The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis

 , , and
, , and
Abstract
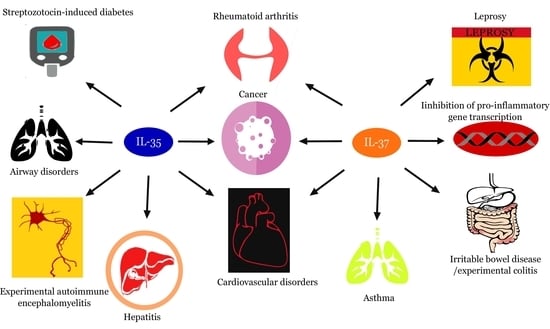
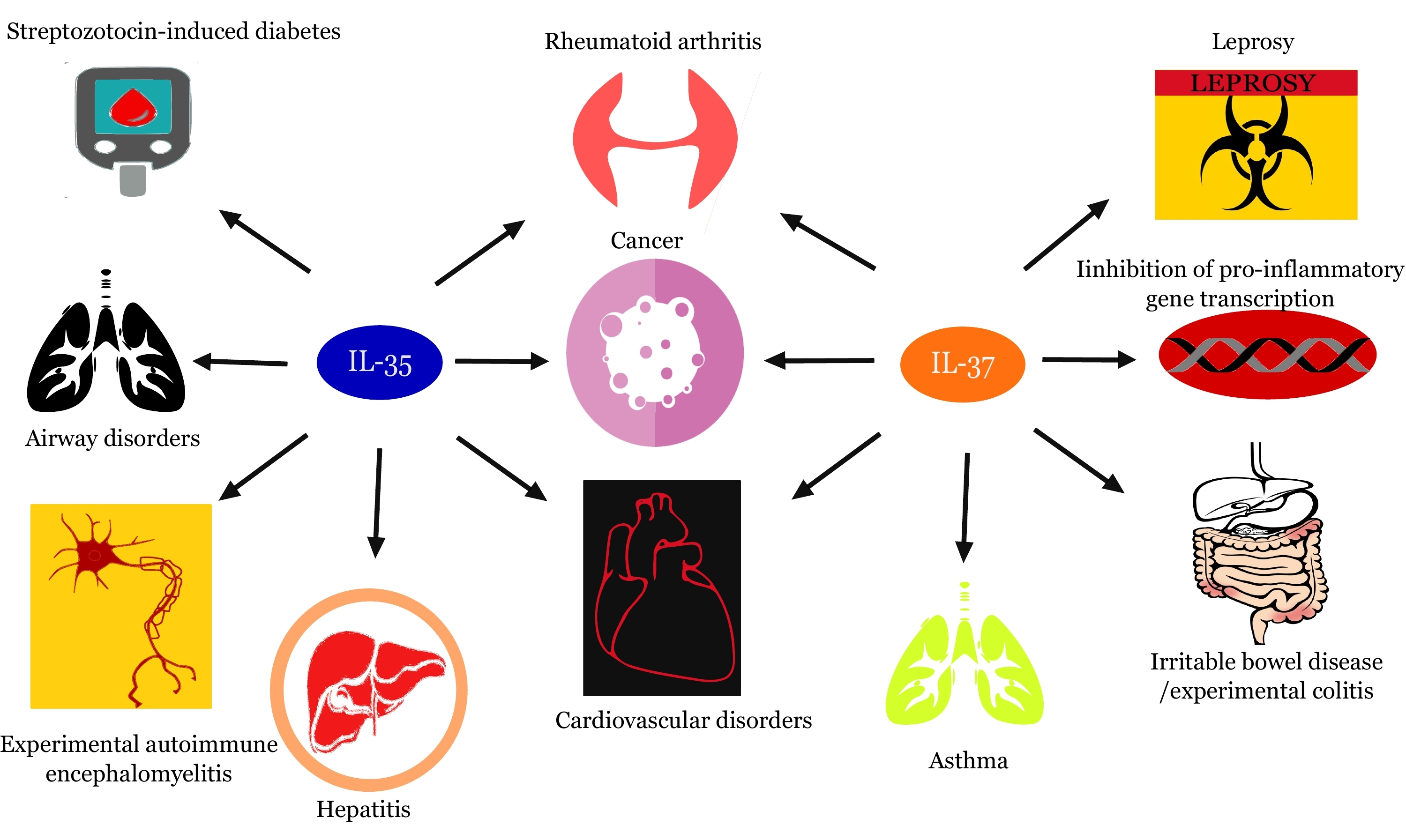
:
1. Introduction
2. Immunobiological Significance of IL-35 in Disease Pathogenesis
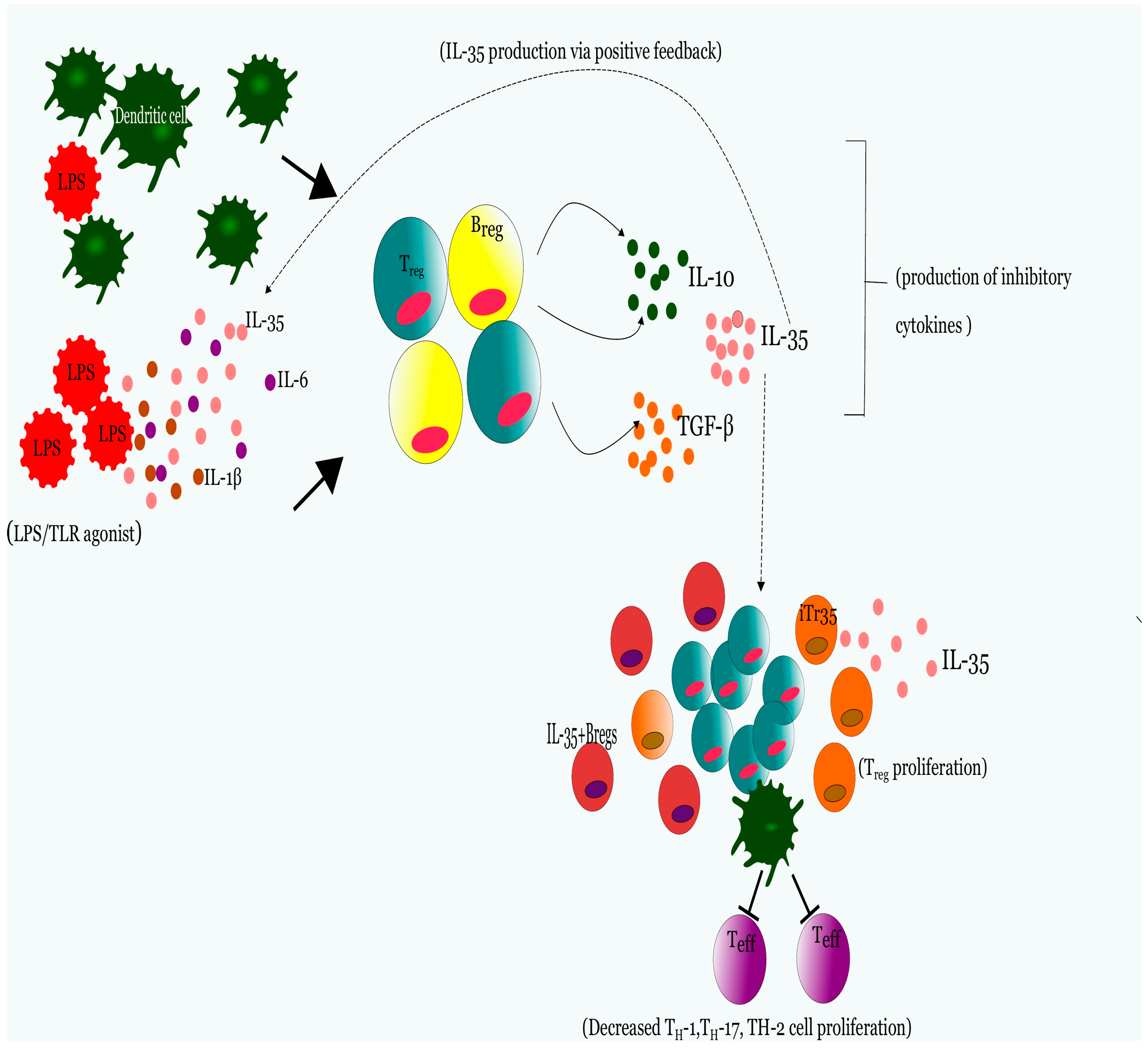
2.1. IL-35 Generates a Lineage of Functionally Suppressive IL-35 Expressing T-cells (iTr35) in Human and Murine Subjects
2.2. Function(s) of IL-35 in Autoimmune Disorders
2.2.1. Rheumatoid Arthritis
2.2.2. Experimental Autoimmune Encephalomyelitis (EAE)
2.2.3. Hashimoto’s Thyroiditis
2.2.4. Multiple Low Dose Streptozotocin (MLDSTZ)
2.3. Role of IL-35 in Respiratory Disorders
2.4. A Role for IL-35 in Cardiovascular Diseases
2.5. IL-35 Mediates Inflammation in Sepsis
2.6. Role for IL-35 in Connective Tissue Disorders
2.6.1. Systemic Sclerosis (Scleroderma)
2.6.2. Actions of IL-35 in a Murine Model of Systemic Lupus Erythematosus (SLE)
2.7. Actions of IL-35 in Hepatitis Infection
2.8. Function(s) of IL-35 in Cancer
3. The Immunobiology and Signaling of IL-37
3.1. Biological Occurrence of IL-37.
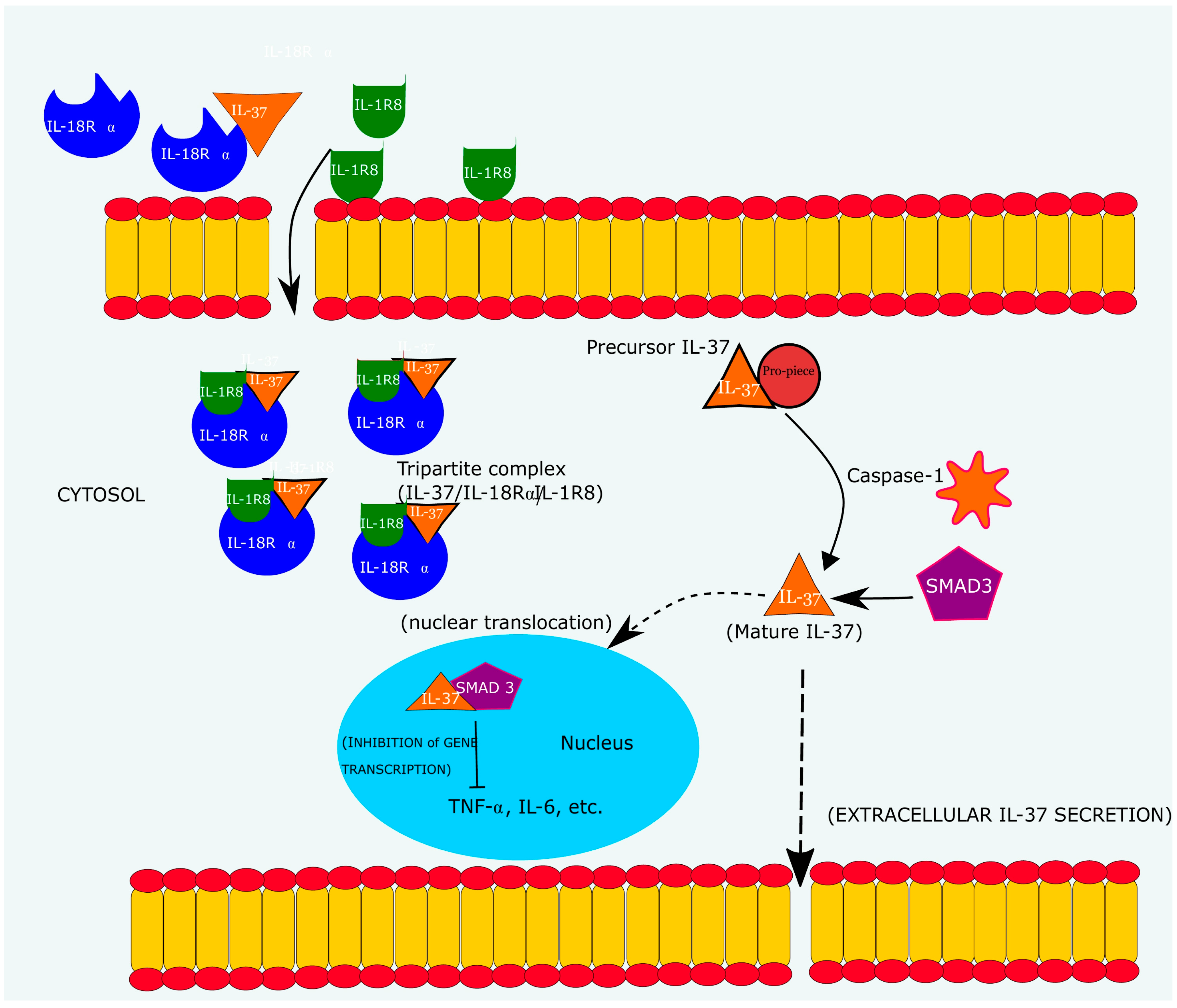
3.2. Mechanism of IL-37 Mediated Immunosuppression
- (a)
- Binding to IL-18Rα/single immunoglobulin-g-interleukin-1-related receptor (IL-18Rα/SIGGIR) complex to form a tripartite ligand complex (IL-18Rα/L-37/SIGGIR)
- (b)
- Binding to SMAD3 receptor (SMAD3/IL-37)
3.3. Biological Actions of Endogenous IL-37
3.4. Signal Transduction of IL-37
4. Immunoregulatory Mechanisms of IL-37 in Disease Pathogenesis
4.1. Actions of IL-37 in Murine (Experimental) Colitis and Irritable Bowel Disease
4.2. A Role for IL-37 in Asthma
4.3. Immunomodulatory Action of IL-37 in Collagen-Induced Arthritis
4.4. IL-37 in Cancer
4.5. Function of IL-37 in Cardiovascular Disorders
4.6. IL-37 and Its Role in Sleep-Wake Cycle
4.7. Immunological Significance of IL-37 in Leprosy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yoshimoto, T.; Yoshimoto, T. (Eds.) Introduction. In Cytokine Frontiers: Regulation of Immune Responses in Health and Disease; Springer: London, UK, 2013; ISBN 978-4-431-54442-5. [Google Scholar]
- Arseniy, E.Y.; Anton, G.K. Interleukins in Cancer Biology: Their Heterogeneous Role; Elsevier: London, UK; Academic Press: Cambridge, MA, USA, 2014; ISBN 9780128013335. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Kaiser, C.A.; Krieger, M.; Scott, M.P.; Bretscher, A.; Ploegh, H.; Matsudaira, P. Molecular Biology of the Cell, 6th ed.; W. H. Freeman: New York, NY, USA, 2000; ISBN-13: 978-0716776017. [Google Scholar]
- Venkatesha, S.H.; Dudics, S.; Acharya, B.; Moudgil, K.D. Cytokine-Modulating Strategies and Newer Cytokine Targets for Arthritis Therapy. Int. J. Mol. Sci. 2015, 16, 887–906. [Google Scholar] [CrossRef] [PubMed]
- Duque, A.G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef]
- Ning, X.; Jian, Z.; Wang, W. Low Serum Levels of Interleukin 35 in Patients with Rheumatoid Arthritis. Tohoku J. Exp. Med. 2015, 237, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, M.; Uyemura, K.; Deans, R.J.; Weinberg, K.; Rea, T.H.; Bloom, B.R.; Modin, R.L. Defining protective responses to pathogens: Cytokine profiles in leprosy lesions. Science 1991, 254, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Moraga, I.; Spangler, J.; Mendoza, J.L.; Garcia, C.K. Multifarious determinants of cytokine receptor signaling specificity. Adv. Immunol. 2014, 121, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Cohen, I. Biologics for Targeting Inflammatory Cytokines, Clinical Uses and Limitations. Int. J. Cell Biol. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
