Oncosuppressors and Oncogenes: Role in Haemangioma Genesis and Potential for Therapeutic Targeting


Department of Physiology, Faculty of Health Sciences, University of Pretoria, 9 Botshelo Road, Pretoria 0007, South Africa
Int. J. Mol. Sci. 2018, 19(4), 1192; https://doi.org/10.3390/ijms19041192
Submission received: 19 February 2018
/
Revised: 26 March 2018
/
Accepted: 4 April 2018
/
Published: 13 April 2018
(This article belongs to the Section Molecular Pathology, Diagnostics, and Therapeutics)
Abstract
:Genetic lesions in proto-oncogenes result in the perturbation of angiogenesis, the formation of neovessels from a pre-existing microvasculature. Similarly, the subversion of tumor suppressor genes promotes tumor vascularization. Excessive neovessel formation is associated with various neoplasms such as infantile hemangiomas (IH). Hemangiomas are the most common tumors in pediatric patients and at present have no definitive treatment. The pathogenesis of IH is not well understood; however, both vasculogenesis and angiogenesis are associated with hemangioma genesis. A number of factors that modulate angiogenesis and vasculogenesis have been shown to be dysregulated in IH. Several of the oncogenes and tumor suppressors linked to the promotion of angiogenesis are also altered in infantile hemangioma. In this review, the roles of oncogenes and tumor suppressor genes during neovascularization and hemangioma genesis are explored. In addition, the potential for targeting these genes in IH therapy is discussed.
1. Introduction
Infantile hemangioma (IH) is deemed the most common tumor of infancy. It is estimated that IH develops in 10% of infants and that the frequency of hemangioma development increases to 22.9% for premature infants with a birth weight below 1 kg [1,2]. The tumors have a life-cycle of three phases. The first phase, known as the proliferative phase, is characterized by an increase in the proliferation of endothelial cells (ECs) [1]. The second or involuting phase is characterized by a decrease in EC turnover. During the third or involuted phase, there is complete resolution of the tumor with replacement of vascular tissue by fibro-fatty tissue [1]. Most IH involute spontaneously; however, a subset of the lesions may be disfiguring, while some may impair function or be potentially life-threatening [3]. Such lesions require treatment. For many years, steroids have been the mainstay treatment for problematic IH [3]. At present, β-blockers represent the first-line treatment for IH [4]. Current treatment modalities are not without limitations, and locally-administered β-blockers are mainly effective in the treatment of superficial lesions [4,5]. Thus, understanding the precise pathways underlying hemangioma genesis will be useful for devising targeted and more effective therapies.
Several theories have been proposed to explain the pathogenesis of IH. Clonal expansion of a stem cell carrying a somatic mutation in a critical gene, tissue hypoxia, as well as an abnormal hormonal milieu, such as an increase in estrogen levels, have been postulated as potential stimuli for hemangioma genesis [6,7]. While IH may develop due to one or several of the postulated defects, the common underlying feature in the pathogenesis of IH is excessive neovascularization [3,8].
There is a strong correlation between the subversion of tumor suppressor genes and angiogenesis, a process that is associated with proliferating IH [9,10]. In addition, oncogenic modifications promote tumor angiogenesis [11]. Many of the altered tumor suppressors and oncogenes promote tumor neovascularization in part by affecting vascular endothelial growth factor (VEGF) signaling [12]. This review explores the genetic alterations associated with the promotion of the neovascularization in IH, as well as possible therapeutic approaches that can be employed to exploit these genetic perturbations.
2. Vascular Endothelial Growth Factor
The vascular endothelial growth factor (VEGF) family is a group of secreted glycoproteins that consists of VEGF-A, -B, -C, -D and placental growth factor (PlGF) [13,14]. The most characterized angiogenic growth factor in this family, VEGF-A (referred to in this review as VEGF), is a required for vasculogenesis, the development of blood vessels from primordial stem cells [14]. Vascular endothelial growth factor is also an important mediator of embryonic and postnatal angiogenesis [14,15].
2.1. Vascular Endothelial Growth Factor Signaling in Angiogenesis
The VEGF family of ligands binds to receptor tyrosine kinases (RTKs), and the availability of the VEGF ligands regulates the activity of the vascular endothelial growth factor receptors (VEGFRs) [14]. Vascular endothelial growth factor-B (VEGF-B) and placental growth factor (PlGF) bind to vascular endothelial growth factor receptor-1 (VEGFR-1) or Fms-like tyrosine kinase 1 (FLT-1). The ligands vascular endothelial growth factor C (VEGF-C) and vascular endothelial growth factor D (VEGF-D) bind to VEGFR-3 to promote lymphangiogenesis, the formation of lymphatic vessels from pre-existing lymphatic vessels. On the other hand, the principal receptors for VEGF are VEGFR-1 and VEGFR-2/fetal liver kinase 1 (Flk-1) or kinase insert domain receptor (KDR) [16]. These receptors are mainly expressed by endothelial cells, although some tumor cells do express VEGF receptors [15].
In endothelial cells, VEGF modulates mitogenic signals by activating both VEGFR-1 and -2 [13,14]. Vascular endothelial growth factor receptor-2 has less affinity for VEGF when compared with VEGFR-1; however, VEGFR-2 presents greater signaling activity [13,15,16]. As such, the mitogenic action of VEGF in endothelial cells is mediated mainly by VEGFR-2 [14,15]. Additionally, VEGFR-2 mediates cell leakage and vascular permeability in response to VEGF, whereas VEGFR-1 has a weak or undetectable response [13]. Furthermore, the signaling pathways elicited by VEGF contribute to pathological vasculogenesis and angiogenesis [15].
2.2. Vascular Endothelial Growth Factor Signaling in Hemangioma
The vascular hyperpermeability associated with VEGF is also observed in IH. Of particular interest is that the increased permeability of the endothelium and the leakage of cells into the tumor interstitium contribute to the swelling and red color observed in most cutaneous lesions. Cells that are found in IH lesions include hemangioma endothelial cells (HemECs), hemangioma endothelial progenitor cells (HemEPCs), hemangioma stem cells (HemSc) and inflammatory cells [17,18,19]. Both HemEPCs and HemSc promote angiogenesis and vasculogenesis, respectively, while inflammatory cells secrete proangiogenic factors such as VEGF [20]. Thus, these inflammatory cells support neovessel formation.
An increase in VEGF promotes IH development. For instance, high levels of VEGF have been measured in the urine samples of patients with proliferating IH [1]. Also, previously, the inhibition of VEGF secretion was shown to correlate with reduced tumor cell growth in human and murine hemangioma cells [21,22]. Furthermore, targeting downstream effectors in the VEGF signaling pathway resulted in the diminished growth of endothelial cells isolated from the murine hemangiomas [23,24]. Recently, sirolimus, a drug that targets the mammalian target of rapamycin (mTOR) downstream of the VEGF pathway, was effective in regressing tumor mass in a patient with segmental IH who had been unresponsive to treatment [25].
Concerning the receptors for VEGF, research has revealed that VEGFR-2 is overexpressed in murine and human hemangioma tissue [20,26]. In contrast, VEGFR-1 expression is relatively low in hemangioma endothelial cells when compared to other endothelial cell types [8,27].
Impaired signaling through VEGFR-2 appears to play an important role in the promotion of hemangioma genesis [8]. Studies have shown that some of the patients with IH have germline mutations in VEGFR-2 or tumor endothelial marker 8 (TEM8) and that such patients appear to be at risk of developing lesions [27]. The C482R mutation in VEGFR-2 results in the inability of the receptor to regulate VEGFR-1 co-expression [27]. On the other hand, TEM8, which has been shown to suppress β1 integrin activity in vitro and to activate nuclear translocation of nuclear factor of activated T cells (NFAT), exhibits reduced activity [27]. Worthy of note is that the transcription of genes coding for VEGFR-1 require NFAT, and the low activity of the pathway that involves NFAT thus has a negative effect on VEGFR-1 levels [8]. Interestingly, endothelial cells isolated from hemangioma tissue show altered expression of NFAT-regulated genes [8], further supporting observations of diminished VEGFR-1 signaling in IH.
In a normal physiological setting, VEGFR-1 sequesters VEGF by binding to it and preventing it from activating VEGFR-2. In the absence of VEGFR-1’s decoy function, the VEGF levels are increased [27]. The high levels of VEGF lead to the constitutive activation of VEGFR-2 and possibly contribute to hemangioma genesis. Indeed a previous study has shown that VEGF confers increased mitogenic potential in stromal cells isolated from human hemangioma biopsies [28]. A more recent study on human IH revealed that propranolol, the first Food and Drug Administration (FDA)-approved drug for the treatment of the tumor, inhibits the growth of hemangioma endothelial cells partly by inhibiting VEGF secretion [21]. Given that these observations show that signaling through VEGF and its receptors, VEGFR-1 and VEGFR-2, is impaired in hemangioma, the pathway may be important in designing therapeutic strategies for IH.
