Excessive Extracellular ATP Desensitizes P2Y2 and P2X4 ATP Receptors Provoking Surfactant Impairment Ending in Ventilation-Induced Lung Injury

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Extracellular Release of ATP by AT I and AT II Cells and Clearance of Extracellular ATP
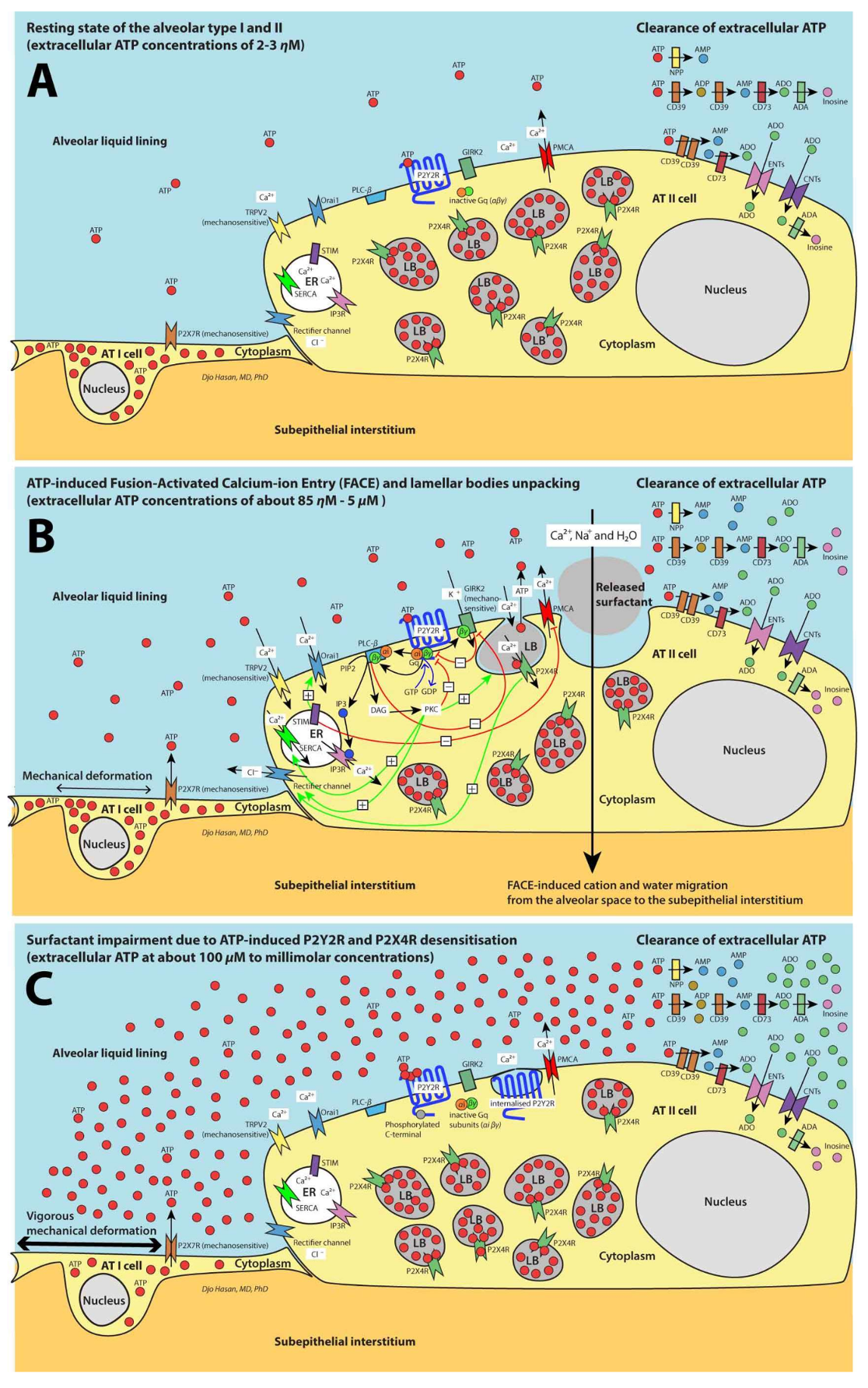
3. Purinergic Signaling Increases the AT II Cytoplasmic Ca2+ Levels by the Entry of Extracellular Ca2+ and Store-Operated Ca2+ Entry (SOCE)
4. Fusion of Lysosomes and LBs with the Plasma Membrane Plays a Role in the Repair of Damaged Plasma Membrane of the AT I and AT II Cells, Respectively
5. FACE Causes a Trans-Epithelial Transport of Na+, Ca2+ and Water Molecules
6. Surfactant Remodeling in the Alveolar Space
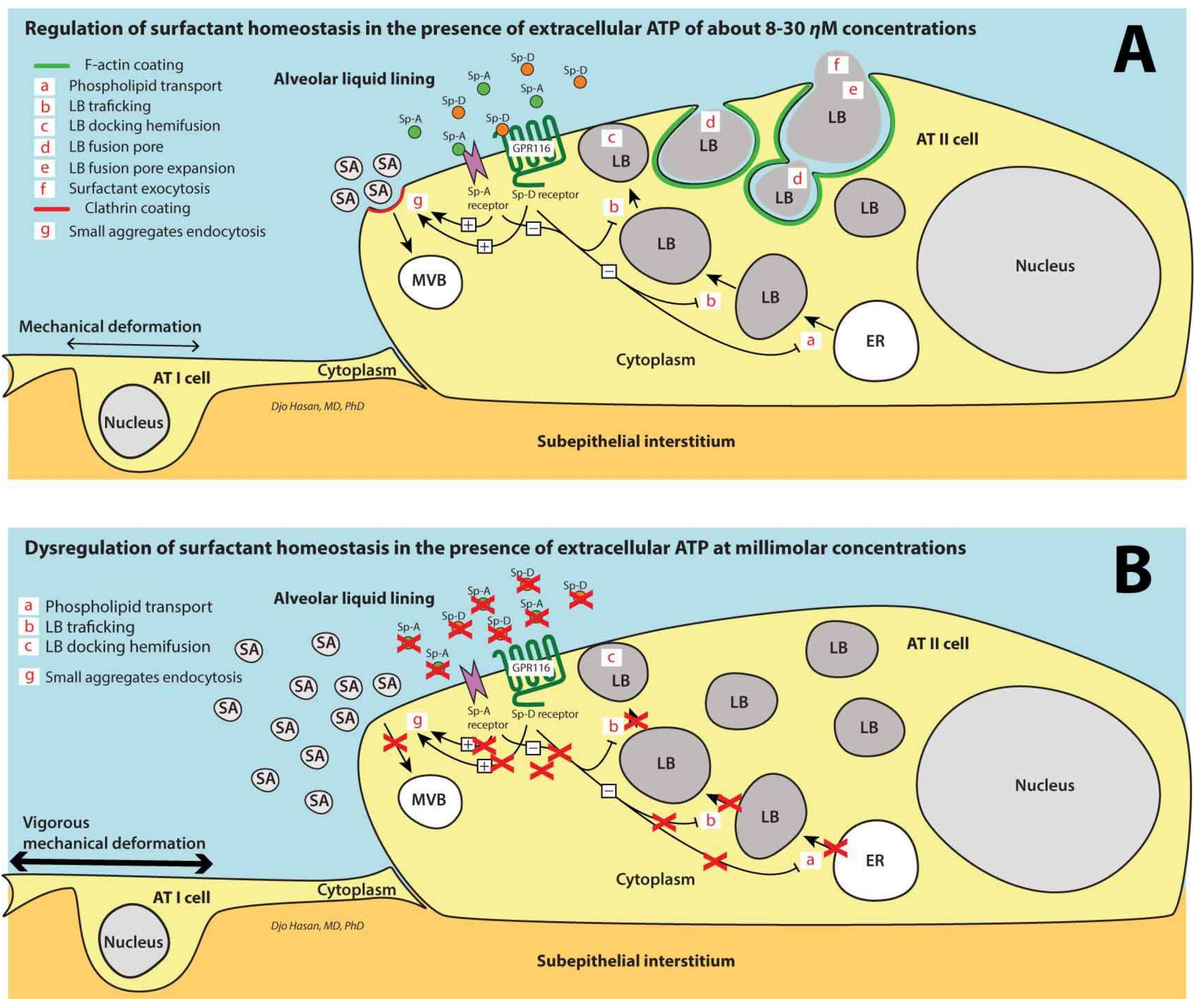
7. Surfactant Homeostasis in the Alveolar Space
8. Clearance of Ca2+ Ions from the Cytoplasm
9. Ventilation-Induced Extracellular ATP: (1) Initially Increases the Surfactant Release, (2) Halts Surfactant Release and Plasma Membrane Repair at >100 μM Concentrations and (3) Triggers the Pro-Inflammatory Response of the Innate Immunity at >300 μM Concentrations
10. Surfactant Deactivation Develops Significantly before Alveolar Space Flooding Caused by Increased Capillary Permeability
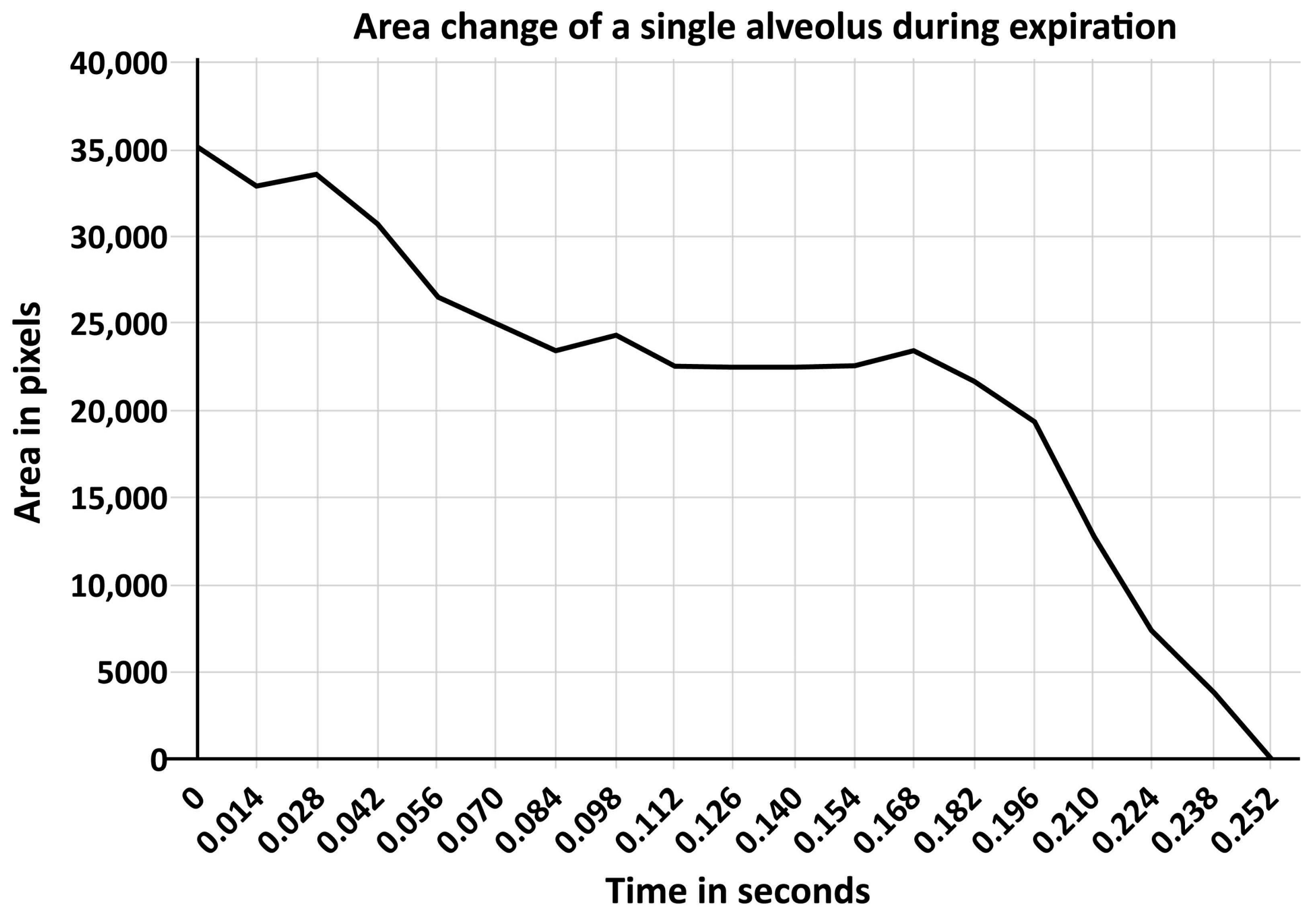
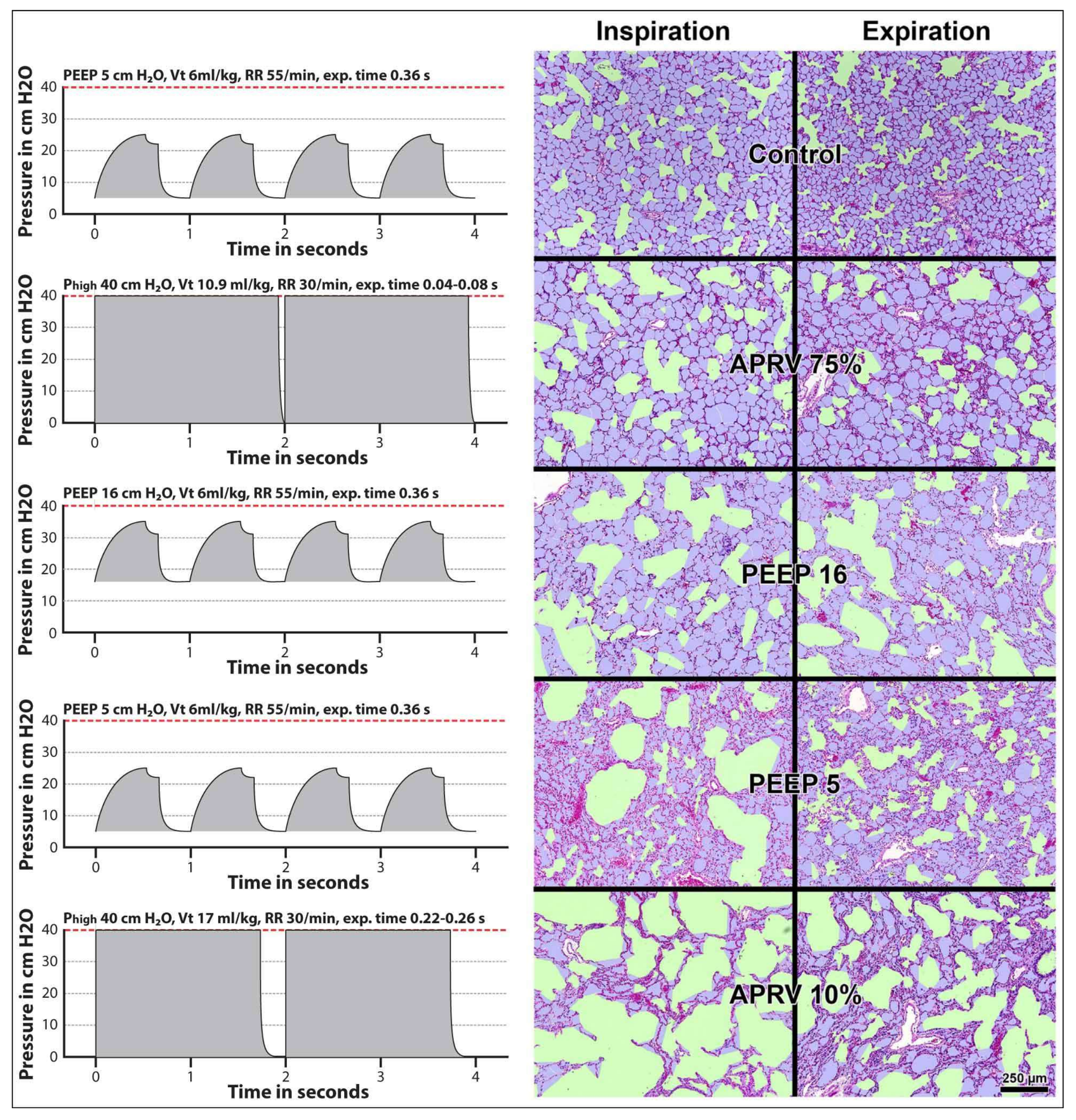
11. Surfactant Impairment Causes Changes in Alveolar Mechanics Exacerbating the Release of Extracellular ATP
12. Summary and Conclusions
Author Contributions
Conflicts of Interest
References
- Lohmann, K. Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 1929, 17, 624–625. [Google Scholar] [CrossRef]
- Langen, P.; Hucho, F. Karl Lohmann and the discovery of ATP. Angew. Chem. Int. Ed. Engl. 2008, 47, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J. The history and the hypotheses concerning ATP-formation by energised protons. FEBS Lett. 1978, 85, 9–19. [Google Scholar] [CrossRef]
- Wikstrom, M.K.; Saari, H.T. Conformational changes in cytochrome aa3 and ATP synthetase of the mitochondrial membrane and their role in mitochondrial energy transduction. Mol. Cell. Biochem. 1976, 11, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Feldberg, W.; Hebb, C. The stimulating action of phosphate compounds on the perfused superior cervical ganglion of the cat. J. Physiol. 1948, 107, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Holton, P. The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J. Physiol. 1959, 145, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar] [PubMed]
- Burnstock, G. Purinergic signalling: Its unpopular beginning, its acceptance and its exciting future. Bioessays 2012, 34, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling: From discovery to current developments. Exp. Physiol. 2014, 99, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Hasan, D.; Blankman, P.; Nieman, G.F. Purinergic signalling links mechanical breath profile and alveolar mechanics with the pro-inflammatory innate immune response causing ventilation-induced lung injury. Purinergic Signal. 2017, 13, 363–386. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Tonetti, T.; Cressoni, M.; Cadringher, P.; Herrmann, P.; Moerer, O.; Protti, A.; Gotti, M.; Chiurazzi, C.; Carlesso, E.; et al. Ventilator-related causes of lung injury: The mechanical power. Intensive Care Med. 2016, 42, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Nieman, G.F.; Gatto, L.A.; Habashi, N.M. Impact of mechanical ventilation on the pathophysiology of progressive acute lung injury. J. Appl. Physiol. 2015, 119, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, A.S.; Ranieri, V.M. Ventilator-induced lung injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Rezoagli, E.; Fumagalli, R.; Bellani, G. Definition and epidemiology of acute respiratory distress syndrome. Ann. Transl. Med. 2017, 5, 282. [Google Scholar] [CrossRef] [PubMed]
- Cressoni, M.; Gotti, M.; Chiurazzi, C.; Massari, D.; Algieri, I.; Amini, M.; Cammaroto, A.; Brioni, M.; Montaruli, C.; Nikolla, K.; et al. Mechanical power and development of ventilator-induced lung injury. Anesthesiology 2016, 124, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.B.; Douillet, C.D.; Mahler, S.A.; Husain, S.A.; Boucher, R.C. Adenosine triphosphate is released during injurious mechanical ventilation and contributes to lung edema. J. Trauma 2003, 55, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Amaya, F.; Hashimoto, S.; Ueno, H.; Beppu, S.; Mizuta, M.; Shime, N.; Ishizaka, A.; Hashimoto, S. Acute lung inflammation and ventilator-induced lung injury caused by ATP via the P2Y receptors: An experimental study. Respir. Res. 2008, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.Y. ARDS and diffuse alveolar damage: A pathologist’s perspective. Semin. Thorac. Cardiovasc. Surg. 2006, 18, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Maruscak, A.A.; Vockeroth, D.W.; Girardi, B.; Sheikh, T.; Possmayer, F.; Lewis, J.F.; Veldhuizen, R.A. Alterations to surfactant precede physiological deterioration during high tidal volume ventilation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L974–L983. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, S.J.; Bohm, S.H.; Gommers, D.; Zimmerman, L.J.; Lachmann, B. Surfactant impairment after mechanical ventilation with large alveolar surface area changes and effects of positive end-expiratory pressure. Br. J. Anaesth. 1998, 80, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Martinez Sarrasague, M.; Cimato, A.; Rubin de Celis, E.; Facorro, G. Influence of serum protein and albumin addition on the structure and activity of an exogenous pulmonary surfactant. Respir. Physiol. Neurobiol. 2011, 175, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Faridy, E.E. Effect of ventilation on movement of surfactant in airways. Respir. Physiol. 1976, 27, 323–334. [Google Scholar] [CrossRef]
