Study of Imidazolium Salt Derivatives as PIK3CA Inhibitors Using a Comprehensive in Silico Method

 ,
,
Abstract
:1. Introduction
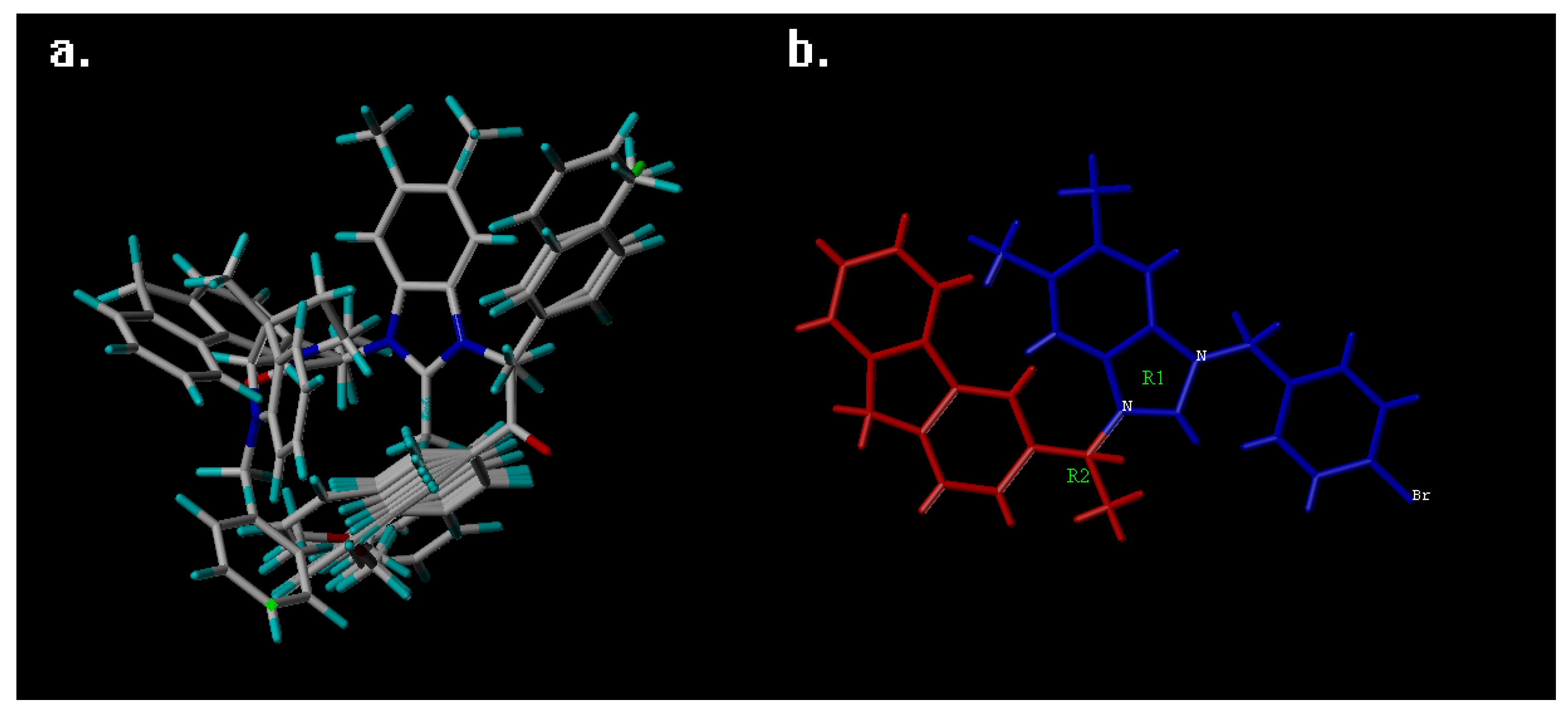
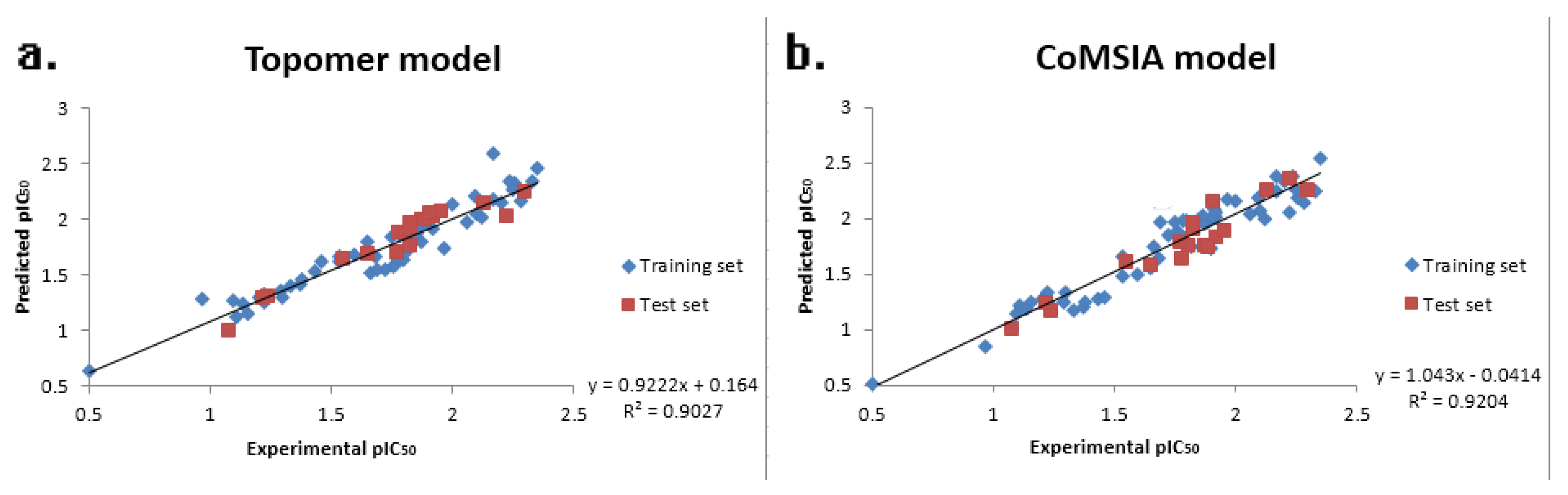
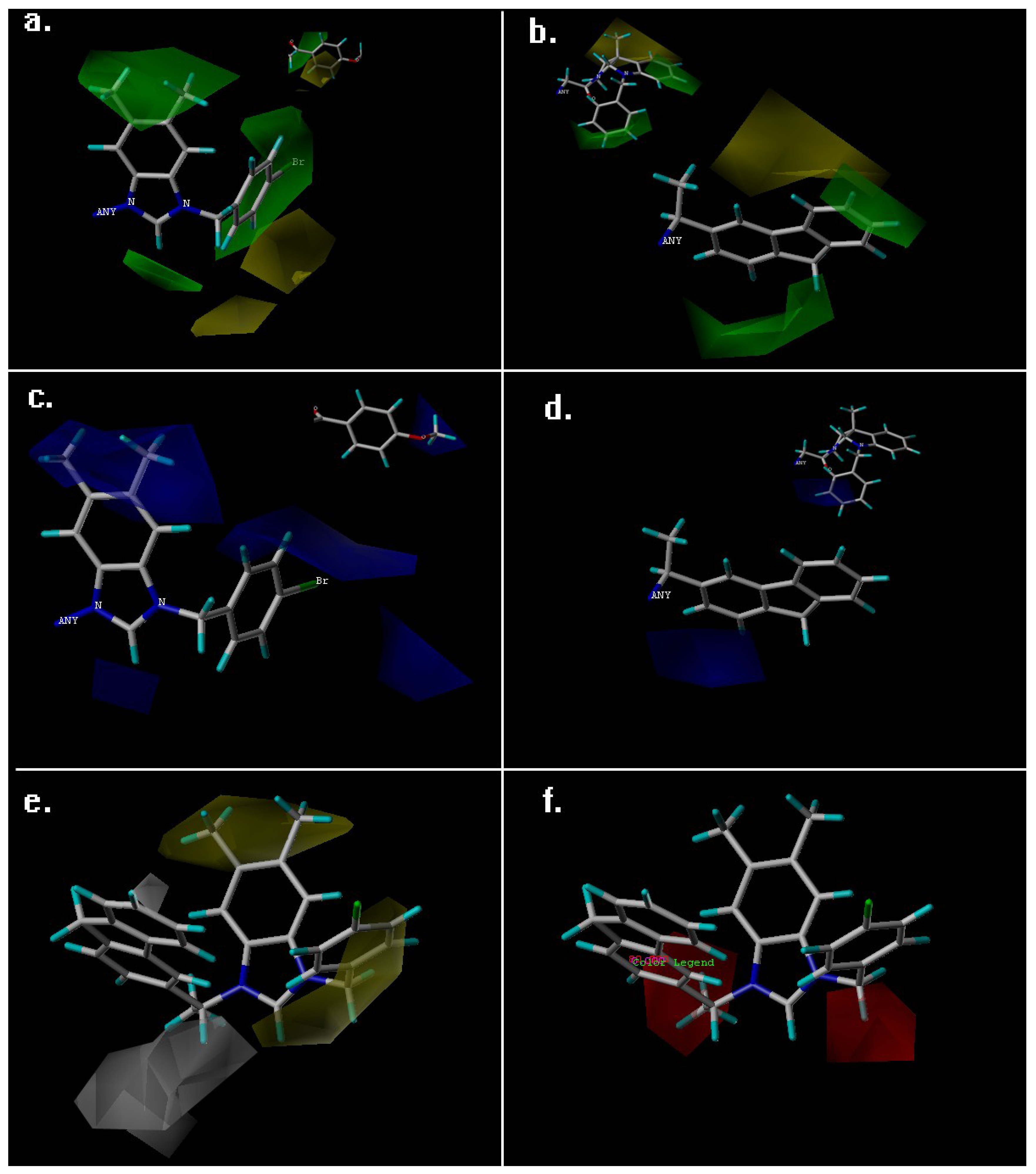
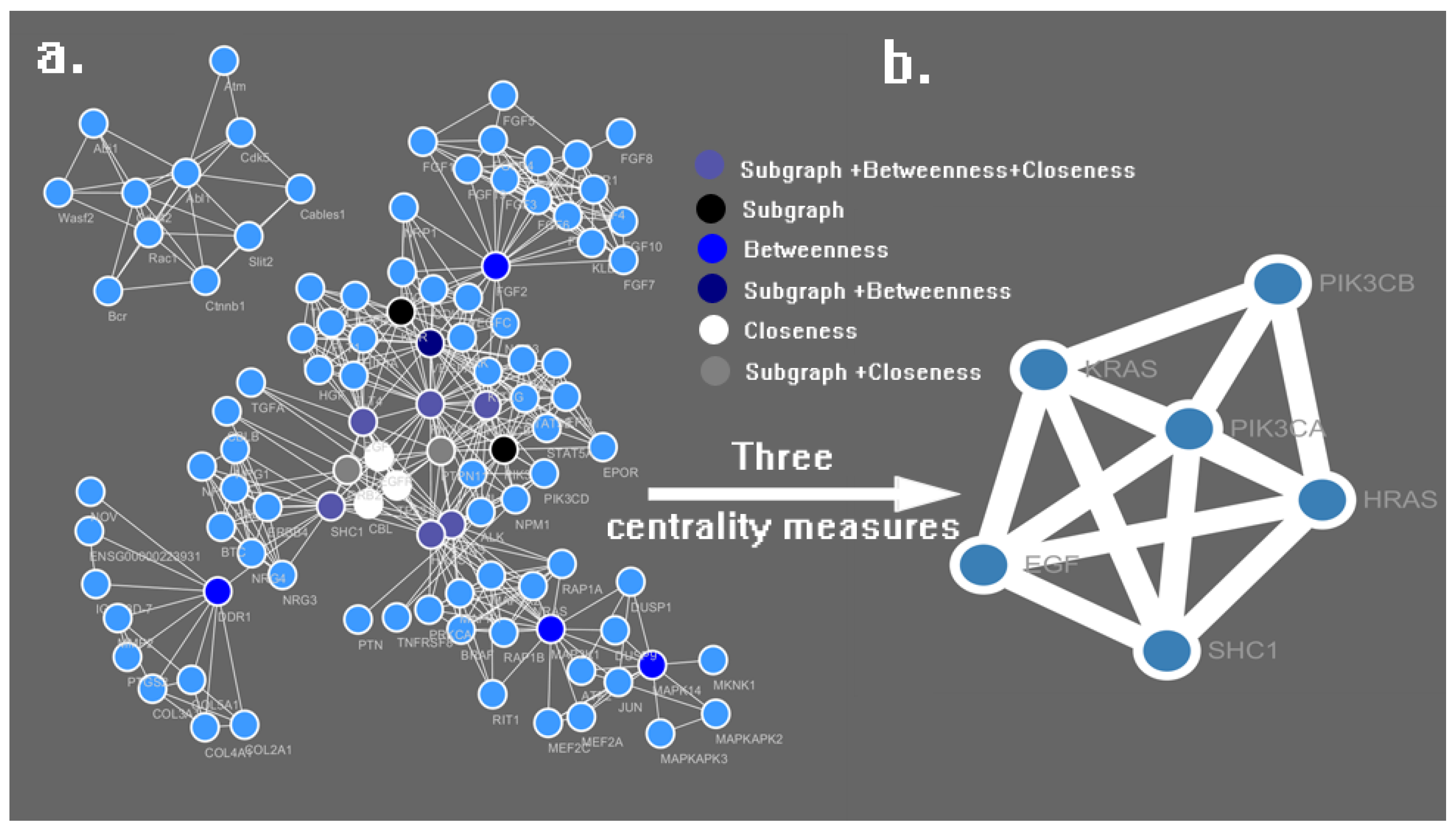
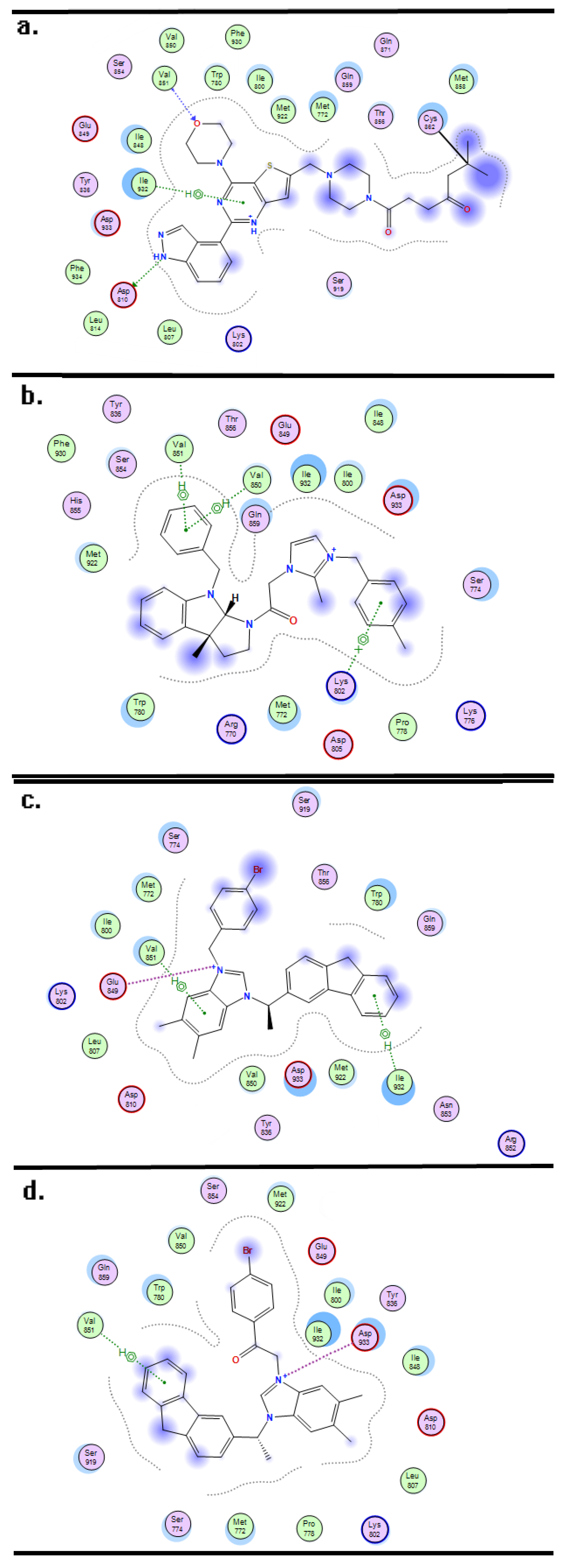
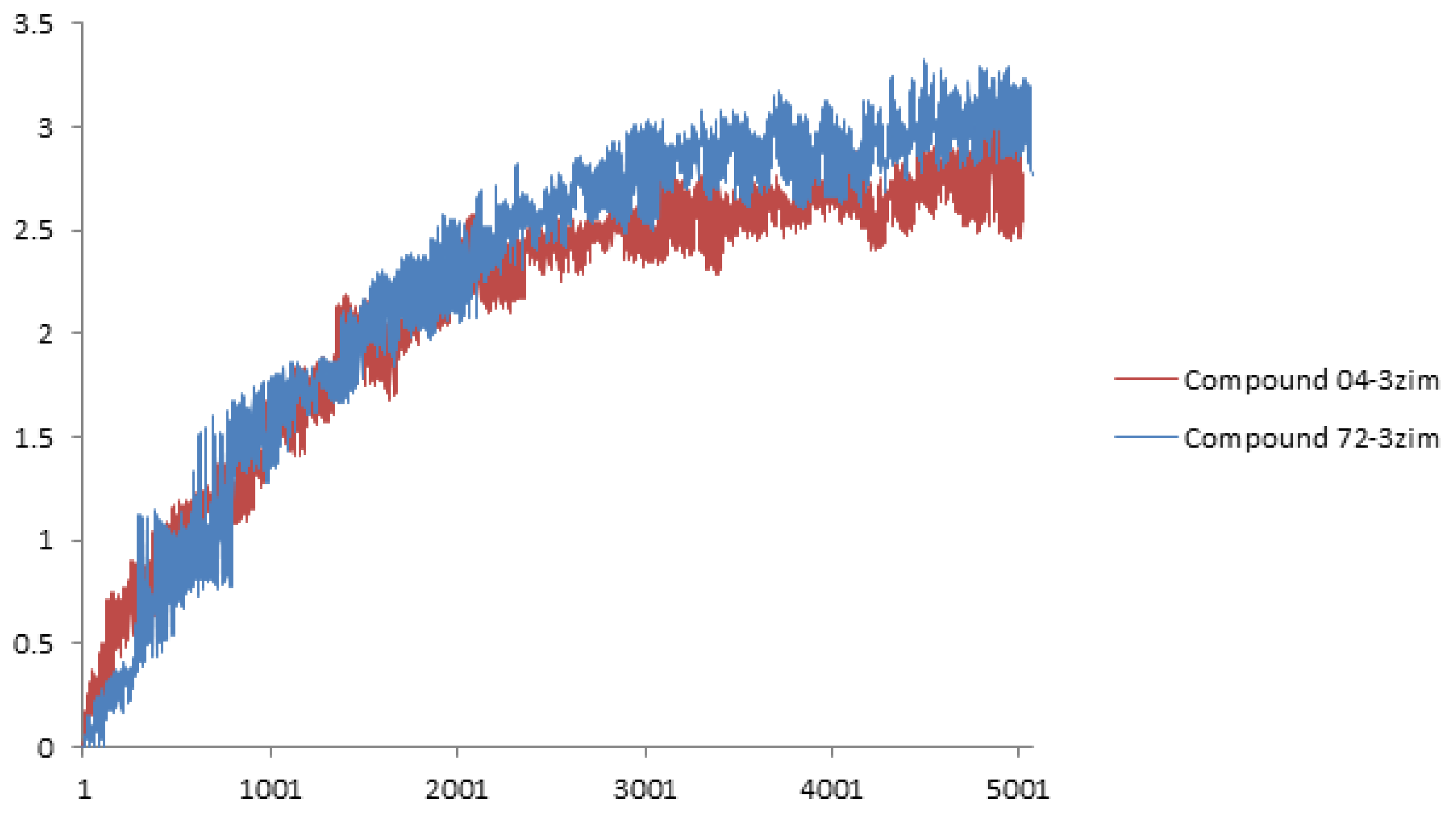
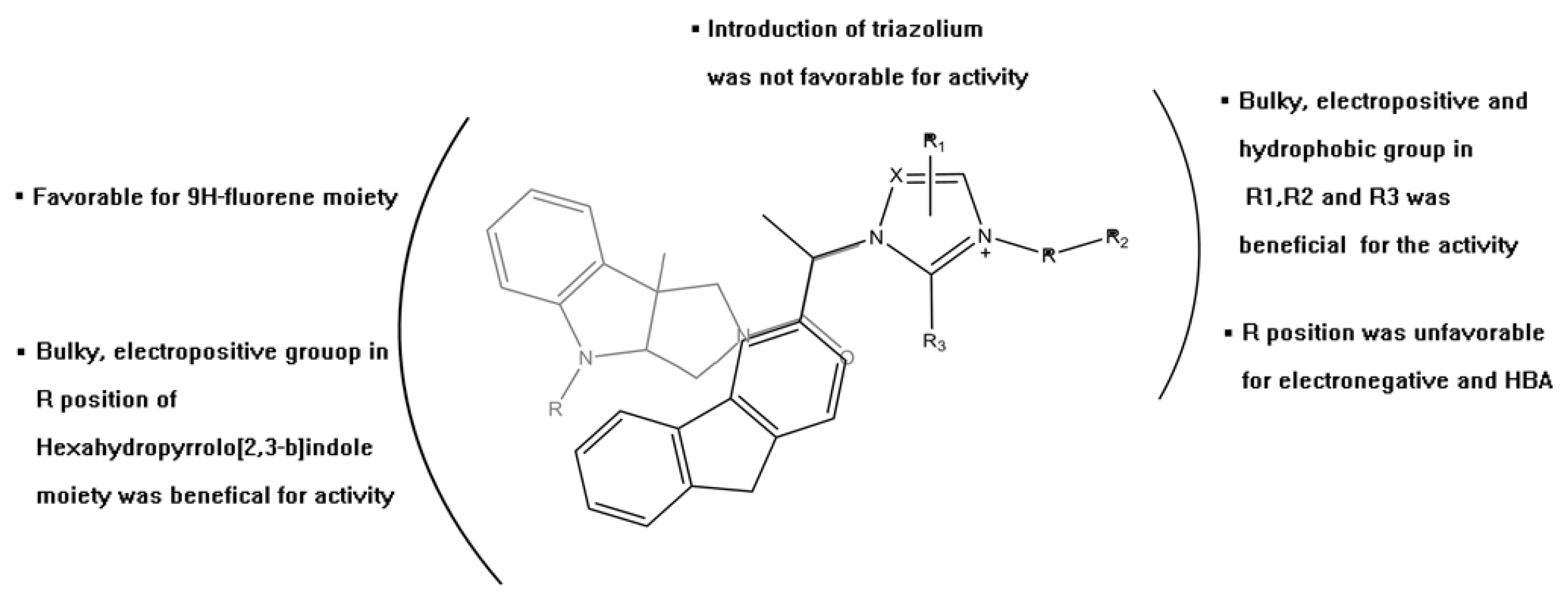
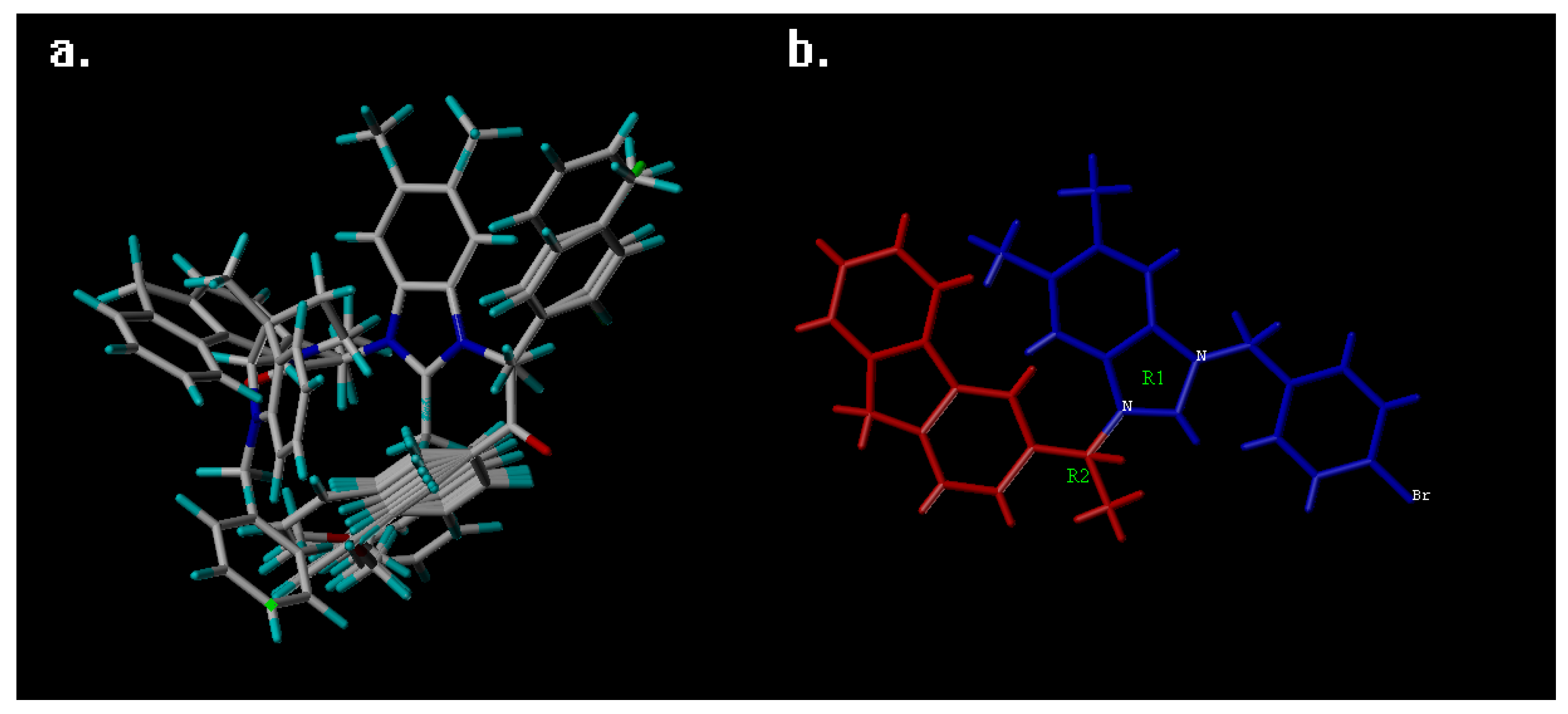
2. Results and Discussion
3. Materials and Methods
3.1. QSAR Study
3.2. PPI Network Construction and Analysis
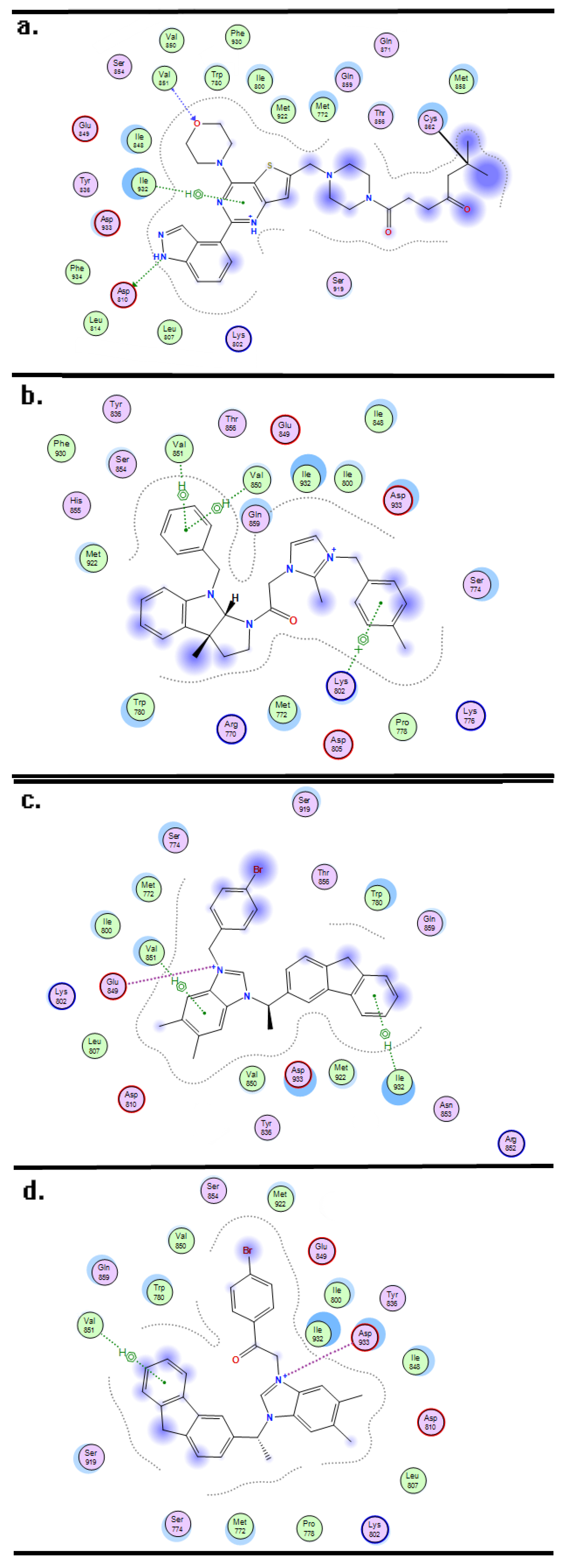
3.3. Molecular Docking and Dynamics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| QSAR | Quantitative Structure-Activity Relationship |
| CoMFA | Comparative of Molecular Field Analysis |
| CoMSIA | Comparative Molecular Similarity Indices Analysis |
| MD | Molecular Dynamics |
References
- Ernst, E.; Cassileth, B.R. The prevalence of complementary/alternative medicine in cancer: A systematic review. Cancer 2015, 83, 777–782. [Google Scholar] [CrossRef]
- Brydøy, M.; Fosså, S.D.; Dahl, O.; Bjøro, T. Gonadal dysfunction and fertility problems in cancer survivors. Acta Oncol. 2007, 46, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Cholewiński, G.; Dzierzbicka, K.; Kołodziejczyk, A.M. Natural and synthetic acridines/acridones as antitumor agents: Their biological activities and methods of synthesis. Pharm. Rep. 2011, 63, 305–336. [Google Scholar] [CrossRef]
- Hijiya, N.; Hudson, M.M.; Lensing, S.; Zacher, M.; Onciu, M.; Behm, F.G.; Razzouk, B.I.; Ribeiro, R.C.; Rubnitz, J.E.; Sandlund, J.T. Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 2007, 297, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.-J.; Nunnensiek, C.; Rüther, U. (Eds.) Secondary Neoplasias Following Chemotherapy, Radiotherapy, and Immunosuppression; Karger: Basel, Switzerland, 2000. [Google Scholar]