2.3. Therapeutic Targeting of Vascular Endothelial Growth Factor
The first antiangiogenic drug to be approved by the Food and Drug Administration was bevacizumab, and it blocks VEGF signaling by neutralizing VEGF [13,15]. Since then, several drugs (Table 1) have been developed to target VEGF and its receptors for the treatment of tumors characterized by excessive angiogenesis [15]. The prevailing theory was that such drugs would not lead to the development of resistance since the target, namely the endothelial cell, was genetically stable [14,16]. However, in the clinic, the therapeutic benefits of these drugs have been modest and transient, and being refractory to disease remains the major drawback [16]. From investigations on the mechanism of resistance to anti-VEGF/VEGFR therapy, it appears that the stimulation of alternate proangiogenic pathways through hypoxia inducible factor-1α promotes the resumption of angiogenesis [13,15,16]. While toxicities observed with traditional chemotherapeutic drugs are not observed when VEGF neutralizing drugs are employed as monotherapy against tumor angiogenesis, the combination of these drugs with chemotherapy results in increased toxicity. Thus, the elaboration of therapies should consider, in addition to the VEGF pathway, other targets that contribute to angiogenesis, as well as combinatorial approaches that will be effective at less toxic doses.
It is also noteworthy that in several tumors, oncogenic transformations promote the angiogenic phenotype [20]. Some of these genetic alterations affect VEGF signaling, as well as other pathways that promote angiogenesis [20], thus making these genes plausible targets in the design of effective therapeutic modalities.
3. Oncogenic Pathways Linked to Tumor Angiogenesis
Historically, genes with the potential to induce cancer development when modified, also known as oncogenes, have been studied extensively in various neoplastic diseases [11]. However, the role of oncogenes in promoting neovascularization has recently gained considerable attention [11,12]. Oncogenic mutations have been implicated in the activation of the ‘angiogenic switch’ in tumors [12]. Some of the oncogenes that promote tumor angiogenesis are altered in IH or they influence the transcription of factors implicated in the pathogenesis of the lesion.
3.1. Altered Expression of Bcl-2 in Angiogenesis and Hemangioma
B-cell lymphoma-2 (Bcl-2) is an oncogene activated due to the t(14;18) chromosome translocation and codes for the Bcl-2 integral membrane protein, which is mainly located on the outer mitochondrial membrane [29,30]. Bcl-2 protects cells from apoptotic stimuli [29]. When overexpressed, it also delays the induction of apoptosis by various chemotherapeutic drugs [30].
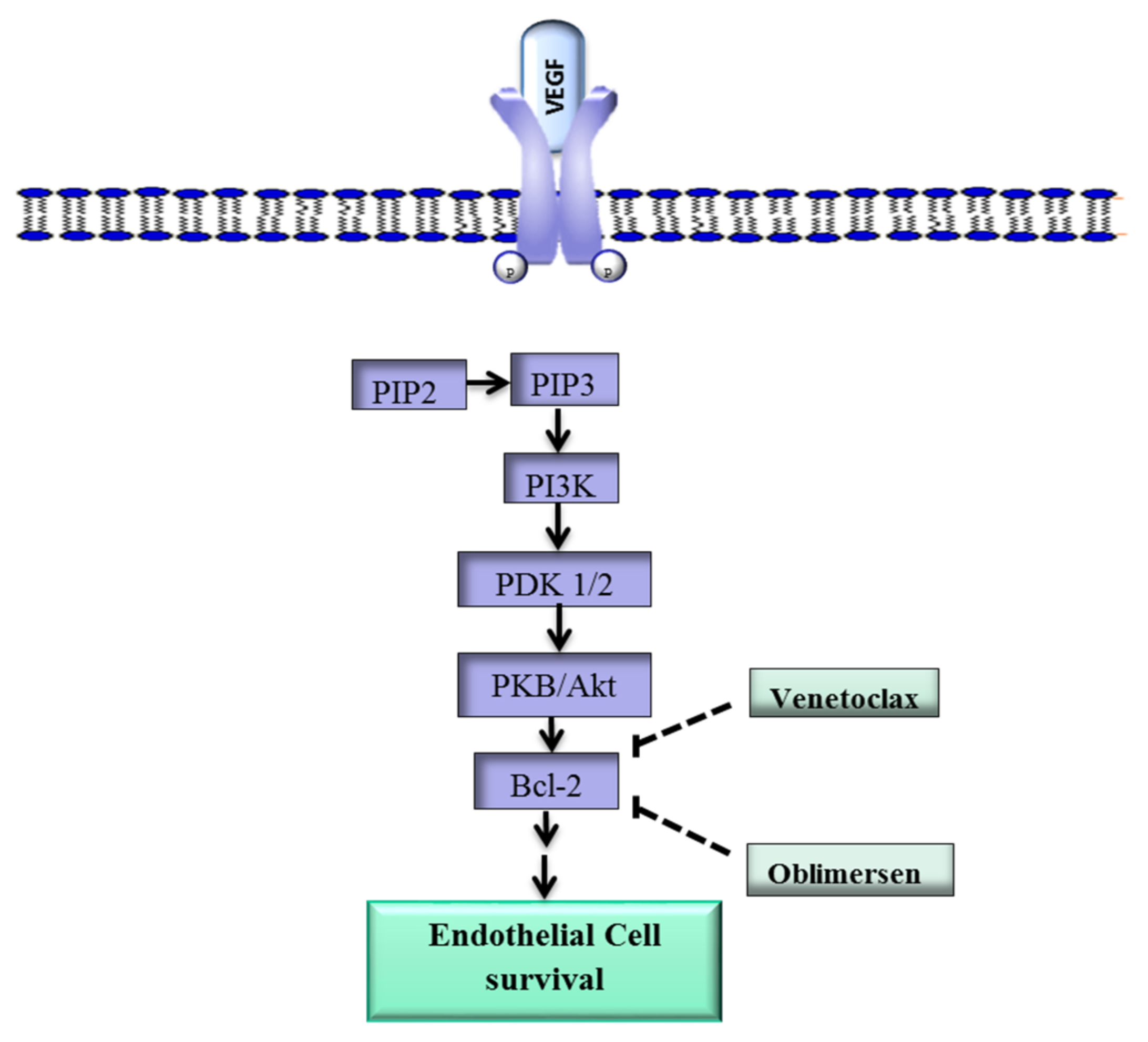
In addition, Bcl-2 plays a role in angiogenesis via a mechanism that involves the proangiogenic factor VEGF [31]. Upon binding of VEGF to VEGFR-2, Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3k), a phospholipid that belongs to a family of enzymes that regulate a network of cellular processes, is recruited to the internal side of the cell membrane and activated through phosphorylation (Figure 1) [32]. Activated PI3k phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to form phosphatidylinositol 3,4,5-trisphosphate (PIP3) [33]. Phosphatidylinositol 3,4,5-trisphosphate initiates a cascade of events that lead to the phosphorylation of protein 3-phosphoinositide-dependent protein kinase-1 and -2 (PDK1/2), which in turn activate protein kinase B (PKB/Akt), a key downstream effector of PI3k [34,35]. Protein kinase B in turn upregulates Bcl-2, and the Bcl-2 protein ultimately promotes the survival of ECs [36,37].
Previous work has shown that HUVECs expressing Bcl-2 were protected from undergoing apoptosis even following the withdrawal of VEGF [36,38]. Nor et al. [38] further showed that the implantation of human microvascular endothelial cells (HDMEC) transfected with Bcl-2 into SCID mice with oral squamous cell carcinoma enhanced tumor growth. Furthermore, microvascular density increased in the mice injected with Bcl-2 transfected cells. A study by Mabeta and Pepper [26] showed that murine hemangioma overexpressed Bcl-2 and that such expression was diminished following antiangiogenic treatment. Another study revealed that Bcl-2 was overexpressed in proliferating IH, and that Bcl-2 expression in involuting IH was similar to that of normal tissue [37]. These findings underscore the importance of Bcl-2 in supporting tumor angiogenesis and possibly contributing to the growth of IH.
Various drugs have been developed to target Bcl-2. The Bcl-2 inhibitor TW37 has exhibited antiangiogenic effects in vivo in preclinical models, while S-055746 and PNT-2258 are in early stage clinical trials [39,40]. Other Bcl-2 inhibitors such as Navitoclax and Venetoclax are in phase I/II studies for several carcinomas both as monotherapy and as part of combination strategies [41,42].