- Pettenazzo, A.; Jobe, A.; Humme, J.; Seidner, S.; Ikegami, M. Clearance of surfactant phosphatidylcholine via the upper airways in rabbits. J. Appl. Physiol. 1988, 65, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Lewis, J.; Copland, I.; Engelberts, D.; Kavanagh, B.P.; Post, M.; Schurch, S.; Belik, J. Mechanical ventilation effect on surfactant content, function, and lung compliance in the newborn rat. Pediatr. Res. 2004, 56, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cressoni, M.; Chiurazzi, C.; Gotti, M.; Amini, M.; Brioni, M.; Algieri, I.; Cammaroto, A.; Rovati, C.; Massari, D.; di Castiglione, C.B.; et al. Lung inhomogeneities and time course of ventilator-induced mechanical injuries. Anesthesiology 2015, 123, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Busch, T.; Gaertner, J.; Haitsma, J.J.; Krabbendam, S.; Ebsen, M.; Lachmann, B.; Kaisers, U.X. Complement activation contributes to ventilator-induced lung injury in rats. J. Physiol. Pharmacol. 2016, 67, 911–918. [Google Scholar] [PubMed]
- Schwiebert, E.M.; Zsembery, A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim. Biophys. Acta 2003, 1615, 7–32. [Google Scholar] [CrossRef]
- Lazarowski, E.R.; Boucher, R.C.; Harden, T.K. Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentrations. J. Biol. Chem. 2000, 275, 31061–31068. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Reigada, D.; Mitchell, C.H.; Bates, S.R.; Margulies, S.S.; Koval, M. Paracrine stimulation of surfactant secretion by extracellular ATP in response to mechanical deformation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L489–L496. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Tan, J.J.; Boudreault, F.; Sokabe, M.; Berthiaume, Y.; Grygorczyk, R. Real-time imaging of inflation-induced ATP release in the ex-vivo rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L956–L969. [Google Scholar] [CrossRef] [PubMed]
- Brandao-Burch, A.; Key, M.L.; Patel, J.J.; Arnett, T.R.; Orriss, I.R. The P2X7 Receptor is an Important Regulator of Extracellular ATP Levels. Front. Endocrinol. 2012, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Chintagari, N.R.; Guo, Y.; Weng, T.; Su, L.; Liu, L. Purinergic P2X7 receptor regulates lung surfactant secretion in a paracrine manner. J. Cell Sci. 2011, 124, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Brunschweiger, A.; Muller, C.E. P2 receptors activated by uracil nucleotides—An update. Curr. Med. Chem. 2006, 13, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol: Two interacting second messengers. Annu. Rev. Biochem. 1987, 56, 159–193. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, R.G.; Heller, S. The mechanosensitive nature of TRPV channels. Pflügers Arch. 2005, 451, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Keselman, I.; Fribourg, M.; Felsenfeld, D.P.; Logothetis, D.E. Mechanism of PLC-mediated Kir3 current inhibition. Channels 2007, 1, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.L.; Harline, M.C.; Otero, M.A.; Glover, G.G.; Garrad, R.C.; Krugh, B.; Walker, N.M.; Gonzalez, F.A.; Turner, J.T.; Weisman, G.A. Desensitization of P2Y2 receptor-activated transepithelial anion secretion. Am. J. Physiol. 1999, 276, C777–C787. [Google Scholar] [CrossRef] [PubMed]
