heterogeneous Ca(2+) signaling in mouse aortic endothelium. Cell. Physiol. Biochem. 2013, 31, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.I.; Kim, J.Y.; Yeon, D.S.; Abramowitz, J.; Birnbaumer, L.; Muallem, S.; Lee, Y.H. Transient receptor potential canonical type 3 channels control the vascular contractility of mouse mesenteric arteries. PLoS ONE 2014, 9, e110413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Mendoza, S.A.; Bubolz, A.H.; Mizuno, A.; Ge, Z.D.; Li, R.; Warltier, D.C.; Suzuki, M.; Gutterman, D.D. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 2009, 53, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C.; Zhang, F.; Xu, X. Role of nitric oxide synthase-containing vascular nerves in cerebrovasodilation elicited from cerebellum. Am. J. Physiol. 1993, 264 (Pt 2), R738–R346. [Google Scholar] [CrossRef] [PubMed]
- De Labra, C.; Rivadulla, C.; Espinosa, N.; Dasilva, M.; Cao, R.; Cudeiro, J. Different sources of nitric oxide mediate neurovascular coupling in the lateral geniculate nucleus of the cat. Front. Syst. Neurosci. 2009, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Fiumana, E.; Parfenova, H.; Jaggar, J.H.; Leffler, C.W. Carbon monoxide mediates vasodilator effects of glutamate in isolated pressurized cerebral arterioles of newborn pigs. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1073–H1079. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, M.; Kato, A.; Lovett, C.; Tonegawa, S.; Watanabe, M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4855–4860. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, W.; Freidl, M.; Szkokan, P.; Berger, M.; Wirth, M.; Winkler, J.; Gabor, F.; Pifl, C.; Noe, C.R. Effects of NMDA receptor modulators on a blood-brain barrier in vitro model. Brain Res. 2011, 1394, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide Epigenetically Attenuates Homocysteine-Induced Mitochondrial Toxicity Mediated Through NMDA Receptor in Mouse Brain Endothelial (bEnd3) Cells. J. Cell. Physiol. 2015, 230, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Busija, D.W.; Bari, F.; Domoki, F.; Louis, T. Mechanisms involved in the cerebrovascular dilator effects of N-methyl-d-aspartate in cerebral cortex. Brain Res. Rev. 2007, 56, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Cholinergic modulation of the cortical microvascular bed. Prog. Brain Res. 2004, 145, 171–178. [Google Scholar] [PubMed]
- Zhang, F.; Xu, S.; Iadecola, C. Role of nitric oxide and acetylcholine in neocortical hyperemia elicited by basal forebrain stimulation: Evidence for an involvement of endothelial nitric oxide. Neuroscience 1995, 69, 1195–1204. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F. Permissive and obligatory roles of NO in cerebrovascular responses to hypercapnia and acetylcholine. Am. J. Physiol. 1996, 271 (Pt 2), R990–R1001. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Lamping, K.G.; Duttaroy, A.; Zhang, W.; Cui, Y.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C.; Deng, C.X.; Faraci, F.M.; et al. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 14096–14101. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Senadheera, S.; Grayson, T.H.; Welsh, D.G.; Murphy, T.V. Calcium and endothelium-mediated vasodilator signaling. Adv. Exp. Med. Biol. 2012, 740, 811–831. [Google Scholar] [PubMed]
- Kasai, Y.; Yamazawa, T.; Sakurai, T.; Taketani, Y.; Iino, M. Endothelium-dependent frequency modulation of Ca2+ signalling in individual vascular smooth muscle cells of the rat. J. Physiol. 1997, 504 (Pt 2), 349–357. [Google Scholar] [CrossRef] [PubMed]
- Weikert, S.; Freyer, D.; Weih, M.; Isaev, N.; Busch, C.; Schultze, J.; Megow, D.; Dirnagl, U. Rapid Ca2+-dependent NO-production from central nervous system cells in culture measured by NO-nitrite/ozone chemoluminescence. Brain Res. 1997, 748, 1–11. [Google Scholar] [CrossRef]
- Radu, B.M.; Osculati, A.M.M.; Suku, E.; Banciu, A.; Tsenov, G.; Merigo, F.; Di Chio, M.; Banciu, D.D.; Tognoli, C.; Kacer, P.; et al. All muscarinic acetylcholine receptors (M1–M5) are expressed in murine brain microvascular endothelium. Sci. Rep. 2017, 7, 5083. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Frost, C.; Berra-Romani, R.; Tanzi, F.; Adams, D.J. Expression and function of neuronal nicotinic ACh receptors in rat microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H486–H491. [Google Scholar] [CrossRef] [PubMed]
- Laforenza, U.; Pellavio, G.; Moccia, F. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. 2017; manuscript in preparation. [Google Scholar]
- Uhlen, P.; Fritz, N. Biochemistry of calcium oscillations. Biochem. Biophys. Res. Commun. 2010, 396, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1046. [Google Scholar] [CrossRef] [PubMed]