Oblimersen, an antisense oligodeoxyribonucleotide that targets Bcl-2 RNA, is showing promising results in clinical studies against various cancers [43]. Oblimersen was also shown to inhibit tumor angiogenesis [43,44]. These drugs may have potential in the treatment of IH lesions that overexpress Bcl-2.
3.2. STAT3 Overexpression Promotes Tumor Angiogenesis
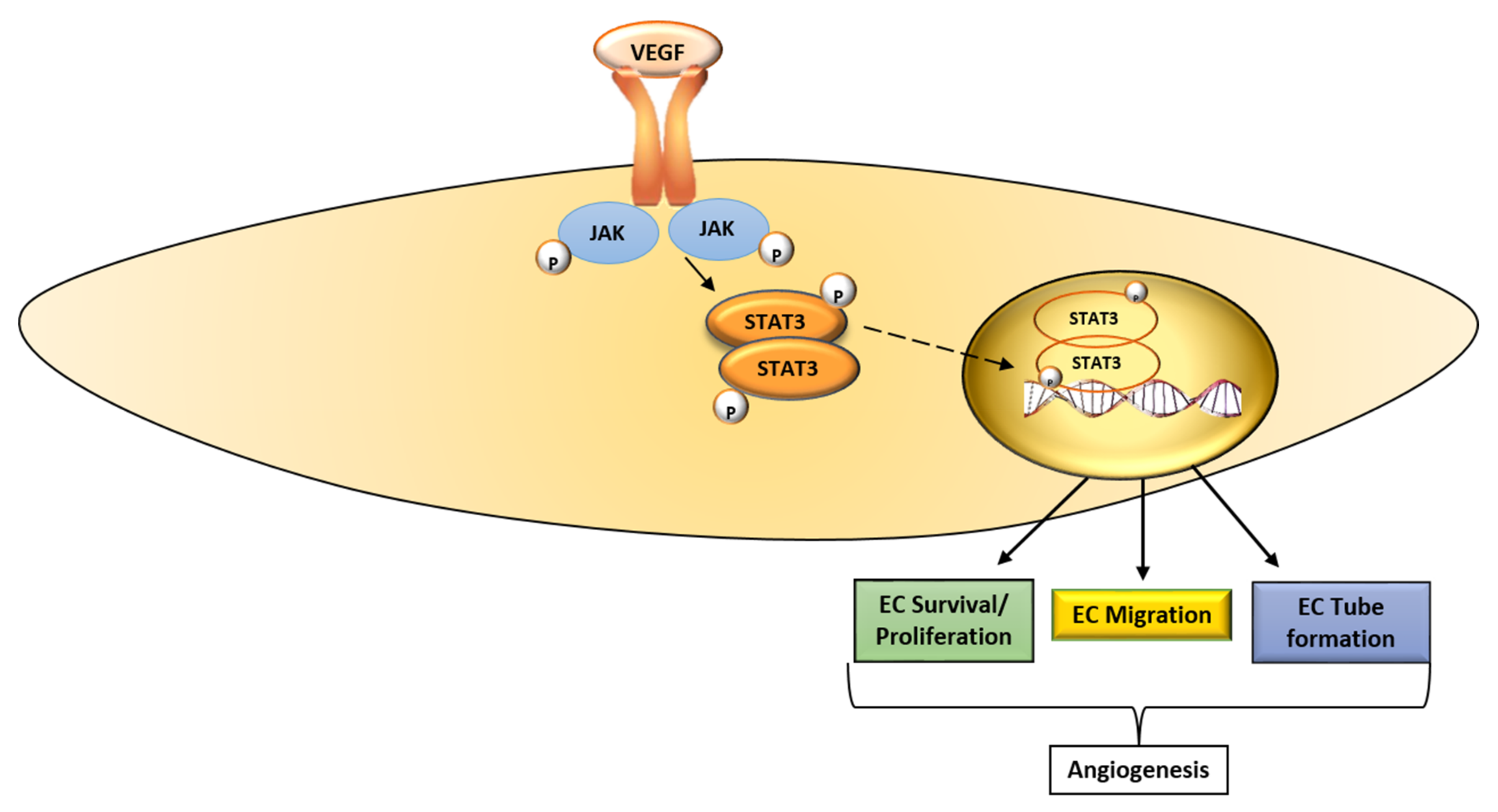
The signaling transducer and activator of transcription (STAT) 3 is a member of the STAT family, and it mediates the expression of genes involved in various cellular processes [45]. Multiple pathways that transduce signals in response to cytokines and growth factors like VEGF converge on STAT3 [45,46]. The binding of VEGF to VEGFR-2 activates Janus kinases (JAK), which in turn phosphorylate STAT3 (tyr 705) [45,47]. This leads to the dimerization of STAT3 monomers through phosphotyrosine-Src homology 2 (SH2) and the translocation of the dimer to the nucleus (Figure 2). In the nucleus, the STAT3 dimer promotes the transcription of target genes involved in various stages of the angiogenic process [45]. Under normal physiological conditions, STAT3 activation is transient; however, in tumors, STAT3 is activated perpetually [45,46]. Activated STAT3 contributes to tumor progression, as well as resistance to treatment [46].
STAT3 appears to play a role in IH as phosphorylated STA3 was found to be highly expressed in human hemangioma endothelial cells [5]. STAT3 was also shown to be overexpressed in the endothelium of IH biopsies [48]. Furthermore, in a meta-analysis study of IH, STAT3 was one of the regulators identified through gene mapping [49].
Another separate study revealed that propranolol suppressed the expression of phospo-STAT3 in hemangioma cells [5], making STAT3 an attractive potential therapeutic target.
The STAT3 pathway can be disrupted at different levels [45]: (i) through the inhibition of upstream receptors that activate signals that converge on STAT3; (ii) through the prevention of dimerization by inhibiting the phosphorylation of the SH2 domain on STAT3; (iii) through the prevention of STAT3 from binding to DNA and (iv) through the downregulation of total STAT3 by inhibiting transcription. The drugs that target STAT3 at different levels are listed in Table 2.
Drugs such as ruxolitinib and fedratinib inhibit upstream receptors in the STAT3 pathway. Both ruxolitinib and fedratinib have been approved by the United States Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis and are in phase I/II studies for the treatment of various cancers [45]. Several other drugs that target STAT3 are at various stages of clinical development, although some of the drugs have not progressed beyond phase I/II due to toxicity or poor efficacy [46]. Effective STAT3 inhibitors may have potential in the treatment of problematic IH.
3.3. Possible Role of K-Ras in Hemangioma Growth
The Kirsten rat sarcoma oncogene homologue (K-ras) encodes the GTP-ase protein K-ras, which regulates the cell cycle and, when mutated, promotes uncontrolled cell division [11]. There is strong evidence to show that K-ras mutations are also associated with alterations in vascular homeostasis [50,51]. Sustained K-ras activation in vascular endothelial cells promotes the by-passing of senescence and supports proliferation [52]. Furthermore, ‘enforced’ expression of K-ras has been shown to contribute to the increased expression of VEGF [53,54].
The proangiogenic effects of K-ras have been attributed in part to the oncogene’s negative regulation of thrombospondin, an endogenous inhibitor of angiogenesis. Studies have revealed a link between K-ras-mediated cellular transformation and the downregulation of thrombospondin-1 (TSP-1), which leads to increased angiogenesis [52]. The K-ras gene is mutated in several benign and malignant neoplasms. Furthermore, a number of vascular tumors carry K-ras mutations [52]. In the context of IH, it has been shown that TSP-1 is downregulated in tissue biopsies of proliferating IH [62]. Such downregulation of TSP-1 was not observed in involuting IH. The suppression of a negative regulator of angiogenesis, namely TSP-1, may contribute to the excessive vascularization observed in proliferating IH. In addition, given the role of K-ras in regulating TSP-1 expression and the association of overexpressed TSP-1 with growing IH, it is plausible that the targeting of K-ras mutations might be beneficial. Further studies are necessary to explore a possible link between mutated K-ras and IH and to determine any possible therapeutic benefit in targeting the mutated form of the gene or the protein, although the development of such drugs is still in its infancy.
4. Tumor Suppressors in Angiogenesis and Hemangioma Development
Tumor suppressor genes, also known as antioncogenes or oncosuppressors, are genes that encode proteins that regulate cell growth and proliferation [63]. A number of tumor suppressor genes have been shown to play a role in the promotion of tumor angiogenesis. Some of the oncosuppressors are mutated or inactivated in IH.