- Varela, D.; Penna, A.; Simon, F.; Eguiguren, A.L.; Leiva-Salcedo, E.; Cerda, O.; Sala, F.; Stutzin, A. P2X4 activation modulates volume-sensitive outwardly rectifying chloride channels in rat hepatoma cells. J. Biol. Chem. 2010, 285, 7566–7574. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.; Isales, C.M.; Calle, R.; Throckmorton, D.; Anderson, M.; Gasalla-Herraiz, J.; McCarthy, R. Diacylglycerol production, Ca2+ influx, and protein kinase C activation in sustained cellular responses. Endocr. Rev. 1995, 16, 649–681. [Google Scholar] [CrossRef] [PubMed]
- Dietl, P.; Haller, T.; Frick, M. Spatio-temporal aspects, pathways and actions of Ca2+ in surfactant secreting pulmonary alveolar type II pneumocytes. Cell Calcium 2012, 52, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Miklavc, P.; Mair, N.; Wittekindt, O.H.; Haller, T.; Dietl, P.; Felder, E.; Timmler, M.; Frick, M. Fusion-activated Ca2+ entry via vesicular P2X4 receptors promotes fusion pore opening and exocytotic content release in pneumocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.E.; Korbmacher, J.P.; Hecht, E.; Hobi, N.; Wittekindt, O.H.; Dietl, P.; Kranz, C.; Frick, M. Fusion-activated cation entry (FACE) via P2X4 couples surfactant secretion and alveolar fluid transport. FASEB J. 2013, 27, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Fois, G.; Winkelmann, V.E.; Bareis, L.; Staudenmaier, L.; Hecht, E.; Ziller, C.; Ehinger, K.; Schymeinsky, J.; Kranz, C.; Frick, M. ATP is stored in lamellar bodies to activate vesicular P2X4 in an autocrine fashion upon exocytosis. J. Gen. Physiol. 2018, 150, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Hiasa, M.; Sakamoto, S.; Omote, H.; Nomura, M. Vesicular nucleotide transporter (VNUT): Appearance of an actress on the stage of purinergic signaling. Purinergic Signal. 2017, 13, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Z.; Cao, Q.; Sun, X.; Dong, X.P. Activation of lysosomal P2X4 by ATP transported into lysosomes via VNUT/SLC17A9 using V-ATPase generated voltage gradient as the driving force. J. Physiol. 2016, 594, 4253–4266. [Google Scholar] [CrossRef] [PubMed]
- Stojilkovic, S.S.; Yan, Z.; Obsil, T.; Zemkova, H. Structural insights into the function of P2X4: An ATP-gated cation channel of neuroendocrine cells. Cell. Mol. Neurobiol. 2010, 30, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Zemkova, H.; Khadra, A.; Rokic, M.B.; Tvrdonova, V.; Sherman, A.; Stojilkovic, S.S. Allosteric regulation of the P2X4 receptor channel pore dilation. Pflügers Arch. 2015, 467, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Parker, I.; Smith, I.F. Communication of Ca2+ signals via tunneling membrane nanotubes is mediated by transmission of inositol trisphosphate through gap junctions. Cell Calcium 2016, 60, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Koval, M. Sharing signals: Connecting lung epithelial cells with gap junction channels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L875–L893. [Google Scholar] [CrossRef] [PubMed]