- Vadhan-Raj, S. Management of chemotherapy-induced thrombocytopenia: Current status of thrombopoietic agents. Semin. Hematol. 2009, 46, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Caruso, F. Particle carriers for combating multidrug-resistant cancer. ACS Nano 2013, 7, 9512–9517. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the pik3ca gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Xu, X.; Davies, M.A.; Gimotty, P.A.; Morrissette, J.J.; Tetzlaff, M.T.; Wani, K.; Liu, S.; Deng, W.; Buckley, M.; et al. A multicenter phase i study evaluating dual PI3K and BRAF inhibition with PX-866 and vemurafenib in patients with advanced BRAF V600 mutant solid tumors. Clin. Cancer Res. 2017, 545. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lucas, J.; Zhu, T.; Zask, A.; Gaydos, C.; Toralbarza, L.; Gu, J.; Li, F.; Chaudhary, I.; Cai, P.; et al. PWT-458, a novel pegylated-17-hydroxywortmannin, inhibits phosphatidylinositol 3-kinase signaling and suppresses growth of solid tumors. Cancer Biol. Ther. 2005, 4, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.A.; Adnan, I.M.; Khadeer, A.M.B.; Abdul, M.A.; Abdul, H.Z.A. Design, synthesis and structural studies ofmeta-xylyl linkedbis-benzimidazolium salts: Potential anticancer agents against ‘human colon cancer’. Chem. Cent. J. 2012, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.A.; Iqbal, M.A.; Asekunowo, P.; Majid, A.M.S.A.; Ahamed, M.B.K.; Umar, M.I.; Al-Rawi, S.S.; Al-Suede, F.S.R. Synthesis, structure, anticancer, and antioxidant activity of para-xylyl linked bis-benzimidazolium salts and respective dinuclear Ag(I) N-heterocyclic carbene complexes (part-II). Med. Chem. Res. 2013, 22, 4663–4676. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Z.F.; Deng, G.G.; Chen, W.; Li, M.Y.; Yang, L.J.; Li, Y.; Yang, X.D.; Zhang, H.B. Synthesis and antitumor activity of novel n-substituted tetrahydro-β-carboline-imidazolium salt derivatives. Org. Biomol. Chem. 2016, 14, 9423–9430. [Google Scholar] [CrossRef] [PubMed]