- Guzman, S.J.; Gerevich, Z. P2Y Receptors in Synaptic Transmission and Plasticity: Therapeutic Potential in Cognitive Dysfunction. Neural Plast. 2016, 2016, 1207393. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.F.; Puebla, M.; Figueroa, X.F. Control of the neurovascular coupling by nitric oxide-dependent regulation of astrocytic Ca(2+) signaling. Front. Cell. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, H.H.; Horiuchi, T.; Xiang, C.; Hongo, K.; Falck, J.R.; Dacey, R.G., Jr. Mechanism of ATP-induced local and conducted vasomotor responses in isolated rat cerebral penetrating arterioles. J. Vasc. Res. 2009, 46, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, H.H.; Kajita, Y.; Dacey, R.G., Jr. Local and conducted vasomotor responses in isolated rat cerebral arterioles. Am. J. Physiol. 1996, 71, H1109–H1116. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.; Tarantini, S.; Davila, A.; Valcarcel-Ares, M.N.; Tucsek, Z.; Varamini, B.; Ballabh, P.; Sonntag, W.E.; Baur, J.A.; Csiszar, A.; et al. Purinergic glio-endothelial coupling during neuronal activity: Role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1837–H1845. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Baruffi, S.; Spaggiari, S.; Coltrini, D.; Berra-Romani, R.; Signorelli, S.; Castelli, L.; Taglietti, V.; Tanzi, F. P2y1 and P2y2 receptor-operated Ca2+ signals in primary cultures of cardiac microvascular endothelial cells. Microvasc. Res. 2001, 61, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, S.P. Mechanisms of endothelial P2Y(1)- and P2Y(2)-mediated vasodilatation involve differential [Ca2+]i responses. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1759–H1766. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Cheng, K.T.; Ma, Y.; Huang, Y.; Tang, N.L.; Yu, S.; Yao, X. CNGA2 contributes to ATP-induced noncapacitative Ca2+ influx in vascular endothelial cells. J. Vasc. Res. 2010, 47, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Duza, T.; Sarelius, I.H. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H26–H37. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Hoyer, J. The endothelium-derived hyperpolarizing factor: Insights from genetic animal models. Kidney Int. 2007, 72, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, J.; Taylor, M.S.; Bonev, A.D.; Hannah, R.M.; Solodushko, V.; Shui, B.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc. Natl. Acad. Sci. USA 2008, 105, 9627–9632. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Gallagher, N.T.; McNeish, A.; Garland, C.J. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ. Res. 2008, 102, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Spreading the signal for vasodilatation: Implications for skeletal muscle blood flow control and the effects of ageing. J. Physiol. 2012, 590, 6277–6284. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Tuning electrical conduction along endothelial tubes of resistance arteries through Ca(2+)-activated K(+) channels. Circ. Res. 2012, 110, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Kapela, A.; Behringer, E.J.; Segal, S.S.; Tsoukias, N.M. Biophysical properties of microvascular endothelium: Requirements for initiating and conducting electrical signals. Microcirculation 2017, 25. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Science Signal. 2015, 8, ra2. [Google Scholar] [CrossRef] [PubMed]
- Hannah, R.M.; Dunn, K.M.; Bonev, A.D.; Nelson, M.T. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J. Cereb. Blood Flow Metab. 2011, 31, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Sokoya, E.M.; Burns, A.R.; Setiawan, C.T.; Coleman, H.A.; Parkington, H.C.; Tare, M. Evidence for the involvement of myoendothelial gap junctions in EDHF-mediated relaxation in the rat middle cerebral artery. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H385–H393. [Google Scholar] [CrossRef] [PubMed]
- Bagher, P.; Beleznai, T.; Kansui, Y.; Mitchell, R.; Garland, C.J.; Dora, K.A. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc. Natl. Acad. Sci. USA 2012, 109, 18174–18179. [Google Scholar] [CrossRef] [PubMed]
- Golding, E.M.; Marrelli, S.P.; You, J.; Bryan, R.M., Jr. Endothelium-derived hyperpolarizing factor in the brain: A new regulator of cerebral blood flow? Stroke 2002, 33, 661–663. [Google Scholar] [PubMed]
- McNeish, A.J.; Dora, K.A.; Garland, C.J. Possible role for K+ in endothelium-derived hyperpolarizing factor-linked dilatation in rat middle cerebral artery. Stroke 2005, 36, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, S.P.; Eckmann, M.S.; Hunte, M.S. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1590–H1599. [Google Scholar] [CrossRef] [PubMed]