4.1. Phosphatase and Tensin Homolog Is Downregulated in Hemangioma
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor protein encoded by the PTEN gene [34]. It dephosphorylates PIP3, thereby suppressing the phosphoinositol-3-kinase (PI3K)/Akt pathway [64]. Given that the PI3K/Akt pathway regulates processes such as cell survival, proliferation and migration, PTEN negatively affects these processes [64,65]. In addition, PTEN regulates angiogenesis by modulating the expression of hypoxia inducible factor 1-α (HIF1-α) [65]. Hypoxia inducible factor 1-α is involved in the tissue’s adaptive response to hypoxia [65]. Indeed, HIF1-α has been reported to be increased in specimens of IH patients [8].
Loss of PTEN promotes the expression of HIF1-α and supports angiogenesis [66], while the restoration of wild-type PTEN results in the reduced expression of HIF1-α, leading to angiogenesis inhibition [66]. Thus, PTEN loss may in part potentiate the excessive angiogenesis observed in IH.
Indeed, PTEN loss of function due to somatic mutations has been observed in some patients with vascular anomalies and also in preclinical models of hemangioma [67]. These observations make the delivery of PTEN to HemECs an attractive therapeutic option to consider.
Although there have been studies that showed effective delivery of functional PTEN into neoplastic cells, the design of therapies that are directed at tumor suppressors poses a challenge. It is conceivable that in the future, the combination of current strategies used to treat IH with PTEN targeting approaches may be of benefit in the treatment of IH.
4.2. p53 Modulates Hypoxia Inducible Factor and Angiogenesis in IH
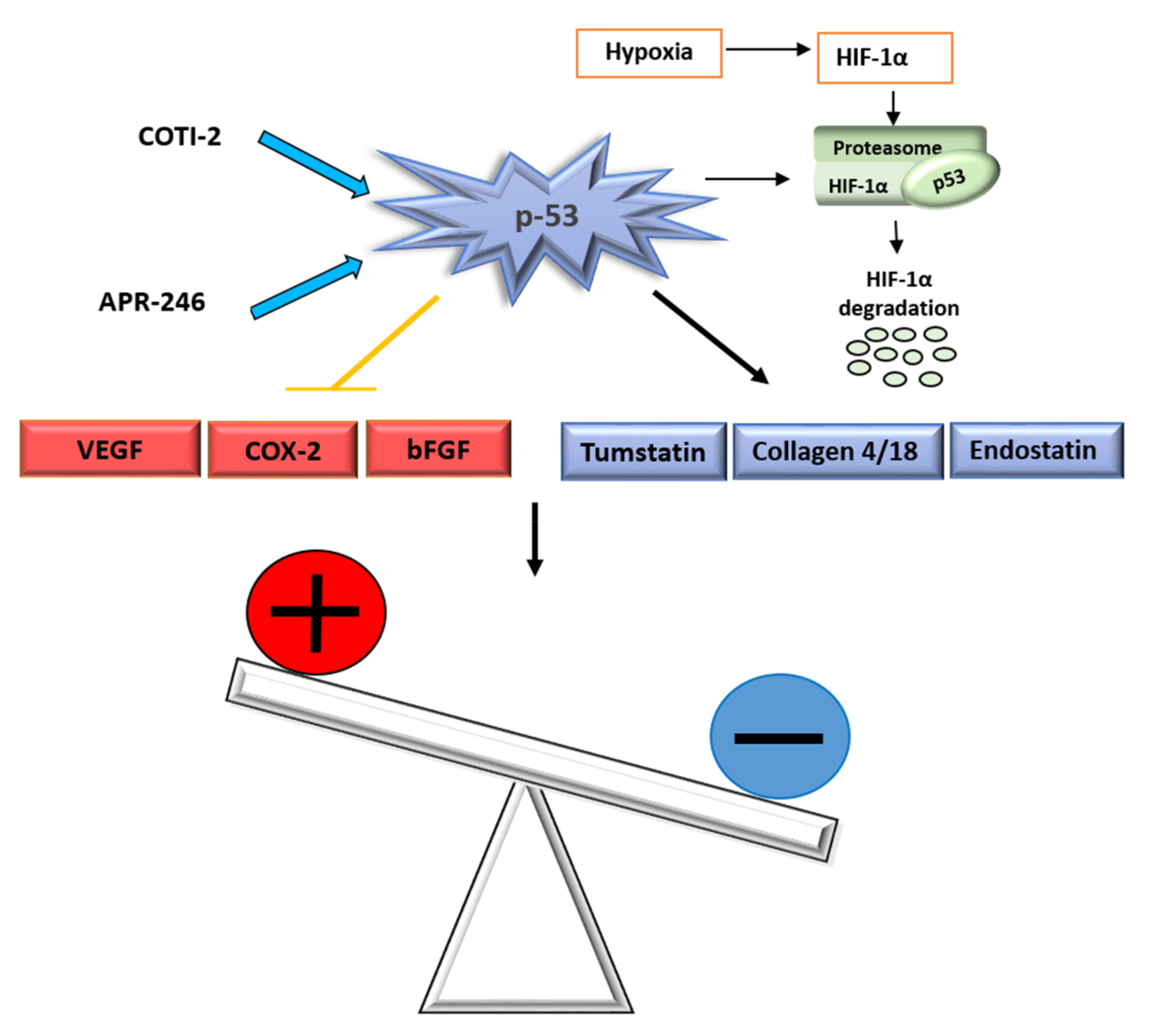
p53 is a tumor suppressor gene that codes for the p53 tumor suppressor protein (Tp53) [68,69]. The p53 protein is activated through phosphorylation in response to cellular stress or insult [69]. In turn, p53 can induce cell growth arrest, activate DNA repair or induce apoptosis [68,70]. p53 also suppresses the hypoxia sensing system (Figure 3) and thus plays a role in the regulation of angiogenesis [71]. Hypoxia is an important driver of angiogenesis and tumor progression. p53 inhibits angiogenesis by suppressing the hypoxia sensing system.
The central regulatory component that responds to oxygen deprivation (hypoxia) and, as a consequence, upregulates the production of new blood vessels is called hypoxia inducible factor (HIF). Hypoxia inducible factor is a heterodimer transcription factor comprised of two subunits, HIF-1α and HIF-1β, and HIF-1α in particular is key in the cell’s response to hypoxia [71]. The biological effects of HIF-1α are derived from its ability to transcriptionally activate a variety of genes that are key to the cell’s response to oxygen deprivation, such as the gene encoding the proangiogenic factor VEGF [71,72]. Hypoxia has been postulated as a significant player in the etiology of IH [1,6,7]. Furthermore, HIF-1α is overexpressed in IH, and such expression diminishes in involuting lesion.
p53 affects the process of angiogenesis at multiple levels [70,71,72,73]. In addition to inhibiting the transcription of HIF-1A, it has a direct effect on a number of proangiogenic molecules. The binding of p53 to the transcription factor Sp1 inhibits VEGF expression during hypoxia by blocking Sp1’s ability to bind the VEGF promoter and to activate VEGF transcription [72]. Thus, p53 inhibits VEGF. Studies have further shown that wild-type p53 inhibits COX-2, ultimately suppressing angiogenesis [73]. p53 also inhibits the proangiogenic factor basic fibroblast growth factor (bFGF) through direct repression of the bFGF basal core promoter, as well as through suppressing the expression of the bFGF-BP gene [74]. It is interesting to note that p53 has evolved redundant mechanisms to inhibit two of the most potent secreted proangiogenic factors, namely VEGF and bFGF, and that these factors are also associated with hemangioma development.
p53 can inhibit angiogenesis by upregulating or activating endogenous angiogenesis inhibitors [72]. Previous studies have revealed that p53 activates the α(II) collagen prolyl-4-hydroxylase gene [75]. The transcriptional activation of this gene leads to the release of the angiogenesis inhibitors tumstatin and endostatin (Figure 3) [75,76]. Furthermore, an association between p53 and the production of type 4 and 18 collagen fragments has been observed (Figure 3) [75]. Both of these fragments have antiangiogenic properties.
Several clinical studies have revealed a link between p53 mutations and tumor vascularization [31,77]. Human prostate cancer expressing mutated p53 is characterized by a marked increase in microvessel density (MVD) [78]. Similar correlations between p53 status and MVD were observed in colon, head and neck, as well as breast cancers [78,79]. Likewise, reduced p53 expression has been observed in proliferating IH [80]. These observations may underscore the importance of p53 in limiting angiogenesis.
There is a low expression of p53, as well as p53-related genes, including p53 inducible gene 3 (PIG3) and p53 upregulated modulator of apoptosis (PUMA) in human hemangioma endothelial cells (HemECs) [80].