- Mustaffa, N.; Leong, R.W.; Katelaris, P.H. Education and imaging. Gastrointestinal: Spigelian hernia; an uncommon cause of longstanding intermittent abdominal pain. J. Gastroenterol. Hepatol. 2013, 28, 202. [Google Scholar] [CrossRef] [PubMed]
- Sweeting, J.G. Bockus gastroenterology, fifth edition. Gastrointest. Endosc. 1995, 42, 498–500. [Google Scholar] [CrossRef]
- Zhao, X.; Ogunwobi, O.O.; Liu, C. Survivin inhibition is critical for Bcl-2 inhibitor-induced apoptosis in hepatocellular carcinoma cells. PLoS ONE 2011, 6, e21980. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Wang, M.; Zhou, Y.J.; Yan, J.M.; Yang, L.J.; Li, Y.; Zhang, H.B.; Yang, X.D. Novel 3-substituted fluorine imidazolium/triazolium salt derivatives: Synthesis and antitumor activity. RSC Adv. 2015, 5, 63936–63944. [Google Scholar] [CrossRef]
- Zhou, Y.; Duan, K.; Liang, Z.; Liu, Z.; Zhang, C.; Yang, L.; Li, M.; Zhang, H.; Yang, X. Synthesis and cytotoxic activity of novel hexahydropyrrolo[2,3-b]indole imidazolium salts. Bioorg. Med. Chem. Lett. 2016, 26, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Moore, M. Anti-Cancer Drug Design; Macmillan Press: London, UK, 1985; p. i3. [Google Scholar]
- Gibbs, J.B. Mechanism-based target identification and drug discovery in cancer research. Science 2000, 287, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Liu, Y.; Zhu, Y.; Liu, Z. Progress of computer-aided drug design (CADD) of proteasome inhibitors. Curr. Top. Med. Chem. 2011, 11, 2931–2944. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. Pharmmapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [PubMed]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Bhattacharyya, D.K.; Kalita, J.K. Centrality analysis in PPI networks. In Proceedings of the 2nd International Conference on Accessibility to Digital World, Assam, India, 27–29 December 2017; pp. 135–140. [Google Scholar]