- McNeish, A.J.; Sandow, S.L.; Neylon, C.B.; Chen, M.X.; Dora, K.A.; Garland, C.J. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke 2006, 37, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Papadopoulos, P.; Hamel, E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: Impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br. J. Pharmacol. 2013, 170, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell. Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Thodeti, C.K.; Matthews, B.; Ravi, A.; Mammoto, A.; Ghosh, K.; Bracha, A.L.; Ingber, D.E. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 2009, 104, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Pan, Q.; Chen, Z.; Sun, C.; Zhang, P.; Mao, A.; Zhu, Y.; Li, H.; Lu, C.; Xie, M.; et al. Treatment of hypertension by increasing impaired endothelial TRPV4-KCa2.3 interaction. EMBO Mol. Med. 2017, 9, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.S.; Zheng, X.; Zhang, D.X. Role of endothelial TRPV4 channels in vascular actions of the endocannabinoid, 2-arachidonoylglycerol. Br. J. Pharmacol. 2015, 172, 5251–5264. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.A.; Fang, J.; Gutterman, D.D.; Wilcox, D.A.; Bubolz, A.H.; Li, R.; Suzuki, M.; Zhang, D.X. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H466–H476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zinkevich, N.S.; Gebremedhin, D.; Gauthier, K.M.; Nishijima, Y.; Fang, J.; Wilcox, D.A.; Campbell, W.B.; Gutterman, D.D.; Zhang, D.X. Arachidonic acid-induced dilation in human coronary arterioles: Convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J. Am. Heart Assoc. 2013, 2, e000080. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Graier, W.F.; Simecek, S.; Sturek, M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. 1995, 482 (Pt 2), 259–274. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R.; et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Pauyo, T.; Drapp, R.; Tavares, M.J.; Liedtke, W.; Brayden, J.E. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1096–H1102. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, S.P.; O’Neil R, G.; Brown, R.C.; Bryan, R.M., Jr. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1390–H1397. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Marrelli, S.P.; Bryan, R.M., Jr. Role of cytoplasmic phospholipase A2 in endothelium-derived hyperpolarizing factor dilations of rat middle cerebral arteries. J. Cereb. Blood Flow Metab. 2002, 22, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Gebremedhin, D.; Hwang, S.H.; Hammock, B.D.; Falck, J.R.; Roman, R.J.; Harder, D.R.; Koehler, R.C. Epoxyeicosatrienoic acid-dependent cerebral vasodilation evoked by metabotropic glutamate receptor activation in vivo. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H373–H381. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, C.; Alkayed, N.J.; Harder, D.R.; Koehler, R.C. Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in rats. J. Cereb. Blood Flow Metab. 2004, 24, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.R.; Kozberg, M.G.; Bouchard, M.B.; Shaik, M.A.; Hillman, E.M. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 2014, 3, e000787. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Francis, M.; Solodushko, V.; Earley, S.; Taylor, M.S. Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 2013, 20, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Senadheera, S.; Kim, Y.; Grayson, T.H.; Toemoe, S.; Kochukov, M.Y.; Abramowitz, J.; Housley, G.D.; Bertrand, R.L.; Chadha, P.S.; Bertrand, P.P.; et al. Transient receptor potential canonical type 3 channels facilitate endothelium-derived hyperpolarization-mediated resistance artery vasodilator activity. Cardiovasc. Res. 2012, 95, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Kochukov, M.Y.; Balasubramanian, A.; Abramowitz, J.; Birnbaumer, L.; Marrelli, S.P. Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium-activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J. Am. Heart Assoc. 2014, 3, e000913. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Sullivan, M.N.; Pritchard, H.A.; Robinson, J.J.; Earley, S. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2031–H2041. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhu, M.X. Trpv3. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2014; Volume 222, pp. 273–291. [Google Scholar]
- Zygmunt, P.M.; Hogestatt, E.D. Trpa1. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2014; Volume 222, pp. 583–630. [Google Scholar]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.R.; Bouchard, M.B.; McCaslin, A.F.; Burgess, S.A.; Hillman, E.M. High-speed vascular dynamics of the hemodynamic response. NeuroImage 2011, 54, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Hillman, E.M.; Devor, A.; Bouchard, M.B.; Dunn, A.K.; Krauss, G.W.; Skoch, J.; Bacskai, B.J.; Dale, A.M.; Boas, D.A. Depth-resolved optical imaging and microscopy of vascular compartment dynamics during somatosensory stimulation. NeuroImage 2007, 35, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, B.; Hutchinson, E.; Yakovleva, V.; Schram, V.; Russell, J.T.; Belluscio, L.; Koretsky, A.P.; Silva, A.C. Functional reactivity of cerebral capillaries. J. Cereb. Blood Flow Metab. 2008, 28, 961–972. [Google Scholar] [CrossRef] [PubMed]