- Miklavc, P.; Wittekindt, O.H.; Felder, E.; Dietl, P. Ca2+-dependent actin coating of lamellar bodies after exocytotic fusion: A prerequisite for content release or kiss-and-run. Ann. N. Y. Acad. Sci. 2009, 1152, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Haller, T.; Dietl, P.; Pfaller, K.; Frick, M.; Mair, N.; Paulmichl, M.; Hess, M.W.; Furst, J.; Maly, K. Fusion pore expansion is a slow, discontinuous, and Ca2+-dependent process regulating secretion from alveolar type II cells. J. Cell Biol. 2001, 155, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Strayer, D.S.; Yang, S.; Jerng, H.H. Surfactant protein A-binding proteins. Characterization and structures. J. Biol. Chem. 1993, 268, 18679–18684. [Google Scholar] [PubMed]
- Wissel, H.; Looman, A.C.; Fritzsche, I.; Rustow, B.; Stevens, P.A. SP-A-binding protein BP55 is involved in surfactant endocytosis by type II pneumocytes. Am. J. Physiol. 1996, 271, L432–L440. [Google Scholar] [CrossRef] [PubMed]
- Kresch, M.J.; Christian, C.; Lu, H. Isolation and partial characterization of a receptor to surfactant protein A expressed by rat type II pneumocytes. Am. J. Respir. Cell Mol. Biol. 1998, 19, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Chroneos, Z.C.; Abdolrasulnia, R.; Whitsett, J.A.; Rice, W.R.; Shepherd, V.L. Purification of a cell-surface receptor for surfactant protein A. J. Biol. Chem. 1996, 271, 16375–16383. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, T.; Ishida, J.; Kato, A.; Ichinose, T.; Ariestanti, D.M.; Takahashi, T.; Ito, K.; Abe, J.; Suzuki, T.; Wakana, S.; et al. Lung surfactant levels are regulated by Ig-Hepta/GPR116 by monitoring surfactant protein D. PLoS ONE 2013, 8, e69451. [Google Scholar] [CrossRef] [PubMed]
- Cooley, J.; McDonald, B.; Accurso, F.J.; Crouch, E.C.; Remold-O’Donnell, E. Patterns of neutrophil serine protease-dependent cleavage of surfactant protein D in inflammatory lung disease. J. Leukoc. Biol. 2008, 83, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Rubio, F.; Cooley, J.; Accurso, F.J.; Remold-O’Donnell, E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax 2004, 59, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Belete, H.A.; Hubmayr, R.D.; Wang, S.; Singh, R.D. The role of purinergic signaling on deformation induced injury and repair responses of alveolar epithelial cells. PLoS ONE 2011, 6, e27469. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.R. P63 (CKAP4) as an SP-A receptor: Implications for surfactant turnover. Cell. Physiol. Biochem. 2010, 25, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Birch, N.P.; Suresh, V. An Optimised Human Cell Culture Model for Alveolar Epithelial Transport. PLoS ONE 2016, 11, e0165225. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, B.; Martinez-Calle, M.; Perez-Gil, J. Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann. Anat. 2017, 209, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.Y.; Veldhuizen, R.A.; Neumann, A.W.; Petersen, N.O.; Possmayer, F. Current perspectives in pulmonary surfactant—Inhibition, enhancement and evaluation. Biochim. Biophys. Acta 2008, 1778, 1947–1977. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.; Perez-Gil, J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 2015, 185, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, B.; Villen, L.; Cruz, A.; Orellana, G.; Perez-Gil, J. Pulmonary surfactant layers accelerate O2 diffusion through the air-water interface. Biochim. Biophys. Acta 2010, 1798, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Baoukina, S.; Tieleman, D.P. Computer simulations of lung surfactant. Biochim. Biophys. Acta 2016, 1858, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Sibug-Aga, R.; Dunn, R.C. High-resolution studies of lung surfactant collapse. Photochem. Photobiol. 2004, 80, 471–476. [Google Scholar] [CrossRef]