- Estrada, E.; Rodríguez-Velázquez, J.A. Subgraph centrality and clustering in complex hyper-networks. Phys. A Stat. Mech. Appl. 2006, 364, 581–594. [Google Scholar] [CrossRef]
- Prountzos, D.; Pingali, K. Betweenness Centrality; ACM SIGPLAN Notices: New York, NY, USA, 2013; Volume Voulme 48. [Google Scholar]
- Okamoto, K.; Chen, W.; Li, X.Y. Ranking of closeness centrality for large-scale social networks. In Proceedings of the 2nd International Workshop on Frontiers in Algorithmics, Changsha, China, 19–21 June 2008; pp. 186–195. [Google Scholar]
- Nacht, M.; Qiao, L.; Sheets, M.P.; St. Martin, T.; Labenski, M.; Mazdiyasni, H.; Karp, R.; Zhu, Z.; Chaturvedi, P.; Bhavsar, D.; et al. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kα. J. Med. Chem. 2013, 56, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (COMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (COMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D. Topomer COMFA: A design methodology for rapid lead optimization. J. Med. Chem. 2003, 46, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Narayanan, R.; Gopalakrishnan, B. 3-D-QSAR COMFA and COMSIA studies on tetrahydrofuroyl-l-phenylalanine derivatives as VLA-4 antagonists. Bioorg. Med. Chem. 2003, 11, 4235–4244. [Google Scholar] [CrossRef]
- Wang, X.; Pan, C.; Gong, J.; Liu, X.; Li, H. Enhancing the enrichment of pharmacophore-based target prediction for the polypharmacological profiles of drugs. J. Chem. Inf. Model. 2016, 56, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The string database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Ji Yin Zu Yan Jiu Za Zhi 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Li, J.; Wurtele, E.S.; Gu, X. A tool for comprehensive analysis of protein interaction networks. In Proceedings of the International Conference on Bioinformatics & Computational Biology (BIOCOMP 2007), Las Vegas, NV, USA, 25–28 June 2007; Volume II, pp. 523–527. [Google Scholar]