- O’Herron, P.; Chhatbar, P.Y.; Levy, M.; Shen, Z.; Schramm, A.E.; Lu, Z.; Kara, P. Neural correlates of single-vessel haemodynamic responses in vivo. Nature 2016, 534, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhou, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Socha, M.J.; Polo-Parada, L.; Segal, S.S. Electrical conduction along endothelial cell tubes from mouse feed arteries: Confounding actions of glycyrrhetinic acid derivatives. Br. J. Pharmacol. 2012, 166, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Vaucher, E.; Hamel, E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: Electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J. Neurosci. 1995, 15, 7427–7441. [Google Scholar] [PubMed]
- Vaucher, E.; Linville, D.; Hamel, E. Cholinergic basal forebrain projections to nitric oxide synthase-containing neurons in the rat cerebral cortex. Neuroscience 1997, 79, 827–836. [Google Scholar] [CrossRef]
- Jensen, L.J.; Holstein-Rathlou, N.H. The vascular conducted response in cerebral blood flow regulation. J. Cereb. Blood Flow Metab. 2013, 33, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Uhrenholt, T.R.; Domeier, T.L.; Segal, S.S. Propagation of calcium waves along endothelium of hamster feed arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1634–H1640. [Google Scholar] [CrossRef] [PubMed]
- Domeier, T.L.; Segal, S.S. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J. Physiol. 2007, 579, 175–186. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Culot, M.; Wang, N.; da Costa, A.; Decrock, E.; Bol, M.; Bultynck, G.; Cecchelli, R.; Leybaert, L. Low extracellular Ca2+ conditions induce an increase in brain endothelial permeability that involves intercellular Ca2+ waves. Brain Res. 2012, 1487, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Waldrup, J.R.; Qian, X.; Solodushko, V.; Meriwether, J.; Taylor, M.S. Functional Tuning of Intrinsic Endothelial Ca2+ Dynamics in Swine Coronary Arteries. Circ. Res. 2016, 118, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Clusters of specialized detector cells provide sensitive and high fidelity receptor signaling in the intact endothelium. FASEB J. 2016, 30, 2000–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, R.; Hoyer, J. Role of TRPV4 in the Mechanotransduction of Shear Stress in Endothelial Cells. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Bryan, R.M., Jr.; Marrelli, S.P.; Steenberg, M.L.; Schildmeyer, L.A.; Johnson, T.D. Effects of luminal shear stress on cerebral arteries and arterioles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2011–H2022. [Google Scholar] [CrossRef] [PubMed]
- Bryan, R.M., Jr.; Steenberg, M.L.; Marrelli, S.P. Role of endothelium in shear stress-induced constrictions in rat middle cerebral artery. Stroke 2001, 32, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Ngai, A.C.; Winn, H.R. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circ. Res. 1995, 77, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.F.; Ledo, A.; Barbosa, R.M.; Laranjinha, J. Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp. Neurol. 2017, 291, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.F.; Ledo, A.; Dias, C.; Barbosa, R.M.; Laranjinha, J. Neurovascular and neurometabolic derailment in aging and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Katakam, P.V.; Domoki, F.; Snipes, J.A.; Busija, A.R.; Jarajapu, Y.P.; Busija, D.W. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R289–R298. [Google Scholar] [CrossRef] [PubMed]
- Mottola, A.; Antoniotti, S.; Lovisolo, D.; Munaron, L. Regulation of noncapacitative calcium entry by arachidonic acid and nitric oxide in endothelial cells. FASEB J. 2005, 19, 2075–2077. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. https://doi.org/10.3390/ijms19040938
Guerra G, Lucariello A, Perna A, Botta L, De Luca A, Moccia F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. International Journal of Molecular Sciences. 2018; 19(4):938. https://doi.org/10.3390/ijms19040938
Chicago/Turabian StyleGuerra, Germano, Angela Lucariello, Angelica Perna, Laura Botta, Antonio De Luca, and Francesco Moccia. 2018. "The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen" International Journal of Molecular Sciences 19, no. 4: 938. https://doi.org/10.3390/ijms19040938