- Schief, W.R.; Antia, M.; Discher, B.M.; Hall, S.B.; Vogel, V. Liquid-crystalline collapse of pulmonary surfactant monolayers. Biophys. J. 2003, 84, 3792–3806. [Google Scholar] [CrossRef]
- Mokra, D.; Kosutova, P. Biomarkers in acute lung injury. Respir. Physiol. Neurobiol. 2015, 209, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Demberg, L.M.; Winkler, J.; Wilde, C.; Simon, K.U.; Schon, J.; Rothemund, S.; Schoneberg, T.; Promel, S.; Liebscher, I. Activation of Adhesion G Protein-coupled Receptors: Agonist specificity of Stachel sequence-derived peptides. J. Biol. Chem. 2017, 292, 4383–4394. [Google Scholar] [CrossRef] [PubMed]
- Burk, S.E.; Lytton, J.; MacLennan, D.H.; Shull, G.E. cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J. Biol. Chem. 1989, 264, 18561–18568. [Google Scholar] [PubMed]
- Allen, B.G.; Katz, S. Phosphorylation of cardiac junctional and free sarcoplasmic reticulum by PKC α, PKC beta, PKA and the Ca2+/calmodulin-dependent protein kinase. Mol. Cell. Biochem. 1996, 155, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Caride, A.J.; Filoteo, A.G.; Enyedi, A.; Verma, A.K.; Penniston, J.T. Detection of isoform 4 of the plasma membrane calcium pump in human tissues by using isoform-specific monoclonal antibodies. Biochem. J. 1996, 316 Pt 1, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Thomas, D.D. Time-resolved FRET reveals the structural mechanism of SERCA-PLB regulation. Biochem. Biophys. Res. Commun. 2014, 449, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.F.; Samakai, E.; Soboloff, J. STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J. 2012, 31, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Garrad, R.C.; Otero, M.A.; Erb, L.; Theiss, P.M.; Clarke, L.L.; Gonzalez, F.A.; Turner, J.T.; Weisman, G.A. Structural basis of agonist-induced desensitization and sequestration of the P2Y2 nucleotide receptor. Consequences of truncation of the C terminus. J. Biol. Chem. 1998, 273, 29437–29444. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.V.; Hernandez-Perez, M.G.; Aquino, E.; Garrad, R.C.; Weisman, G.A.; Gonzalez, F.A. Agonist-induced phosphorylation and desensitization of the P2Y2 nucleotide receptor. Mol. Cell. Biochem. 2005, 280, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.M.; Lou, X.; Erik, A.; Persson, G.; Ring, A. Growth hormones reverse desensitization of P2Y2 receptors in rat mesangial cells. Biochem. Biophys. Res. Commun. 2000, 270, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Sromek, S.M.; Harden, T.K. Agonist-induced internalization of the P2Y2 receptor. Mol. Pharmacol. 1998, 54, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Stokes, L. Rab5 regulates internalisation of P2X4 receptors and potentiation by ivermectin. Purinergic Signal. 