- Andersson, C.D.; Thysell, E.; Lindström, A.; Bylesjö, M.; Raubacher, F.; Linusson, A. A multivariate approach to investigate docking parameters’ effects on docking performance. J. Chem. Inf. Model. 2007, 47, 1673–1687. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.Y.; Guo, S. Concise applications of molecular modeling software-MOE. Comput. Appl. Chem. 2005. [Google Scholar] [CrossRef]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Aghaee, E.; Ghasemi, J.B.; Manouchehri, F.; Balalaie, S. Combined docking, molecular dynamics simulations and spectroscopic studies for the rational design of a dipeptide ligand for affinity chromatography separation of human serum albumin. J. Mol. Model. 2014, 20, 2446. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Luck, L.A. Molecular modeling of estrogen receptor using molecular operating environment. Biochem. Mol. Biol. Educ. 2007, 35, 238–243. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
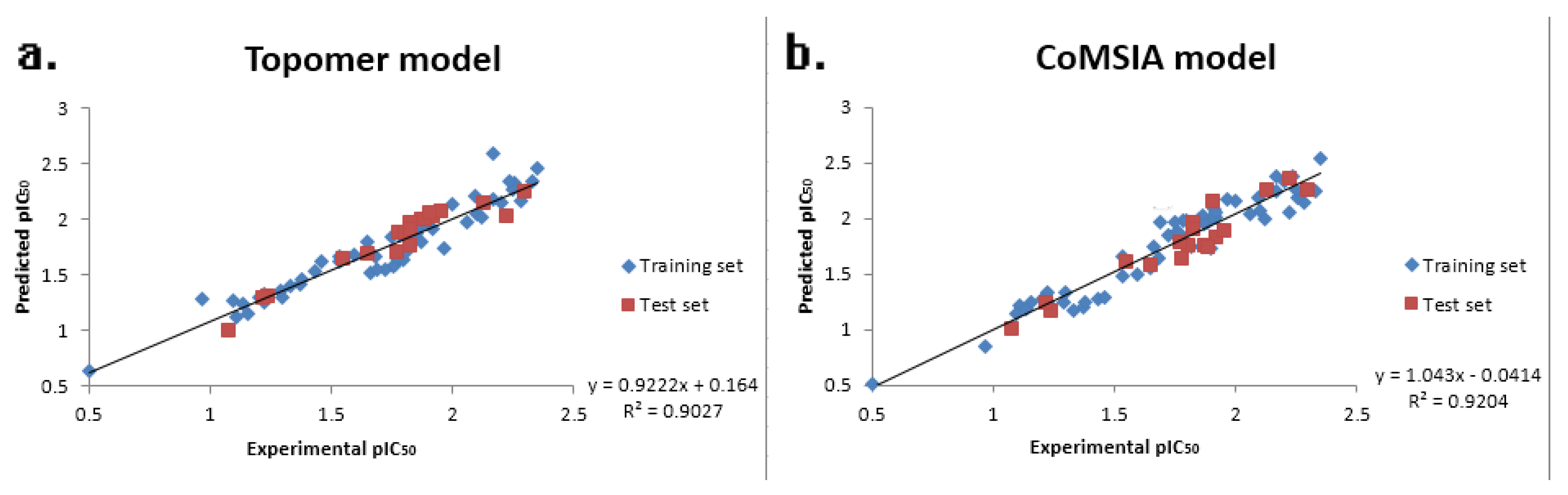
| PLS Statistical Parameters | Topomer CoMFA | CoMSIA |
|---|---|---|
| q2 a | 0.648 | 0.714 |
| r2 b | 0.896 | 0.925 |
| ONC c | 6 | 7 |
| SEE d | 0.107 | 0.094 |
| F e | - | 255.417 |
| MAE f | 0.085 | 0.111 |
| rpred2 g | 0.914 | 0.947 |
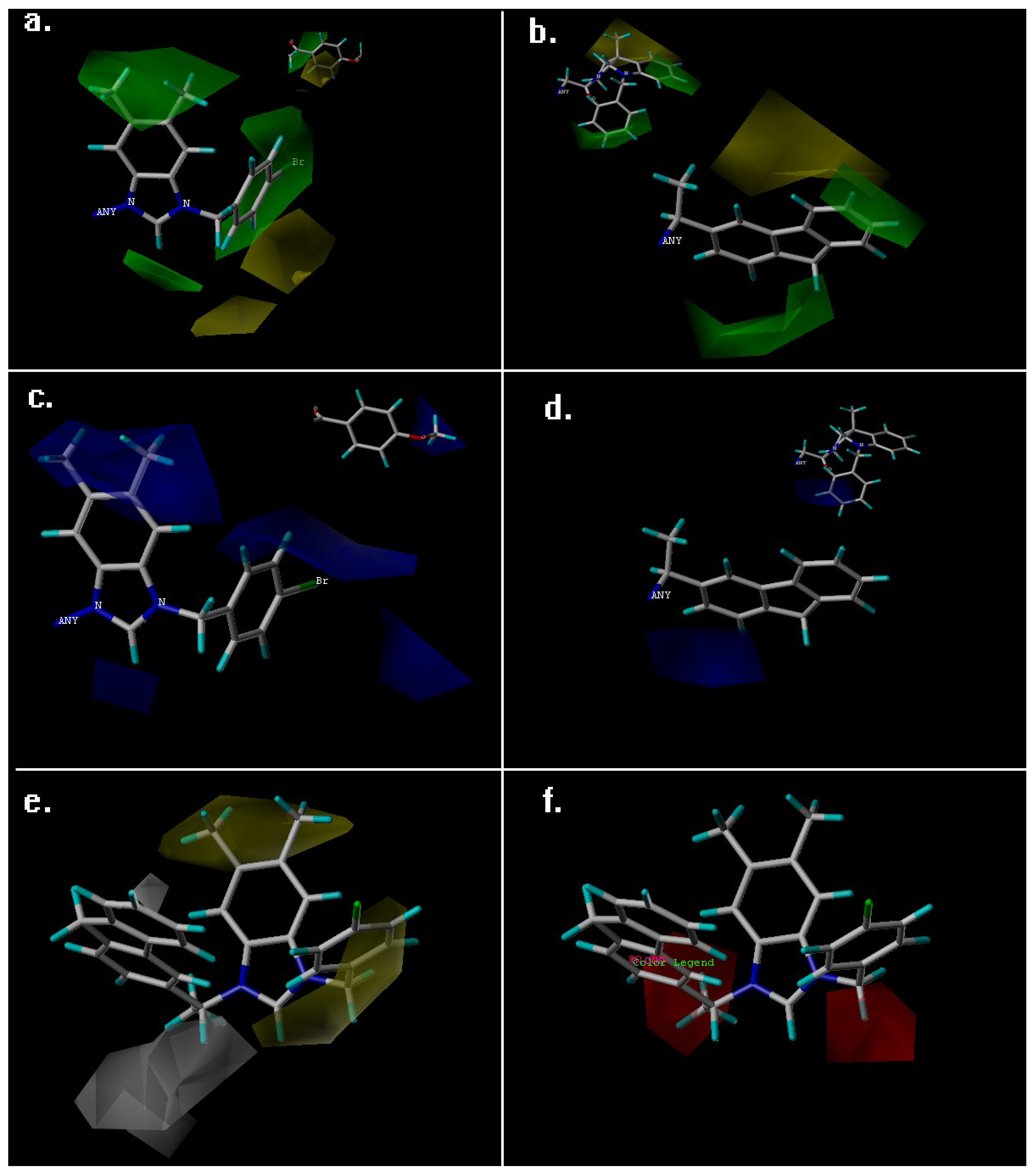
| Fraction of Field contribution h | ||
| steric | 0.691 | 0.264 |
| Electrostatic | 0.309 | 0.196 |
| Hydrophobic | - | 0.221 |
| H-bond acceptor | - | 0.319 |
| H-bond donor | - | 0 |
| PDB ID | Gene Names |
|---|---|
| 1UWH 1UWJ | BRAF BRAF1 RAFB1 |
| 3BBT | ERBB4 HER4 |
| 3GCS 3HEG | MAPK14 CSBP CSBP1 CSBP2 CSPB1 MXI2 SAPK2A |
| 3ZOS | DDR1 CAK EDDR1 NEP NTRK4 PTK3A RTK6 TRKE |
| 3WZD 3WZE 4ASD | KDR FLK1 VEGFR2 |
| 3ZIM | PIK3CA |
| 4MKC | ALK |
| 4UOI | KIT SCFR |
| 4TYJ 4UXQ | FGFR4 JTK2 TKF |
| 4VO4 | FGFR1 BFGFR CEK FGFBR FLG FLT2 HBGFR |
| 1M17 1XKK 2ITO 3UG2 4G5J 4G5P 4HJO 4I1Z 4I22 4WKQ | EGFR ERBB ERBB1 HER1 |
| 5FV1 | VEGFA VEGF |