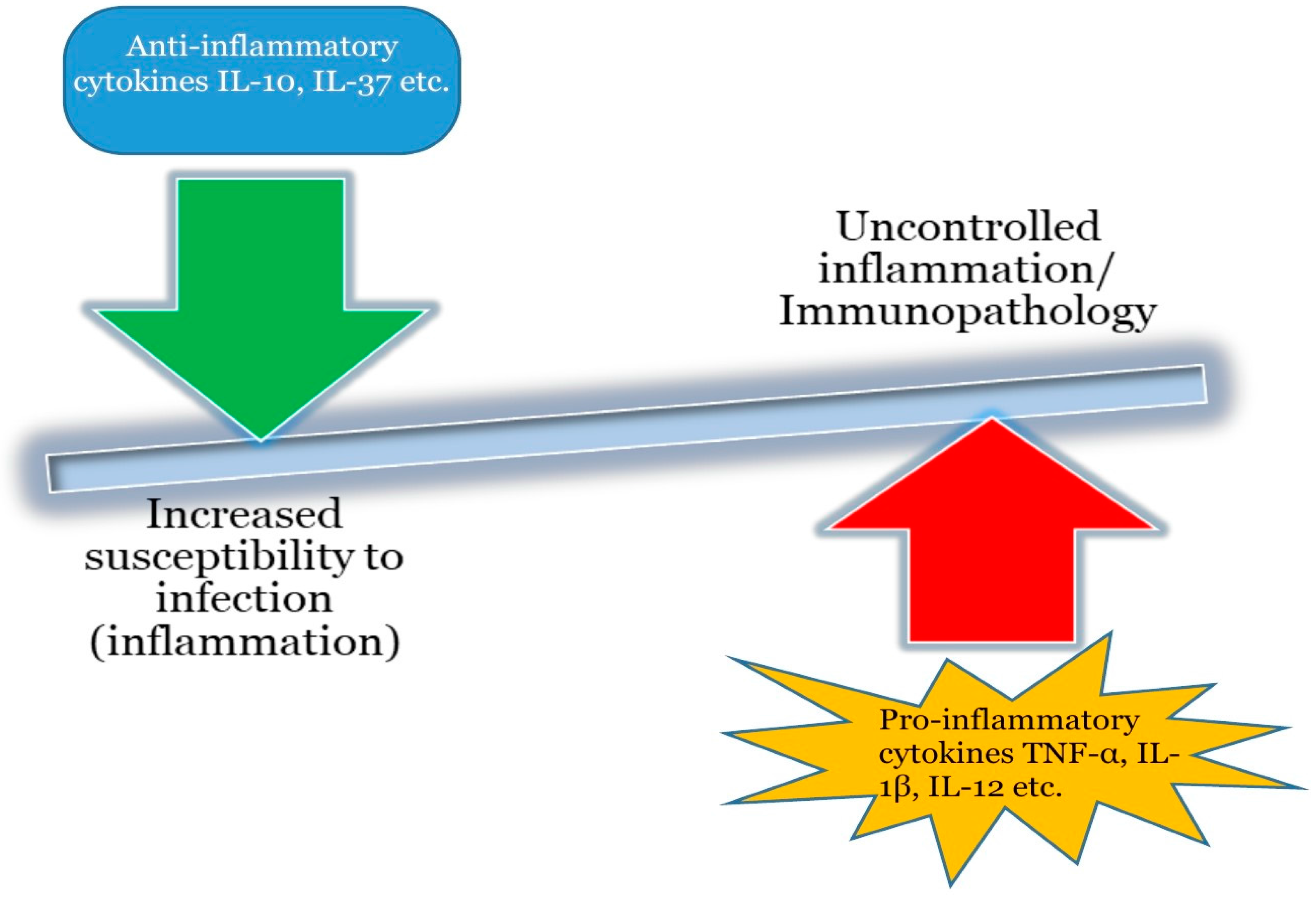
- Warrington, R.; Watson, W.; Kim, H.L.; Antonetti, F.R. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2011, 7, S1. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Bachmann, F.M.; Marsland, J.B. Averting inflammation by targeting the cytokine environment. Nat. Rev. Drug Discov. 2010, 9, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Hamilton, K.; Vignali, D.A.A. Interleukin-35: Expanding Its Job Profile. J. Interferon Cytokine Res. 2015, 35, 499–512. [Google Scholar] [CrossRef] [PubMed]
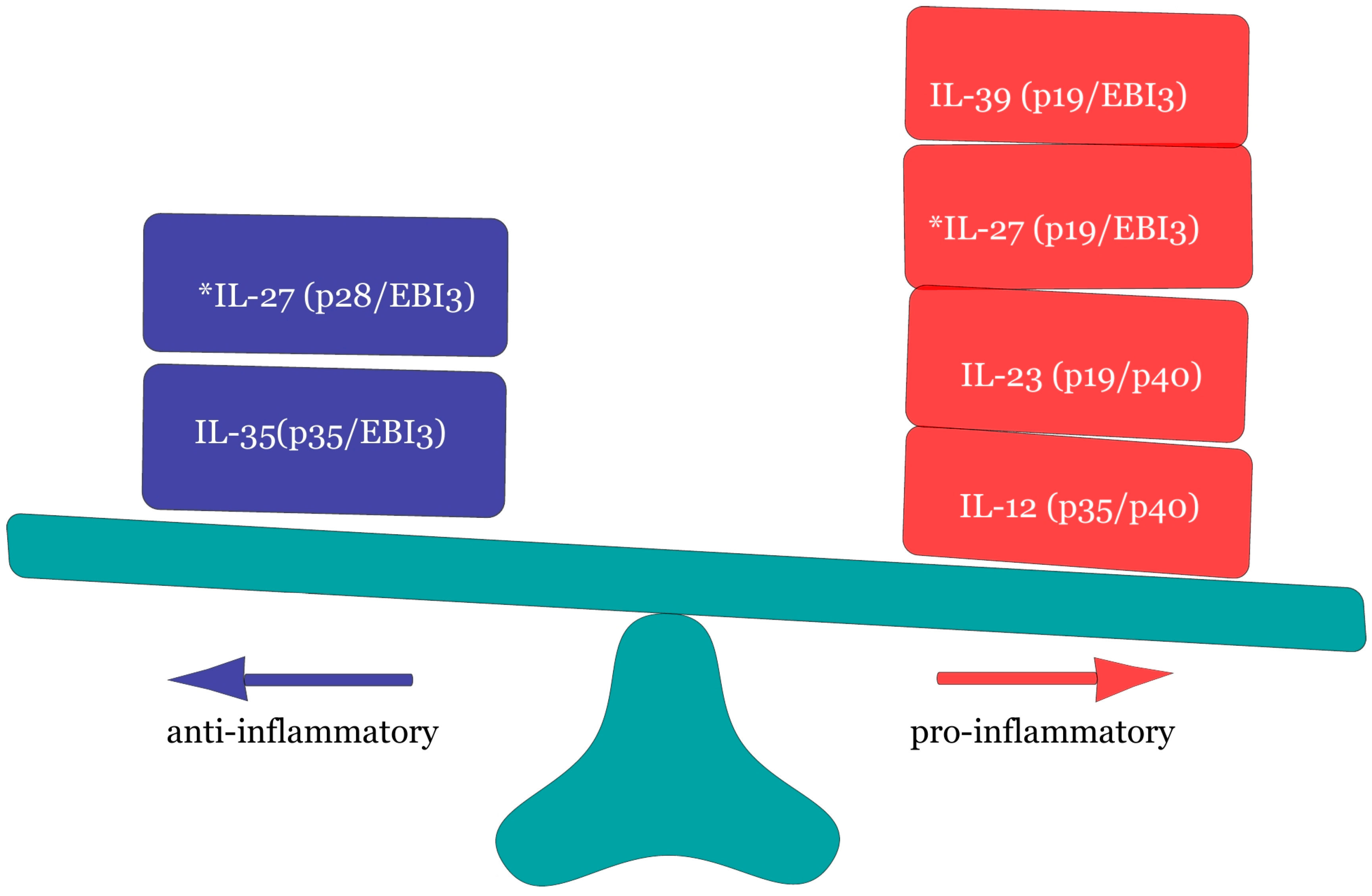
- Hasegawa, H.; Mizoguchi, I.; Chiba, Y.; Ohashi, M.; Xu, M.; Yoshimoto, T. Expanding Diversity in Molecular Structures and Functions of the IL-6/IL-12 Heterodimeric Cytokine Family. Front. Immunol. 2016, 7, 479. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Grohmann, U. Bioengineering heterodimeric cytokines: Turning promiscuous proteins into therapeutic agents. Biotechnol. Genet. Eng. Rev. 2013, 29, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Egwuagu, C.E.; Yu, C.R.; Sun, L.; Wang, R. Interleukin 35: Critical regulator of immunity and lymphocyte-mediated diseases. Cytokine Growth Factor Rev. 2015, 26, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Devergne, O.; Hummel, M.; Koeppen, H.; Le Beau, M.M.; Nathanson, E.C.; Kieff, E.; Birkenbach, M. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J. Virol. 1996, 70, 1143–1153. [Google Scholar] [PubMed]
- Song, M.; Ma, X. The Immunobiology of IL-35 and its Regulation and Gene Expression. Adv. Exp. Med. Biol. 2016, 941, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mai, J.; Virtue, A.; Yin, Y.; Gong, R.; Sha, X.; Gutchigian, S.; Frisch, A.; Hodge, I.; Jiang, X.; et al. IL-35 is a novel responsive anti-inflammatory cytokine—A new system of categorizing anti-inflammatory cytokines. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Huan, E.; Hunter, C.A. Understanding the pro- and anti-inflammatory properties of IL-27. J. Immunol. 2004, 173, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Siegmund, S.; Gerbers, C. The biology of interleukin-27 reveals unique pro and anti-inflammatory functions in immunity. Cytokine Growth Factor Rev. 2015, 26, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Gao, W.; Ma, C.; Sun, J.; Liu, J.; Shao, Q.; Song, B.; Qu, X. Human placental trophoblasts express the immunosuppressive cytokine IL-35. Hum. Immunol. 2013, 74, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Leung, P.S.; Bowlus, C.; Gershwin, M.E. IL-35 and Autoimmunity: A Comprehensive Perspective. Clin. Rev. Allergy Immunol. 2015, 49, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Sha, X.; Meng, S.; Li, X.; Xi, H.; Maddaloni, M.; Pascual, D.W.; Shan, H.; Jiang, X.; Wang, H.; Yang, X.-F. Interleukin-35 Inhibits Endothelial Cell Activation by Suppressing MAPK-AP-1 Pathway. J. Biol. Chem. 2015, 290, 19307–19318. [Google Scholar] [CrossRef] [PubMed]
- Xinyuan, L.; Pu, F.; William, Y.Y.; Hong, W.; Xiaofeng, Y. IL-35, as a newly proposed homeostasis-associated molecular pattern, plays three major functions including anti-inflammatory initiator, effector and blocker in cardiovascular diseases. Cytokine 2017, 6, 003. [Google Scholar] [CrossRef]
- Collison, L.W.; Vignali, D.A.A. Interleukin-35: Odd one out or part of the family? Immunol. Rev. 2008, 226, 248–262. [Google Scholar] [CrossRef] [PubMed]
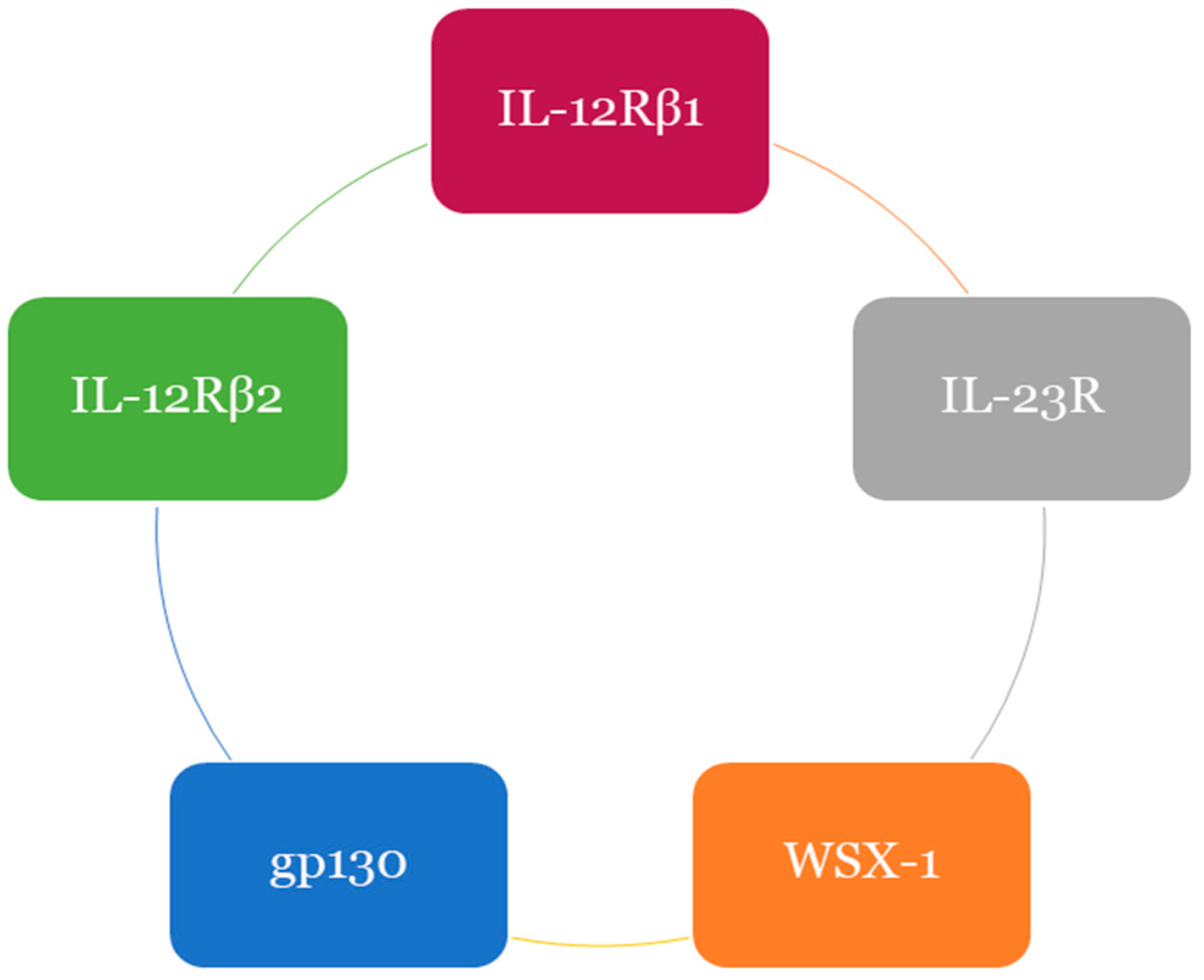