Further, propranolol has been shown to induce apoptosis in human hemangioma endothelial cells partly by activating p53 [81]. Another chemotherapeutic used to treat IH, bleomycin A-5, was also shown to upregulate p53 in human hemangioma cells [80]. Earlier clinical trials targeting the p53 pathway have been disappointing; however, as a result there is increasing effort to design therapeutic strategies that will yield a beneficial outcome. At present COTI-2, a small molecule which restores mutant, APR-246 an activator of mutant p53, are in clinical trials for various malignancies [79]. It is likely that these approaches may be of benefit to IH.
4.3. Deregulation of Kiss1 Metastasis-Suppressor Is Associated with IH
In a previous study, Kiss1 metastasis-suppressor (Kiss1) was underexpressed in endothelial cells isolated from human hemangioma tissue [82]. The Kiss1 gene is a tumor suppressor that codes for a protein that regulates cancer cell metastasis by suppressing matrix metalloproteinase 9 (MMP9) [83]. The gene also suppresses angiogenesis. Concerning IH, Stiles et al. [82] further showed that the low expression of Kiss1 associated with haemangioma endothelial cells, was not observed in other endothelial cell types. Perhaps Kiss1 may serve as a predictive biomarker to monitor IH response to therapy. Further studies are required to determine its expression in the various phases of the IH life cycle.
4.4. Cyclin-Dependent Kinase Inhibitor 2A
Cyclin-dependent kinase inhibitor 2A (CDKN2A) is a gene that codes for two proteins, the INK4 family member p16 (or p16INK4a) and p14arf [84]. These proteins regulate the cell cycle and function as tumor suppressors. The gene was shown to be downregulated in hemangioma cells. These observations correlated with alterations in the cell cycle of hemangioma cells when compared to normal dermal microvascular endothelial cells [82]. Similar to kiss1, the expression of CDKN2A in the various phases of IH requires investigation in order to determine a possible link with disease progression. However, given the selective expression of these genes in IH compared to normal endothelial cells, they may be useful as predictive biomarkers to determine the effectiveness of therapeutic approaches employed in IH, those modalities that are in the developmental phase.
5. Conclusions
There is strong evidence to show that the acquisition of mutations in proto-oncogenes contributes to the onset of the ‘angiogenic switch’, especially by enhancing VEGF signaling. In addition, loss of function due to genetic lesions in oncosuppressors promotes angiogenesis. Vascular endothelial growth factor signaling is a requirement for vasculogenesis and is also key during the process of angiogenesis. These two process underlie the development of hemangiomas in juveniles. Some of the oncogenes and oncosuppressors that are associated with defective VEGF signaling are altered also in IH, while some genes regulate factors linked to the neoplasm. As such, an in-depth understanding of the roles of oncogenes and oncosuppressors in hemangioma genesis may provide the basis for therapeutic targeting. Furthermore, understanding the profile of the lesions in each context is necessary and may lead to a more sustainable approach as treatment can be adapted depending on the genetic profile of the tumors. Indeed, some of the drugs in this review may have future application in IH either as monotherapies or as part of combination therapeutic strategies.
Acknowledgments
The author thanks the National Research Foundation and the University of Pretoria for funding.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| Bcl-2 | B-cell lymphoma 2 |
| bFGF | Basic fibroblast growth factor |
| COX-2 | Cyclooxygenase 2 |
| EC | Endothelial cell |
| GTP-ase | Guanosine triphosphatase |
| HemECs | Hemangioma endothelial cells |
| HemEPCs | Hemangioma endothelial progenitor cells |
| HemSc | Hemangioma stem cells |
| HIF-1α | Hypoxia inducible factor-1α |
| IH | Infantile hemangioma |
| JAK | Janus kinases |
| K-ras | Kirsten rat sarcoma oncogene homologue |
| mTOR | Mammalian target of rapamycin |
| NFAT | Nuclear translocation of nuclear factor of activated T cells |
| PDK1/2 | Protein 3-phosphoinositide-dependent protein kinase-1 and -2 |
| PI3k | Phosphatidylinositide 3-kinases |
| PKB | Protein kinase B |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 1 |
| TSP-1 | Thrombospondin-1 |
| STAT3 | Signaling transducer and activator of transcription |
| VEGF | Vascular endothelial growth factor |
| VEGFR-1 | Vascular endothelial growth factor receptor-1 |
| VEGFR-2 | Vascular endothelial growth factor receptor-2 |
References
- Mulliken, J.B.; Glowacki, J. Classification of pediatric vascular lesions. Plast. Reconstr. Surg. 1982, 70, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Blei, F. Basic science and clinical aspects of vascular anomalies. Curr. Opin. Pediatr. 2005, 17, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Pepper, M.S. Hemangiomas-current therapeutic strategies. Int. J. Dev. Biol. 2011, 55, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, S. Infantile Hemangioma: New Insights on Pathogenesis and β Blockers Mechanisms of Action. In Angiogenesis-Based Dermatology, 1st ed.; Abiser, J.L., Ed.; Springer: Berlin, Germany, 2017; pp. 27–39. ISBN 978-1-4471-7314-4. [Google Scholar]
- Li, P.; Guo, Z.; Gao, Y.; Pan, W. Propranolol represses infantile hemangioma cell growth through the B2-adrenergic receptor in a HIF-1α dependent manner. Oncol. Rep. 2015, 33, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Boye, E.; Yu, Y.; Paranya, G.; Mulliken, J.B.; Olsen, B.R.; Bischoff, J. Clonality and altered behavior of endothelial cells from hemangiomas. J. Clin. Investig. 2001, 107, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, S.; Bischoff, J. Pathogenesis of infantile haemangioma. Br. J. Dermatol. 2013, 169, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Boye, E.; Olsen, B.R. Signaling mechanisms in infantile hemangioma. Curr. Opin. Hematol. 2009, 16, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. The p53 tumor suppressor gene inhibits angiogenesis by stimulating the production of thrombospondin. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1994; Volume 59, pp. 483–489. [Google Scholar]
- Rodriguez, S.; Huynh-Do, U. The role of PTEN in tumor angiogenesis. J. Oncol. 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Yu, J.; Klement, G.; Kerbel, R. Oncogenes and angiogenesis: Signaling three-dimensional tumor growth. J. Investig. Dermatol. Symp. Proc. 2000, 5, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Bardelli, A. Oncogenes and angiogenesis: A way to personalize anti-angiogenic therapy? Cell. Mol. Life Sci. 2013, 70, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Ye, W. The complexity of translating anti-angiogenesis therapy from basic science to the clinic. Dev. Cell 2016, 37, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Ambler, C.A.; Schmunk, G.M.; Bautch, V.L. Stem cell-derived endothelial cells/progenitors migrate and pattern in the embryo using the VEGF signaling pathway. Dev. Biol. 2003, 257, 205–219. [Google Scholar] [CrossRef]
- Yu, Y.; Flint, A.F.; Mulliken, J.B.; Wu, J.K.; Bischoff, J. Endothelial progenitor cells in infantile hemangioma. Blood 2004, 103, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mulliken, J.B.; Kozakewich, H.P.; Rogers, R.A.; Folkman, J.; Ezekowitz, R.A. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J. Clin. Investig. 1994, 93, 2357–2364. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Li, K.; Li, L.; Xu, C.; Xiang, B. Signaling pathways in the development of infantile hemangioma. J. Hematol. Oncol. 2014, 7, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Li, P.; Guo, Z.; Huang, Q.; Gao, Y. Propranolol induces regression of hemangioma cells via the down-regulation of the PI3K/Akt/eNOS/VEGF pathway. Pediatr. Blood Cancer 2015, 62, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P. Inhibition of phosphoinositide 3-kinase is associated with reduced angiogenesis and an altered expression of angiogenic markers in endothelioma cells. Biomed. Pharmacother. 2014, 68, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Tang, W.; Zhang, Y.; Li, X. Rapamycin inhibits the proliferation of endothelial cells in hemangioma by blocking the mTOR-FABP4 pathway. Biomed. Pharmacother. 2017, 85, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P. PF573, 228 inhibits vascular tumor cell growth, migration as well as angiogenesis, induces apoptosis and abrogates PRAS40 and S6RP phosphorylation. Acta Pharm. 2016, 66, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Kaylani, S.; Theos, A.J.; Pressey, J.G. Treatment of infantile hemangiomas with Sirolimus in a patient with PHACE syndrome. Pediatr. Dermatol. 2013, 30, e194–e197. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Pepper, M.S. Inhibition of hemangioma development in a syngeneic mouse model correlates with Bcl-2 suppression and the inhibition of Akt kinase activity. Angiogenesis 2012, 15, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Jinnin, M.; Medici, D.; Park, L.; Limaye, N.; Liu, Y.; Boscolo, E.; Bischoff, J.; Vikkula, M.; Boye, E.; Olsen, B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 2008, 14, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Berard, M.; Sordello, S.; Ortega, N.; Carrier, J.L.; Peyri, N.; Wassef, M.; Bertrand, N.; Enjolras, O.; Drouet, L.; Plouet, J. Vascular endothelial growth factor confers a growth advantage in vitro and in vivo to stromal cells cultured from neonatal hemangiomas. Am. J. Pathol. 1997, 150, 1315–1326. [Google Scholar] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Candiloro, A.; Mottolese, M.; Sapora, O.; Albini, A.; Zupi, G.; Del Bufalo, D. Bcl-2 overexpression and hypoxia synergistically act to modulate vascular endothelial growth factor expression and in vivo angiogenesis in a breast carcinoma cell line. FASEB J. 2000, 14, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Fontanini, G.; Boldrini, L.; Vignati, S.; Chinè, S.; Basolo, F.; Silvestri, V.; Lucchi, M.; Mussi, A.; Angeletti, C.A.; Bevilacqua, G. Bcl2 and p53 regulate vascular endothelial growth factor (VEGF)-mediated angiogenesis in non-small cell lung carcinoma. Eur. J. Cancer 1998, 34, 718–723. [Google Scholar] [CrossRef]