2013, 9, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Fullbier, L.; Wehrmann, M.; Khoury, J.; Mittelbronn, M.; Ibla, J.; Rosenberger, P.; Eltzschig, H.K. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J. Immunol. 2007, 178, 8127–8137. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Vaneker, M.; Halbertsma, F.J.; van Egmond, J.; Netea, M.G.; Dijkman, H.B.; Snijdelaar, D.G.; Joosten, L.A.; van der Hoeven, J.G.; Scheffer, G.J. Mechanical ventilation in healthy mice induces reversible pulmonary and systemic cytokine elevation with preserved alveolar integrity: An in vivo model using clinical relevant ventilation settings. Anesthesiology 2007, 107, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Morad, H.O.; Belete, S.C.; Read, T.; Shaw, A.M. Time-course analysis of C3a and C5a quantifies the coupling between the upper and terminal Complement pathways in vitro. J. Immunol. Methods 2015, 427, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J. Vascular permeability changes induced by complement-derived peptides. Agents Act. 1983, 13, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wittekindt, O.H. Tight junctions in pulmonary epithelia during lung inflammation. Pflügers Arch. 2017, 469, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Barth, K.; Blasche, R.; Neisser, A.; Bramke, S.; Frank, J.A.; Kasper, M. P2X7R-dependent regulation of glycogen synthase kinase 3β and claudin-18 in alveolar epithelial type I cells of mice lung. Histochem. Cell Biol. 2016, 146, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Wray, C.; Mao, Y.; Pan, J.; Chandrasena, A.; Piasta, F.; Frank, J.A. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L219–L227. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, K.; Zhang, X.; Zhang, J.; Xu, B. ATP induces disruption of tight junction proteins via IL-1 β-dependent MMP-9 activation of human blood-brain barrier in vitro. Neural Plast. 2016, 2016, 8928530. [Google Scholar] [CrossRef] [PubMed]
- Sera, T.; Yokota, H.; Tanaka, G.; Uesugi, K.; Yagi, N.; Schroter, R.C. Murine pulmonary acinar mechanics during quasi-static inflation using synchrotron refraction-enhanced computed tomography. J. Appl. Physiol. 2013, 115, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.P.; DiRocco, J.; Allen, G.B.; Bates, J.H.; Lafollette, R.; Kubiak, B.D.; Fischer, J.; Maroney, S.; Nieman, G.F. The role of time and pressure on alveolar recruitment. J. Appl. Physiol. 2009, 106, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Suki, B.; Bates, J.H. Lung tissue mechanics as an emergent phenomenon. J. Appl. Physiol. 2011, 110, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Kollisch-Singule, M.; Emr, B.; Smith, B.; Ruiz, C.; Roy, S.; Meng, Q.; Jain, S.; Satalin, J.; Snyder, K.; Ghosh, A.; et al. Airway pressure release ventilation reduces conducting airway micro-strain in lung injury. J. Am. Coll. Surg. 2014, 219, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Ochs, M.; Nyengaard, J.R.; Jung, A.; Knudsen, L.; Voigt, M.; Wahlers, T.; Richter, J.; Gundersen, H.J. The number of alveoli in the human lung. Am. J. Respir. Crit. Care Med. 2004, 169, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Barre, S.F.; Haberthur, D.; Stampanoni, M.; Schittny, J.C. Efficient estimation of the total number of acini in adult rat lung. Physiol. Rep. 2014, 2, e12063. [Google Scholar] [CrossRef] [PubMed]