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, R.A.; Garcia, C.K.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Hunter, C.A. Review gp130 at the nexus of inflammation, autoimmunity and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.M.; Sullivan, J.A.; Burlingham, W.J. Interleukin 35: A Key Mediator of Suppression and the Propagation of Infectious Tolerance. Front. Immunol. 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, X.; Gadina, M.; O’Shea, J.J.; Presky, D.H.; Magram, J. IL-12 receptor β 2 (IL-12Rβ2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J. Immunol. 2000, 165, 6221–6228. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, D.V.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.G.; Haribhai, D.; Williams, J.B.; Aggarwal, P.; Jia, S.; Charbonnier, L.M.; Yan, K.; Lorier, R.; Turner, A.; Ziegelbauer, J.; et al. IL-10 produced by iTreg cells controls colitis and pathogenic ex-iTreg cells during immunotherapy. J. Immunol. 2012, 189, 5638–5648. [Google Scholar] [CrossRef] [PubMed]
- Niedbala, W.; Wei, X.Q.; Cai, B.; Hueber, A.; Leung, B.P.; McInnes, I.B.; Liew, F.Y. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur. J. Immunol. 2007, 37, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekara, S. The treatment strategies of autoimmune disease may need a different approach from conventional protocol. A review. Indian J. Pharmacol. 2012, 44, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, P. Cytokines and Chemokines in Autoimmune Disease: An Overview. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2000–2013; Available online: https://www.ncbi.nlm.nih.gov/books/NBK6453 (accessed on 21 December 2017).
- Kochetkova, I.; Golden, S.; Holderness, K.; Callis, G.; Pascual, D.W. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J. Immunol. 2010, 184, 7144–7153. [Google Scholar] [CrossRef] [PubMed]
- Shams, R.; Ali, E.M. IL-35 and IL-10 dependent immunoregulatory lymphocytes in Rheumatoid Arthritis. Sci. J. Immunol. Immunother. 2017, 2, 11–17. [Google Scholar]
- Clavel, G.; Bessis, N.; Boissier, M.C. Recent data on the role for angiogenesis in rheumatoid arthritis. Joint Bone Spine 2003, 70, 321–326. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Y.; Lin, T.; Yuan, L.; Li, Y.; Wu, S.; Xia, L.; Shen, H.; Lu, J. IL-35 inhibits Angiogenesis through VEGF/Ang2/Tie2 Pathway in Rheumatoid Arthritis. Cell Physiol. Biochem. 2016, 40, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse Models of Multiple Sclerosis: Experimental Autoimmune Encephalomyelitis and Theiler’s Virus-Induced Demyelinating Disease. Methods Mol. Biol. 2012, 900, 381–401. [Google Scholar] [CrossRef] [PubMed]
- Ajjan, R.A.; Weetman, A.P. The pathogenessis of Hashimotos Thyroiditis: Further Developments in our understanding. Horm. Metab. Res. 2015, 47, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, H.; Cakmak, M.; Ceydilek, B.; Demir, C.; Aktas, A. Role of interleukin-35 as a biomarker in patients with newly diagnosed Hashimoto’s Thyroidtis. Endocr. Regul. 2016, 50, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kadesjo, E.; Lindroos, J.; Hjort, M.; Lundberg, M.; Espes, D.; Carlsson, P.O.; Sandler, S.; Thorvaldson, L. Interleukin-35 administration counteracts established murine type 1 diabetes—Possible involvement of regulatory T cells. Sci. Rep. 2015, 5, 12633. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Liu, Y.; Wang, Y.; Zuo, X.; Li, Y.; Lu, X. The Possible Role of the Novel Cytokines IL-35 and IL-37 in Inflammatory Bowel Disease. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Camarillo, F.G.; Carballeda, F.J.; Yamamoto-Furusho, J.K. Interleukin 35 (IL-35) and IL-37: Intestinal and peripheral expression by T and B regulatory cells in patients with Inflammatory Bowel Disease. Cytokine 2015. [Google Scholar] [CrossRef]