- Dituri, F.; Mazzocca, A.; Giannelli, G.; Antonaci, S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin. Dev. Immunol. 2011, 947858–947958. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, S.I.; Zvelebi, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2008, 102, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Nesterova, A.; Sable, C.; Heidenreich, K.A.; Boxer, L.M.; Heasley, L.E.; Reusch, J.E. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J. Biol. Chem. 2000, 275, 10761–10766. [Google Scholar] [CrossRef] [PubMed]
- Karl, E.; Zhang, Z.; Dong, Z.; Neiva, K.G.; Soengas, M.S.; Koch, A.E.; Polverini, P.J.; Núñez, G.; Nör, J.E. Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ. 2007, 14, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Zhang, D. Detection of p53 and Bcl-2 expression in cutaneous hemangioma through the quantum dot technique. Oncol. Lett. 2017, 13, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Nor, J.E.; Christensen, J.; Liu, J.; Peters, M.; Mooney, D.J.; Strieter, R.M.; Polverini, P.J. Up-regulation of Bcl-2 in microvascular endothelial cell enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001, 61, 2183–2188. [Google Scholar] [PubMed]
- Zeitlin, B.D.; Joo, E.; Dong, Z.; Warner, K.; Wang, G.; Nikolovska-Coleska, Z.; Wang, S.; Nor, J.E. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res. 2006, 66, 8698–8706. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, B.D.; Zeitlin, I.J.; Nor, J.E. Expanding circle of inhibition: Small-molecule inhibitors of Bcl-2 as anticancer cell and antiangiogenic agents. J. Clinl. Oncol. 2008, 26, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Peterson, T.; Horvat, T.; Rodriguez, M.; Tang, L. Venetoclax: A First-in-Class Oral BCL-2 Inhibitor for the Management of Lymphoid Malignancies. Ann. Pharmacother. 2017, 51, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Mordant, P.; Brown, B.D.; Bourhis, J.; Soria, J.C.; Deutsch, E. Inhibition of Bcl-2 in small cell lung cancer cell lines with oblimersen, an antisense Bcl-2 oligodeoxynucleotide (ODN): In vitro and in vivo enhancement of radiation response. Anticancer Res. 2010, 30, 3869–3878. [Google Scholar] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.-C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ong, C.; Hur, G.; Shen, H. Inhibition of the JAK-STAT3 pathway by andrographolide enhances chemosensitivity of cancer cells to doxorubicin. Biochem. Pharmacol. 2010, 79, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Itinteang, T.; Tan, S.T.; Brasch, H.D.; Steel, R.; Best, H.A.; Vishvanath, A.; Jia, J.; Day, D.J. Infantile haemangioma expresses embryonic stem cell markers. J. Clin. Pathol. 2012, 65, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Tran, D.; Bryan, B.A.; Mitchell, D.C. Meta-analysis of infantile hemangioma endothelial cell microarray expression data reveals significant aberrations of gene networks involved in cell adhesion and extracellular matrix composition. Angiol. Open Access 2013, 1, 107. [Google Scholar] [CrossRef]
- Lim, Y.H.; Douglas, S.R.; Ko, C.J.; Antaya, R.J.; McNiff, J.M.; Zhou, J.; Choate, K.A.; Narayan, D. Somatic Activating RAS Mutations Cause Vascular Tumors Including Pyogenic Granuloma. J. Investig. Dermatol. 2015, 135, 1698–1700. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Gebbink, M.F.B.G.; Voest, E.E. Stimulation of angiogenesis by Ras proteins. Biochim Biophys. Acta Rev. Cancer 2004, 1654, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, A.; Zheng, Q.; Adam, A.; Vincent, P.; Pumiglia, K. Activation of endothelial ras signaling bypasses senescence and causes abnormal vascular morphogenesis. Cancer Res. 2010, 70, 3803–3812. [Google Scholar] [CrossRef] [PubMed]
- Grugel, S.; Finkenzeller, G.; Weindel, K.; Barleon, B.; Marmé, D. Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. J. Biol. Chem. 1995, 270, 25915–25919. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.N.; Bryant, P.; Pumiglia, K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 2001, 276, 49289–49298. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A. Bcl-2 Inhibition in Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, S112–S114. [Google Scholar] [CrossRef]
- Nakajima, W.; Sharma, K.; Hicks, M.A.; Le, N.; Brown, R.; Krystal, G.W.; Harada, H. Combination with vorinostat overcomes ABT-263 (navitoclax) resistance of small cell lung cancer. Cancer Biol. Ther. 2016, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H.M.; Saglio, G.; Steegmann, J.L.; Shah, N.P.; Boque, C.; Chuah, C.; Pavlovsky, C.; Mayer, J.; Cortes, J.; et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 2014, 123, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Ouedraogo, Z.G.; Biau, J.; Kemeny, J.; Morel, L.; Verrelle, P.; Chautard, E. Role of STAT3 in genesis and progression of human malignant gliomas. Mol. Neurobiol. 2017, 54, 5780–5797. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Calicchio, M.L.; Collins, T.; Kozakewich, H.P. Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am. J. Pathol. 2009, 174, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.S. The molecular biology of cancer. Mol. Asp. Med. 2000, 21, 167–223. [Google Scholar] [CrossRef]
- Jiang, B.H.; Zheng, J.Z.; Aoki, M.; Vogt, P.K. Phosphotidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Manzano, C.; Fueyo, J.; Jiang, H.; Glass, T.L.; Lee, H.; Hu, M.; Liu, J.; Jasti, S.L.; Liu, T.; Conrad, C.A. Mechanisms underlying PTEN regulation of vascular endothelial growth factor and angiogenesis. Ann. Neurol. 2003, 53, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Tan, W.H.; Baris, H.N.; Burrows, P.E.; Robson, C.D.; Alomari, A.I.; Mulliken, J.B.; Fishman, S.J.; Irons, M.B. The spectrum of vascular anomalies in patients with PTEN mutations: Implications for diagnosis and management. J. Med. Genet. 2007, 44, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 2001, 29, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Michiels, C.M. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar] [PubMed]
- De Moraes, E.; Dar, N.A.; de Moura Gallo, C.V.; Hainaut, P. Cross-talks between cyclooxygenase-2 and tumor suppressor protein p53: Balancing life and death during inflammatory stress and carcinogenesis. Int. J. Cancer 2007, 121, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Creancier, L.; Zanibellato, C.; Prats, A.; Prats, H. Tumour suppressor p53 inhibits human fibroblast growth factor 2 expression by a post-transcriptional mechanism. Oncogene 2001, 20, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.G.; Parker, A.E.; Zhu, X.; Green, M.R. P53-Mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 2006, 313, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Koukourakis, M.I.; Kakolyris, S.; Turley, H.; O’Byrne, K.; Scott, P.A.E.; Pezzella, F.; Georgoulias, V.; Harris, A.L.; Gatter, K.C. Vascular endothelial growth factor, wild type p53, and angiogenesis in early operable non-small cell lung cancer. Clin. Cancer Res. 1998, 4, 3017–3024. [Google Scholar] [PubMed]
- Perrone, G.; Santini, V.D.; Verzi, A.; Tonini, G.; Vetrani, A.; Rabitti, V.C. Correlation of p53 and Bcl-2 expression with vascular endothelial growth factor (VEGF), microvessel density (MVD) and clinico-pathological features in colon cancer. Cancer Lett. 2004, 208, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tahaney, W.M.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecularly targeted therapies for p53-mutant cancers. Cell. Mol. Life Sci. 2017, 74, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.-B.; Li, Q.-Y.; Jiang, F.; Hu, X.-Y.; Ma, R.-Z.; Dong, Q.; Zhang, H.; Pattar, P.; Li, S.-X. Pingyangymycin stimulates apoptosis in human hemangioma-derived endothelial cells through activation of the p53 pathway. Mol. Med. Rep. 2014, 10, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, K.; Xiao, X.; Zheng, S.; Xu, T.; Chen, S. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J. Pediatr. Surg. 2012, 47, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.M.; Rowntree, R.K.; Amaya, C.; Diaz, D.; Kokta, V.; Mitchell, D.C.; Bryan, B.A. Gene expression analysis reveals marked differences in the transcriptome of infantile hemangioma endothelial cells compared to normal dermal microvascular endothelial cells. Vasc. Cell 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Al-Odaini, A.A.; Wang, Y.; Korah, J.; Dai, M.; Xiao, L.; Ali, S.; Lebrun, J.J. KiSS1 gene as a novel mediator of TGFβ-mediated cell invasion in triple negative breast cancer. Cell Signal. 2018, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alhejaily, A.; Day, A.G.; Feilotter, H.E.; Baetz, T.; LeBrun, D.P. Inactivation of the CDKN2A tumor-suppressor gene by deletion or methylation is common at diagnosis in follicular lymphoma and associated with poor clinical outcome. Clin. Cancer Res. 2014, 20, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of the VEGF-Bcl-2-mediated cell survival pathway. The VEGF ligand binds to VEGFR-2 and activates PI3K/Akt, which leads to the upregulation of Bcl-2 and ultimately promotes cell survival. The drugs Venetoclax and oblimersen inhibit Bcl-2. VEGF, vegfr-2, vascular endothelial growth factor; VEGFR-2, vascular endothelial growth factor-2; Bcl-2, B-cell lymphoma 2; PKB, protein kinase B; PDK1/2, protein 3-phosphoinositide-dependent protein kinase-1 and -2; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; ![Ijms 19 01192 i001]() inhibit; →, activate.