| 5L2I | CDK6 CDKN6 |
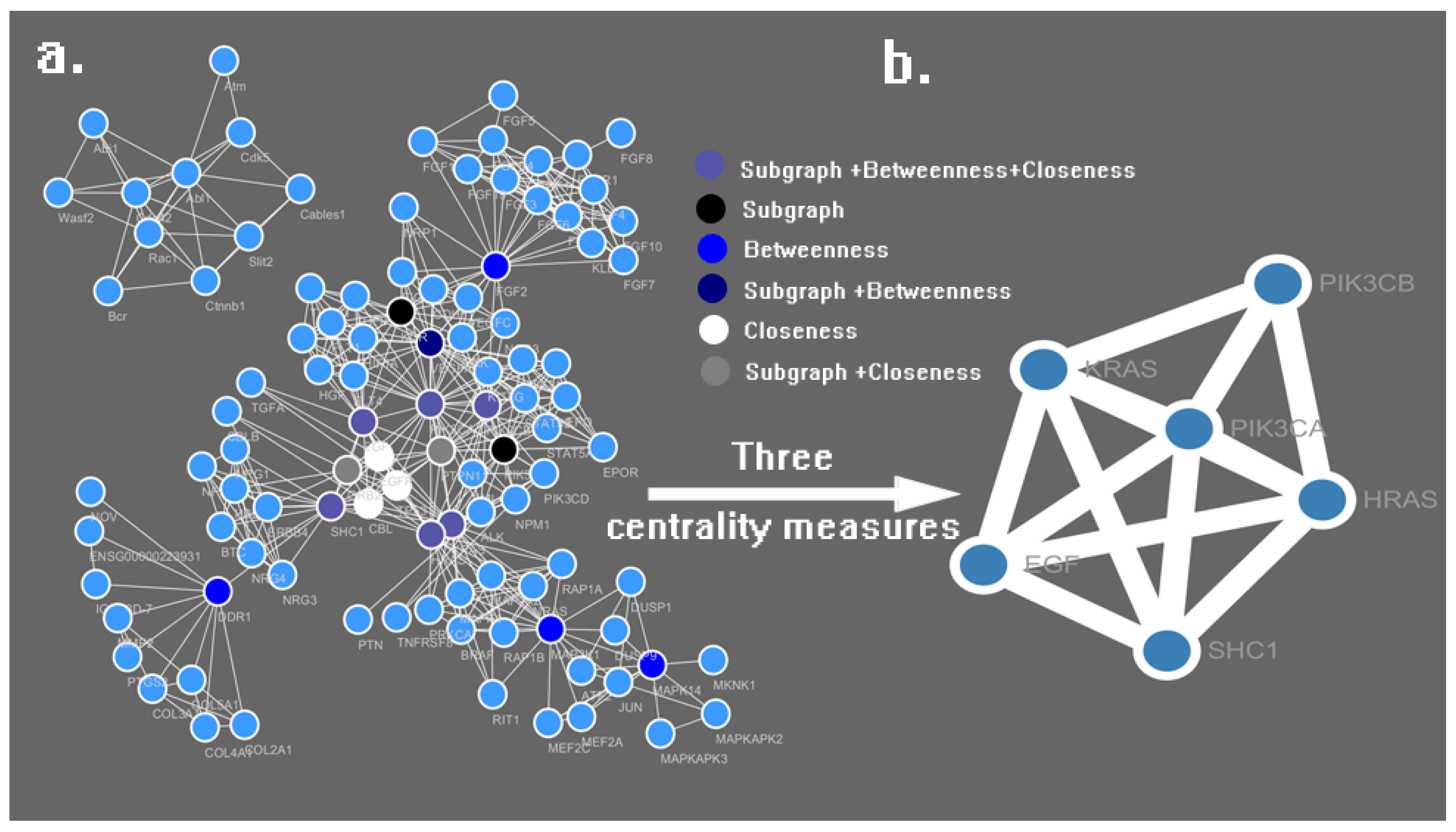
| No. | Name | Subgraph | Betweenness | Closeness |
|---|---|---|---|---|
| 1 | PIK3CA | 112,381.20 | 3161.31 | 0.0781 |
| 2 | HRAS | 43,006.22 | 1533.64 | 0.0772 |
| 3 | KRAS | 40,547.32 | 1445.97 | 0.0771 |
| 4 | SHC1 | 18,803.34 | 2494.36 | 0.0766 |
| 5 | EGF | 42,816.57 | 474.41 | 0.0763 |
| 6 | PTPN11 | 42,259.02 | 376.02 | 0.0762 |
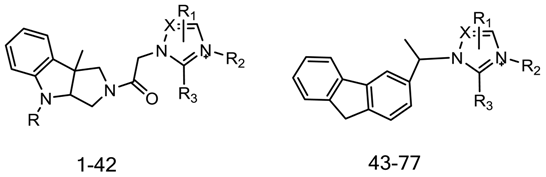
| Compound | IC50 | Imidazole/Triazole Ring | R2 | R |
|---|---|---|---|---|
| 1 | 7.75 | 2-Methyl-imidazole | 4-Bromophenacyl | Bn |
| 2 | 1.3 | 2-Methyl-imidazole | 4-Methoxyphenacyl | Bn |
| 3 * | 2.2 | 2-Methyl-imidazole | 4-Methylbenzyl | Bn |
| 4 | 0.47 | 2-Methyl-imidazole | 4-Methylbenzyl | Bn |
| 5 * | 5.72 | Benzimidazole | Phenacyl | Bn |
| 6 | 2.93 | Benzimidazole | 4-Bromophenacyl | Bn |
| 7 | 5.94 | Benzimidazole | 4-Methoxyphenacyl | Bn |
| 8 * | 1.64 | Benzimidazole | 2-Naphthylacy | Bn |
| 9 | 1.21 | Benzimidazole | 2-Bromobenzyl | Bn |
| 10 | 1.42 | Benzimidazole | 4-Bromobenzyl | Bn |
| 11 * | 1.2 | Benzimidazole | 4-Methylbenzyl | Bn |
| 12 | 4.64 | 2-Methyl-benzimidazole | Phenacyl | Bn |
| 13 | 4.25 | 2-Methyl-benzimidazole | 4-Bromophenacyl | Bn |
| 14 | 4.18 | 2-Methyl-benzimidazole | 4-Methoxyphenacyl | Bn |
| 15 * | 1.33 | 2-Methyl-benzimidazole | 2-Bromobenzyl | Bn |
| 16 | 1.35 | 2-Methyl-benzimidazole | 4-Bromobenzyl | Bn |
| 17 | 1.25 | 2-Methyl-benzimidazole | 4-Methylbenzyl | Bn |
| 18 | 1.38 | 2-Methyl-benzimidazole | 2-Naphthylmethyl | Bn |
| 19 * | 1.47 | 5,6-Dimethyl-benzimidazole | Phenacyl | Bn |
| 20 * | 1.55 | 5,7-Dimethyl-benzimidazole | 4-Bromophenacyl | Bn |
| 21 | 1.64 | 5,8-Dimethyl-benzimidazole | 4-Methoxyphenacyl | Bn |
| 22 | 1.27 | 5,9-Dimethyl-benzimidazole | 2-Bromobenzyl | Bn |
| 23 * | 1.29 | 5,10-Dimethyl-benzimidazole | 4-Bromobenzyl | Bn |
| 24 | 1.23 | 5,11-Dimethyl-benzimidazole | 4-Methylbenzyl | Bn |