- Barnes, P.J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Investig. 2008, 118, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Park, A.M.; Yoshida, H.; Tsunoda, I.; Yoshie, O. IL-35 suppresses Lipopolysaccharide induced airway eosinophilia in Ebi3-deficient mice. J. Immunol. 2017, 198, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Faffe, S.D.; Flynt, L.; Mellema, M.; Moore, E.P.; Silverman, S.E.; Subramaniam, V.M.; Jones, R.M.; Mizgerd, J.P.; Whitehead, T.; Imrich, A.; et al. Oncostatin M causes eotaxin-1 release from aairway smooth muscle: Synergy with IL-4 and IL-13. J. Allergy Clin. Immunol. 2005, 115, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Yokata, M.; Nakaamura, Y.; Ozaki, S.; Murakami, S. Intranasal administration of IL-35 inhibits allergic responses and symptoms in mice with allergic rhinitis. Allergol. Int. 2017, 66, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Deng, Y.; Chen, H.; Wu, X.; Cheng, S.; Xu, Y.; Xiong, W.; Xie, J. Decreased concentration of IL-35 in plasma of patients with asthma and COPD. Asian Pac. J. Allergy Immunol. 2014, 32, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.K. Mechanisms of disease: Inflammation, atherosclerosis and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.R.; Hernandez, P.N.; Martines, A.J.; Bautista, L.; Molina, V.T.; Rodriguez-Perez, M.J.; Fragoso, M.J.; Posadas-Romero, C.; Vargas-Alarcon, C. Interleukin 35 polymorphisms are associated with decreased risk of premature coronary artery disease, metabolic parameters and IL-35 levels: The Genetics of Atherosclerotic Disease (GEA) Study. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, Y.; Lu, Z.; Luo, C.; Shi, Y.; Zeng, Q.; Cao, Y.; Liu, L.; Wang, X.; Ji, Q. Decreased plasma IL-35 are related to the Left Ventricular Ejection Fraction in Coronary Artery Diseases. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.; McGuire, F.; Nagarkatti, P.; Nagarkatti, M. Role of cytokines as a double edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar]
- Bosman, M.; Russkamp, N.F.; Strobl, B.; Roewe, J.; Balouzian, L.; Pache, F.; Radsak, M.P.; van Rooijen, N.; Zetoune, F.S.; Sarma, J.V.; et al. Interruption of macrophage-derived IL-279p(28) production by IL-10 during sepsis requires STAT3 but not SOCS3. J. Immunol. 2014, 193, 5668–5677. [Google Scholar] [CrossRef] [PubMed]
- Du, W.X.; He, Y.; Jiang, H.Y.; Ai, Q.; Yu, J.L. Interleukin 35: A novel candidate biomarker to diagnose early onset sepsis in neonates. Clin. Chim. Acta 2016, 462, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Tomcik, M.; Zerr, P.; Plumbo-Zerr, K.; Storkanova, H.; Hulejova, H.; Spiritnovic, M.; Kodett, O.; Stork, J.; Becva, R.; Vencovsky, J.; et al. Interleukin-35 is upregulated in systemic sclerosis and its serum levels are associated with early disease. Rheumatology 2015, 54, 2273–2282. [Google Scholar] [CrossRef] [PubMed]
- Dantas, A.T.; Gonclaves, S.M.; Pereira, M.C.; Gonclaves, R.S.; Marques, C.D.; Rego, M.J.; Pitta, I.R.; Duarte, A.L.; Pitta, M.G. Increased IL-35 serum levels in systemic sclerosis and association with pulmonary interstitial involvement. J. Clin. Rheumatol. 2015, 34, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wong, C.K.; Dong, J.; Chu, M.; Jiao, D.; Kam, N.W.; Lam, C.W.K.; Tam, L.S. Remission of systemic lupus erythematosus disease activity with regulatory cytokine interleukin (IL)-35 in Murphy Roths Large (MRL)/lpr mice. Clin. Exp. Immunol. 2015, 181, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Liu, C.-J.; Su, T.-H.; Fang, Y.-J.; Yang, H.-C.; Chen, P.-J.; Chen, D.-S.; Kao, J.