inhibit; →, activate.
 inhibit; →, activate.
inhibit; →, activate.
Figure 1.
Overview of the VEGF-Bcl-2-mediated cell survival pathway. The VEGF ligand binds to VEGFR-2 and activates PI3K/Akt, which leads to the upregulation of Bcl-2 and ultimately promotes cell survival. The drugs Venetoclax and oblimersen inhibit Bcl-2. VEGF, vegfr-2, vascular endothelial growth factor; VEGFR-2, vascular endothelial growth factor-2; Bcl-2, B-cell lymphoma 2; PKB, protein kinase B; PDK1/2, protein 3-phosphoinositide-dependent protein kinase-1 and -2; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; ![Ijms 19 01192 i001]() inhibit; →, activate.
inhibit; →, activate.
inhibit; →, activate.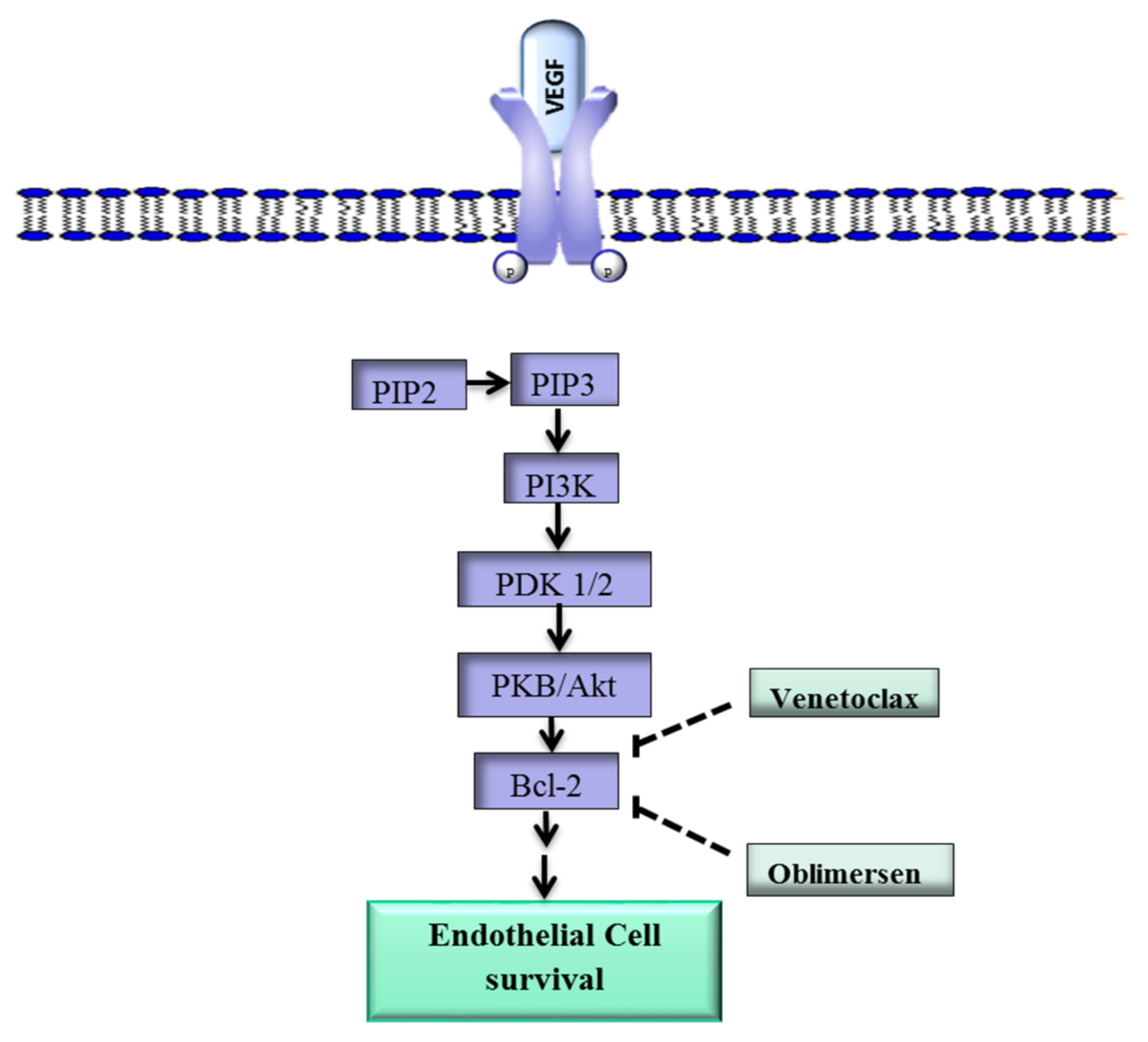
Figure 2.
Schematic diagram of the VEGF-STAT3 signaling pathway. The binding of VEGF to VEGFR-2 activates JAK, which in turn phosphorylates STAT3. STAT3 is translocated to the nucleus where it promotes the transcription of molecules that promote endothelial cell proliferation, migration and tube formation, key EC functions that underlie angiogenesis. VEGF, vascular endothelial growth factor; JAK, Janus kinase; STAT3, signaling transducer and activator of transcription 3; EC, endothelial cell; →, activate; ![Ijms 19 01192 i004]() , translocate into the nucleus.
, translocate into the nucleus.
 , translocate into the nucleus.
, translocate into the nucleus.
Figure 2.
Schematic diagram of the VEGF-STAT3 signaling pathway. The binding of VEGF to VEGFR-2 activates JAK, which in turn phosphorylates STAT3. STAT3 is translocated to the nucleus where it promotes the transcription of molecules that promote endothelial cell proliferation, migration and tube formation, key EC functions that underlie angiogenesis. VEGF, vascular endothelial growth factor; JAK, Janus kinase; STAT3, signaling transducer and activator of transcription 3; EC, endothelial cell; →, activate; ![Ijms 19 01192 i004]() , translocate into the nucleus.
, translocate into the nucleus.