| 25 | 1.21 | 5,12-Dimethyl-benzimidazole | 2-Naphthylmethyl | Bn |
| 26 * | 5.98 | Benzimidazole | 2-Naphthylacyl | Me |
| 27 | 5.07 | Benzimidazole | 2-Bromobenzyl | Me |
| 28 | 6.96 | Benzimidazole | 4-Bromobenzyl | Me |
| 29 | 5.13 | Benzimidazole | 4-Methylbenzyl | Me |
| 30 * | 2.8 | Benzimidazole | 2-Naphthylmethyl | Me |
| 31 | 5.95 | 2-Methyl-benzimidazole | 2-Bromobenzyl | Me |
| 32 | 3.72 | 3-Methyl-benzimidazole | 2-Bromobenzyl | Me |
| 33 | 2.25 | 4-Methyl-benzimidazole | 4-Methylbenzyl | Me |
| 34 * | 1.49 | 5-Methyl-benzimidazole | 2-Naphthylmethyl | Me |
| 35 | 8.03 | 5,6-Dimethyl-benzimidazole | Phenacyl | Me |
| 36 | 7.31 | 5,6-Dimethyl-benzimidazole | 4-Bromophenacyl | Me |
| 37 | 6.23 | 5,6-Dimethyl-benzimidazole | 4-Methoxyphenacyl | Me |
| 38 | 1.6 | 5,6-Dimethyl-benzimidazole | 2-Naphthylacyl | Me |
| 39 * | 1.67 | 5,6-Dimethyl-benzimidazole | 2-Bromobenzyl | Me |
| 40 | 1.73 | 5,6-Dimethyl-benzimidazole | 4-Bromobenzyl | Me |
| 41 | 1.52 | 5,6-Dimethyl-benzimidazole | 4-Methylbenzyl | Me |
| 42 | 1.35 | 5,6-Dimethyl-benzimidazole | 2-Naphthylmethyl | Me |
| 43 | 10.75 | 2-Methyl-benzimidazole | - | - |
| 44 | 31.5 | 5,6-Dimethyl-benzimidazole | - | - |
| 45 | 2.17 | Imidazole | 4-Bromobenzyl | - |
| 46 | 3.49 | Imidazole | Phenacyl | - |
| 47 | 1.75 | Imidazole | 4-Bromophenacyl | - |
| 48 | 2.92 | Imidazole | 4-Fluorophenacyl | - |
| 49 * | 1.1 | Imidazole | 4-Methoxyphenacyl | - |
| 50 | 1.01 | Imidazole | Naphthylacyl | - |
| 51 | 1.09 | 2-Methyl-imidazole | 4-Bromobenzyl | - |
| 52 * | 1.47 | 2-Methyl-imidazole | Phenacyl | - |
| 53 | 1.9 | 2-Methyl-imidazole | 4-Bromophenacyl | - |
| 54 | 0.52 | 2-Methyl-imidazole | 4-Methoxyphenacyl | - |
| 55 | 0.79 | 2-Methyl-imidazole | Naphthylacyl | - |
| 56 | 2.05 | Triazole | 4-Bromobenzyl | - |
| 57 * | 8.29 | Triazole | Phenacyl | - |
| 58 | 2.07 | Triazole | 4-Bromophenacyl | - |
| 59 | 2.55 | Triazole | 4-Methoxyphenacyl | - |
| 60 | 1.7 | Triazole | Naphthylacyl | - |