-H. Hepatitis B virus Reactivation in Patients Receiving Interferon-free Direct-Acting Antiviral Agents for Chronic Hepatitis C virus Infection. Open Forum Infect. Dis. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dai, M.; Wu, G.; Zhou, P.; Fang, Y.; Yan, X.B. Levels of Interleukin-35 and its relationship with regulatory T-cells in chronic Hepatitis B patients. Viral Immunol. 2015, 28, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A.A. Interleukin-35 limits anti-tumor immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in Cancer immunotherapy. Curr. Opin. Immuol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, J.-Q.; Liu, Z.; Shen, R.; Zhang, G.; Xu, J.; Basau, S.; Bai, X.-F. Tumor-derived IL-35 promotes tumor growth by enhancing myeloid cell accumulation and angiogenesis. J. Immunol. 2013, 190, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Regulatory T cells and immune tolerance to tumors. Immunol. Res. 2010, 46, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Borsellino, N.; Bonavida, B.; Ciliberto, G.; Toniatti, C.; Travali, S.; D’Alessandro, N. Blocking signaling through the Gp130 receptor chain by interleukin-6 and oncostatin M inhibits PC-3 cell growth and sensitizes the tumor cells to etoposide and cisplatin-mediated cytotoxicity. Cancer 1999, 85, 134–144. [Google Scholar] [CrossRef]
- Zeng, J.-C.; Zhang, Z.; Li, T.-Y.; Liang, Y.-F.; Wang, H.-M.; Bao, J.-J.; Zhang, J.-A.; Wang, W.-D.; Xiang, W.-Y.; Kong, B.; et al. Assessing the role of IL-35 in colorectal cancer progression and prognosis. Int. J. Clin. Exp. Pathol. 2013, 6, 1806–1816. [Google Scholar] [PubMed]
- Zhang, Y.; Sun, H.; Wu, H.; Tan, Q.; Xiang, K. Interleukin 35 is an independent prognostic factor and a therapeutic target for nasopharyngeal carcinoma. Contemp. Oncol. 2015, 119, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Nishino, R.; Takano, A.; Oshita, H.; Ishikawa, N.; Akiyama, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; Tsuchiya, E.; Kohno, N.; et al. Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin. Cancer Res. 2011, 17, 6272–6286. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Guo, H.; Cui, S.; Zhang, H.; Liu, X.; Li, D.; Ding, Y. IL-35 expression in hepatocellular carcinoma cells is associated with tumor progression. Oncotarget 2017, 7, 45678–45686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mao, T.; Wang, S.; Wang, D.; Niu, Z.; Sun, Z.; Zhang, J. Inteleukin-35 expression is associated with colon cancer progression. Oncotarget 2017, 8, 71563–71573. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wu, C.; Wang, Y.; He, X.; Zhao, N. Cytokine profiles in papillary thyroid carcinoma, with or without Hashimoto’s thyroiditis. Eur. J. Inflamm. 2017, 1–5. [Google Scholar] [CrossRef]
- Liao, K.L.; Bai, X.F.; Friedman, A. Mathematical Modeling of Interleukin-35 Promoting Tumor Growth and Angiogenesis. PLoS ONE 2014, 9, e110126. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Netea, M.G. New insights in the immunobiology of IL-1 family members. Front. Immunol. 2013, 4, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abulkhir, A.; Samarani, S.; Amre, D.; Duval, M.; Haddad, E.; Sinnett, D.; Leclerc, J.; Diorio, C.; Ahmad, A. A protective role of IL-37 in cancer: A new hope for cancer patients. J. Leukoc. Biol. 2016, 101, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Nold, M.F.; Nold-petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, M.A.; Nold-Petry, C.A.; Laura, D.; Cho, S.X.; Lao, C.J.; Rudloff, I.; Ngo, D.; Lo, C.Y.; Soares da Costa, T.P.; Perugini, M.A.; et al. Homodimerization attenuates the anti-inflammatory activity of interleukin-37. Sci. Immunol. 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 6, 685–700. [Google Scholar] [CrossRef]