, translocate into the nucleus.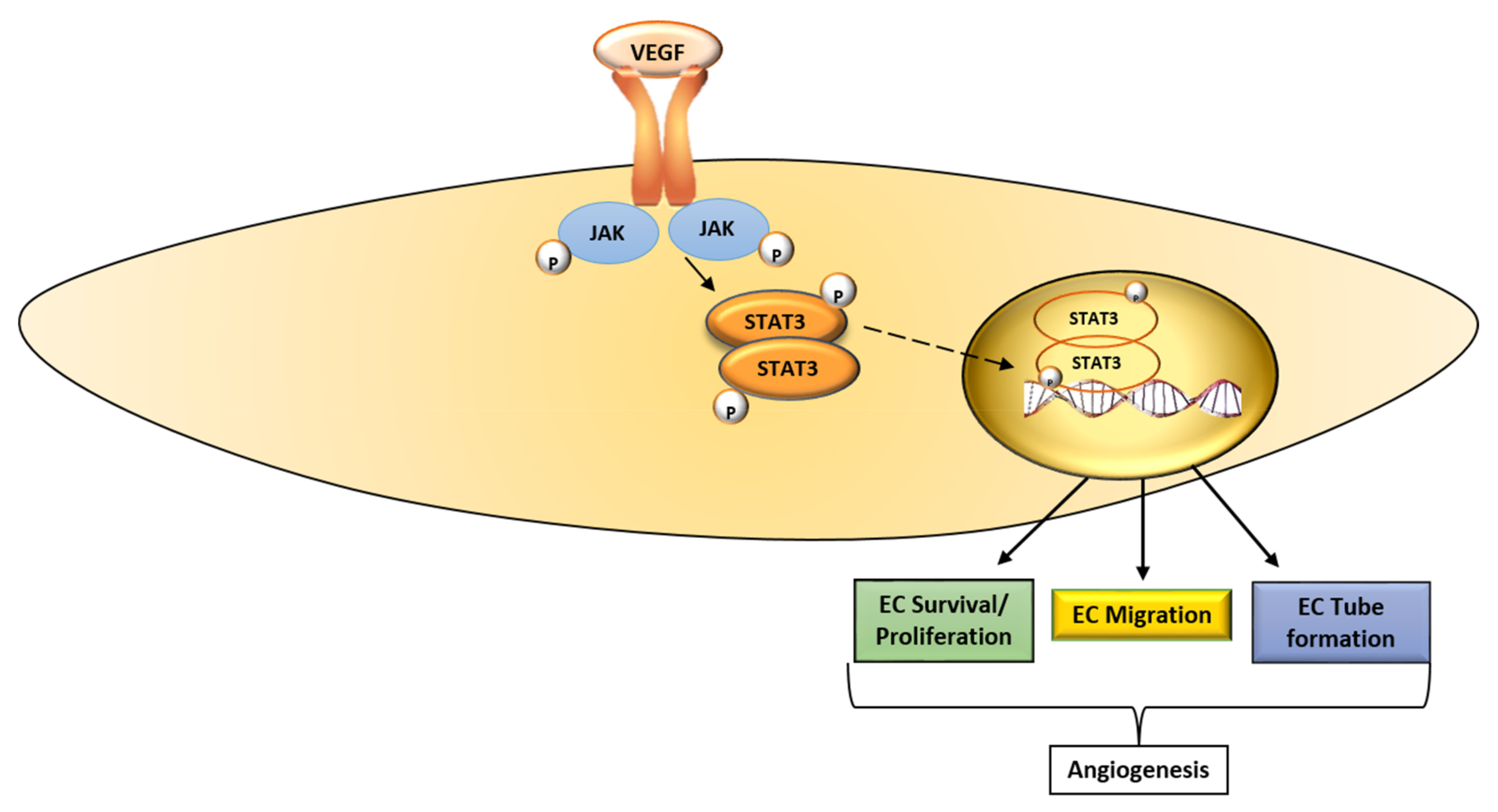
Figure 3.
p53 negatively regulates angiogenesis. It inhibits proangiogenic proteins VEGF, COX-2 and bFGF and stimulates angiogenesis inhibitors tumstatin, endostatin and type 4 and type 8 collagen, thus tilting the scale in favor of anti-angiogenesis. HIF-1α, hypoxia inducible factor-1α VEGF, vascular endothelial growth factor; COX-2, cyclooxygenase; bFGF, basic fibroblast growth factor, −, anti-angiogenesis; +, proangiogenesis; ![Ijms 19 01192 i002]() , drug inhibition;
, drug inhibition; ![Ijms 19 01192 i003]() , suppress; →, activate/stimulate.
, suppress; →, activate/stimulate.
 , drug inhibition;
, drug inhibition;  , suppress; →, activate/stimulate.
, suppress; →, activate/stimulate.
Figure 3.
p53 negatively regulates angiogenesis. It inhibits proangiogenic proteins VEGF, COX-2 and bFGF and stimulates angiogenesis inhibitors tumstatin, endostatin and type 4 and type 8 collagen, thus tilting the scale in favor of anti-angiogenesis. HIF-1α, hypoxia inducible factor-1α VEGF, vascular endothelial growth factor; COX-2, cyclooxygenase; bFGF, basic fibroblast growth factor, −, anti-angiogenesis; +, proangiogenesis; ![Ijms 19 01192 i002]() , drug inhibition;
, drug inhibition; ![Ijms 19 01192 i003]() , suppress; →, activate/stimulate.
, suppress; →, activate/stimulate.
, drug inhibition; , suppress; →, activate/stimulate.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Drugs targeting the vascular endothelial growth factor family of ligands and their receptors.
Table 1.
Drugs targeting the vascular endothelial growth factor family of ligands and their receptors.
| Drug | Target | Disease Indication | Reference |
|---|---|---|---|
| Aflibercept | VEGF-A, VEGF-B, PlGF | Broad spectrum of malignancies | [13,14] |
| Axitinib | VEGFR-1, VEGFR-2, VEGFR-3 | Renal cell carcinoma | [13] |
| Bevacizumab | VEGF-A | Breast, ovarian, metastatic colorectal and non-small cell lung cancers, recurrent glioblastoma | [13,15] |
| Lenvatinib | VEGF-A | Thyroid cancer | [16] |
| Pegaptanib | VEGF-A | Age-related macular degeneration | [14] |
| Pazopanib | VEGFR1, VEGFR2, VEGFR3 | Advanced soft tissue sarcoma | [13] |
| Sorafenib | VEGFR1, VEGFR2, VEGFR3 | Renal cell cancer, hepatocellular carcinoma | [13,14] |
| Sunitinib | VEGFR1, VEGFR2, VEGFR3 | Renal cell carcinoma, pancreatic neuroendocrine | [16] |
| Regorafenib | Refractory metastatic colorectal cancer | [14] | |
| Ramucirumab | VEGFR-2 | Metastatic colorectal, gastric/gastro-esophageal, non-small-cell lung cancer | [14] |
| Vatalanib | VEGFR1, VEGFR2, VEGFR3 | Solid tumors | [14] |
Table 2.
Oncogenes linked to IH and the drugs that inhibit these genes.
| Target | Drug | Phase of Development | Reference |
|---|---|---|---|
| Bcl-2 | |||
| S-055746 | I | [55] | |
| PNT-2258 | II | [41] | |
| Navitoclax | I/II and III | [56] | |
| Venetoclax | I/II | [41,42] | |
| Oblimersen | I/II and III | [43,44] | |
| STAT3 | |||
| Upstream tyrosine kinase inhibitors | Ruxolitinib | I–III | [45] |
| Dasatinib | III | [57] | |
| Fedratinib | I/II and III | [58] | |
| Tofacitinib | I/II | [45] | |
| SH2 domain inhibitors | OPB-31121 | I/II | [46] |
| OBP-51602 | I/II | [59] | |
| STAT3 DNA-binding domain inhibitors | CPA-1 | Preclinical | [46] |
| Cyclic STAT3 decoy | Preclinical | [45] | |
| STAT3 transcription inhibitors | AZD9150 | I/II | [60] |
| STAT3 decoy oligonucleotide | 0 | [61] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mabeta, P. Oncosuppressors and Oncogenes: Role in Haemangioma Genesis and Potential for Therapeutic Targeting. Int. J. Mol. Sci. 2018, 19, 1192. https://doi.org/10.3390/ijms19041192
AMA Style
Mabeta P. Oncosuppressors and Oncogenes: Role in Haemangioma Genesis and Potential for Therapeutic Targeting. International Journal of Molecular Sciences. 2018; 19(4):1192. https://doi.org/10.3390/ijms19041192
Chicago/Turabian StyleMabeta, Peace. 2018. "Oncosuppressors and Oncogenes: Role in Haemangioma Genesis and Potential for Therapeutic Targeting" International Journal of Molecular Sciences 19, no. 4: 1192. https://doi.org/10.3390/ijms19041192
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.