| 61 * | 0.74 | Benzimidazole | 4-Bromobenzyl | - |
| 62 | 0.76 | Benzimidazole | Phenacyl | - |
| 63 | 1.38 | Benzimidazole | 4-Bromophenacyl | - |
| 64 | 0.56 | Benzimidazole | 4-Methoxyphenacyl | - |
| 65 * | 1.23 | Benzimidazole | Naphthylacyl | - |
| 66 | 0.6 | 2-Methyl-benzimidazole | 4-Bromobenzyl | - |
| 67 | 0.63 | 2-Methyl-benzimidazole | Phenacyl | - |
| 68 | 0.81 | 2-Methyl-benzimidazole | 4-Bromophenacyl | - |
| 69 | 0.68 | 2-Methyl-benzimidazole | 4-Fluorophenacyl | - |
| 70 * | 0.59 | 2-Methyl-benzimidazole | 4-Methoxyphenacyl | - |
| 71 | 0.57 | 2-Methyl-benzimidazole | Naphthylacyl | - |
| 72 | 0.45 | 5,6-Dimethyl-benzimidazole | 4-Bromobenzyl | - |
| 73 | 0.68 | 5,6-Dimethyl-benzimidazole | Phenacyl | - |
| 74 | 0.58 | 5,6-Dimethyl-benzimidazole | 4-Bromophenacyl | - |
| 75 | 1.78 | 5,6-Dimethyl-benzimidazole | 4-Fluorophenacyl | - |
| 76 * | 0.5 | 5,6-Dimethyl-benzimidazole | 4-Methoxyphenacyl | - |
| 77 | 0.87 | 5,6-Dimethyl-benzimidazole | Naphthylacyl | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.-y.; Liang, J.-w.; Li, X.-y.; Olounfeh, K.M.; Li, S.-l.; Wang, S.; Wang, L.; Meng, F.-h. Study of Imidazolium Salt Derivatives as PIK3CA Inhibitors Using a Comprehensive in Silico Method. Int. J. Mol. Sci. 2018, 19, 896. https://doi.org/10.3390/ijms19030896
Wang M-y, Liang J-w, Li X-y, Olounfeh KM, Li S-l, Wang S, Wang L, Meng F-h. Study of Imidazolium Salt Derivatives as PIK3CA Inhibitors Using a Comprehensive in Silico Method. International Journal of Molecular Sciences. 2018; 19(3):896. https://doi.org/10.3390/ijms19030896
Chicago/Turabian StyleWang, Ming-yang, Jing-wei Liang, Xin-yang Li, Kamara Mohamed Olounfeh, Shi-long Li, Shan Wang, Lin Wang, and Fan-hao Meng. 2018. "Study of Imidazolium Salt Derivatives as PIK3CA Inhibitors Using a Comprehensive in Silico Method" International Journal of Molecular Sciences 19, no. 3: 896. https://doi.org/10.3390/ijms19030896