- Taylor, S.L.; Renshaw, B.R.; Garka, K.E.; Smith, D.E.; Sims, J.E. Genomic organization of the interleukin-1 locus. Genomics 2002, 79, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Quirk, S.; Agrawal, D.K. Immunobiology of Interleukin-37: Mechanism of Action and Clinical Perspectives. Expert Rev. Clin. Immunol. 2014, 10, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Nold-petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.D.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18R-α and IL-1R8 (SIGGIR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Lucchesi, D.; Hainzl, S.; Leitner, M.; Maier, E.; Mangelberger, D.; Oostingh, G.J.; Pfaller, T.; Pixner, C.; Posselt, G.; et al. IL-37: A new anti-inflammatory cytokine of the IL-1 family. Eur. Cytokine Netw. 2011, 22, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Bulau, A.-M.; Fink, M.; Maucksch, C.; Kappler, R.; Mayr, D.; Wagner, K.; Buffler, P. In vivo expression of interleukin-37 reduces local and systemic inflammation in concanavalin A-induced hepatitis. Sci. World J. 2011, 11, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Hanning, C.R.; Brigham-Burke, M.R.; Rieman, D.J.; Lehr, R.; Khandekar, S.; Kirkpatrick, R.B.; Scott, G.F.; Lee, J.C.; Lynch, F.J.; et al. Interleukin-1F7b is processed by caspase-1 and mature IL-1F7b binds to the IL-18 receptor but does not induce IFN-γ production. Cytokine 2002, 2, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Nold, M.F.; Nielsen, J.W.; Bustamante, A.; Zepp, J.A.; Storm, K.A.; Hong, J.-W.; Kim, S.-H.; Dinarello, C.A. Increased cytokine production in interleukin-18 receptor α-deficient cells is associated with dysregulation of suppressors of cytokine signaling. J. Biol. Chem. 2009, 284, 25900–25911. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Niel, L.; Goldbach-Mansky, R.; Pizzarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 family nomenclature. Nat. Immunol. 2010, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Rivera-Nieves, J. Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [PubMed]
- Hu, D. Role of anti-inflammatory cytokines IL-35 and IL-37 in Asthma. Int. J. Inflamm. 2017, 40, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Jiang, B.; Deng, J.; Du, J.; Xiong, W.; Guan, Y.; Wen, Z.; Huang, K.; Huang, Z. IL-37 Alleviates Rheumatoid Arthritis by Suppressing IL-17 and IL-17–Triggering Cytokine Production and Limiting Th17 Cell Proliferation. J. Immunol. 2015, 194, 5110–5119. [Google Scholar] [CrossRef] [PubMed]
- Ballak, D.B.; Van Diepen, J.A.; Moschen, A.R.; Jansen, H.J.; Hijmans, A.; Groenhof, G.J.; Leenders, F.; Bufler, P.; Boekschoten, M.V.; Müller, M.; et al. IL-37 protects against obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, G.; Justice, J.N.; Boyle, K.E.; D’Alessandro, A.; Eissenmsser, E.Z.; Jonathan, J.H.; Hansen, K.C.; Nemkov, T.; Stienstra, R.; Garlanda, C.; et al. Interleukin 37 reverses the metabolic cost of inflammation, increases oxidative respiration and improves exercise tolerance. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Kumar, S.; Lotze, M.T.; Hanning, C.; Robbins, P.D.; Gambotto, A. Innate immunity mediated by the cytokine IL-1 homologue 4 (IL-1H4/IL-1F7) induces IL-12-dependent adaptive and profound antitumor immunity. J. Immunol. 2003, 170, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Pan, Q.Z.; Pan, K.; Weng, D.-S.; Wang, Q.-J.; Li, J.-J.; Lv, L.; Wang, D.-D.; Zheng, H.-X.; Jiang, S.-S.; et al. Interleukin-37 mediates the antitumor activity in hepatocellular carcinoma: Role for CD57+ NK cells. Sci. Rep. 2014, 4, 5177–5178. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, D.; Zhou, Y.; Hu, X.; Sun, Z.; Yu, G.; Wu, W.-T.; Chen, S.; Kuang, J.-L.; Xu, G.-G.; et al. Transfer of the IL-37b gene elicits anti-tumor responses in mice bearing 4T1 breast cancer. Acta Pharmacol. Sin. 2015, 36, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Shu, Y.; Luo, J.; Liu, D.; Huang, D.S.; Han, Y.; Chen, C.; Li, Y.C.; Zou, J.M.; Qin, J.; et al. Intracellular IL-37b interacts with Smad3 to suppress multiple signaling pathways and the metastatic phenotype of tumor cells. Oncogene 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Datta, P.K. Alterations in the Smad pathway in human cancers. Front. Biosci. 2012, 17, 1281–1293. [Google Scholar] [CrossRef]
- Kaabachi, W.; Kacem, O.; Belhaj, R.; Hamzaoui, A.; Hamzaoui, K. Interleukin-37 in Endometriosis. Immunol. Lett. 2017, 185, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.S.; Shanaham, C. Vascular calcification and hypertension: Cause and effect. Ann. Med. 2012, 44, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, O. Cross talk between the bone and immune system: Osteoclasts function as antigen presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Madhava, V.M.; Tarigopula, M.; Mintz, S.G.; Maehara, A.; Stone, W.G.; Genereuk, P. Coronary artery calcification: Pathogenesis and prognostic implications. J. Am. Coll. Cardiol. 2014, 63, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Ji, Q.; Zhang, H.; Zhou, Y.; Yang, Q.; Zhou, Y.; Guo, G.; Liu, W.; Han, W.; Yang, L.; et al. The protective effect of interleukin-37 on vascular calcification and atherosclerosis in Apolipoprotein E-deficient mice with diabetes. J. Interferon Cytokine Res. 2015, 35, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, A.; Jiang, F.; Hu, J.; Zhang, X. Effects of interleukin-37 on cardiac function after myocardial infarction in mice. Int. J. Clin. Exp. Pathol. 2015, 8, 5247–5251. [Google Scholar] [PubMed]
- Krueger, J.M.; Walter, J.; Dinarello, C.A.; Wolff, S.M.; Chedid, L. Sleep promoting effects of endogenous pyrogen (Interleukin-1). Am. J. Physiol. 1984, 246, R994–R999. [Google Scholar] [CrossRef] [PubMed]
- Jewett, K.A.; Taishi, P.; Sengupta, P.; Roy, S.; Davis, C.J.; Krueger, J.M. Tumor necrosis factor enhances the sleep-like state and electrical stimulation induces a wake like state in co-cultures of neurons and glia. Eur. J. Neurosci. 2015, 42, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.J.; Zielinski, M.R.; Dunbrasky, D.; Taishi, P.; Dinarello, C.A.; Krueger, J.M. Interleukin 37 expression in mice alters sleep responses to inflammatory agents and influenza virus infection. Neurobiol. Sleep Circadian Rhythm. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Degang, Y.; Nakamura, K.; Akama, T.; Ishido, Y.; Lu, Y.; Ishi, N.; Suzuki, K. Leprosy as a model of immunity. Future Microbiol. 2014, 9, 43–54. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, R.J.; Prudente, R.R.; Dias Junior, L.B.; Oliveira Carneiro, F.R.; Sotto, M.N.; Simoes Quaresma, J.A. IL-37 and Leprosy: A novel cytokine involved in the host response to Mycobacterium Leprae infection. Cytokine 2017, S1043–S4666. [Google Scholar] [CrossRef] [PubMed]
- Shuai, X.; Wei-min, L.; Tong, Y.L.; Dong, N.; Sheng, Z.Y.; Yao, Y.M. Expression of IL-37 contributes to immunosuppressive property of human CD4+, CD25+ regulatory T cells. Sci. Rep. 2015, 5, 14478. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Disorder | Biological Action | References |
|---|---|---|---|
| 1. | Rheumatoid arthritis | ↑ Promotes Treg expansion | [6,35,38] |
| ↑ Teff cell suppression | |||
| ↓ TH1 differentiation | |||