- Satalin, J.; Jain, S.V.; Kollisch-Singule, M.C.; Andrews, P.L.; Searles, Q.; Sweeney, T.; Gatto, L.A.; Habashi, N.M.; Nieman, G.F. Discerning expiratory time vs. pressure in preventing in-vivo alveolar collapse. Respir. Care 2016, 61, OF41. [Google Scholar]
- Roy, S.; Habashi, N.; Sadowitz, B.; Andrews, P.; Ge, L.; Wang, G.; Roy, P.; Ghosh, A.; Kuhn, M.; Satalin, J.; et al. Early airway pressure release ventilation prevents ARDS—A novel preventive approach to lung injury. Shock 2013, 39, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.L.; Cruz, F.F.; Samary, C.D.S.; Moraes, L.; de Magalhaes, R.F.; Fernandes, M.V.S.; Bose, R.; Pelegati, V.B.; Carvalho, H.F.; Capelozzi, V.L.; et al. Biological response to time-controlled adaptive ventilation depends on acute respiratory distress syndrome etiology. Crit. Care Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bellingan, G.; Maksimow, M.; Howell, D.C.; Stotz, M.; Beale, R.; Beatty, M.; Walsh, T.; Binning, A.; Davidson, A.; Kuper, M.; et al. The effect of intravenous interferon-β-1a (FP-1201) on lung CD73 expression and on acute respiratory distress syndrome mortality: An open-label study. Lancet Respir. Med. 2014, 2, 98–107. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.; Suzumura, E.A.; Laranjeira, L.N.; Paisani, D.M.; Damiani, L.P.; Guimaraes, H.P.; Romano, E.R.; Regenga, M.M.; Taniguchi, L.N.T.; Teixeira, C.; et al. Effect of lung recruitment and titrated positive end-expiratory pressure (PEEP) vs low PEEP on mortality in patients with acute respiratory distress syndrome: A randomized clinical trial. JAMA 2017, 318, 1335–1345. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, D.; Satalin, J.; Van der Zee, P.; Kollisch-Singule, M.; Blankman, P.; Shono, A.; Somhorst, P.; Den Uil, C.; Meeder, H.; Kotani, T.; et al. Excessive Extracellular ATP Desensitizes P2Y2 and P2X4 ATP Receptors Provoking Surfactant Impairment Ending in Ventilation-Induced Lung Injury. Int. J. Mol. Sci. 2018, 19, 1185. https://doi.org/10.3390/ijms19041185
Hasan D, Satalin J, Van der Zee P, Kollisch-Singule M, Blankman P, Shono A, Somhorst P, Den Uil C, Meeder H, Kotani T, et al. Excessive Extracellular ATP Desensitizes P2Y2 and P2X4 ATP Receptors Provoking Surfactant Impairment Ending in Ventilation-Induced Lung Injury. International Journal of Molecular Sciences. 2018; 19(4):1185. https://doi.org/10.3390/ijms19041185
Chicago/Turabian StyleHasan, Djo, Joshua Satalin, Philip Van der Zee, Michaela Kollisch-Singule, Paul Blankman, Atsuko Shono, Peter Somhorst, Corstiaan Den Uil, Han Meeder, Toru Kotani, and et al. 2018. "Excessive Extracellular ATP Desensitizes P2Y2 and P2X4 ATP Receptors Provoking Surfactant Impairment Ending in Ventilation-Induced Lung Injury" International Journal of Molecular Sciences 19, no. 4: 1185. https://doi.org/10.3390/ijms19041185