| ↓ TH17 differentiation | |||
| ↑ IL-10 production | |||
| ↓ angiogenesis | |||
| 2. | Experimental autoimmune encephalomyelitis | ↑ IL-10 producing Bregs | [32,33] |
| ↑ IL-35+ Bregs | |||
| ↓ IFN-γ expression | |||
| ↓ IL-17 expression | |||
| 3. | Experimental autoimmune uveitis | ↑ IL-35+ Breg expansion | [32] |
| ↑ iTr35 expansion | |||
| Inhibition of pathogenic TH17 cells differentiation | |||
| Inhibition of pathogenic TH1 cells differentiation | |||
| 4. | Hashimoto’s thyroiditis | ↑ Treg proliferation (↑ immune tolerance) | [44] |
| ↓ thyroid cell apoptosis/auto destruction of thyroid tissue | |||
| ↓ TPoAb producing B cells | |||
| 5. | Multiple low dose streptozotocin | ↑ IL-35 expression | [45] |
| ↑ IL-10 expression | |||
| ↑ Suppressive potential of Tregs | |||
| 6. | Asthma | ↓ airway CCL11 and CCL24 recruitment | [49] |
| ↓ IL-6 and TH1 cytokine expression | |||
| ↓ STAT1 and STAT3 phosphorylation | |||
| 7. | Allergic rhinitis | ↑ Treg expansion | [51] |
| ↑ IL-10 expression | |||
| ↓ Teff cell proliferation | |||
| ↓ TH2 cytokine expression | |||
| 8. | Cardiovascular diseases | rs2243115 and rs428253 linked (↓) decrease in type-2 diabetes mellitus and metabolic syndrome | [54] |
| 9. | Systemic lupus erythematosus | ↓ TH17 differentiation | [61] |
| ↑ peripheral and thymic IL-10+ Bregs | |||
| 10. | Cancer | ↓ CTL responses | [28,64,65,67,68] |
| ↑ recruitment of CD4+ CD25+ Treg | |||
| ↑ gp130 expression by tumor cells |
| No | Disorder | Actions of IL-37 | References |
|---|---|---|---|
| 1. | Experimental colitis | ↓ TNF-α expression | [91] |
| ↓ IL-1β expression | |||
| ↓ Leukocyte recruitment | |||
| ↑ Amplification of anti-inflammatory responses mediated by TGF-β | |||
| 2. | Asthma | ↓ TH1 cytokine production | [92] |
| ↓ IL-17 production | |||
| ↓ TH2 cytokine expression | |||
| 3. | Collagen induced arthritis | ↓ TH17 cell proliferation | [93] |
| ↓ IL-17 triggering cytokines (viz IL-6, IL-1β, etc.) | |||
| ↑ tolerogenic DCs (↓ DC-MAPK signal transduction) | |||
| 4. | Cancer | ↓ tumor cell invasiveness and metastases | [77,96,99] |
| ↓ Angiogenesis | |||
| ↓ IL-6, IL-8 and MMP-9 | |||
| ↑ NK cell recruitment | |||
| ↑ PTP expression | |||
| ↑ STAT3 inactivation | |||
| 5. | Cardiovascular disorders | ↓ IL-18 expression | [85,104,105,106] |
| ↓ TNF-α expression | |||
| ↓ BMP-2 and ALP expression | |||
| ↑ IL-10 production | |||
| ↑ OPG titers | |||
| 6. | Leprosy | ↓ TNF and IL-1β expression | [110,111,112] |
| ↓ TH17 cell differentiation | |||
| ↑ Treg proliferation | |||
| ↑ IL-10 expression | |||
| ↑ TGF-β1 signaling | |||
| 7. | Sleep | Attenuation of pro-inflammatory cytokine mediated sleep deprivation | [109] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello, R.O.; Chin, V.K.; Abd Rachman Isnadi, M.F.; Abd Majid, R.; Atmadini Abdullah, M.; Lee, T.Y.; Amiruddin Zakaria, Z.; Hussain, M.K.; Basir, R. The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1149. https://doi.org/10.3390/ijms19041149
Bello RO, Chin VK, Abd Rachman Isnadi MF, Abd Majid R, Atmadini Abdullah M, Lee TY, Amiruddin Zakaria Z, Hussain MK, Basir R. The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis. International Journal of Molecular Sciences. 2018; 19(4):1149. https://doi.org/10.3390/ijms19041149
Chicago/Turabian StyleBello, Ramatu Omenesa, Voon Kin Chin, Mohammad Faruq Abd Rachman Isnadi, Roslaini Abd Majid, Maizaton Atmadini Abdullah, Tze Yan Lee, Zainul Amiruddin Zakaria, Mohd Khairi Hussain, and Rusliza Basir. 2018. "The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis" International Journal of Molecular Sciences 19, no. 4: 1149. https://doi.org/10.3390/ijms19041149